
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a crystallochromic indolizine dye by a base- and catalyst-free photochemical route†
Christoph
Dohmen
a,
Heiko
Ihmels
 *a,
Rouven
Kreienmeier
a and
Brian O.
Patrick
b
*a,
Rouven
Kreienmeier
a and
Brian O.
Patrick
b
aDepartment Chemie-Biologie, Organische Chemie II, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: ihmels@chemie.uni-siegen.de
bDepartment of Chemistry, Structural Chemistry Facility, The University of British Columbia, 2036 Main Mall, Vancouver, V6T 1Z1, BC, Canada
First published on 20th August 2019
Abstract
Indolizine derivatives are obtained by irradiation of 2-benzoyl-N-benzylpyridinium derivatives and dimethyl acetylene dicarboxylate, thus providing a competitive and complementary base- and catalyst-free synthesis. With this method, the first example of a crystallochromic indolizine is presented, whose color in the solid state depends on the out-of-plane torsion of the benzoyl substituent.
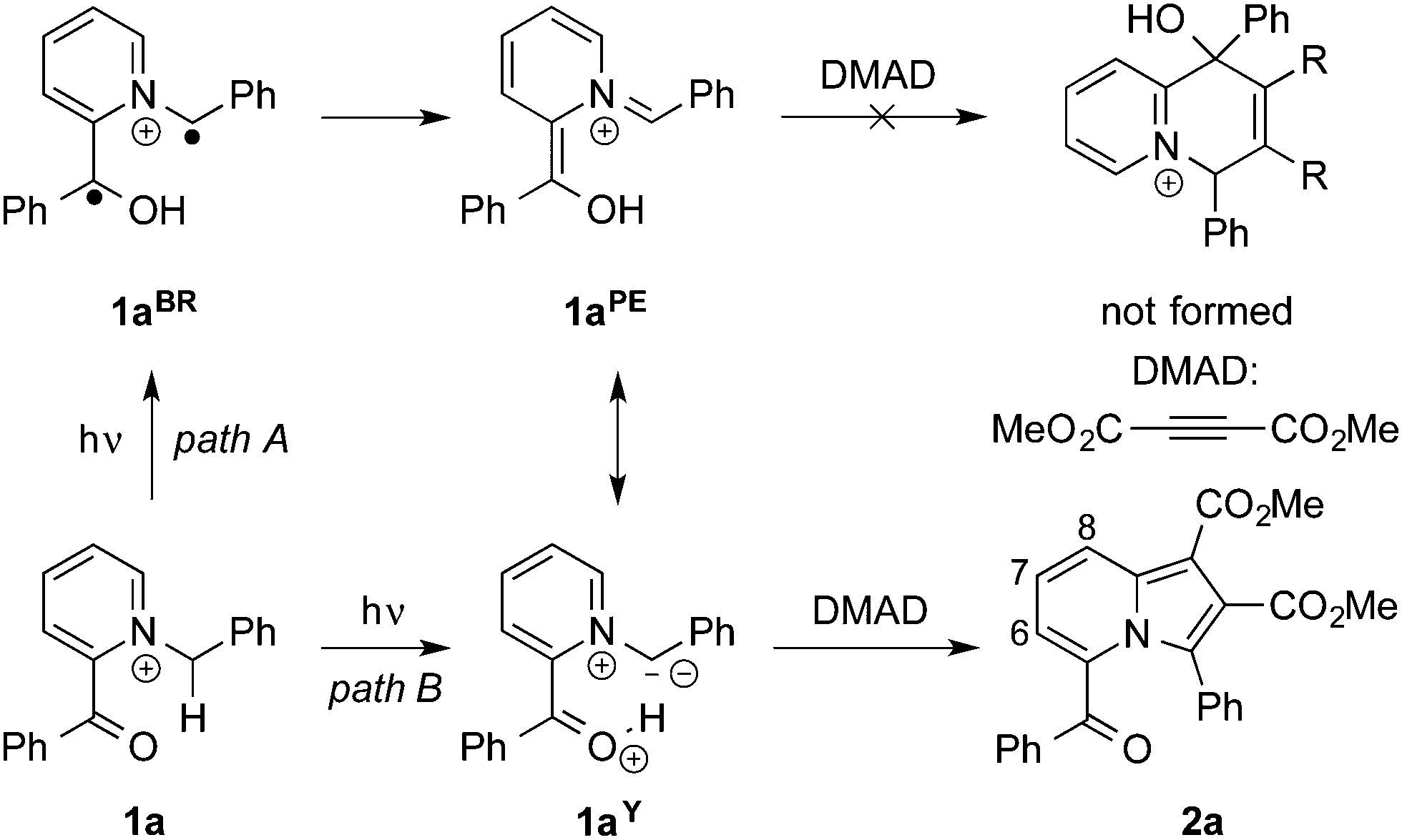
Indolizine and its derivatives constitute an important class of bicyclic hetarenes.1 Specifically, the indolizine unit is found in several bioactive natural products and represents a promising lead structure in pharmaceutical chemistry and drug design.2 In addition, indolizines usually exhibit favorable fluorescent and photophysical properties and may be applied as functional dyes,3 fluorescent probes4 or organic light-emitting diodes (OLED).5 As a result, the development of new synthetic strategies toward indolizine derivatives with defined substitution patterns has become a challenging and appealing research topic,6 leading to synthetic approaches such as the generally applicable 1,3-dipolar cycloaddition between N-alkylpyridinium ylides and dipolarophiles,7 oxidative cyclization,8 cross coupling,9 transition-metal catalysis,10 cycloisomerization reactions,11 along with a variety of specific reactions.12 Notably, photochemical syntheses of indolizines are rather rare despite the obvious benefits and advantages of light as controllable and traceless reagent. To the best of our knowledge, only two photochemical indolizine syntheses have been reported, so far. Hence, it has been shown that 2-vinylpyridine adds to photochemically generated singlet carbenes to form pyridinium ylides, which subsequently cyclize to the respective indolizines.13 More recently, a light-mediated synthesis from bromomethylpyridine derivatives and enol carbamates was reported.14 In this context, we coincidentally discovered the unexpected formation of indolizines in a photoreaction of N-alkylpyridinium derivatives and dimethyl acetylene-dicarboxylate (DMAD). And herein, we demonstrate that this photoreaction represents a useful complementary base-free synthesis of indolizine derivatives, and we describe that this method led to the discovery of the first crystallochromic indolizine derivative.
Initially, we investigated the benzoyl-N-benzylpyridinium 1a as substrate for photochemical synthesis of quinolizinium derivatives by initial γ-H abstraction and subsequent Diels–Alder reaction of the resulting photoenol 1aPE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 with DMAD (Scheme 1). But instead of the anticipated product, the dimethyl-5-benzoyl-3-phenylindolizine-1,2-dicarboxylate (2a) was isolated in low yield as the only product. The formation of product 2a was indicated by the characteristic 1H-NMR spectroscopic shifts and multiplets of the protons of the pyridine unit (6-H: 7.09 ppm, 7-H: 7.34 ppm, 8-H: 8.45 ppm) and further confirmed by 1D- and 2D-NMR spectroscopic analysis, mass spectrometry and elemental analysis data (cf. ESI†). After optimization of the reaction conditions (Table 1), the product 2a was isolated in 18% yield after chromatographic work-up and crystallization. As indolizines are known to be formed also by the ground-state reaction of N-alkylpyridinium derivatives and acetylenes in the presence of base,7 it was also tested whether 2a can be synthesized by this complementary route. Indeed, the reaction of 1a with DMAD under alkaline conditions also gave the product 2a in a slightly lower yield of 11% under identical work-up conditions (Table 1). These low yields are in line with the commonly reported observation that pyridinium ylides require a strong electron-withdrawing substituent at the N-methylene fragment for an efficient 1,3-dipolar cycloaddition.7 In agreement with this trend, the reaction of the ester-substituted substrate 1b gave the corresponding indolizine 2b in significantly increased yields of 46% in the photoreaction and 51% in the base-promoted reaction (Table 1). To test the limit of this method with regard to the CH acidity of the N-methylene group, we examined whether the N-methylpyridinium 1c can still be used for the photoinduced indolizine synthesis. Hence, the formation of 2c was observed on irradiation of 1c in the presence of DMAD, albeit in a very low yield of 4%. In turn, product 2c could only be isolated in less than 1% yield by the reaction of 1c with DMAD under alkaline conditions (Table 1).16 Furthermore, the irradiation of N-benzylpyridinium 1d, which was employed as control to assess the relevance of the ketone chromophore in the photoreaction, in the presence of DMAD also gave the resulting indolizine 2d only in very low yield (<1%).
15 with DMAD (Scheme 1). But instead of the anticipated product, the dimethyl-5-benzoyl-3-phenylindolizine-1,2-dicarboxylate (2a) was isolated in low yield as the only product. The formation of product 2a was indicated by the characteristic 1H-NMR spectroscopic shifts and multiplets of the protons of the pyridine unit (6-H: 7.09 ppm, 7-H: 7.34 ppm, 8-H: 8.45 ppm) and further confirmed by 1D- and 2D-NMR spectroscopic analysis, mass spectrometry and elemental analysis data (cf. ESI†). After optimization of the reaction conditions (Table 1), the product 2a was isolated in 18% yield after chromatographic work-up and crystallization. As indolizines are known to be formed also by the ground-state reaction of N-alkylpyridinium derivatives and acetylenes in the presence of base,7 it was also tested whether 2a can be synthesized by this complementary route. Indeed, the reaction of 1a with DMAD under alkaline conditions also gave the product 2a in a slightly lower yield of 11% under identical work-up conditions (Table 1). These low yields are in line with the commonly reported observation that pyridinium ylides require a strong electron-withdrawing substituent at the N-methylene fragment for an efficient 1,3-dipolar cycloaddition.7 In agreement with this trend, the reaction of the ester-substituted substrate 1b gave the corresponding indolizine 2b in significantly increased yields of 46% in the photoreaction and 51% in the base-promoted reaction (Table 1). To test the limit of this method with regard to the CH acidity of the N-methylene group, we examined whether the N-methylpyridinium 1c can still be used for the photoinduced indolizine synthesis. Hence, the formation of 2c was observed on irradiation of 1c in the presence of DMAD, albeit in a very low yield of 4%. In turn, product 2c could only be isolated in less than 1% yield by the reaction of 1c with DMAD under alkaline conditions (Table 1).16 Furthermore, the irradiation of N-benzylpyridinium 1d, which was employed as control to assess the relevance of the ketone chromophore in the photoreaction, in the presence of DMAD also gave the resulting indolizine 2d only in very low yield (<1%).
 | ||
| Scheme 1 Photochemical syntheses of indolizine derivative 2a. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| R1 | R2 | Cond. | Solvent | t/h | Yield/% | |
| a Counterion: X = Br. b Counterion: X = BF4. | ||||||
| 1a | COPh | Ph | hν | MeCN | 15.5 | 18 (2a) |
| 1b | COPh | CO2Et | hν | MeCN | 15.5 | 46 (2b) |
| 1c | COPh | H | hν | MeCN | 15.5 | 4 (2c) |
| 1a | COPh | Ph | ΔT, K2CO3 | THF | 24 | 11 (2a) |
| 1b | COPh | CO2Et | ΔT, K2CO3 | THF | 24 | 51 (2b) |
| 1c | COPh | H | ΔT, Na2CO3 | MeCN | 24 | <1 (2c) |
| 1d | H | Ph | hν | MeCN | 19.5 | <1 (2d) |
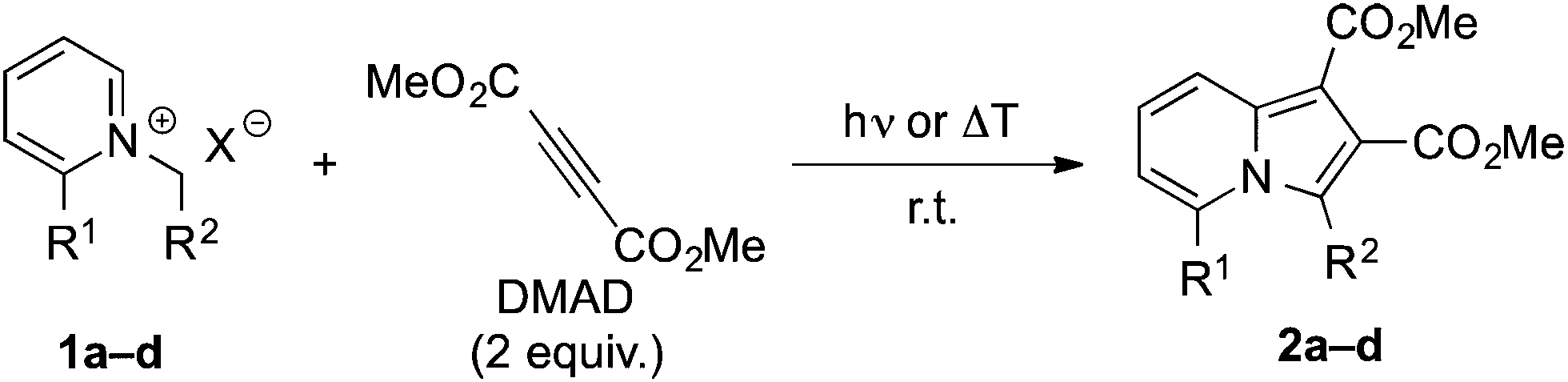
The mechanism of the photoinduced indolizine synthesis may be introduced with the γ-H abstraction at the N-methylene group by the excited ketone followed by photoenol formation (Scheme 1, path A). Although azonia-photoenols are not known, at least aza-photoenols have been generated and subsequently trapped with dienophils.17 However, in the case of pyridinium substrates 1a–d, the resonance structure of a pyridinium ylide, e.g.1aY, should also be considered that contributes significantly to the overall electron density and thus to the reactivity. Pyridinium ylides are well known to add in a Michael addition to acceptor-substituted alkynes to give indolizine derivatives after subsequent cyclization and oxidation.7b Presumably due to significant steric hinderance, the competing Diels–Alder reaction of photoenol 1aPE with DMAD did not occur, as indicated by the lack of characteristic NMR signals of the resulting quinolizinium derivative in the reaction mixture. Even though the unambiguous proof of the photoinduced formation of the pyridinium ylides is pending – and requires detailed photophysical studies – some evidence for their formation is provided by the observation that the independently base-generated pyridinium ylides give the same products on reaction with DMAD (Table 1). At the same time, the pyridinium ylides may also be formed in an excited-state proton transfer (ESPT)18 reaction between the carbonyl and the N-methylene protons (Scheme 1, path B). Carbonyls are known to have an increased basicity in the excited state19 and the photoacidity of benzylic positions has been reported already,20 but not for ylides, so far. But the strong electron withdrawing effect of the pyridinium unit21 should lead to a significant acidity in the excited state. Such an ESPT reaction, e.g. with the solvent or DMAD as proton acceptor, would explain why the substrate 1d, which lacks the benzoyl substituent, also gives an indolizine product in very low yield. It should be noted also that a characteristic photoreaction of pyridinium derivatives is the formation of bicyclic aziridines.22 While the formation of a complex reaction mixture does not allow to exclude this reaction pathway, we were not able to detect unambiguously the corresponding reaction products.
For the indolizine derivatives 2a–c, broad absorption bands were detected with long-wavelength maxima at 410 nm (2a), 375 nm (2b), and 415 nm (2c) in acetonitrile solution, respectively (Fig. 1, cf. ESI†). These derivatives also exhibit emission bands in acetonitrile solution with maxima at 563 nm (2a, Φfl = 0.07), 540 nm (2b, Φfl = 0.01) and 529 nm (2c, Φfl = 0.20). Both absorption and emission maxima are significantly red-shifted relative to the parent indolizine,23 which indicates a donor–acceptor interplay between the electron-donating indolizine ring and the electron-acceptor substituents.24 The emission quantum yields of derivatives 2a–c are relatively low, in particular when compared with the one of the parent compound (Φfl = 0.84 in hexane),23 which may be explained by the quenching effect of the carbonyl groups.25
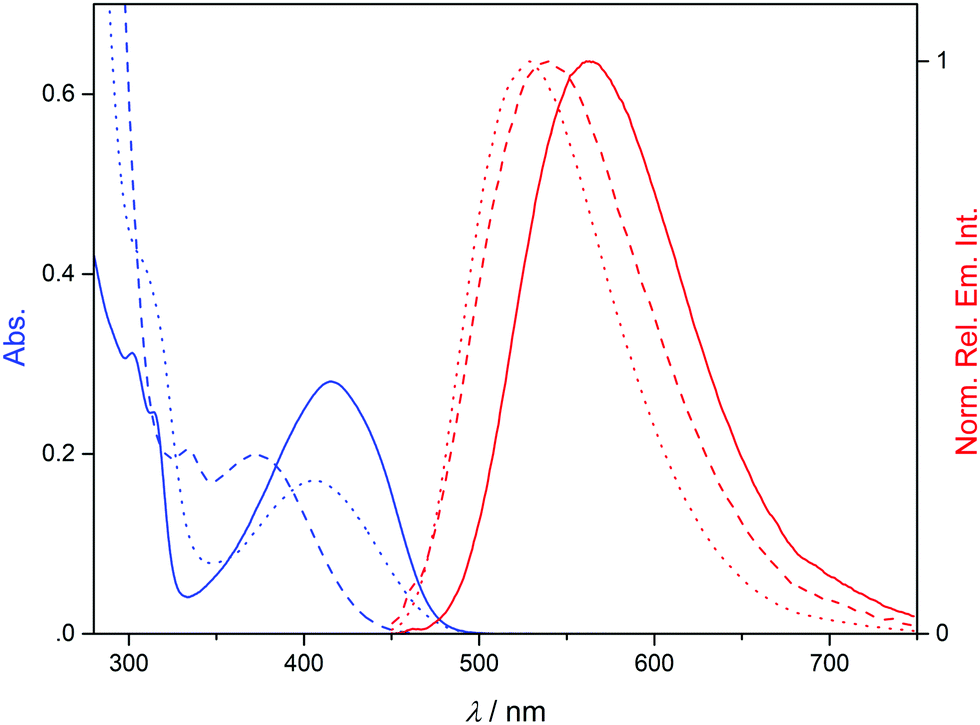 | ||
| Fig. 1 Absorption (blue, c = 40 μM) and emission spectra (red, Abs. = 0.10 at λex = 405 nm) of 2a (solid ine), 2b (dashed line) and 2c (dotted line) in MeCN. | ||
Most notably, we discovered during workup that the indolizine 2a gave two different types of crystals with distinctly different colors on recrystallization (Fig. 2D), a property that is often referred to as crystallochromism or color polymorphism.26 Namely, the compound 2a initially crystallized as orange-colored cubes from cyclohexane or hexane/ethyl acetate, whereas after longer crystallization time in cyclohexane or upon cooling in hexane/ethyl acetate a second fraction of yellow-green needles was formed. X-ray diffraction analysis showed that the two crystal forms are polymorphs of compound 2a, hereafter assigned 2an (n for needles) and 2ac (c for cubes) (Fig. 2A and 2B). The yellow needles, 2an, crystallized in the space group C2/c, whereas the orange cubes, 2ac, crystallized in the space group P21/n (cf. ESI†). In each crystal lattice, pairs of indolizine molecules are arranged in an anti-head-to-tail orientation with a distance between the molecular planes (2an: 320–340 pm; 2ac: 340–350 pm), that is in the range of the sum of the van der Waals radii of two carbon atoms (340 pm).27 However, the sterically demanding substituents impede an extended π-stacking interaction. Hence, the molecules exhibit large tranverse shifts relative to the long molecular axes (2an: 140–150 pm; 2ac: 170 pm), so that the actual overlap of the π systems is very small in each case (cf. ESI†). Therefore, the different color of the crystals is likely not caused by differently pronounced π-stacking interactions, as shown for example in polymorphs of perylene derivatives.28
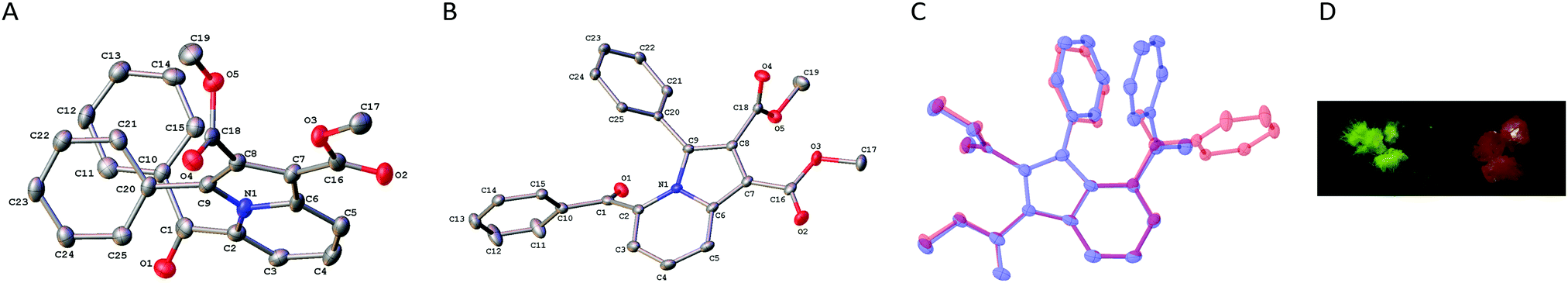 | ||
| Fig. 2 ORTEP-style structures of the polymorphs 2an (A) and 2ac (B) in the solid state (50% probability ellipsoids). C: Overlay of structures of 2an (blue) and 2ac (red). Hydrogen atoms are omitted for clarity. D: Inherent color of crystals of 2an (left) and 2ac (right). | ||
In general, the bond length and angles are essentially the same in 2an and 2ac (cf. ESI†). In both polymorphs the indolizine unit is slightly twisted out of planarity, whereby in 2ac the deviation from the plane is slightly more pronounced as seen from the torsion angles C2–N1–C6–C7 and C5–C6–N1–C9 (2an: 176°/176°; 2ac: 171°/175). This out-of-plane distortion is obviously caused by the strong repulsive peri-interactions between the phenyl and the benzoyl substituents as also indicated by the dihedral angle C1–C2–C9–C20 (2an: 18°; 2ac: 36°). Most notably, the orientation of the benzoyl functionality relative to the indolizine is completely different in the polymorphs 2an and 2ac, as shown by an overlay of both structures (Fig. 2C). In particular, the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond points away from the indolizine core in 2an, whereas in 2ac it is aligned in the opposite direction. The particular arrangement of the benzoyl substituent in 2an is likely caused by its π stacking interaction with the phenyl group as indicated by very small distances between the slightly tilted aromatic rings, e.g. C10–C20: 300 pm; C11–C25 = 353 pm; C12–C24 = 404 pm, C15–C21 = 366 pm. This significant and most pronounced difference between the two molecular structures may explain the distinct color of the polymorphs because it causes a different degree of π conjugation of the carbonyl functionality with the indolizine chromophore, as shown by the dihedral angles N1–C2–C1 O1 of 31° in 2ac and C3–C2–C1–O1 of 51° in 2an. The different degree of conjugation of the ketone functionality of 2an and 2ac was confirmed by slightly different shifts of the IR absorptions in the solid state (2an: 1666 cm−1; 2ac: 1661 cm−1). Thus, specifically in 2an this out-of-plane displacement of the carbonyl group in combination with the deviation of the C1–C2 bond from the indolizine plane hamper the overlap of pz orbitals of these separate units, thus suppressing resonance interaction. Even though the carbonyl functionality in 2ac is also twisted from coplanarity with the indolizine, it still enables sufficiently more overlap with the latter. As a result, a pronounced donor–acceptor interplay with the strong electron-donating indolizine is possible that results in the red shift of the absorption, i.e. the orange color of the crystal. This dependency of the color of crystals on the torsional angle between different parts of the π-conjugated chromophore has been demonstrated already, for example with polymorphs of thiophenecarbonitrile,26f,29p-chloroaniline,30 or p-toluidine31 derivatives. Herein, it is demonstrated for the first time that indolizine derivatives may develop this type of crystallochromsim as well. And based on the analysis and interpretation of the solid-state data, this property is a direct consequence of the steric hinderance between the carbonyl functionality and the peri-substituted phenyl group. Thus, it may be proposed that indolizine or indolizine-type fluorophores with the same substitution pattern have the same propensity to form crystallochromic polymorphs.
O bond points away from the indolizine core in 2an, whereas in 2ac it is aligned in the opposite direction. The particular arrangement of the benzoyl substituent in 2an is likely caused by its π stacking interaction with the phenyl group as indicated by very small distances between the slightly tilted aromatic rings, e.g. C10–C20: 300 pm; C11–C25 = 353 pm; C12–C24 = 404 pm, C15–C21 = 366 pm. This significant and most pronounced difference between the two molecular structures may explain the distinct color of the polymorphs because it causes a different degree of π conjugation of the carbonyl functionality with the indolizine chromophore, as shown by the dihedral angles N1–C2–C1 O1 of 31° in 2ac and C3–C2–C1–O1 of 51° in 2an. The different degree of conjugation of the ketone functionality of 2an and 2ac was confirmed by slightly different shifts of the IR absorptions in the solid state (2an: 1666 cm−1; 2ac: 1661 cm−1). Thus, specifically in 2an this out-of-plane displacement of the carbonyl group in combination with the deviation of the C1–C2 bond from the indolizine plane hamper the overlap of pz orbitals of these separate units, thus suppressing resonance interaction. Even though the carbonyl functionality in 2ac is also twisted from coplanarity with the indolizine, it still enables sufficiently more overlap with the latter. As a result, a pronounced donor–acceptor interplay with the strong electron-donating indolizine is possible that results in the red shift of the absorption, i.e. the orange color of the crystal. This dependency of the color of crystals on the torsional angle between different parts of the π-conjugated chromophore has been demonstrated already, for example with polymorphs of thiophenecarbonitrile,26f,29p-chloroaniline,30 or p-toluidine31 derivatives. Herein, it is demonstrated for the first time that indolizine derivatives may develop this type of crystallochromsim as well. And based on the analysis and interpretation of the solid-state data, this property is a direct consequence of the steric hinderance between the carbonyl functionality and the peri-substituted phenyl group. Thus, it may be proposed that indolizine or indolizine-type fluorophores with the same substitution pattern have the same propensity to form crystallochromic polymorphs.
In summary, we present a photoinduced, base- and catalyst-free synthetic route to polysubstituted indolizine derivatives. Although the yields are not high for the present examples, it is shown that this mild route is generally competitive and complementary to the established synthesis with base-generated pyridinium ylides. In addition, we discovered the first example of a crystallochromic indolizine derivative whose analysis revealed some principles that may be employed for the rational design of further crystallochromic indolizine derivatives.
Generous support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. We thank Ms Jennifer Hermann and Ms Sandra Uebach for technical assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Sandeep, K. N. Venugopala, M. A. Khedr, M. Attimarad, B. Padmashali, R. S. Kulkarni, R. Venugopala and B. Odhav, J. Basic Clin. Pharm., 2017, 8, 49–60 CAS; (b) J. J. Vaquero and J. Alvarez-Builla, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, 2011, vol. 4, p. 1989 Search PubMed.
- (a) V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 CrossRef CAS; (b) R. C. Oslund and M. H. Gelb, Biochemistry, 2012, 51, 8617–8626 CrossRef CAS PubMed; (c) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237–5257 CrossRef CAS PubMed; (d) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- (a) É. Lévesque, W. S. Bechara, L. Constantineau-Forget, G. Pelletier, N. M. Rachel, J. N. Pelletier and A. B. Charette, J. Org. Chem., 2017, 82, 5046–5067 CrossRef; (b) Y. R. Song, C. W. Lim and T. W. Kim, Luminescence, 2016, 31, 364–371 CrossRef CAS PubMed.
- (a) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed; (b) X. Zheng, R. Ji, X. Cao and Y. Ge, Anal. Chim. Acta, 2017, 978, 48–54 CrossRef CAS PubMed.
- (a) A. A. Kalinin, M. A. Smirnov, L. N. Islamova, G. M. Fazleeva, T. A. Vakhonina, A. I. Levitskaya, O. D. Fominykh, N. V. Ivanova, A. R. Khamatgalimov and I. R. Nizameev, Dyes Pigm., 2017, 147, 444–454 CrossRef CAS; (b) J. Wan, C.-J. Zheng, M.-K. Fung, X.-K. Liu, C.-S. Lee and X.-H. Zhang, J. Mater. Chem., 2012, 22, 4502–4510 RSC; (c) M. K. Bayazita and K. S. Coleman, J. Am. Chem. Soc., 2009, 131, 10670–10676 CrossRef.
- D. Čerņaks, Chem. Heterocycl. Compd., 2016, 52, 524–526 CrossRef.
- (a) Y. Liu, H. Hu, J. Zhou, W. Wang, Y. He and C. Wang, Org. Biomol. Chem., 2017, 15, 5016–5024 RSC; (b) S. Bonte, I. O. Ghinea, R. Dinica, I. Baussanne and M. Demeunynck, Molecules, 2016, 21, 332 CrossRef; (c) T. Gao, X. Chen, L. Jiang, M. Wu, H. Guo, J. Wang, S. Sun, J. Oiler and Y. Xing, Eur. J. Org. Chem., 2016, 4957–4960 CrossRef; (d) Y. Shang, M. Zhang, S. Yu, K. Ju, C. Wang and X. He, Tetrahedron Lett., 2009, 50, 6981–6984 CrossRef CAS.
- (a) L. Xiang, Y. Yang, X. Zhou, X. Liu, X. Li, X. Kang, R. Yan and G. Huang, J. Org. Chem., 2014, 79, 10641–10647 CrossRef CAS PubMed; (b) T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868–9878 CrossRef CAS PubMed; (c) B. Yan and Y. Liu, Org. Lett., 2007, 9, 4323–4326 CrossRef CAS.
- (a) L. Zhang, X. Li, Y. Liu and D. Zhang, Chem. Commun., 2015, 51, 6633–6636 RSC; (b) H. Kim, K. Lee, S. Kim and P. H. Lee, Chem. Commun., 2010, 46, 6341–6343 RSC.
- (a) Y. Shi and V. Gevorgyan, Chem. Commun., 2015, 51, 17166–17169 RSC; (b) X. Li, X. Xie and Y. Liu, J. Org. Chem., 2016, 81, 3688–3699 CrossRef CAS PubMed; (c) T. Wu, M. Chen and Y. Yang, J. Org. Chem., 2017, 82, 11304–11309 CrossRef CAS.
- (a) B. Yuan, R. He, W. Shen and M. Li, Tetrahedron, 2017, 73, 6092–6100 CrossRef CAS; (b) L.-X. Wang and Y.-L. Tang, Eur. J. Org. Chem., 2017, 2207–2213 CrossRef CAS.
- (a) H. Liu, D. He, Z. Sun, W. He, J. Han, J. Chen, H. Deng, M. Shao, H. Zhang and W. Cao, Tetrahedron, 2018, 74, 135–141 CrossRef CAS; (b) I. Yavari, K. Ghafouri, M. Naeimabadi and M. Halvagar, Synlett, 2018, 243–245 CAS; (c) T. Lepitre, R. Le Biannic, M. Othman, A. M. Lawson and A. Daïch, Org. Lett., 2017, 19, 1978–1981 CrossRef CAS; (d) G. Martinez-Ariza, B. T. Mehari, L. A. G. Pinho, C. Foley, K. Day, J. C. Jewett and C. Hulme, Org. Biomol. Chem., 2017, 15, 6076–6079 RSC; (e) W. Zhong, H. Zhu and H. Zou, Tetrahedron, 2017, 73, 3181–3187 CrossRef CAS; (f) D. He, Y. Xu, J. Han, H. Deng, M. Shao, J. Chen, H. Zhang and W. Cao, Tetrahedron, 2017, 73, 938–944 CrossRef CAS; (g) S. Park and I. Kim, Tetrahedron, 2015, 71, 1982–1991 CrossRef CAS.
- R. Bonneau, Y. N. Romashin, M. T. H. Liu and S. E. MacPherson, J. Chem. Soc., Chem. Commun., 1994, 509–510 RSC.
- B. Sahoo, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 15545–15549 CrossRef CAS.
- P. J. Wagner and B. S. Park, in OrganicPhotochemistry, ed. A. Padwa, Dekker, New York, 1991, vol. 11, p. 227 Search PubMed.
- For the only report about an indolizine synthesis that starts with the deprotonation of an otherwise unsubstituted N-methylpyridinium substrate, see: B.-X. Wang, Y.-M. Shen, J. Shen, C. Li and H.-W. Hu, Huaxue Xuebao, 2004, 17, 1649–1652 Search PubMed.
- O. A. Mukhina, N. N. B. Kumar, T. M. Arisco, R. A. Valiulin, G. A. Metzel and A. G. Kutateladze, Angew. Chem., 2011, 123, 9595–9600 CrossRef.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- J. F. Ireland and P. A. Wyatt, Adv. Phys. Org. Chem., 1976, 12, 131–221 CrossRef CAS.
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
- Z. Wang, Y. Zheng, Y. Zheng, X.-S. Xue and P. Ji, J. Phys. Chem. A, 2018, 122, 5750–5755 CrossRef CAS PubMed.
- J. Zou and P. S. Mariano, Photochem. Photobiol. Sci., 2008, 7, 393–404 RSC.
- (a) D. A. Lerner, P. M. Horowitz and E. M. Evleth, J. Phys. Chem., 1977, 81, 12–17 CrossRef CAS; (b) D. A. Lerner and E. M. Evleth, Chem. Phys. Lett., 1972, 15, 260–262 CrossRef CAS.
- A. I. Levitskaya, A. A. Kalinin, O. D. Fominykh, I. V. Vasilyev and M. Y. Balakina, Comput. Theor. Chem., 2016, 1094, 17–22 CrossRef CAS.
- B. Valeur, Molecular Fluorescence Principles and Applications, Wiley-VCH, Weinheim, 2002 Search PubMed.
- (a) D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC; (b) B. Swapna, K. Suresh and A. Nangia, Chem. Commun., 2016, 52, 4037–4040 RSC; (c) E. Sangtani, S. K. Sahu, S. H. Thorat, R. L. Gawade, K. K. Jha, P. Munshi and R. G. Gonnade, Cryst. Growth Des., 2015, 15, 5858–5872 CrossRef CAS; (d) S. Md. Pratik, A. Nijamudheen, S. Bhattacharya and A. Datta, Chem. – Eur. J., 2014, 20, 3218–3224 CrossRef; (e) C. Kitamura, A. Takenaka, T. Kawase, T. Kobayashi and H. Naito, Chem. Commun., 2011, 47, 6653–6655 RSC; (f) L. Yu, Acc. Chem. Res., 2010, 43, 1257–1266 CrossRef CAS; (g) C. Kitamura, Y. Abe, T. Ohara, A. Yoneda, T. Kawase, T. Kobayashi, H. Naito and T. Komatsu, Chem. – Eur. J., 2010, 16, 890–898 CrossRef CAS PubMed.
- A. J. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) F. Würthner, C. Thalacker and A. Sautter, Adv. Mater., 1999, 11, 754–758 CrossRef; (b) F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef; (c) P. Kazmaier and R. Hoffmann, J. Am. Chem. Soc., 1994, 116, 9684–9691 CrossRef CAS; (d) C. Aakeroy, Acta Crystallogr., 1997, B53, 569–586 CrossRef CAS.
- L. Yu, J. Phys. Chem. A, 2002, 106, 544–550 CrossRef CAS.
- J. Bernstein and A. T. Hagler, J. Am. Chem. Soc., 1978, 100, 673–681 CrossRef CAS.
- D. E. Braun, T. Gelbrich, R. K. R. Jetti, V. Kahlenberg, S. L. Price and U. J. Griesser, Cryst. Growth Des., 2008, 8, 1977–1989 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic protocols, absorption and emission spectra, crystallographic data, NMR spectra. CCDC 1923160 and 1923161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc04730a |
| This journal is © The Royal Society of Chemistry 2019 |