
Phase transfer-driven rapid and complete ligand exchange for molecular assembly of phospholipid bilayers on aqueous gold nanocrystals†
Youngjae
Lee‡
a,
Jungwoo
Jang‡
a,
Jinho
Yoon
a,
Jeong-Woo
Choi
a,
Inhee
Choi
 b and
Taewook
Kang
*a
b and
Taewook
Kang
*a
aDepartment of Chemical and Biomolecular Engineering, Sogang University, Seoul 04107, Korea. E-mail: twkang@sogang.ac.kr
bDepartment of Life Science, University of Seoul, Seoul 02504, Korea
First published on 30th January 2019
Abstract
A phase transfer-mediated ligand exchange method is developed for highly selective and rapid synthesis of colloidal phospholipid bilayer-coated gold nanocrystals. The complete replacement of strongly bound surface ligands such as cetyltrimethylammonium bromide (CTAB) and citrate by phospholipid bilayer can be quickly achieved by water–chloroform phase transfer.
Tailoring the surface of inorganic nanoparticles with biologically relevant molecules is considered as an essential step toward in vivo applications because pre-existing ligands on the nanoparticles have been found to be toxic.1 Phospholipids, which are a constituent of cell membranes, are one of the ideal molecules for this purpose since, in addition to biocompatibility, they can impart colloidal stability to inorganic nanoparticles in physiological solutions.2 Unfortunately, because of their low water solubility, molecular phospholipids are introduced to form a surface layer on iron oxide nanoparticles and quantum dots only in organic solvents.3 In aqueous solutions, instead of the molecular phospholipids, they tend to form large and well-organized structures such as bilayer films and liposomes by themselves, which generally hinders their binding to the surface of colloidal nanoparticles.4 Moreover, the surface of the colloidal nanoparticles is densely covered by stabilizing ligands such as cetyltrimethylammonium bromide (CTAB) and citrate ions, which also makes tailoring the surface with phospholipids more challenging.5
Hydration of a thin lipid film is the most common method by which colloidal nanoparticles happen to be encapsulated within liposomes.6 Alternatively, colloidal metal and upconversion nanoparticles can be coated with phospholipid bilayers by mechanical disruption of liposomes.7 Upon the mechanical disruption, the nanoparticles and the liposomes thermodynamically favor core–shell structures to be formed together.8 However, these methods suffer from low yield and poor selectivity.9 More importantly, stabilizing ligands which can be cytotoxic remain on the surface, rendering the nanoparticles less biocompatible.10 Solvent extraction can be used to replace CTAB with a phosphatidylcholine layer.11 However, this method is very time-consuming (∼days) since multiple extraction processes need to be employed to ensure the removal of CTAB.5b,12 On the other hand, gold nanoparticles can be coated with lipid bilayers by using direct reduction of the gold precursor in water–chloroform emulsion and subsequent self-assembly of a lipid layer.13 In addition, metal nanoparticles can be encapsulated by liposomes by utilizing the diffusion of metal ions and subsequent reduction to the nanoparticles inside the liposomes.14 However, the yield of these methods is very low. Therefore, the ligand exchange method that enables the rapid production of colloidal nanoparticles with an outer phospholipid bilayer with high yield and selectivity without toxic stabilizing ligands is urgently needed for practical in vivo applications.
Here, we propose a highly selective and rapid surface tailoring for various gold nanoparticles with a phospholipid bilayer via phase transfer-mediated ligand exchange. The step-by-step procedure for our method is schematically outlined in Fig. 1a. Colloidal gold nanorods (GNRs) are chosen since their surface modification is considered to be difficult due to the presence of a strongly bound CTAB bilayer.15 In a typical procedure, a small amount of thiolated polyethylene glycol (PEG-SH) is added to an aqueous GNR solution to partially replace CTAB. Chloroform in which thiolated lipid molecule 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (DPPTE) is dissolved beforehand is poured into the aqueous solution to form two phases. Note that the thiolated lipid molecule is chosen to quickly replace CTAB.7a,16 Once the two phases form, the surface PEG-SH molecule would help the GNRs move to the chloroform phase because of their amphiphilic nature.17 PEG-SH would also prevent aggregation of the GNRs because of a steric effect.18 Then, DPPTE is expected to completely replace CTAB mainly due to the elevated critical micelle concentration of CTAB in chloroform and stronger Au–S bonding (∼44 kcal mol−1).19 After the chloroform phase is separated, the DPPTE modified GNRs are re-dispersed in water with another lipid molecule, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), is dissolved. Finally, a lipid bilayer is expected to be spontaneously formed at the surface of the GNRs because of hydrophobic interaction between DPPTE and DSPC (Fig. 1b).
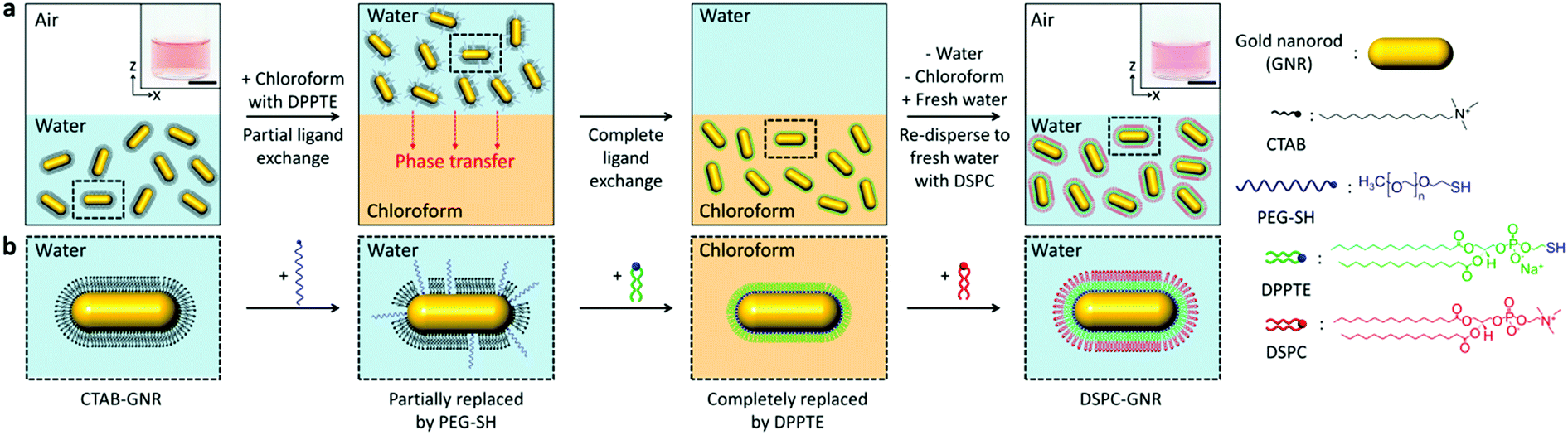 | ||
| Fig. 1 Phase transfer-mediated selective ligand exchange of various gold nanoparticles with a phospholipid bilayer. (a) Schematic illustration of the step-by-step experimental procedures of our method with gold nanorods (GNRs). Scale bars, 1 cm. (b) Representative surface tailoring of GNRs that corresponds to each experimental step. | ||
Our method is systematically characterized to ensure that GNRs with outer lipid bilayers are successfully produced, free of CTAB. Fig. 2a shows that the aqueous phase is initially colored as red due to surface plasmon resonance (SPR) scattering of GNRs and the chloroform phase is transparent. These colors remain unchanged over time. After the addition of a small amount of PEG-SH to the aqueous phase, the mixture is vortexed for 2 min. Interestingly, the color of the chloroform phase rapidly changes to reddish brown as the aqueous phase turns transparent. UV-vis measurement also shows that a new SPR band appears in the chloroform with a concomitant decrease in the SPR band in the aqueous phase (Fig. S1, ESI†). Note that the reddish brown color is because the SPR band is red-shifted in chloroform. These observations indicate that with the aid of PEG-SH, GNRs are transferred to the chloroform. Without the addition of PEG-SH, the GNRs remain in the aqueous phase. In order to avoid both GNR aggregation and incomplete ligand exchange, it is important to optimize the concentration of PEG-SH. At a lower concentration, after the phase transfer, the color of the chloroform gradually turns from reddish brown to violet and the SPR band in the chloroform is significantly decreased due to GNR aggregation (Fig. S2, ESI†). As the concentration of PEG-SH is increased, the color and SPR band in the chloroform remain unchanged. The SPR band of GNRs in the chloroform is more red-shifted than that without DPPTE, and its intensity increases over time (Fig. S3, ESI†). This indirectly suggests that the DPPTE molecule is introduced to most of the surface of the GNRs. It is well known that large thiolated molecules like PEG-SH form a less dense layer on the surface of GNRs and smaller thiolated molecules such as 11-mercaptoundecanoic acid which are similar to DPPTE in size can exchange or backfill PEG-SH.20
 | ||
| Fig. 2 Step-by-step characterization for complete ligand exchange with a phospholipid bilayer on the surface of GNRs. (a) Schematic illustration of surface modification of the GNRs during the first phase transfer and corresponding photographs. Scale bar, 1 cm. (b) Raman spectra of the GNRs (i.e., CTAB-GNR, black line) after PEG-SH modification (red line) and DPPTE-coated GNRs in chloroform (blue line). (c) Representative absorbance spectra of CTAB-GNRs and phospholipid bilayer-coated GNRs (e.g., DSPC-GNR). (d) Representative transmission electron microscopy (TEM) images of CTAB-GNRs and DSPC-GNRs. Inset scale bar, 20 nm. (e) Distribution of interparticle distance of CTAB-GNRs and DSPC-GNRs. (f) Measured zeta-potentials of CTAB-GNRs, DOPG-GNRs and DSPC-GNRs, respectively. | ||
To further elaborate the surface ligand exchange of the GNRs, surface-enhanced Raman spectroscopy (SERS) is performed. After the surface modification with PEG-SH, Raman transition of Au–Br bonding at 176 cm−1 is decreased (Fig. 2b, left).21 Three characteristic peaks for CTAB at 760, 958, and 1442 cm−1 are also decreased.22 Instead, two SERS peaks at 865 and 1468 cm−1, which are originated from PEG-SH, are newly observed (Fig. 2b, right).23 This suggests that the surface of the GNRs is partially modified with PEG-SH. After the phase transfer of the GNRs to the chloroform, a Raman transition of Au–S bonding newly appears at 254 cm−1.21 Simultaneously, the Raman transitions that include Au–Br bonding at 176 cm−1 and an additional three characteristic peaks for CTAB disappeared. Moreover, two SERS peaks for PEG-SH are not observed at 865 and 1468 cm−1. This is directly indicative of the complete removal of CTAB by DPPTE.
After the separation of the chloroform, the DPPTE-coated GNRs are collected from the chloroform by centrifugation and re-dispersed in a small amount of acetone. During this solvent exchange, the SPR band slightly shifts because of the difference in the dielectric constants of the solvents and no GNR aggregation occurs (Fig. S4, ESI†).17 The acetone solution is then added dropwise to fresh water where another lipid molecule (e.g., DSPC or 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG)) is dissolved with the aid of ethanol. After the evaporation of acetone and ethanol by stirring for 2 h, the resulting GNR solution is washed twice with deionized water by centrifugation. Compared to the GNRs (CTAB-GNRs), the SPR band of the phospholipid bilayer-coated GNRs (DSPC-GNRs) is red-shifted. This also indicates the ligand exchange (Fig. 2c).
Transmission electron microscopy (TEM) images are taken before and after the ligand exchange. Representative TEM images show that the shape and size of the GNRs are unchanged during the ligand exchange (Fig. 2d). The interparticle distance (side to side) appears to be slightly increased (magnified images in Fig. 2d and e) because of the longer phospholipid molecule. The difference in theoretical side-to-side interparticle distances is about 2.8 nm.24
To further ensure the self-assembly of the second lipid molecule (i.e., DSPC) with the DPPTE layer of the GNRs, zeta potential measurement is carried out with lipid molecules having opposite charges. DOPG is selected because it carries a negative charge on its head group.25 When the second lipid molecule is changed from DSPC to DOPG, the measured zeta-potential of the resulting particle is also changed from positive to negative (Fig. 2f). This result strongly supports the formation of the lipid bilayer on the surface of the GNRs via self-assembly.
To prove the versatility of our method, gold nanospheres (GNSs) with diameters of 20 and 50 nm (Fig. S5, ESI†) are also tested. Note that GNSs are stabilized by citrate ions (citrate-GNSs). The increased interparticle distance between neighboring GNSs can be attributed to the ligand exchange from the citrate ions to the lipid bilayer (Fig. 3a). Magnified TEM images in Fig. 3a clearly show the outer lipid layer on the surface of the GNSs. Upon drying, the citrate-GNSs are prone to aggregate; however, the lipid bilayer-coated GNSs are well dispersed individually. This can be attributed to the size difference between the citrate and the lipid bilayer. The SPR bands of the GNSs are commonly red-shifted after the ligand exchange (Fig. 3b). The zeta potential measurement in Fig. 3c shows that the surface charge of the GNSs changes from negative to neutral or positive after the ligand exchange. These results consistently support that the surface citrate ligand is successfully replaced by the phospholipid bilayer using our method.
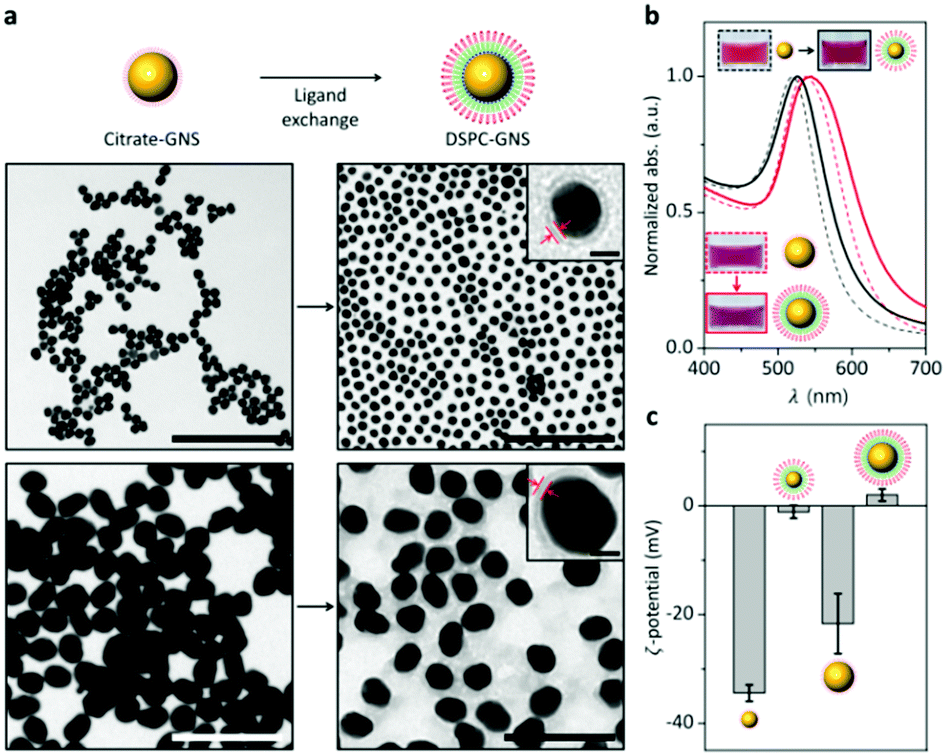 | ||
| Fig. 3 Complete ligand exchange for gold nanospheres (GNSs) from citrate ions to phospholipid bilayer. (a) TEM images of 20 nm- (upper) and 50 nm-GNSs (lower) before and after the ligand exchange, respectively. Scale bars correspond to 200 nm and 10 nm (inset). (b) Representative absorbance spectra of 20 nm- and 50 nm-GNSs before and after the ligand exchange, respectively. (c) Zeta-potentials of 20 nm- and 50 nm-GNSs before and after the ligand exchange, respectively. | ||
To explore the benefit of our method, the colloidal stability and cytotoxicity are investigated. When citrate-GNSs with different sizes are exposed to biologically relevant solutions such as phosphate buffered saline (PBS) and sodium chloride (NaCl) solutions, the colors are immediately changed to dark blue due to aggregation, which can be attributed to weakened electrostatic repulsion in the electrolyte solutions. In contrast, the lipid bilayer-coated GNSs retain their colors upon exposure to PBS and NaCl solutions for 1 h, as shown in Fig. 4a. The same tendency is also observed for the GNRs. The CTAB-GNRs significantly aggregated in PBS and NaCl solutions, whereas the DSPC-GNRs remain well-dispersed (Fig. 4b). In order to examine the cytotoxicity, CTAB-GNRs and DSPC-GNRs are separately treated with Homo sapiens bone marrow neuroblastoma SH-SY5Y cells which have been widely used as a human neuronal cell model and gold nanoparticles have been exploited to treat these cells for imaging and therapy (Fig. 4c).26 The viabilities of the cells treated with CTAB-GNRs and DSPC-GNRs (ca. 5 × 1011 particles per ml) are examined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. As shown in Fig. 4c, more than 96% of the cells treated with DSPC-GNRs remain viable after 24 h. However, the cell viability dramatically decreases to about 60% for CTAB-GNRs.
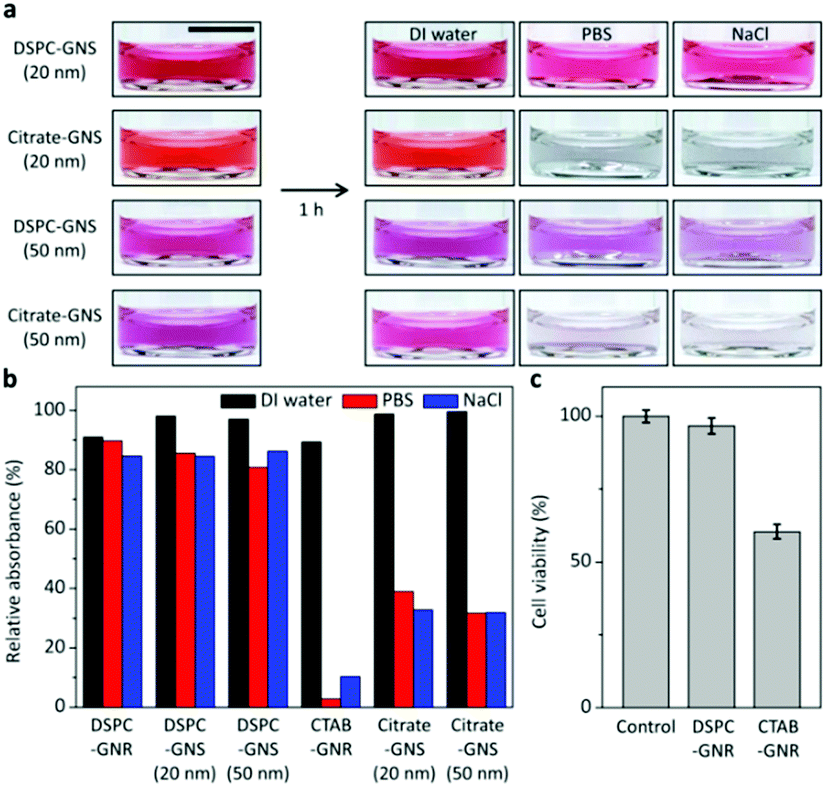 | ||
| Fig. 4 Enhanced colloidal stability and cell viability of phospholipid-coated gold nanoparticles. (a) Photographs for each gold nanoparticle in DI water, PBS and NaCl, respectively. Scale bar, 1 cm. (b) Relative absorbance of GNR and GNS solution (GNR after 1 day, GNS after 1 hour). (c) Cell viability test using the MTT assay. | ||
We have demonstrated a selective and facile method of phase transfer-mediated rapid and complete ligand exchange for various gold nanoparticles. Our method utilizes phase transfer in order to overcome low water solubility of a lipid molecule as well as completely remove the surface-bound ligand in a few minutes. The phase transfer is mediated by PEG-SH. DPPTE and DSPC are sequentially introduced to the surface of the gold nanoparticles in chloroform and water phases, respectively. We show that phospholipid bilayer-coated gold nanorods and nanospheres with high yield are produced free of CTAB and citrate ions by using our method. Better stability in PBS and NaCl solutions is commonly observed for the lipid bilayer-coated particles and higher cell viability is confirmed, based on the MTT assay. We believe that our method greatly improves the practical utility of gold nanoparticles in many in vivo applications ranging from biological imaging and molecular detection to drug delivery.27
This work was supported by the Mid-Career Researcher Support Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (MSIP) (no. 2016R1A2B3014157), by the Basic Science Research Program through the NRF funded by the Ministry of Education (no. 2016R1A6A1A03012845) to T. K., and by the Basic Science Research Program through the NRF funded by the MSIP (no. 2017R1A2B4003267) to I. C. The authors are thankful to Professor Dan Huh for helpful discussion.
Conflicts of interest
There are no conflicts to declare.References
- (a) E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS PubMed; (b) M. Sheikhpour, L. Barani and A. Kasaeian, J. Controlled Release, 2017, 253, 97–109 CrossRef CAS PubMed.
- (a) E. Alipour, D. Halverson, S. McWhirter and G. C. Walker, Annu. Rev. Phys. Chem., 2017, 68, 261–283 CrossRef CAS PubMed; (b) P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- (a) S. Tong, S. Hou, B. Ren, Z. Zheng and G. Bao, Nano Lett., 2011, 11, 3720–3726 CrossRef CAS PubMed; (b) Y. Duan, Y. Xu, D. Mao, W. H. Liew, B. Guo, S. Wang, X. Cai, N. Thakor, K. Yao, C. J. Zhang and B. Liu, Small, 2018, 14, 1800652 CrossRef PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- (a) M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141 CrossRef CAS PubMed; (b) S. Zhou, D. Huo, S. Goines, T. H. Yang, Z. Lyu, M. Zhao and Y. Xia, J. Am. Chem. Soc., 2018, 140, 11898–11901 CrossRef CAS PubMed.
- (a) X. Su, Y. Wang, W. Wang, K. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 10201–10211 CrossRef CAS PubMed; (b) A. Y. Rwei, B. Y. Wang, T. Ji, C. Zhan and D. S. Kohane, Nano Lett., 2017, 17, 7138–7145 CrossRef CAS PubMed.
- (a) E. T. Castellana, R. C. Gamez and D. H. Russell, J. Am. Chem. Soc., 2011, 133, 4182–4185 CrossRef CAS PubMed; (b) L. Rao, L. L. Bu, B. Cai, J. H. Xu, A. Li, W. F. Zhang, Z. J. Sun, S. S. Guo, W. Liu, T. H. Wang and X. Z. Zhao, Adv. Mater., 2016, 28, 3460 CrossRef CAS PubMed; (c) R. Mei, Y. Wang, W. Liu and L. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 23605–23616 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 1706759 CrossRef PubMed.
- T. Guan, W. Shang, H. Li, X. Yang, C. Fang, J. Tian and K. Wang, Bioconjugate Chem., 2017, 28, 1221–1228 CrossRef CAS PubMed.
- L. Su, S. Hu, L. Zhang, Z. Wang, W. Gao, J. Yuan and M. Liu, Small, 2017, 13, 1602809 CrossRef PubMed.
- H. Takahashi, Y. Niidome, T. Niidome, K. Kaneko, H. Kawasaki and S. Yamada, Langmuir, 2006, 22, 2–5 CrossRef CAS PubMed.
- M. R. Dewi, G. Laufersky and T. Nann, RSC Adv., 2014, 4, 34217–34220 RSC.
- M. R. Mackiewicz, B. R. Ayres and S. M. Reed, Nanotechnology, 2008, 19, 115607 CrossRef PubMed.
- J. H. Lee, Y. Shin, W. Lee, K. Whang, D. Kim, L. P. Lee, J. Choi and T. Kang, Sci. Adv., 2016, 2, e1601838 CrossRef PubMed.
- C. Kinnear, H. Dietsch, M. J. Clift, C. Endes, B. Rothen-Rutishauser and A. Petri-Fink, Angew. Chem., Int. Ed., 2013, 52, 1934–1938 CrossRef CAS PubMed.
- M. Wan, X. Li, L. Gao and W. Fang, Nanotechnology, 2016, 27, 465704 CrossRef PubMed.
- A. B. Serrano-Montes, D. Jimenez de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzán, Langmuir, 2015, 31, 9205–9213 CrossRef CAS PubMed.
- M. G. Soliman, B. Pelaz, W. J. Parak and P. Del Pino, Chem. Mater., 2015, 27, 990–997 CrossRef CAS.
- (a) A. S. D. Indrasekara, R. C. Wadams and L. Fabris, Part. Part. Syst. Charact., 2014, 31, 819–838 CrossRef CAS; (b) B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS.
- A. M. Smith, L. E. Marbella, K. A. Johnston, M. J. Hartmann, S. E. Crawford, L. M. Kozycz, D. S. Seferos and J. E. Millstone, Anal. Chem., 2015, 87, 2771–2778 CrossRef CAS PubMed.
- Z. Zhang and M. Lin, RSC Adv., 2014, 4, 17760–17767 RSC.
- J. R. Matthews, C. M. Payne and J. H. Hafner, Langmuir, 2015, 31, 9893–9900 CrossRef CAS PubMed.
- A. Z. Samuel and S. Umapathy, Polym. J., 2014, 46, 330–336 CrossRef CAS.
- (a) T. K. Sau and C. J. Murphy, Langmuir, 2005, 21, 2923–2929 CrossRef CAS PubMed; (b) E. Amstad, J. Kohlbrecher, E. Müller, T. Schweizer, M. Textor and E. Reimhult, Nano Lett., 2011, 11, 1664–1670 CrossRef CAS PubMed.
- F. Versluis, D. M. van Elsland, S. Mytnyk, D. L. Perrier, F. Trausel, J. M. Poolman, C. Maity, V. A. A. le Sage, S. I. Kasteren, J. H. van Esch and R. Eelkema, J. Am. Chem. Soc., 2016, 138, 8670–8673 CrossRef CAS PubMed.
- (a) H. Xicoy, B. Wieringa and G. J. Martens, Mol. Neurodegener., 2017, 12, 10 CrossRef PubMed; (b) M. Mohammadniaei, T. Lee, B. G. Bharate, J. Yoon, H. K. Choi, S. J. Park, J. Kim, J. Kim and J. W. Choi, Small, 2018, 14, 1802934 CrossRef PubMed.
- (a) M. Moros, S. G. Mitchell and V. Grazu, Curr. Med. Chem., 2013, 20, 2759–2778 CrossRef CAS PubMed; (b) L. A. Lane, X. Qian, A. M. Smith and S. Nie, Annu. Rev. Phys. Chem., 2015, 66, 521–547 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc10037c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |