
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polyoxometalate clusters using carbohydrates as reducing agents leads to isomer-selection†
Eric
Janusson
 ,
Noël
de Kler
,
Laia
Vilà-Nadal
,
De-Liang
Long
and
Leroy
Cronin
*
,
Noël
de Kler
,
Laia
Vilà-Nadal
,
De-Liang
Long
and
Leroy
Cronin
*
School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: Lee.Cronin@glasgow.ac.uk
First published on 26th April 2019
Abstract
By using sugars as the reducing agents, we demonstrate that it is possible to control the self-assembly of polyoxomolybdates through selective preparation of a single heteropolyanion isomer. D-(−)-Fructose has been proved to be an effective reducing sugar compared to the chemically similar carbohydrate D-(+)-glucose. The gentle reduction results in favourable formation of the Wells–Dawson type gamma isomer in 6-fold reduced form at room temperature.
Polyoxometalates (POMs) are inorganic metal-oxide clusters which self-assemble into larger, organised, discreet molecular structures from platonic subunits. Olation and condensation of the anionic transition metal subunits in aqueous solution occurs spontaneously with exposure to acidic media.1 A closer examination of this process is important and necessary as the study of this area brings forth mechanistic information of self-assembling chemical systems; this information is valuable in the design of promising new materials.2–6 Reduced polyoxometalate species are of considerable interest lately as is evidenced by newfound applications in diverse fields including medicine, molecular magnetism, and catalysis.7 In order to explore the physicochemical properties of these materials, it is important to consider alternative routes in producing reduced polyoxometalates. However, the sizeable chemical space employed in POM synthesis means that structural and mechanistic repercussions of several process variables, including the impact of various reducing agents used, remain largely unexplored. Control over the spatial configuration of the many metal centres of polyoxometalate clusters is desirable for fine-tuning of bulk electronic properties for tailored catalyst activity, energy storage material applications, or simply improved mechanistic understanding of complex self-assembling systems.7,8 While POM synthesis is simple, the self-assembly process is extremely sensitive to reaction conditions including pH, solvent composition, metal, heteroatom, cations, reducing agents, temperature, pressure, reagent concentrations, and other variables (including type of reaction vessel). The intricacies of self-assembly naturally lead to several empirically-designed synthetic conditions in the literature which must be carefully controlled to produce one out of thousands of possible structures.9–11
The Wells–Dawson POM cluster consists of two {XM6} moieties that form the belt which are both capped by two {M3} units forming a symmetrical cage. Rotating the caps and belts by specific angles yields six theoretically possible rotational isomers: α, β, γ, α*, β* and γ*. Fig. 1 illustrates the structural difference between the alpha and gamma isomers discussed herein. Computational studies of {P2W18} isomers with DFT calculations indicate the relative stability between isomers follow the order: α > β > γ > γ* > β* > α*.12,13 Thermolysis of α-H6P2Mo18O62 reveals the β and γ isomers form at high temperatures;14 however, full characterization of these isomers has not yet been reported. The difficulty in obtaining this complex is very likely due to the fact that the {P2Mo18} anion is thought to readily decompose in aqueous solutions at mild temperatures over a broad pH range.15
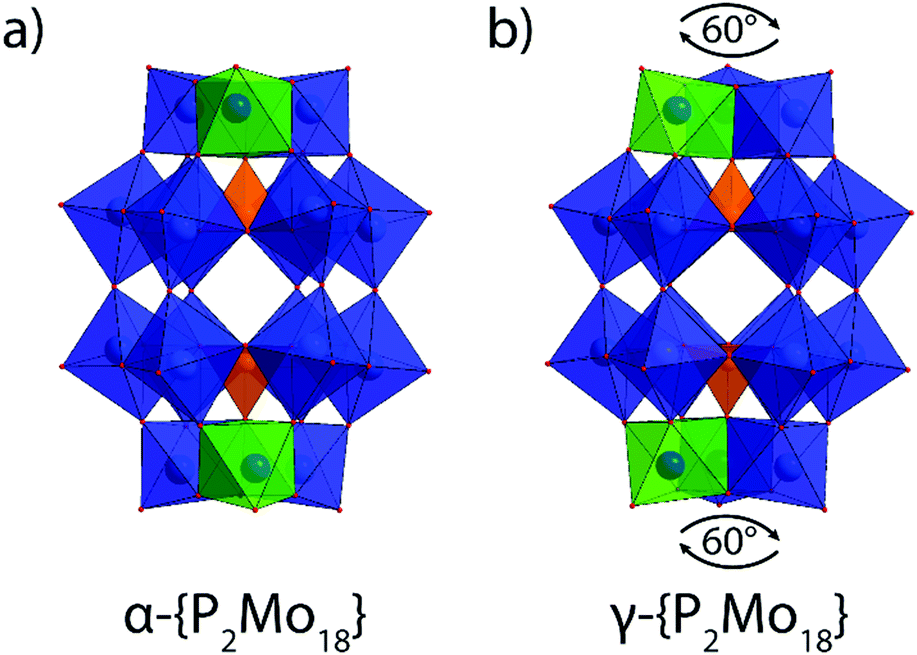 | ||
| Fig. 1 Single crystal XRD structures of molybdenum Wells–Dawson structural isomers (a) α-{P2Mo18} and (b) γ-{P2Mo18} isolated in this work. Blue polyhedra and spheres: Mo, orange polyhedra and spheres: P; red spheres: O. Note one pair of Mo polyhedra are highlighted green to visualize cap rotation difference between isomers. | ||
Chemical reduction is common in polyoxomolybdate syntheses including procedures for {Mo154} and {Mo132} which involve sodium dithionite (Na2S2O4) and hydrazine (N2H4·HCl or N2H4·HSO4), respectively.10,16,17 We employed sugars as organic reducing agents to explore new POM synthetic strategies. Two chemically similar monosaccharides, D-(+)-glucose and D-(−)-fructose (both with the formula C6H12O6), as non-standard reductants since they demonstrated signs of reduction following initial experiments via an obvious colour change on addition.
The alpha isomer of the molybdenum Wells–Dawson cluster, (NH4)6[α-P2Mo18O62]·14(H2O) (referred to as α-{P2Mo18} in this work), was prepared using a scaled-down version of Wu's 1920 procedure.18 This was modified to separately incorporate D-(+)-glucose, D-(−)-fructose, and sodium dithionite for comparison, as reducing agents. Initial screening efforts were focused on low-volume (5–10 mL) reaction mixtures. Aliquots of crude mixtures were examined by electrospray ionization mass spectrometry (ESI-MS) and 31P-NMR in order to optimise synthetic conditions required for single {P2Mo18} isomer selectivity. Generally, the sodium molybdate solutions (330 mM) containing no reducing agent remained clear and colourless when stirred at room temperature overnight at 20 °C while those treated with 1 eq. of dithionite rapidly change to dark brown. Similar samples treated with an equimolar amount of either glucose or fructose develop an intense blue colour as a result of intervalence charge transfer indicative of reduced molybdenum.19 The blue colour that develops in these solutions is easily identifiable through a prominent absorption at 866 nm in the UV-Vis-NIR spectrum (see ESI†).
Samples treated with a very low concentration (0.03 eq.) of fructose yielded needle-shaped crystals of generally poor quality due to co-crystallization of isomers; however, increasing the fructose concentration to 0.1 eq. and above increased a far-upfield 31P-NMR signal chemical shift at −13.1 ppm with few impurities present. Fructose concentrations above 0.1 eq. enhanced this upfield signal. Crystallization of these mixtures was facile following counter cation metathesis with ammonium chloride. Large block-shaped crystals of these samples were isolated within one day and enabled positive identification of the reduced species, (NH4)4H8[γ-P2Mo18O62] (referred to as γ-{P2Mo18} in this work), by single crystal XRD supported by ion mobility mass spectrometry confirming the assignment of the −13.1 ppm 31P-NMR signal to γ-{P2Mo18}. Further increasing the quantity of fructose promotes conversion to the gamma isomer and the application of 80 °C heating overnight with 0.3 eq. yields this isomer with very good conversion observed by 31P-NMR spectra (see ESI†). Following purification via recrystallization and washing (see ESI† for synthetic details) of isolated crystals, one singlet was observed at −13.1 ppm attributed to the 6e− reduced form of γ-{P2Mo18}.
Fig. 2 outlines the result of treatment with the reducing agents used in this study. The 31P-NMR spectra of glucose-containing reactions did not exhibit signs of Dawson type POMs or encapsulated phosphate. Further, the slow development of a comparatively weak blue colour implied the reduction with glucose to be far less effective than with fructose. Despite signs of Mo reduction, samples treated with 0.1–2.0 eq. of glucose rarely produced either the alpha or gamma isomer of {P2Mo18} in significant quantities and crystallization was poor. Despite their similarities, the substantial difference between fructose and glucose likely lies in the structure of the two monosaccharides. Because of their similarity this is likely caused by the limited interaction of glucose with molybdenum as a weaker bidentate ligand compared to the energetically-favoured tridentate interaction with fructose.20 In their cyclic form both sugars have multiple hydroxyl groups near their anomeric carbon centres. The two hydroxyl moieties of glucose are available for a bidentate interaction with transition metal atoms whereas fructose has three close-proximity hydroxyl groups available for a tridentate interaction. This promotes interaction with, and efficient reduction of, solubilized MoVI atoms. This was quickly apparent by comparing the rate and intensity of colour change in small-scale samples with equimolar quantities of either sugar (Fig. 2).
 | ||
| Fig. 2 31P-NMR in D2O of isolated products of reactions containing sodium molybdate (100 mg mL−1), 1 eq. H3PO4, and 1.2 eq. of (a) D-(−)-fructose, (b) D-(+)-glucose, (c) sodium dithionite, or (d) no reducing agent at pH 1. All reactions stirred for 24 h at room temperature (20 °C). | ||
Experiments performed with similar quantities of sodium dithionite as a reducing agent were less selective for one isomer and produced isomeric mixtures of {P2Mo18} evidenced by both the UV-Vis and NMR. A low concentration of sodium dithionite (0.03 equivalents) resulted in two signals in the 31P-NMR at −3.1 and −4.7 ppm. The peak at −3.1 ppm corresponds to the α Dawson while the second peak at −4.7 ppm is likely due to a partially reduced isomer (see ESI†).21 The reduced species, γ-{P2Mo18}, was detected by NMR when used in increased quantity (0.2 eq.) of dithionite; however, the lack of selectivity is clear based on the shallow UV-Vis spectra and numerous signals apparent in 31P-NMR spectra. These solutions did not yield quality crystals for examination by single crystal XRD. UV-vis-NIR spectroscopic characterization of γ-{P2Mo18} prepared with fructose exhibit a bathochromic shift and strong absorption relative to α-{P2Mo18}. Samples which were treated with 0.2 eq. Na2S2O4 absorb across a wide range of the visible region with maxima centred at 680 nm and 863 nm. Scaled-up syntheses with 0.13 eq. of fructose yielded high quality crystals of γ-{P2Mo18}. The degree of reduced molybdenum cores of γ-{P2Mo18} was determined with electrochemical tests on purified crystalline product via bulk electrolysis and redox titration (see ESI†). Electrochemical oxidation indicated γ-{P2Mo18} was reduced by approximately 6 electrons and agrees with the range of 6–8 electron reduction determined by cerimetry. These results are supported by a combination of mass spectrometry, a significant downfield shift of the 31P-NMR signal, the hyperchromicity of UV-Vis-NIR absorbance, and elemental analysis.
The gamma isomer, γ-{P2Mo18}, is similar in structure and symmetry to α-{P2Mo18} as the two complexes both have D3h point group. Both clusters are composed of two {PMo6} belt subunits which are capped on either end by {Mo3O13} octahedral caps. Relative to the α-{P2Mo18}, the γ-{P2Mo18} caps are both rotated by 60°. Overall, molybdenum-oxygen bond lengths in both alpha and gamma structures are similar and range between 1.8 to 2.1 Å for the outer Mo–O bonds and 2.3 to 2.4 Å for the Mo–O interior oxygens. The capping Mo–Mo distances are also only slightly different with an average of 3.38 Å for α-{P2Mo18} and 3.40 Å for γ-{P2Mo18}. The molybdenum atoms forming the {PMo6} belts of α-{P2Mo18} alternate in position thus forming a “zig-zag” pattern whereas the {PMo6} rings of γ-{P2Mo18} are expectedly more planar due to reduction.22,23 The equatorial belt distortion is apparent in the average Mo–O–Mo angle of 137° for α-{P2Mo18} compared to the wider angles of γ-{P2Mo18} resulting in an average of 143°. The planarity of the gamma isomer belts results in an average belt angle which is considerably greater in γ-{P2Mo18} compared to α-{P2Mo18}. Bond valence sum analysis of γ-{P2Mo18} shows the capping molybdenum oxidation states are significantly higher than those of the belt metal atoms (see ESI†). Conversely, the calculated valency of belt Mo atoms is consistent with those of the capping ones in α-{P2Mo18}. Therefore, the reduced electrons in γ-{P2Mo18} preferentially reside delocalized about the belt of the molecule as was observed computationally with the isovalent tungsten-based POM, [α-P2W18O62]6−.24 Additionally, the calculated oxidation state of half of the µ2-oxo atoms between the caps and belts is near unity in both complexes which clearly indicates protonation at these sites.
Electrospray ionization mass spectrometry proved useful for speciation of crude mixtures and isolated clusters. The broad and poorly resolved signals apparent in mass spectra of samples of γ-{P2Mo18} and α-{P2Mo18} (see ESI†) are indicative of a family of ions with a range of counter cations, solvent association, and cluster aggregates. This type of aggregation and solvation is known to further complicate standard ESI-MS assignment of POMs even with well-optimised source conditions.25,26 For this reason, ion mobility separation (IMS) is desirable, if not essential, to observe isotope patterns of the naked parent ion present in complexes γ-{P2Mo18} and α-{P2Mo18}. Optimization of ESI and travelling wave ion mobility separation (TWIMS) settings (see ESI† for details) serves to greatly clarify solution-phase speciation in complex mixtures. For example, the singly-charged, fully protonated, [H5(P2Mo18O62)]− anion centred around m/z 2786 is well-resolved with a drift time (td) of 18.85 ms under the TWIMS settings used for all samples. While the protonated, singly-charged parent ion is obvious in samples of α-{P2Mo18} the analogous anion expected of the 6e− reduced [H11(P2Mo18O62)]− is much more elusive, but detectable, in IMS-mass spectra of γ-{P2Mo18} and appears around m/z 2792 with td = 19.63 ms (Fig. 3). The small difference in collisional cross section distinguishes the gamma isomer from the protonated ion of the pure alpha isomer which normally overlaps significantly with the naked gamma isomer, [H11(P2Mo18O62)]−, in a standard ESI-MS spectrum. The increase in m/z and td support the assignment of z = −12 determined electrochemically with pure samples. A noteworthy feature in mobilograms of γ-{P2Mo18} and α-{P2Mo18} is evidence of cluster self-aggregation. This is especially apparent in TWIMS-MS spectra of α-{P2Mo18} in which aggregates are separated by charge state, easily distinguished by shortened drift times. Higher order aggregates with z = −4 are resolvable with IMS. Aggregates above this charge state are also apparent though signal intensity for these high-charge aggregates was generally insufficient to resolve the isotope patterns.
 | ||
| Fig. 3 ESI-TWIMS-MS of γ-{P2Mo18} demonstrating the naked ion [H11(P2Mo18O62)]− at drift time of 19.63 ms. Note the multiply-charged cluster aggregates in mobilogram. | ||
To summarize, we have demonstrated a single synthetic variable in POM synthesis, the reducing agent, exerts isomeric control in formation of the molybdenum Wells–Dawson. Further, we have selectively prepared, isolated, and fully characterised the reduced molybdenum Wells–Dawson, γ-{P2Mo18}, for the first time. The relatively low fructose concentration required for the 6e− reduction of {P2Mo18} suggests that fructose is an efficient and effective organic reducing agent. The striking contrast between similar reducing sugars indicates careful selection, and quantity, of reducing agent is extremely important in polyoxometalate syntheses. Fructose is an economically and environmentally sustainable reagent which alters polyoxometalate self-assembly through mild reduction and, potentially, stabilization of intermediate building blocks during the stepwise assembly. This effect has been demonstrated via real time monitoring with other alkoxo ligands in the preparation of {Mo132}.27 The evidence in this study suggests fructose acts as a “soft-coordinating” ligand which stabilizes intermediate metal cluster transition states prior to reduction and effectively modifies the assembly process in a unique way. However, additional studies are required for a closer examination of synthetic process variables in POM synthesis. The multiple roles of organic reducing agents (which may serve as both ligands and reductants) will require further exploration to develop a comprehensive toolkit for the synthetic chemist to manage cluster formation and produce new materials by design.
This work was supported by the EPSRC grants (No. EP/S017046/1; EP/S019472/1; EP/R020914/1; EP/R009902/1; EP/R01308X/1; EP/R020892/1; EP/P029329/1; EP/L023652/1; EP/K038885/1) and the ERC for an Advanced Grant (ERC-ADG, 670467 SMART-POM).
Conflicts of interest
The authors declare no conflicts.Notes and references
- L. Vilà-Nadal, S. G. Mitchell, A. Rodríguez-Fortea, H. N. Miras, L. Cronin and J. M. Poblet, Phys. Chem. Chem. Phys., 2011, 13, 20136–20145 RSC.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS PubMed.
- Y. Ji, L. Huang, J. Hu, C. Streb and Y.-F. Song, Energy Environ. Sci., 2015, 8, 776–789 RSC.
- M. Genovese and K. Lian, Curr. Opin. Solid State Mater. Sci., 2015, 19, 126–137 CrossRef CAS.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- S. Omwoma, C. T. Gore, Y. Ji, C. Hu and Y. F. Song, Coord. Chem. Rev., 2014, 286, 17–29 CrossRef.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- W. Xuan, R. Pow, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2017, 56, 9727–9731 CrossRef CAS PubMed.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- W. Xuan, A. J. Surman, H. N. Miras, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2014, 136, 14114–14120 CrossRef CAS PubMed.
- F. Q. Zhang, W. Guan, L. K. Yan, Y. T. Zhang, M. T. Xu, E. Hayfron-Benjamin and Z. M. Su, Inorg. Chem., 2011, 50, 4967–4977 CrossRef CAS PubMed.
- L. Vilà-Nadal, S. G. Mitchell, D.-L. Long, A. Rodríguez-Fortea, X. López, J. M. Poblet and L. Cronin, Dalton Trans., 2012, 41, 2264–2271 RSC.
- R. I. Maksimovskaya, G. M. Maksimov and G. S. Litvak, Russ. Chem. Bull., 2003, 52, 103–108 CrossRef CAS.
- L. E. Briand, G. M. Valle and H. J. Thomas, J. Mater. Chem., 2002, 12, 299–304 RSC.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3359–3363 CrossRef.
- A. Müller, S. K. Das, V. P. Fedin, E. Krickemeyer, C. Beugholt, H. Bogge, M. Schmidtmann and B. Hauptfleisch, Z. Anorg. Allg. Chem., 1999, 625, 1187–1192 CrossRef.
- H. Wu, J. Biol. Chem., 1920, 43, 189–220 CAS.
- L. C. W. Baker and D. C. Glick, Chem. Rev., 1998, 98, 3–50 CrossRef CAS.
- D. H. Brown and J. MacPherson, J. Inorg. Nucl. Chem., 1970, 32, 3309–3313 CrossRef CAS.
- R. I. Maksimovskaya, G. M. Maksimov and G. S. Litvak, Russ. Chem. Bull., 2003, 52, 103–108 CrossRef CAS.
- R. Strandberg, H. Hjersing, A. Kjekshus, A. F. Andresen, J. T. Southern, K. Edlund, M. Eliasen, C. Herskind, T. Laursen and P. M. Pedersen, Acta Chem. Scand., Ser. A, 1975, 29, 350–358 CrossRef.
- L. Yan, X. López, J. J. Carbó, R. Sniatynsky, D. C. Duncan and J. M. Poblet, J. Am. Chem. Soc., 2008, 130, 8223–8233 CrossRef CAS.
- L. Vilà-Nadal, K. Peuntinger, C. Busche, J. Yan, D. Lüders, D.-L. Long, J. M. Poblet, D. M. Guldi and L. Cronin, Angew. Chem., Int. Ed., 2013, 52, 9695–9699 CrossRef PubMed.
- N. M. Al Hasan, G. E. Johnson and J. Laskin, J. Am. Soc. Mass Spectrom, 2013, 24, 1385–1395 CrossRef CAS.
- D.-L. Long, C. Streb, Y. F. Song, S. Mitchell and L. Cronin, J. Am. Chem. Soc., 2008, 130, 1830–1832 CrossRef CAS.
- P. Yin, B. Wu, T. Li, P. V. Bonnesen, K. Hong, S. Seifert, L. Porcar, C. Do and J. K. Keum, J. Am. Chem. Soc., 2016, 138, 10623–10629 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectra, and crystallographic data. CCDC 1903343 and 1903344. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02361e |
| This journal is © The Royal Society of Chemistry 2019 |