
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow knitting of a triptycene hypercrosslinked polymer†
Cher Hon
Lau
 *a,
Tian-dan
Lu
b,
Shi-Peng
Sun
b,
Xianfeng
Chen
a,
Mariolino
Carta
c and
Daniel M.
Dawson
d
*a,
Tian-dan
Lu
b,
Shi-Peng
Sun
b,
Xianfeng
Chen
a,
Mariolino
Carta
c and
Daniel M.
Dawson
d
aSchool of Engineering, The University of Edinburgh, Robert Stevenson Road, Edinburgh EH9 3FB, UK. E-mail: cherhon.lau@ed.ac.uk; Tel: +44 (0)1316507813
bState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech. University, Nanjing 210009, China
cDepartment of Chemistry, College of Science, Swansea University, Grove Building, Singleton Park, Swansea SA2 8PP, UK
dSchool of Chemistry, EaStCHEM and Centre of Magnetic Resonance, University of St. Andrews, KY16 9ST, UK
First published on 27th June 2019
Abstract
By replacing Lewis acids with Brønsted acids as catalysts, continuous flow synthesis of hypercrosslinked polymers is achieved within 10% of the time required for a typical batch reaction. Compared with batch-synthesised polymers, the flow-produced materials take up 24% more CO2, precluding the need for lengthy reaction protocols to yield high-performance hypercrosslinked polymers for carbon capture.
Hypercrosslinked polymers (HCPs) are insoluble, amorphous, microporous materials widely deployed in environmental remediation via ion exchange, toxicant sequestration and carbon capture.1,2 HCPs are typically produced from time-consuming (at least 24 hours) batch Friedel–Crafts alkylation reactions where Lewis acid catalysts such as FeCl3 drive the post-crosslinking of aromatic polymers,3 direct polycondensation of halogenated substrates4 or external crosslinking of aromatic substrates with reactive linkers such as formaldehyde dimethyl acetate (FDA)5 in halogenated solvents under reflux conditions. During the initial stage of Friedel–Crafts alkylation, newly-formed HCPs will adsorb solvents required for nucleophilic substitutions, forming a gel that inhibits further reactions.1 Hence long synthesis durations are mandatory for complete reagent consumption, which is also key for obtaining HCPs with high Brunauer–Emmett–Teller (BET) surface areas.6 The molecular uptake of HCPs depends on their microporosity i.e. specific surface areas that can be enhanced with increasing solvent to substrate ratios7–9 and prolonging reaction times.10 However, these approaches exacerbate energy and material consumption.
Herein we overcome the disadvantages of batch HCP synthesis with continuous flow synthesis, (Fig. 1) a disruptive technology employed to produce active pharmaceutical ingredients,11 low molecular weight polymers12 and metal organic frameworks.13 Flow synthesis has also been deployed to produce microparticles of HCPs14 that are known to possess BET surface areas between 300–500 m2 g−1,15 while high surface area HCPs (>1000 m2 g−1) have yet to be produced in flow reactors.
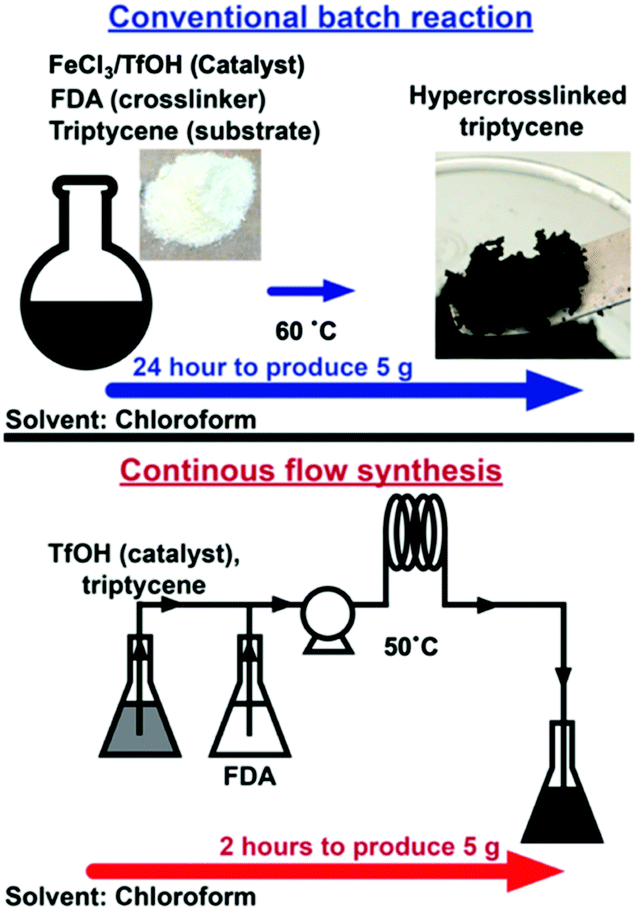 | ||
| Fig. 1 Triptycene HCPs can be produced in flow reactors within 2 hours, a 90% reduction in synthesis duration when compared to batch reactions required to produce HCPs of similar quality. | ||
Using a conventional HCP batch synthesis protocol1 (Scheme 1), we observed that FeCl3 clogged the microreactor during flow synthesis of hypercrosslinked triptycene, a substrate known to yield hierarchical porosity that benefits CO2 adsorption.16 Hence, trifluoromethanesulfonic acid (TfOH) – a liquid Brønsted acid catalyst miscible with chloroform was chosen here to replace FeCl3. TfOH was preferred as it catalysed the direct polycondensation and external crosslinking of aromatic substrates.6 Despite the better catalytic activities of TfOH,17 the BET surface area of externally-crosslinked HCPs produced with this catalyst was 65% lower, leading to lower molecular uptake. This could be due to incomplete consumption of reagents within the short synthesis duration.18
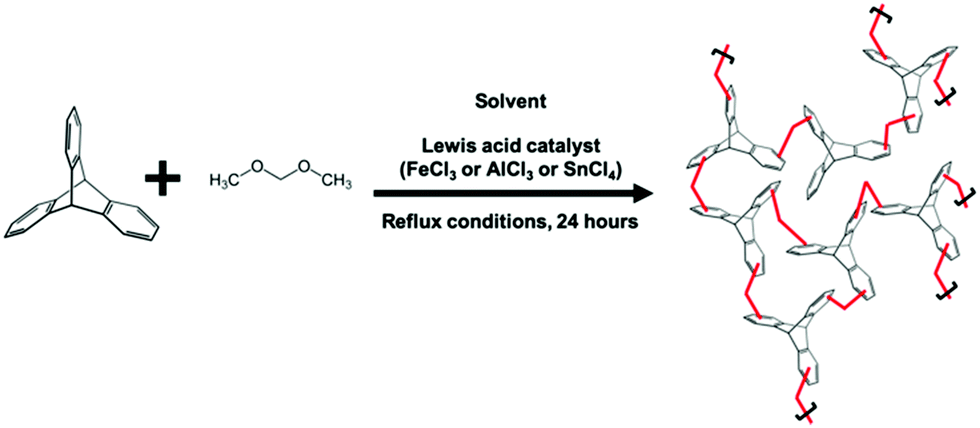 | ||
| Scheme 1 Typical batch reaction protocol to yield hypercrosslinked polymers. In this case, the substrate used was triptycene. | ||
To elucidate the role of synthesis time on HCP microporosity, we performed external crosslinking of 30 mmol of triptycene with 90 mmol of FDA and 90 mmol of catalysts (TfOH or FeCl3) in batch reactions at 60 °C over 24 hours in 250 mL of chloroform. To ensure complete reagent consumption, additional chloroform was added when a black gel was formed within the first two hours of reaction. It is important to point out here that gel formation is not equivalent to a complete crosslinking reaction. Triptycene HCPs were also produced in a 10 mL microreactor made up a 12 m long perfluoroalkoxyl polymer tube with inner diameter of 1 mm coiled in a figure of 8 configuration on a Vapourtec E-series flow synthesis set-up. Separate chloroformic solutions containing 10 mM of triptycene, and 15 mM of TfOH, and 30 mM of FDA were pumped at a flow rate of 5 mL min−1 into the microreactor with a temperature set at 50 °C, producing triptycene HCPs in 2 hours. Upon entry into the microreactor, the chloroformic solutions, turned black with microparticles within 30 seconds (see ESI†). FTIR and NMR analyses revealed that these microparticles were triptycene HCPs (Fig. 2), indicating that HCPs were produced within the first metre of the 12 m long polymer tube. Further work is required to customize the flow reactor to suit HCP synthesis.
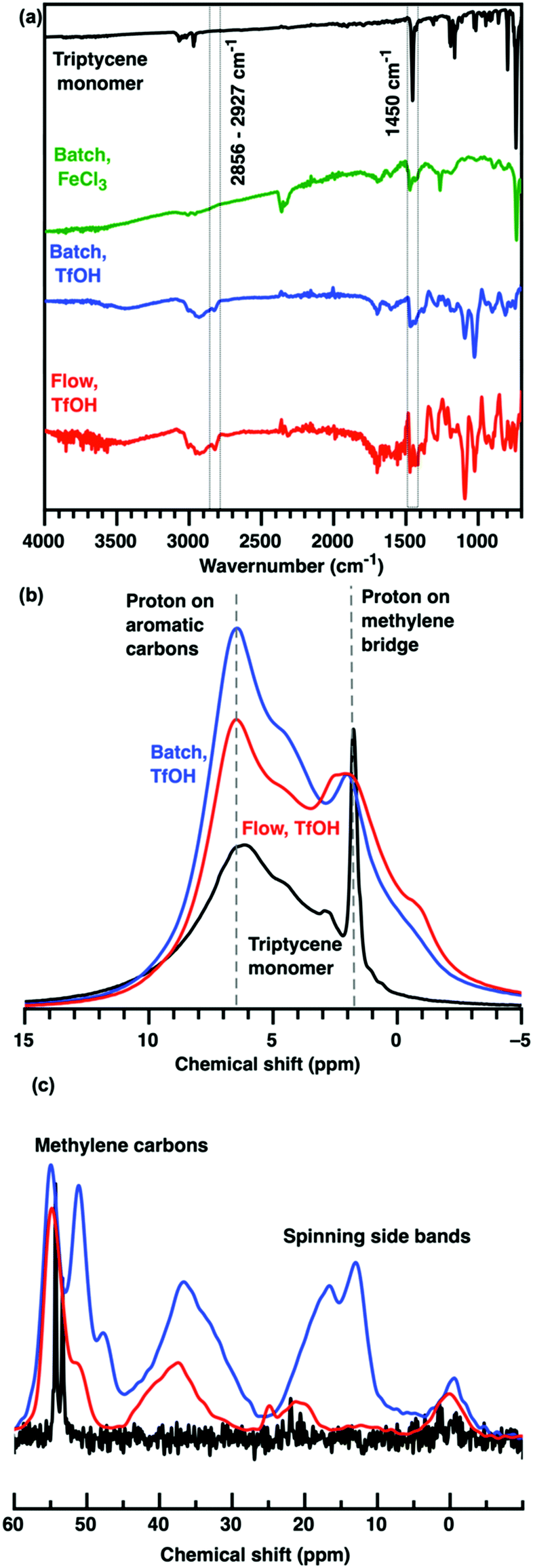 | ||
| Fig. 2 (a) FTIR spectra of triptycene monomer (black), triptycene HCPs produced in a batch reactor using FeCl3 (green) or TfOH (blue) as catalysts and in a flow reactor (red). The presence of methylene bridge crosslinks in triptycene HCPs was validated by the broadening of the peak between (b) 1–2 and 6–7 ppm in the 1H NMR spectra and (c) 25–55 ppm in the 13C NMR spectra. | ||
The chemical structures of triptycene HCPs produced using different catalysts (TfOH vs. FeCl3) and TfOH-catalysed protocols (batch vs. flow) were very similar (Fig. 2). FTIR analyses revealed that new peaks centred at 2856 and 2927 cm−1 were formed upon crosslinking of triptycene, while the intensity of the peak centred at 1450 cm−1 (absorption band of a non-substituted benzene ring) was reduced. These corresponded to the substitution of protons on aromatic rings with methylene bridge crosslinks during Friedel–Crafts reactions.16 This was validated by the broadening of chemical shifts between 1–2 ppm (methylene bridge proton), 6–7 ppm (protons of aromatic rings) and 25–55 ppm (methylene bridge carbon) in the 1H and 13C solid state NMR spectra.19 The NMR spectra of TfOH-catalysed HCPs could be collected after 5–7 water wash cycles but not for those produced using FeCl3 exhibited very rapid relaxation, which precluded acquisition of high-quality NMR spectra. This is most likely due to entrapped Fe ions in the HCPs. The removal of entrapped Fe ions from HCPs typically requires multiple washings using harsher solvents such as hydrochloric acid, chloroform, methanol and acetone.20,21 Other than requiring more benign wash cycles, the use of TfOH catalysts here also enhanced microporosity and CO2 uptake.
The specific surface areas of triptycene HCPs produced here were characterised using nitrogen adsorption isotherm measurements at 77 K (Fig. 3). A type 1 adsorption profile was observed in batch-produced triptycene HCPs, similar to those reported elsewhere.1,4,16,22 The steep slope at P/P0 < 0.01 indicated a sharp increase in N2 uptake, confirming the microporous nature of these HCPs. The BET surface area of the triptycene HCPs produced from TfOH-catalysed external crosslinking in batch reactors over a 24 hour period was 1637 m2 g−1, 36% higher than that produced using FeCl3 catalysts. This differs from the work of Schute and Rose where BET surface areas of their TfOH-catalysed HCPs produced via a 2 hour external crosslinking protocol were 65% lower than those produced using FeCl3 catalysts.6 Clearly, longer synthesis times in batch reactions are mandatory for producing HCPs with high surface areas using TfOH catalysts. The lack of entrapped metal ions in the pores of HCPs from TfOH-catalysed external crosslinking reactions also contributed to the higher BET surface area. Meanwhile, the BET surface area of TfOH-catalysed, flow-produced triptycene HCPs was 1590 cm2 g−1, comparable to that of HCPs produced from batch reactions catalysed by TfOH and 32% higher than those produced using FeCl3 catalysts in 24 hour batch reactions. More importantly, we demonstrated that flow microtechnology is capable of reducing production time of triptycene HCPs by 90% whilst enhancing BET surface areas i.e. molecular uptake.
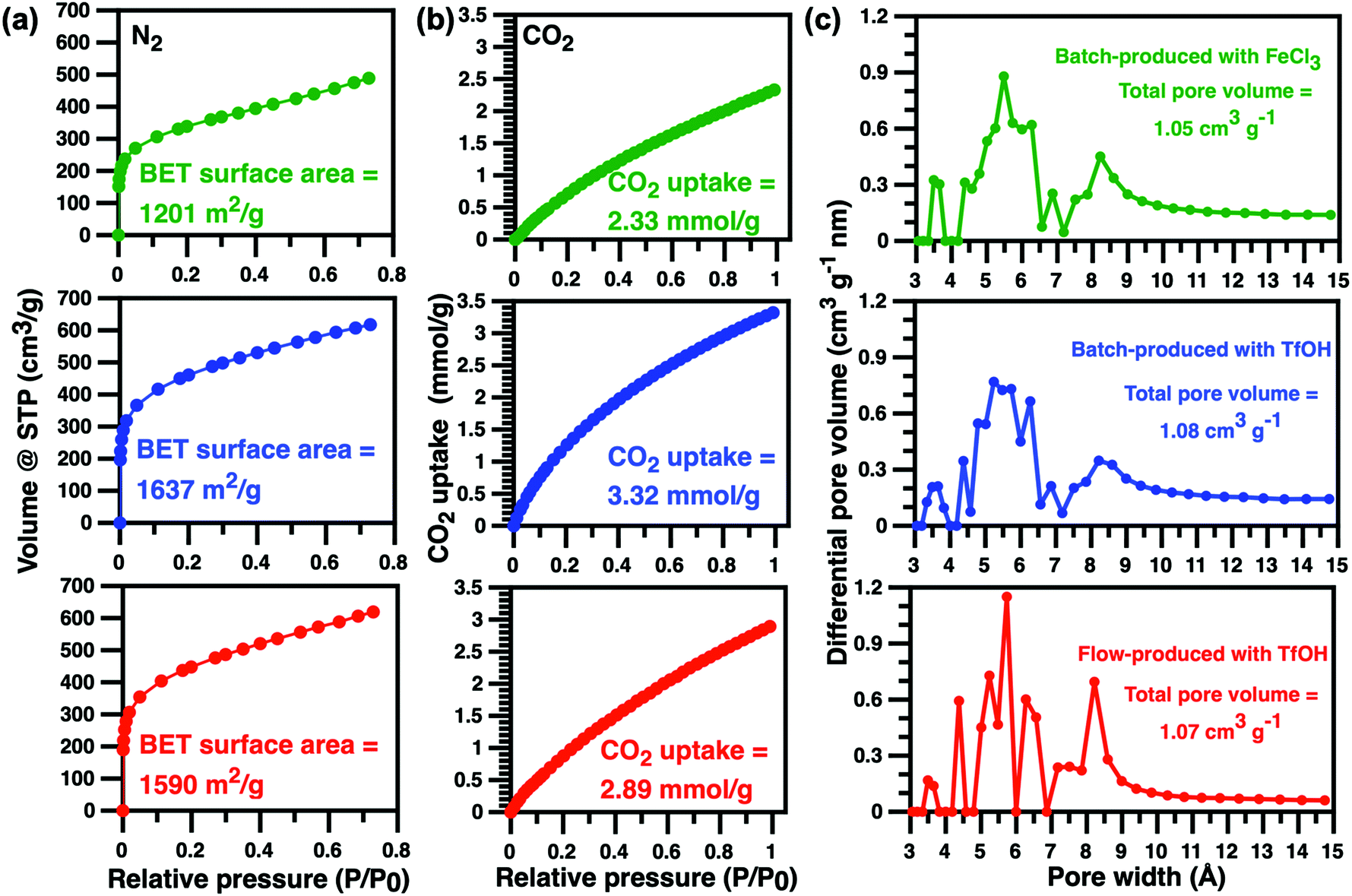 | ||
| Fig. 3 (a) N2 (77 K), (b) CO2 adsorption isotherms at 1.0 bar (273 K) and (c) pore size distributions calculated from 273 K CO2 adsorption isotherms using DFT theory of triptycene HCPs produced in a batch reactor using FeCl3 (green) and TfOH (blue) as catalyst and in a flow reactor with TfOH (red). | ||
CO2 adsorption isotherms obtained at 273 K provided information on pore size distribution and CO2 uptake. The total pore volume for TfOH- and FeCl3 catalysed batch-produced, and flow-produced triptycene HCPs were identical. The amount of ultra-micropores (pore width 7 Å) in flow-produced HCPs was higher than those produced from batch reactions while batch-produced HCPs contained more pores that were larger than 9 Å. The differences in micropore type and quantity could be ascribed to faster reaction kinetics in the significantly smaller confined spaces within the polymer tubes of the flow reactor where such environments typically ensure higher consumption of monomers and reagents.12,13 The CO2 uptake of batch-produced triptycene HCPs at 1.0 bar and 273 K was ∼3.3 mmol g−1, comparable to other organic microporous polymers1 and triptycene HCPs produced from batch reactions using dichloroethane.16 Meanwhile, the CO2 uptake of flow-produced triptycene HCPs at 1.0 bar and 273 K reached 2.9 mmol g−1, 12% lower than that of triptycene HCPs produced in batch reactions.
In summary, we showed that Brønsted acid catalysts could be deployed to produce HCPs with BET surface areas that were 27% higher than those produced using Lewis acid catalysts in batch reactions. The key to achieving this significant increase in BET surface area was prolonging synthesis duration to 24 hours. This finding underpinned the necessity of producing HCPs with similarly high BET surface areas via continuous flow synthesis, using less than 10% of the time required, potentially reducing energy consumption. Catalysed by Brønsted acids in confined spaces with higher surface area to volume ratio that consequently facilitated better heat transfer to reagents, the CO2 uptake of flow-produced HCPs was 24% higher than those produced in batch reactions. To further exploit these findings to scale up the production of HCPs whilst affording better control over product quality in a faster time scale, there is a need to further investigate the impact of system parameters such as reaction temperature, flow rates, catalyst type and reagent concentration in reactant solutions to optimize the advantages of this disruptive polymer production technology.
Commercially available reagents were used without purification. Triptycene, FeCl3, FDA and chloroform were purchased from Sigma Aldrich while TfOH was purchased from Alfa Aesar. Infrared spectra were recorded in the range 4000–600 cm−1 using a PerkinElmer 1600 series FTIR instrument as powder. The positions of absorption bands are quoted in cm−1. The N2 adsorption isotherms at 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K were measured using a Quantachrome Autosorb iQ2. CO2 (273K) sorption data were acquired using a Quantachrome Autosorb iQ. The powder samples were degassed for 600min at 120°C before the experiment. The adsorption isotherm was used to calculate the pore volume and the pore-size distribution. Solid-state NMR spectra were recorded using a Bruker Avance III spectrometer equipped with a 9.4 T wide-bore superconducting magnet (Larmor frequencies of 400.1 and 100.9 MHz for 1H and 13C, respectively). Samples were packed into standard 4 mm rotors and rotated at the magic angle at a rate of 12.5 kHz. The spectra were recorded with cross polarisation (CP) from 1H using a contact pulse (ramped for 1H) of 1.5 ms. High-power TPPM-15 decoupling of 1H (ν1 = 100 kHz) was carried out during acquisition. For the triptycene monomer, signal averaging as carried out for 128 transients with a recycle interval of 120 s, and for the polymer samples, signal averaging was carried out for 4096–14504 transients with a recycle interval of 3 s. Chemical shift are reported in ppm relative to trimethylsilane, using the CH3 resonance of L-alanine (δ = 20.5 ppm) as a secondary solid reference.
K were measured using a Quantachrome Autosorb iQ2. CO2 (273K) sorption data were acquired using a Quantachrome Autosorb iQ. The powder samples were degassed for 600min at 120°C before the experiment. The adsorption isotherm was used to calculate the pore volume and the pore-size distribution. Solid-state NMR spectra were recorded using a Bruker Avance III spectrometer equipped with a 9.4 T wide-bore superconducting magnet (Larmor frequencies of 400.1 and 100.9 MHz for 1H and 13C, respectively). Samples were packed into standard 4 mm rotors and rotated at the magic angle at a rate of 12.5 kHz. The spectra were recorded with cross polarisation (CP) from 1H using a contact pulse (ramped for 1H) of 1.5 ms. High-power TPPM-15 decoupling of 1H (ν1 = 100 kHz) was carried out during acquisition. For the triptycene monomer, signal averaging as carried out for 128 transients with a recycle interval of 120 s, and for the polymer samples, signal averaging was carried out for 4096–14504 transients with a recycle interval of 3 s. Chemical shift are reported in ppm relative to trimethylsilane, using the CH3 resonance of L-alanine (δ = 20.5 ppm) as a secondary solid reference.
The authors are grateful for the support of The University of Edinburgh Chancellor Fellowship Scheme and State Key Laboratory for Membrane Separations, China (NTU-KL17-03). We are also grateful for the fruitful discussions with Professor Neil McKeown and the use of characterization equipment within his labs.
Conflicts of interest
There are no conflicts to declare.References
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3481 RSC.
- J. Huang and S. R. Turner, Polym. Rev., 2018, 58, 1–41 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672, 10.1039/b604625h.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- K. Schute and M. Rose, ChemSusChem, 2015, 8, 3419–3423 CrossRef CAS PubMed.
- V. Davankov, M. Tsyurupa and L. Pavlova, US Pat., US20030027879A1, 2001 Search PubMed.
- V. Davankov, V. Rogozhin and M. Tsjurupa, US Pat., US3729457A, 1970 Search PubMed.
- S. F. Reed and R. K. Pinschmidt, Jr, US Pat., US4191813A, 1978 Search PubMed.
- Y. Yang, B. Tan and C. D. Wood, J. Mater. Chem. A, 2016, 4, 15072–15080 RSC.
- D. R. Snead and T. F. Jamison, Angew. Chem., Int. Ed., 2015, 54, 983–987 CrossRef CAS.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4, 5443 CrossRef CAS.
- P. J. Dowding, J. W. Goodwin and B. Vincent, Colloid Polym. Sci., 2000, 278, 346–351 CrossRef CAS.
- R. Wang, Y. Zhang, G. Ma and Z. Su, Colloids Surf., B, 2006, 51, 93–99 CrossRef CAS PubMed.
- C. Zhang, P.-C. Zhu, L. Tan, J.-M. Liu, B. Tan, X.-L. Yang and H.-B. Xu, Macromolecules, 2015, 48, 8509–8514 CrossRef CAS.
- P. Trillo, A. Baeza and C. Nájera, Eur. J. Org. Chem., 2012, 2929–2934 CrossRef CAS.
- N. Fontanals, R. M. Marcé, F. Borrull and P. A. G. Cormack, Polym. Chem., 2015, 6, 7231–7244 RSC.
- K. G. Kidd, G. Kotowycz and T. Schaefer, Can. J. Chem., 1967, 45, 2155–2162 CrossRef CAS.
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed.
- K. J. Msayib and N. B. McKeown, J. Mater. Chem. A, 2016, 4, 10110–10113 RSC.
- R. Bera, S. Mondal and N. Das, Microporous Mesoporous Mater., 2018, 257, 253–261 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc03731d |
| This journal is © The Royal Society of Chemistry 2019 |