
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStraightforward access to biocompatible poly(2-oxazoline)-coated nanomaterials by polymerization-induced self-assembly†
Dao
Le‡
 ab,
Friederike
Wagner‡
ab,
Masanari
Takamiya
a,
I-Lun
Hsiao
ac,
Gabriela
Gil Alvaradejo
ab,
Uwe
Strähle
a,
Carsten
Weiss
a and
Guillaume
Delaittre
*ab
ab,
Friederike
Wagner‡
ab,
Masanari
Takamiya
a,
I-Lun
Hsiao
ac,
Gabriela
Gil Alvaradejo
ab,
Uwe
Strähle
a,
Carsten
Weiss
a and
Guillaume
Delaittre
*ab
aInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: guillaume.delaittre@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany
cSchool of Food Safety, College of Nutrition, Taipei Medical University, No. 250, Wuxing St., Taipei 11031, Taiwan
First published on 25th February 2019
Abstract
We report the synthesis of poly(2-ethyl-2-oxazoline)-based (PEtOx) nanoobjects by polymerization-induced self-assembly (PISA). First, well-defined PEtOx macromolecular chain transfer agents were synthesized by cationic ring-opening polymerization and click chemistry. The photoinitiated PISA of 2-hydroxypropyl methacrylate mediated by these PEtOx produced nanoobjects spanning the full range of core–shell morphologies. The nanoparticles exhibited high biocompatibility and stealth properties in vitro or in vivo, as well as thermoresponsive behavior.
Poly(2-alkyl/aryl-2-oxazoline)s (PAOx), synthesized by cationic ring-opening polymerization (CROP), are valuable polymers in the biomedical field, thanks to their biocompatibility, stealth behavior, and chemical and physical versatility.1–4 Coinciding with the recently renewed interest in PAOx, the first poly(2-ethyl-2-oxazoline) (PEtOx) conjugate has already entered phase II clinical trials for the treatment of Parkinson's disease.5 Several studies have focused on the synthesis of functional PAOx to achieve functional hydrogels,6–8 surface coatings,9 and polymer–peptide/protein/drug conjugates.10,11 Besides, the interest in PAOx-based complex structures is also increasing.12 PEtOx brush-arm star and comb-shaped polymers incorporating nitroxide radicals and perfluorinated segments, respectively, have been reported for magnetic resonance imaging.13,14 Patterned PEtOx bottlebrush copolymer brushes on diamond were reported for biosensor design.15 Notably, PAOx-based core–shell nanoparticles (NPs) are promising nanocarriers for biomedicine.16 Until now, these have been exclusively prepared by classic self-assembly of pre-synthesized amphiphilic block copolymer (ABCP).
Polymerization-induced self-assembly (PISA), developed in the last decade, has become an important approach for the fabrication of ABCP NPs with elaborate and precise macromolecular architecture, colloidal morphology, and functionality.17–23 In a nutshell, PISA rests upon two events: (i) chain extension of a solvophilic macromolecular precursor with a monomer forming a solvophobic block and (ii) synchronous self-assembly of the so-formed amphiphilic block copolymer leading to core–shell NPs. As opposed to the classic nanoprecipitation–solvent exchange/evaporation method, PISA can be performed at a high polymer concentration (up to 50 vs. 1–2 wt%) and gives in situ access to morphologies as diverse as plain spheres, rods/fibers, and hollow vesicles. Reversible-deactivation radical polymerization, mostly reversible addition–fragmentation transfer (RAFT) polymerization, is the major underlying polymerization mechanism for PISA, occurring under either dispersion or emulsion polymerization conditions with styrene, (meth)acrylate, or (meth)acrylamide monomers. Other polymerization mechanisms are slowly making an appearance in the PISA realm, such as ring-opening metathesis polymerization,24–27 in order to expand the chemistry and functionality of NPs.28 However, because of the interest in greener solvents and the general requirement for polar solvents to produce biocompatible nanoparticles (i.e., alcohols and water), employment of ionic polymerization is still limited.29 Therefore, many interesting biocompatible polymers such as PAOx have not yet been included in PISA nanoobjects.
In this study, we describe the first combination of CROP and RAFTPISA to obtain PAOx-based nanoobjects spanning a wide range of morphologies. First, dithiobenzoate-functionalized PAOx were prepared by CROP of a 2-oxazoline monomer in an aprotic solvent, followed by capping to introduce the chain transfer agent (CTA) moiety necessary for the RAFT polymerization. The functional PAOx were then used as a macromolecular CTA (macroCTA) in photoinitiated aqueous RAFTPISA (photoRAFTPISA). The influence of several parameters on the morphology was investigated, i.e., mode of synthesis of the macroCTAs, chain length ratio between the solvophilic and solvophobic blocks, and concentration. Besides, we demonstrate that the obtained NPs are biocompatible in vitro and in vivo, and hence are suitable for potential bioapplications.
In particular, PEtOx was used in this study for its great hydrophilicity and its potential as an alternative to the established polyethylene glycol (PEG).30 PEtOx was synthesized by CROP using methyl tosylate as an initiator. As shown in Scheme 1, the CTA end group could be incorporated either by direct capping with a deprotonated carboxylic acid-functionalized CTA (Route A)31,32 or by a two-step sequence consisting of a nucleophilic substitution with sodium azide and a subsequent copper-catalyzed azide–alkyne cycloaddition (CuAAC) (Route B). Both routes showed similar kinetic features and fulfilled elementary criteria for a controlled/living polymerization, i.e., linear increase of pseudo-first order plot, linear increase of number-average molar mass with conversion, and a clear shift of size-exclusion chromatography (SEC) traces (Fig. S2, ESI†). However, the nature of the capping reaction impacted the dispersity of the PEtOx (Đ); the Đ values were systematically slightly higher when capping with the carboxyl CTA, as compared with NaN3, concomitant with the presence of a significant shoulder at higher molar masses on the SEC traces (Fig. S2C, ESI†). In Route B, reaction conditions to introduce the CTA moiety by CuAAC (time, solvent) also influenced the shape of the molar mass distribution (MMD) and Đ (Fig. S7, ESI†). Performing CuAAC for 4 hours in dichloromethane (DCM) led to the smallest broadening of the MMD. This shouldering phenomenon may be – at least to some extent – due to partial decomposition of the CTA moiety to a thiol, which can be readily oxidized to disulfide and produce high molar mass coupled species (Fig. S5B and C, ESI†).33 Indeed, CuAAC of PEtOx-N3 with propargyl alcohol in similar conditions led to a significant reduction of MMD broadening (Fig. S8, ESI†). Overall, Route B required one more step but provided well-defined polymers with Đ ≤ 1.2 (Fig. S9, ESI†). For each route, two batches of PEtOx-CTAs were synthesized for the PISA experiments, with Mn of approx. 5500 and 9500 g mol−1. The characteristics of these polymers are listed in Table S1 (ESI†). 1H NMR and SEC coupled to electrospray ionization mass spectrometry (SEC-ESI-MS) data ascertained high RAFT functionality in all polymers (Fig. S3, S4, S10–S12, ESI†). The chain extension experiments with benzyl methacrylate in solution confirmed the previous trends: while macroCTAs from Route A underwent only partial chain extension (Fig. S13, ESI†), Route B led to clean block copolymers (Fig. S14 and S15, ESI†). Nevertheless, as a living character is not mandatory to obtain nanoparticles by PISA,34 we further employed all four macroCTAs.
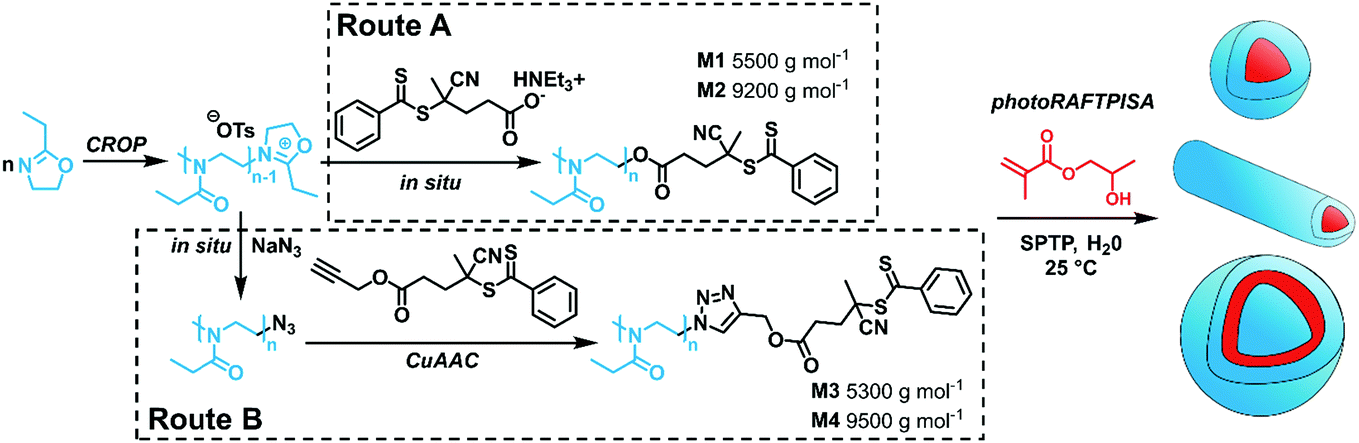 | ||
| Scheme 1 Synthetic routes towards PEtOx-stabilized NPs by merging CROP and photoRAFTPISA. SPTP = sodium phenyl-2,4,6-trimethylbenzoylphosphinate. | ||
2-Hydroxypropyl methacrylate (HPMA) was used as a core-forming monomer in water. Since PEtOx exhibits a lower critical solution temperature in water at approx. 60 °C,35 photopolymerization at 25 °C was applied to maintain good solubility and the ability for steric stabilization. Firstly, PISA experiments with the short PEtOx-CTA M1 were performed with varying targeted degrees of polymerization of HPMA (DPHPMA) and total solids content (Table S3, ESI†). All PISA experiments were conducted for 3 h and led to complete conversion of HPMA, as verified by 1H NMR spectroscopy (Fig. S16, ESI†). As observed by TEM, only pure spherical NPs were obtained in the DPHPMA = 30–100 range at 5 wt% solids (Fig. S18, left, ESI†). At 10 wt%, a mixture of spheres and nanofibers was obtained for DPHPMA = 50 (Fig. S18, right, ESI†). Higher DPHPMA or solids concentration led to precipitation. Similar results were achieved for the CuAAC-based PEtOx-CTA M3 with mainly plain spherical morphology but also large, possibly multicompartmented particles (Table S5 and Fig. S22, ESI†). While both short PEtOx-CTAs seemed to provide insufficient stabilization and led to aggregation at high DPHPMA and high concentration, PISA with longer analogs allowed the formation of nanoobjects spanning the full range of morphologies. Pure spheres, worm-like nanofibers, vesicles as well as morphology mixtures were obtained with M2 (Table S4 and Fig. S20, ESI†). Higher-order morphologies evolved with increasing DPHPMA and solid contents. However, a significant rise in dispersity was also observed, which is most probably due to non-reinitiating PEtOx-CTA rather than to a loss of control. Indeed, SEC traces of PISA samples showed important amounts of low-molecular-mass species consistent with non-functionalized PEtOx and unreacted POx-CTA (Fig. S19, ESI†). By using the better-defined PEtOx-CTA M4, strongly improved MMDs were obtained with a total consumption of POx-CTA (Fig. S23, ESI†). Concomitantly, the PISA NPs displayed well-defined morphologies, as well as further more intricate shapes such as donut- and jellyfish-like (Fig. 1). At low solids content (5 wt%) and with DPHPMA up to 200, only spheres or donuts were observed, as for higher solids content with DPHPMA ≤ 100. However, a range of nanoobjects from spheres to vesicles was achieved when targeting higher DPHPMA above 5 wt%. In particular, conditions for the preparation of pure phases were established, even for jellyfish-like objects, an intermediate stage of the nanofiber-to-vesicle morphological transformation. PISA with higher-molar-mass PEtOx-CTAs, therefore, gave better results in terms of colloidal stability and morphology control. There is, however, no clear influence of the degree of livingness of the macromolecular CTAs on the morphological control.
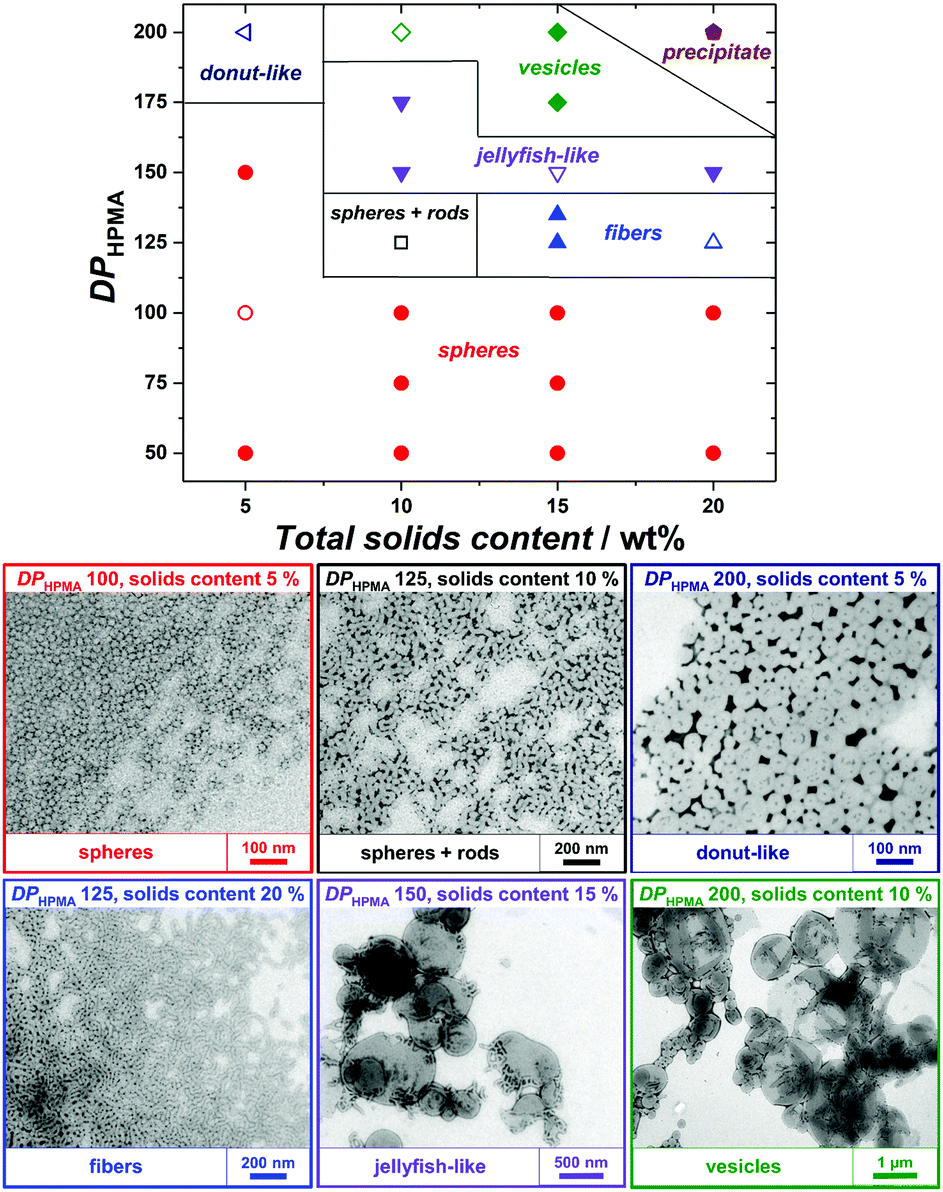 | ||
| Fig. 1 (top) Phase diagram for RAFTPISA of HPMA using PEtOx-CTA M4 as macroRAFT agent and (bottom) TEM images of representative samples (empty symbols from above). Larger TEM images are provided as Fig. S25 (ESI†). | ||
In analogy to systems reported by Armes and co-workers,36 phase transition of the final dispersions could be triggered by changing temperature. At high concentration, the vesicle-to-nanofiber-to-sphere transitions were reversible, irrespective of the initial state, e.g., nanofibers and vesicles for M4-b-PHPMA125 and M4-b-PHPMA175 at 25 °C, respectively (Fig. 2A). Such order–order transitions in a narrow temperature range are due to not only the weakly hydrophobic nature of the core-forming PHPMA block37 but also the temperature-dependent solubility of PEtOx. However, at low concentration, e.g., 0.1 wt%, the transitions became irreversible (Fig. 2B) because the fusion of spheres or fibers is less likely under these conditions. For instance, vesicles of M4-b-PHPMA125 obtained by heating a concentrated solution at 40 °C and carefully diluted at the same temperature irreversibly led to nanofibers, then spheres by gradual cooling.
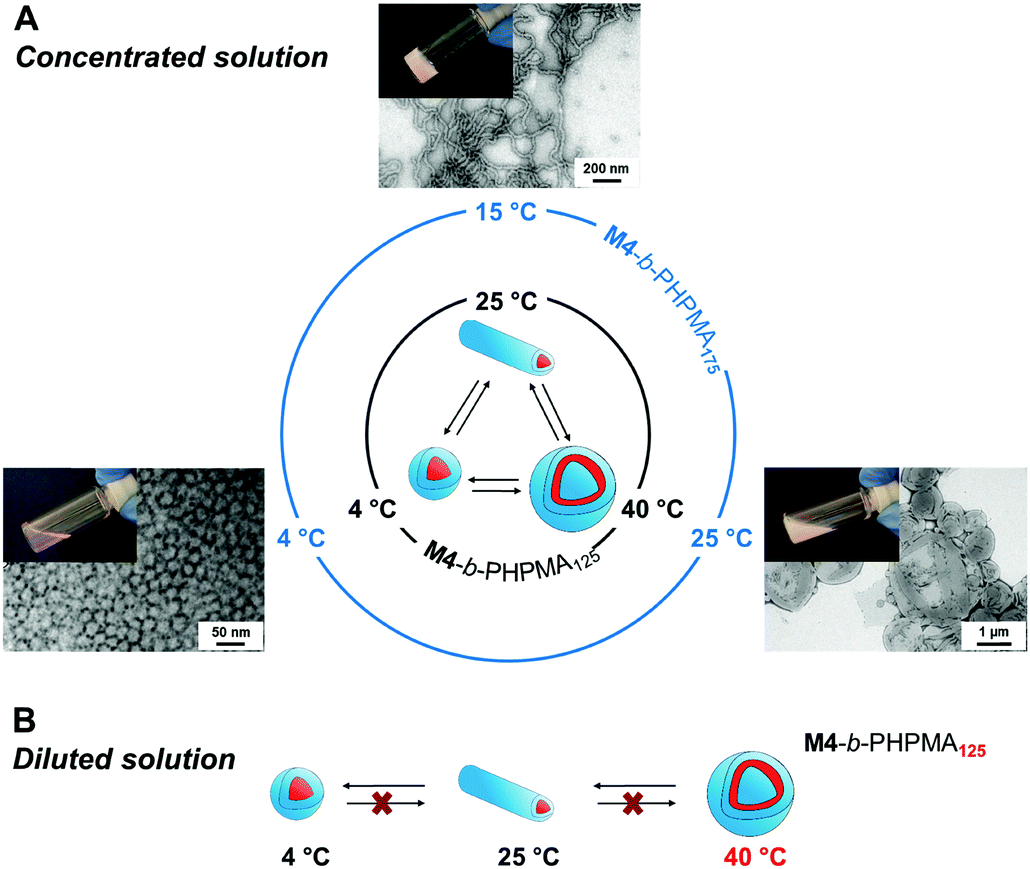 | ||
| Fig. 2 Temperature-induced morphological transformation of (A) original nanofibers (M4-b-PHPMA125, 20 wt% at 25 °C) and vesicles (M4-b-PHPMA175, 15 wt% at 25 °C) in as-synthesized dispersions, and (B) vesicles (M4-b-PHPMA125, 0.1 wt% at 40 °C) at low concentration. Representative solutions and TEM images in (A) from the original nanofiber sample. | ||
Since PEtOx is considered as an attractive alternative to PEG for in vivo applications, we sought to preliminarily assess the biocompatibility and blood circulation behavior of the present PEtOx-coated NPs. For imaging purposes, fluorescent NPs were thus produced by simply adding a small amount of fluorescein methacrylate (FMA) or rhodamine 4-vinylbenzyl ester (RVB) comonomer to a PISA recipe (Scheme S2, ESI†). Spherical NPs with intensity-average hydrodynamic diameters of 40 and 60 nm were obtained (Table S7 and Fig. S27, ESI†) and expected maxima absorption and emission for fluorescein and rhodamine B (Fig. S28, ESI†).
To assess the biocompatibility and ability of the FMA-labeled PEtOx NPs to evade clearance by the reticuloendothelial system (RES), murine macrophages (RAW 264.7), human umbilical vein endothelial cells (HUVEC), and human epithelial lung cancer cells (A549) were incubated for 24 h, using fluorescein-labeled carboxylated polystyrene (PS-COOH) NPs as a reference material.38 The RVB-labeled NPs were administered to zebrafish embryos and their biocompatibility, biodistribution, and clearance were examined for 24 h post-injection (hpi). As controls, PS-COOH NPs as well as rhodamine-labeled dextran were used in parallel. Both in vitro and in vivo studies are reported in more detail in the ESI.†
Briefly, as expected for the NPs covered with stealth polymers, the PEtOx NPs show none-to-very low uptake in all cell types and did not reduce cell viability up to 100 μg mL−1 (Fig. S31–S33, ESI†). In the zebrafish embryos, no noticeable morphological or behavioral changes were detected after intravenous injection of a 1 mg mL−1 dispersion of the RVB-labeled NPs (Fig. 3). In contrast to dextran, which was rapidly cleared and only slightly accumulated in the endothelial cells in the caudal vein (CV) after 24 h (C, F, C′, and F′), the RVB-PEtOx NPs mainly circulated in the bloodstream (B, E and B′, E′). For comparison, the PS-COOH NPs were not detected in the circulation but were, as expected, entrapped in the RES (macrophages and endothelial cells) in the CV (D, and D′). However, contrary to the more simplistic in vitro experiments employing murine macrophages, a small fraction of RVB-PEtOx NPs could be detected in macrophages in vivo (E, and E′). In summary, our PEtOx NPs are biocompatible and display enhanced retention in the blood circulation. Furthermore, to accurately monitor clearance by the RES, the zebrafish model seems to be better suited than the conventional cell culture studies.
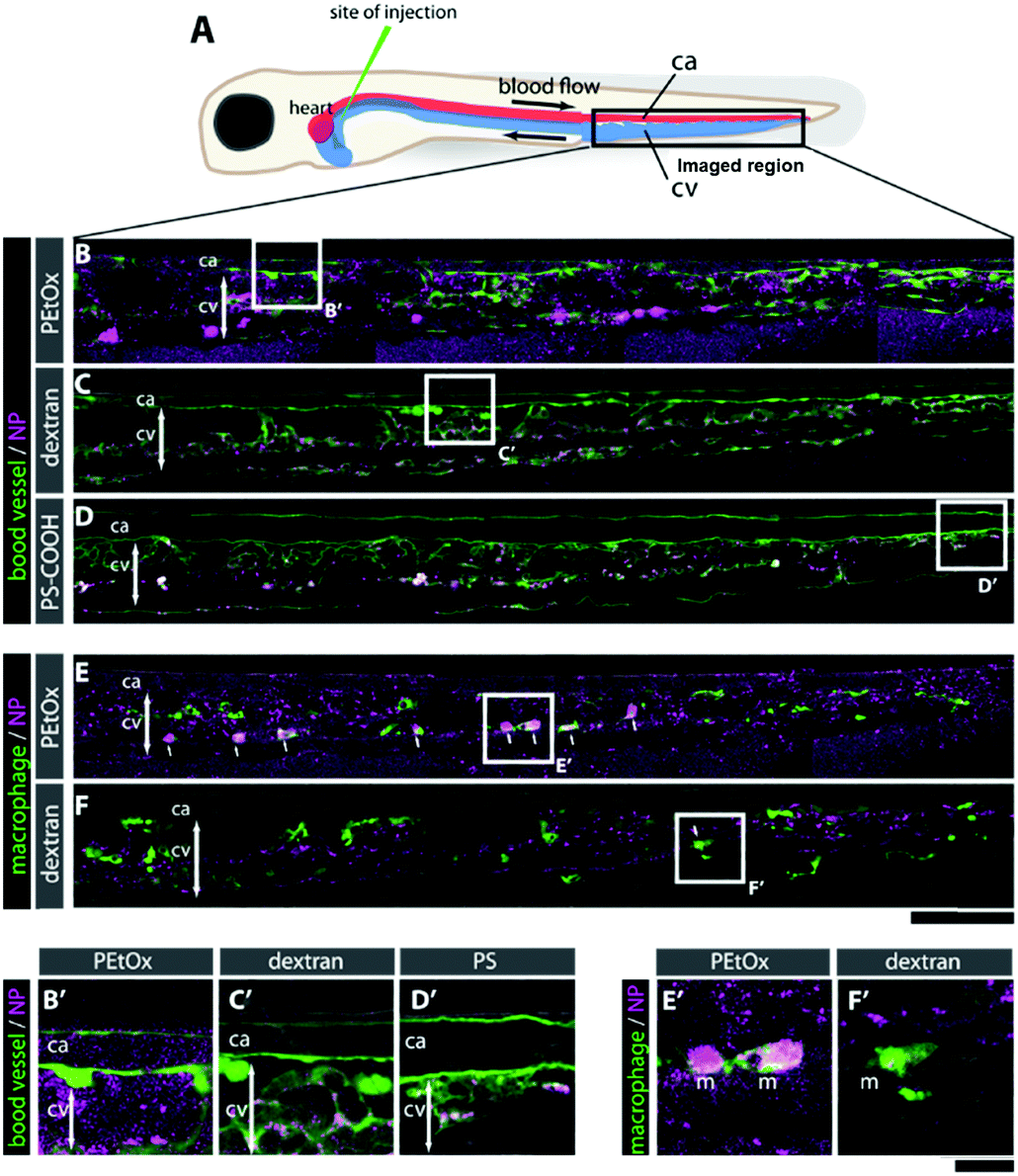 | ||
| Fig. 3 (A) Schematic of a 3 day-post-fertilization zebrafish embryo highlighting the artery (red), vein (blue), and site of NP injection, as well as the imaged region (rectangular region) shown in panels (B–F). The orientation of the embryos is dorsal up, anterior left. (B–D) Caudal region of zebrafish embryos at 24 hpi injected with either PEtOx NPs (magenta, B), rhodamine-conjugated 3 kDa dextran (magenta, C) or PS-COOH NPs (magenta, D). Blood vessels are demarcated with transgenic fluorescence (green). (E and F) The same region of the embryos highlighting the location of macrophages (green, arrows) injected either with PEtOx NPs (E) or dextran (F). ca: caudal artery; cv: caudal vein; m: macrophage; n: notochord. Scale bar: 100 μm (B–F) and 20 μm (B′–F′). | ||
In the present communication, we described for the first time the synthesis of biocompatible PAOx-based diblock copolymer NPs by PISA. Control of morphology was achieved by varying the lengths of each block as well as the reaction mixture concentration. Dispersions based on pure phases of spheres, fibers, or vesicles were obtained. The nanoobjects display a thermosensitive behavior, enabling morphological transformation, which could be an asset for the preparation of bioactive materials. Importantly, the present PEtOx-based nanoparticles are non-toxic and exhibit prolonged circulation in the bloodstream. The versatility of PISA combined with the high potential of polyoxazolines in drug delivery and bioscaffolding promises rapid access to corresponding functional (nano)materials.
D. L. thanks the Alexander von Humboldt Foundation for his postdoctoral fellowship. G. G. A. thanks the Mexican National Council for Science and Technology (CONACyT). G. D. thanks the German Federal Ministry of Education and Research (BMBF, Grant No. 031A095C) for funding (Molecular Interaction Engineering program, Biotechnologie 2020+). The Macroarc group (ITCP, KIT) and Levkin lab (KIT, ITG) are thanked for access to SEC and DLS, respectively. D. Keller and Edgar Molle (ITG, KIT) are acknowledged for the synthesis of RVB and SPTP, respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2016, 88, 451–469 CrossRef.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- R. W. Moreadith, T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, B. P. Rae, X. Li, C. Rader, D. Standaert and W. Olanow, Eur. Polym. J., 2017, 88, 524–552 CrossRef CAS.
- A. M. Kelly and F. Wiesbrock, Macromol. Rapid Commun., 2012, 33, 1632–1647 CrossRef CAS PubMed.
- M. Hartlieb, K. Kempe and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 526–538 RSC.
- T. R. Dargaville, J.-R. Park and R. Hoogenboom, Macromol. Biosci., 2018, 18, 1800070 CrossRef PubMed.
- R. Konradi, B. Pidhatika, A. Mühlebach and M. Textor, Langmuir, 2008, 24, 613–616 CrossRef CAS PubMed.
- M. Schmidt, L. K. Bast, F. Lanfer, L. Richter, E. Hennes, R. Seymen, C. Krumm and J. C. Tiller, Bioconjugate Chem., 2017, 28, 2440–2451 CrossRef CAS PubMed.
- G. Gil Alvaradejo, M. Glassner, R. Hoogenboom and G. Delaittre, RSC Adv., 2018, 8, 9471–9479 RSC.
- P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS.
- G. Gil Alvaradejo, H. V.-T. Nguyen, P. Harvey, N. M. Gallagher, D. Le, M. F. Ottaviani, A. Jasanoff, G. Delaittre and J. A. Johnson, submitted.
- C. Zhang, R. J. P. Sanchez, C. Fu, R. Clayden-Zabik, H. Peng, K. Kempe and A. K. Whittaker, Biomacromolecules, 2018, 20, 365–374 CrossRef PubMed.
- N. A. Hutter, M. Steenackers, A. Reitinger, O. A. Williams, J. A. Garrido and R. Jordan, Soft Matter, 2011, 7, 4861–4867 RSC.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Chem. Commun., 2005, 614 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2018, 1800551 Search PubMed.
- J. Lesagedelahaye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D’Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- J. Liu, Y. Liao, X. He, J. Yu, L. Ding and M. Xie, Macromol. Chem. Phys., 2011, 212, 55–63 CrossRef CAS.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 CrossRef CAS.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O’Reilly, Angew. Chem., Int. Ed., 2018, 10672–10676 CrossRef CAS PubMed.
- O. L. Torres-Rocha, X. Wu, C. Zhu, C. M. Crudden and M. F. Cunningham, Macromol. Rapid Commun., 2019, 40, 1800326 CrossRef PubMed.
- D. Le, M. Dilger, V. Pertici, S. Diabaté, D. Gigmes, C. Weiss and G. Delaittre, Angew. Chem., Int. Ed. DOI:10.1002/anie.201813434.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- A. Krieg, C. Weber, R. Hoogenboom, C. R. Becer and U. S. Schubert, ACS Macro Lett., 2012, 1, 776–779 CrossRef CAS.
- A.-K. Trützschler, M. N. Leiske, M. Strumpf, J. C. Brendel and U. S. Schubert, Macromol. Rapid Commun., 2019, 40, 1800398 CrossRef PubMed.
- H. Willcock and R. K. O’Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- D. Zhou, R. P. Kuchel and P. B. Zetterlund, Polym. Chem., 2017, 8, 4177–4181 RSC.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- N. J. Warren, J. Rosselgong, J. Madsen and S. P. Armes, Biomacromolecules, 2015, 16, 2514–2521 CrossRef CAS PubMed.
- S. R. Mane, I. L. Hsiao, M. Takamiya, D. Le, U. Straehle, C. Barner-Kowollik, C. Weiss and G. Delaittre, Small, 2018, 14, 1801571 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, characterization methods, experimental procedures, additional data, and in-depth discussion of biological data (PDF). See DOI: 10.1039/c9cc00407f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |