
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConnecting a carbonyl and a π-conjugated group through a p-phenylene linker by (5+1) benzene ring formation†
Tatsuya
Morofuji
 *,
Hanae
Kinoshita
and
Naokazu
Kano
*
*,
Hanae
Kinoshita
and
Naokazu
Kano
*
Department of Chemistry, Faculty of Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan. E-mail: tatsuya.morofuji@gakushuin.ac.jp; naokazu.Kano@gakushuin.ac.jp; Tel: +81-3-5904-9394
First published on 5th July 2019
Abstract
A benzene ring was formed to connect a carbonyl group of various methyl ketones with a π-conjugated group through a p-phenylene linker. Methyl ketones and streptocyanines were used as the C1 and C5 sources, respectively, in the (5+1) annulation, which could form donor–π–acceptor molecules.
Connecting two different molecules with a linker is a fundamental design principle of functional organic molecules such as organic devices,1 supramolecular materials,2 bioactive compounds,3 and ligands for metal–organic frameworks.4 The p-phenylene group is one of the most widely used linkers for the following reasons: (1) the rigid structure of the benzene ring defines the relative position of two substituents on the benzene ring,5 and (2) the p-phenylene group can connect two substituents with π-conjugation, which affects the electronic state of the molecule.6 Compared with o- or m-phenylene groups, (3) the p-phenylene group can extend π-conjugation more effectively because of steric or electronic reasons, and (4) the planar structure and π-electrons of the benzene ring induce π–π stacking, which can affect the secondary structure of the molecules.7
Utilizing these characteristics, various p-phenylene-containing molecules were designed and synthesized as functional organic molecules. Particularly, carbonyl-p-phenylene-π molecules (Fig. 1a) are used for a wide range of applications such as receptor antagonists,8 aggregation-induced emission luminogens,9 and photoinitiators for free radical polymerization.10 In addition, when a π-conjugated substituent acts as a donor, such a carbonyl-p-phenylene-π molecule could be regarded as a donor–acceptor molecule, which is an important core structure of thermally activated delayed fluorescent emitters11 and photocatalysts mediating proton-coupled electron transfer.12
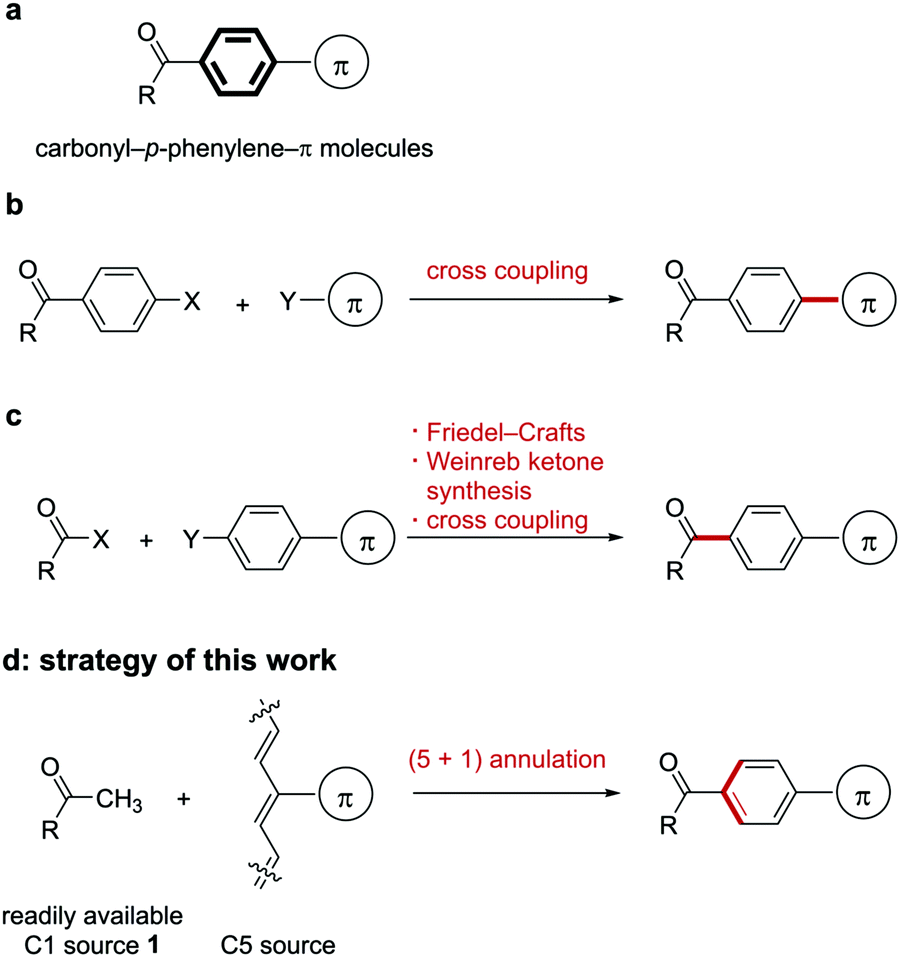 | ||
| Fig. 1 Strategies for synthesis of carbonyl-p-phenylene-π molecules. | ||
One approach to synthesizing carbonyl-p-phenylene-π molecules is the introduction of a π-conjugated substituent to a benzene ring bearing a carbonyl group via cross-coupling reactions13 (Fig. 1b). Another method is the connection of a carbonyl group to a benzene ring to form a ketone (Fig. 1c). The most popular classical approach with respect to the latter method is the Friedel–Crafts reaction of acid chlorides and electron-rich aromatic compounds.14 The reaction of the Weinreb amide or its analogues with organometallic reagents is an alternative method for ketone formation.15 Transition-metal-catalyzed ketone formation is also popular.16 These approaches are valuable, but the scope of the Friedel–Crafts reaction is limited and some aromatic substrates show undesired regioselectivity.
The introduction of a π-conjugated group needs to use both aryl halides and arylmetal reagents that require multistep preparation in most cases. A more convenient regiospecific method for synthesizing carbonyl-p-phenylene-π molecules is still highly desired.
Benzene ring formation would be an alternative approach to synthesize benzene-containing molecules. Various approaches for [2+2+2] and [4+2] benzene ring formation have been developed in the past up to the present.17 However, it is difficult to synthesize carbonyl-p-phenylene-π molecules using the [2+2+2] and [4+2] annulation because of their inherent 1,4-disubstitution pattern. In contrast, (5+1) benzene ring formation18,19 could be expected to give 1,4-disubstituted benzene derivatives regioselectively.
Based on this background, we envisioned that (5+1) benzene ring formation using a methyl ketone 1 as the C1 source and a C5 source bearing a π-conjugated substituent would be an attractive approach to obtaining carbonyl-p-phenylene-π molecules because starting methyl ketone 1 is readily available (Fig. 1d). This approach would give the p-phenylene-linked products regiospecifically. In relation to the approach, conversion of an acetyl group into an unsubstituted benzoyl group using a conjugated methine compound (streptocyanine) as a C5 source was reported by Jutz.20 However, the mere formation of an unsubstituted phenyl group, which restricted applicability of this reaction, has not been applied to connecting two fragments through the benzene ring formed. We report here a new method for connecting a carbonyl and a π-conjugated group through a p-phenylene linker by benzene ring formation using a methyl ketone as the C1 source and a streptocyanine bearing a π-conjugated substituent as the C5 source (eqn (1)).
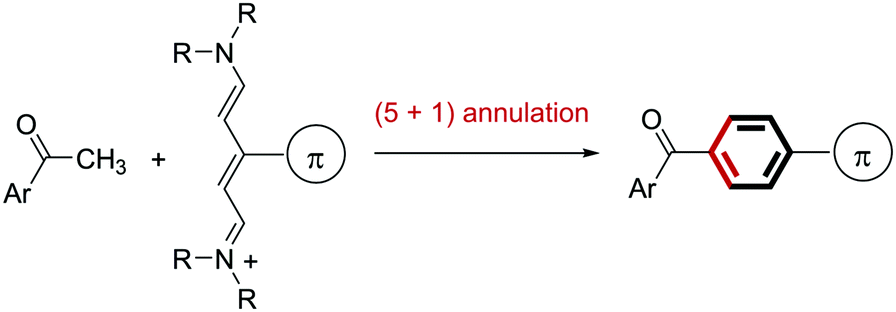 | (1) |
A working hypothesis is that the (5+1) annulation proceeds following a pathway such as that shown in Scheme 1. The enolate generated from methyl ketone 1 and a base attacks an iminium group in streptocyanines 2, and following the elimination of an amine gives triene intermediate 3. Triene 3 could be converted thermally to E/Z isomer 4 because of the push–pull electronic structure.21 Then, 6π electrocyclization22 of 4 gives cyclohexadiene 5, and following the second elimination of the amine produces the desired carbonyl-p-phenylene-π molecule 6. It follows that the base, nitrogen substituents, and reaction temperature could be critical for the successful (5+1) annulation.
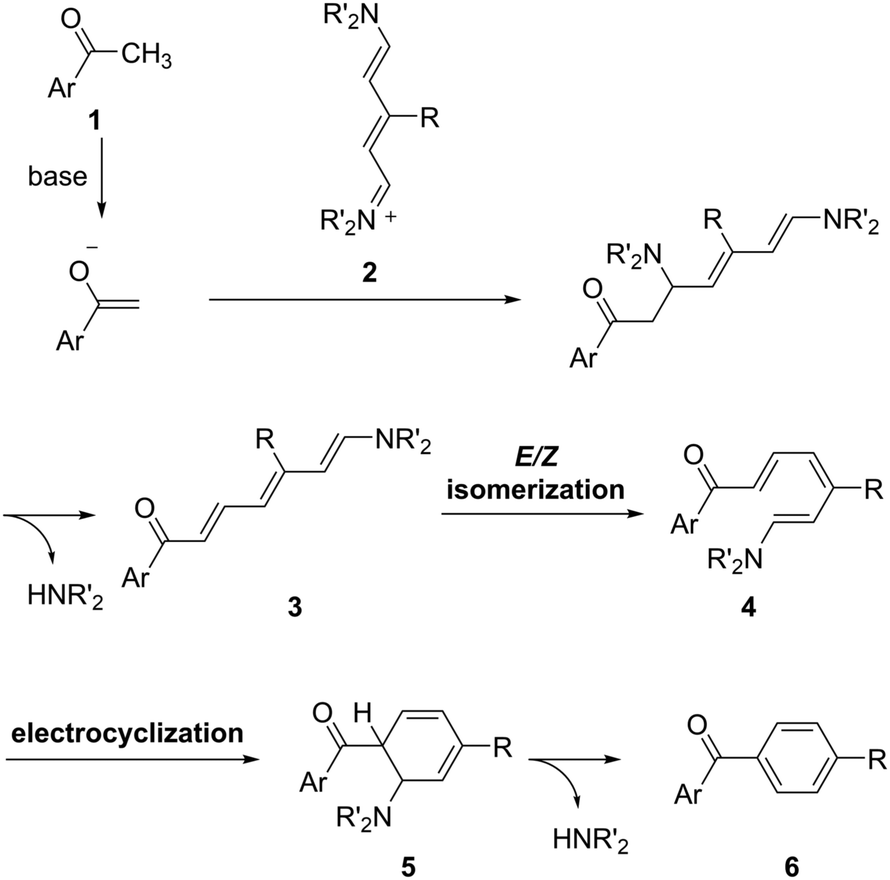 | ||
| Scheme 1 Proposed reaction mechanism of (5+1) benzene ring formation. | ||
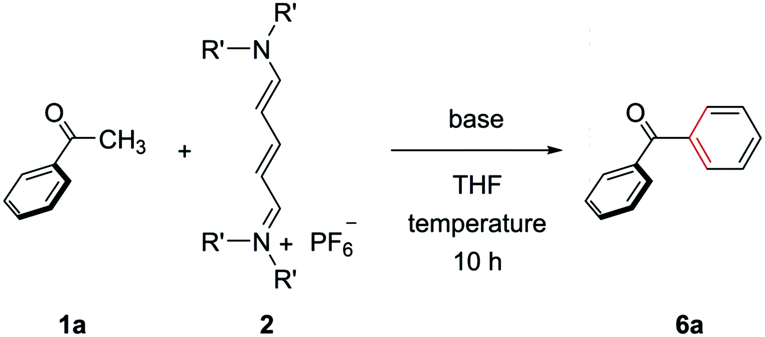
We first optimized the reaction conditions for the (5+1) annulation using streptocyanines bearing no internal substituent (Table 1).20 Acetophenone (1a), streptocyanine 2, and KOtBu were mixed and stirred in THF at 80 °C for 10 h in a pressure tube to give benzophenone (6a) in a 30% yield (entry 1). Higher reaction temperatures increased the yield of the product (entries 2 and 3). The nitrogen substituent in streptocyanine 2 was found to affect the yield, and the piperidinyl substituent turned out to be the best (entries 3–5). The use of NaOtBu instead of KOtBu as a base gave the product in an almost quantitative yield (entry 6). The (5+1) benzene ring formation was achieved in one pot without isolating intermediates 3.
|
|
||||
|---|---|---|---|---|
| Entry | R′, R′ | Base | Temp. (°C) | Yieldb (%) |
| a Acetophenone (1a) (0.2 mmol), streptocyanine 2 (1.5 equiv.), and base (1.5 equiv.) were stirred at the indicated temperature in THF (4 mL) for 10 h. b Yield of the products was determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. c Isolated yield. | ||||
| 1 | Me, Me | KOtBu | 80 | 30 |
| 2 | Me, Me | KOtBu | 100 | 53 |
| 3 | Me, Me | KOtBu | 120 | 84 |
| 4 | –(CH2)2O(CH2)2– | KOtBu | 120 | 29 |
| 5 | –(CH2)5– | KOtBu | 120 | 89 |
| 6 | –(CH2)5– | NaOtBu | 120 | 99 (94)c |
Next, we prepared streptocyanines bearing a π-conjugated substituent at the 3-position, 2, for the (5+1) annulation synthesizing carbonyl-p-phenylene-π molecules. The procedure for synthesizing streptocyanines 2 is illustrated in Fig. 2.23N-Arylation of 4-substituted pyridine 7 with 2,5-dinitrochlorobenzene gave the corresponding N-arylpyridinium salt 8 (Fig. 2, A). The resulting N-arylpyridinium 8 was treated with piperidine, and following salt exchange gave the desired streptocyanine 2 (Fig. 2, B). Streptocyanines bearing a π-conjugated substituent such as a phenyl group, a biphenyl group, a naphthyl group, a benzothiophenyl group, and a pyrenyl group were prepared (2b–2f). Streptocyanines bearing a diphenylamino group, a carbazolyl group, and a tert-butyl group were also synthesized in the same manner (2g–2i). It should be noted that streptocyanines 2a–2i could be purified by filtration and washing and did not require purification by column chromatography.
 | ||
Fig. 2 Synthesis of streptocyanines bearing a substituent. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NH4PF6 was used. bKPF6 was used. cNaSbF6 was used. NH4PF6 was used. bKPF6 was used. cNaSbF6 was used. | ||
The synthesized streptocyanines 2 were used for (5+1) annulation under the optimized reaction conditions described in Table 1. Some of the scope of the reaction is shown in Fig. 3. The functional group compatibility of the present transformation is remarkable. Acetophenones bearing an electron-donating group and an electron-withdrawing group gave the corresponding products (6b–6h) in excellent to moderate yields. Particularly, iodo- or bromo-substituted products (6e, 6f) are valuable because further transformation of the products is easy using a transition-metal catalyst. Additionally, benzylacetone, which is an aliphatic ketone, could also be used for (5+1) annulation to give the corresponding product 6i.
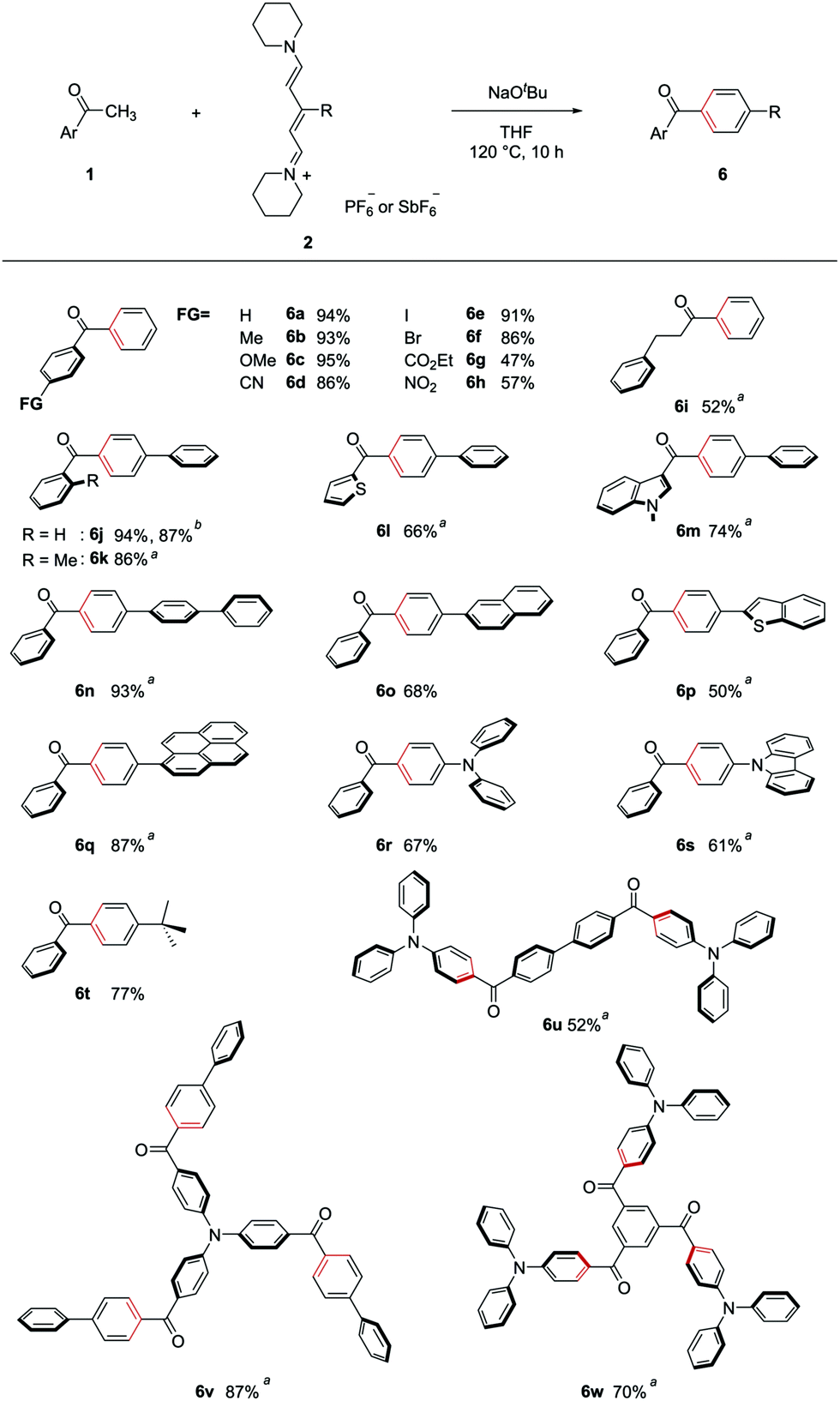 | ||
| Fig. 3 Scope of (5+1) benzene ring formation. Aryl methyl ketone 1 (0.5 mmol), streptocyanine 2 (1.5–6 equiv.), and NaOtBu (1.5–18 equiv.) were stirred at 120 °C in THF for 10 h. aReaction run on 0.2 mmol scale. bReaction run on a gram scale (12 mmol scale). See ESI† for details. | ||
This annulation reaction was applied to the synthesis of carbonyl-p-phenylene-π molecules. Reactions of acetophenone (1a) with streptocyanines bearing a phenyl substituent 2b gave corresponding product 6j in excellent yield. The reaction also worked well on a gram scale. Sterically hindered o-methylacetophenone was also a good substrate for (5+1) annulation (6k). Heteroaryl methyl ketones such as 2-acetyl thiophene and 1-methyl-3-acetylindole gave the corresponding heteroaryl–carbonyl-p-phenylene-π molecules (6l, 6m).
(5+1) benzene ring formation using acetophenone and streptocyanines bearing various π-conjugated substituents was carried out. Biphenyl-, naphthyl-, and 2-benzothiophenyl-substituted streptocyanines were found to be good substrates for the annulation (6n–6p). Use of pyrenyl-substituted streptocyanine 2f gave corresponding product 6q, which was used as a photoinitiator.10 Using diphenylamino- or carbazolyl-substituted streptocyanines (2g, 2h), the present method could be applied to the synthesis of carbonyl-p-phenylene-donor molecules (6r, 6s, respectively). The reaction of acetophenone with tert-butyl-substituted streptocyanine 2i gave p-tert-butylphenyl phenyl ketone (6t).
An advantage of this method is the availability of versatile acetyl compounds as a counterpart to streptocyanines. Even di- or tri-acetyl-substituted compounds are also readily available. By using such compounds as a starting material, we applied this method to the synthesis of symmetric molecules bearing p-phenylene groups connecting the triphenylamine moiety as the donor and carbonyl groups as the acceptor. C2h symmetric donor–acceptor molecule 6u was synthesized from 4,4′-diacetylbiphenyl and 2gvia double (5+1) benzene ring formation. Using 4,4′,4′′-triacetyltriphenylamine and 2b as starting materials, triple-p-phenylene group formation was achieved to give the corresponding C3 symmetric donor–acceptor molecule 6v. The reaction of 1,3,5-triacetylbenzene with 2g also gave the C3 symmetric donor–acceptor molecule 6w.
In conclusion, we have developed an efficient method for synthesizing carbonyl-p-phenylene-π molecules from readily available methyl ketones and easily synthesized streptocyanines in one pot. This convenient method was applied to the synthesis of symmetric donor–acceptor molecules that have two or three such linkages.
This research was partially supported by the MEXT-supported program for the Strategic Research Foundation at Private Universities and the Moritani Scholarship Foundation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- (a) G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed; (b) D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- A. K. Ghosh and S. Gemma, Structure-based Design of Drugs and Other Bioactive Molecules: Tools and Strategies, Wiley, New York, 2014 Search PubMed.
- F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- (a) S. I. Yang, R. K. Lammi, J. Seth, J. A. Riggs, T. Arai, D. Kim, D. F. Bocian, D. Holten and J. S. Lindsey, J. Phys. Chem. B, 1998, 102, 9426–9436 CrossRef CAS; (b) L. Yu and J. Lindsey, Tetrahedron, 2001, 57, 9285–9298 CrossRef CAS.
- (a) M. Taniguchi and J. S. Lindsey, Tetrahedron, 2010, 66, 5549–5565 CrossRef CAS; (b) S. Nishizawa, J.-y. Hasegawa and K. Matsuda, Chem. Phys. Lett., 2013, 555, 187–190 CrossRef CAS.
- E. R. T. Tiekink and J. Zukerman-Schpector, The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering, Wiley, New York, 2012 Search PubMed.
- D. R. Davies, B. Mamat, O. T. Magnusson, J. Christensen, M. H. Haraldsson, R. Mishra, B. Pease, E. Hansen, J. Singh, D. Zembower, H. Kim, A. S. Kiselyov, A. B. Burgin, M. E. Gurney and L. J. Stewart, J. Med. Chem., 2009, 52, 4694–4715 CrossRef CAS PubMed.
- Y. Qi, Y. Wang, Y. Yu, Z. Liu, Y. Zhang, Y. Qi and C. Zhou, J. Mater. Chem. C, 2016, 4, 11291–11297 RSC.
- M.-A. Tehfe, F. Dumur, B. Graff, F. Morlet-Savary, D. Gigmes, J.-P. Fouassier and J. Lalevée, Polym. Chem., 2013, 4, 2313–2324 RSC.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- J. Luo and J. Zhang, J. Org. Chem., 2016, 81, 9131–9137 CrossRef CAS PubMed.
- (a) S. P. Stanforth, Tetrahedron, 1998, 54, 263–303 CrossRef CAS; (b) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed.
- (a) N. O. Calloway, Chem. Rev., 1935, 17, 327–392 CrossRef CAS; (b) P. H. Gore, Chem. Rev., 1955, 55, 229–281 CrossRef CAS.
- (a) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707–3738 CAS; (b) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697–3736 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS PubMed; (b) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed; (c) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC.
- (a) T. Zimmermann and G. W. Fischer, J. Prakt. Chem., 1987, 329, 975–984 CrossRef CAS; (b) J. Steinbach, P. Mäding, F. Füchtner and B. Johannsen, J. Labelled Compd. Radiopharm., 1995, 36, 33–41 CrossRef CAS; (c) S. P. Gromov and N. A. Kurchavov, Eur. J. Org. Chem., 2002, 4123–4126 CrossRef CAS; (d) X. Bi, D. Dong, Q. Liu, W. Pan, L. Zhao and B. Li, J. Am. Chem. Soc., 2005, 127, 4578–4579 CrossRef CAS PubMed; (e) S. S. B. Daniels, J. M. Brown, M. Gayral, Y. Xu and M. I. Stewart, Synlett, 2009, 1387–1390 Search PubMed; (f) Z. Fu, M. Wang, Y. Dong, J. Liu and Q. Liu, J. Org. Chem., 2009, 74, 6105–6110 CrossRef CAS PubMed; (g) G. Ohlendorf, C. W. Mahler, S.-S. Jester, G. Schnakenburg, S. Grimme and S. Höger, Angew. Chem., Int. Ed., 2013, 52, 12086–12090 CrossRef CAS PubMed; (h) H.-Y. Zhao, F.-S. Wu, L. Yang, Y. Liang, X.-L. Cao, H.-S. Wang and Y.-M. Pan, RSC Adv., 2018, 8, 4584–4587 RSC; (i) G. Liang, J. Rong, W. Sun, G. Chen, Y. Jiang and T.-P. Loh, Org. Lett., 2018, 20, 7326–7331 CrossRef CAS PubMed.
- For (5+1) annulation reactions constructing 6-membered carbocycles, see (a) W. M. Akhtar, R. J. Armstrong, J. R. Frost, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2018, 140, 11916–11920 CrossRef CAS PubMed; (b) P. V. Chouthaiwale and F. Tanaka, Chem. Commun., 2014, 50, 14881–14884 RSC.
- C. Jutz, R.-W. Wargner, A. Kraatz and H.-G. Löbering, Liebigs Ann. Chem., 1975, 874–900 CrossRef CAS.
- (a) S. E. Steinhardt, J. S. Silverston and C. D. Vanderwal, J. Am. Chem. Soc., 2008, 130, 7560–7561 CrossRef CAS PubMed; (b) R. S. Paton, S. E. Steinhardt, C. D. Vanderwal and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 3895–3905 CrossRef CAS PubMed.
- (a) I. W. Davies, J.-F. Marcoux, J. T. Kuethe, M. D. Lankshear, J. D. O. Taylor, N. Tsou, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2004, 69, 1298–1308 CrossRef CAS PubMed; (b) L. Viteva, T. Gospodova, Y. Stefanovsky, K. Petrova, I. Timtcheva, M.-R. Mazières and J.-G. Wolf, Eur. J. Org. Chem., 2004, 385–394 CrossRef CAS; (c) L. Bianchi, C. Dell’Erba, M. Maccagno, G. Petrillo, E. Rizzato, F. Sancassan, E. Severi and C. Tavani, J. Org. Chem., 2005, 70, 8734–8738 CrossRef CAS PubMed; (d) M. R. Tatton, I. Simpson and T. J. Donohoe, Org. Lett., 2014, 16, 1920–1923 CrossRef CAS PubMed; (e) X. Li, H. Yu and Y. Huang, Adv. Synth. Catal., 2017, 359, 1379–1387 CrossRef CAS.
- (a) I. Yamaguchi, S. Shingai and M. Sato, Macromolecules, 2008, 41, 6292–6298 CrossRef CAS; (b) A. Colombo, C. Dragonetti, S. Righetto, D. Roberto, A. Valore, T. Benincori, F. Colombo and F. Sannicolò, J. Mater. Chem., 2012, 22, 19761–19766 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR data. See DOI: 10.1039/c9cc04012a |
| This journal is © The Royal Society of Chemistry 2019 |