
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic hydrogenation of α,β-unsaturated carboxylic acid derivatives using copper(I)/N-heterocyclic carbene complexes†
Birte M.
Zimmermann
 ,
Sarah C. K.
Kobosil
and
Johannes F.
Teichert
*
,
Sarah C. K.
Kobosil
and
Johannes F.
Teichert
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: johannes.teichert@chem.tu-berlin.de
First published on 4th February 2019
Abstract
A simple and air-stable copper(I)/N-heterocyclic carbene complex enables the catalytic hydrogenation of enoates and enamides, hitherto unreactive substrates employing homogeneous copper catalysis and H2 as a terminal reducing agent. This atom economic transformation replaces commonly employed hydrosilanes and can also be carried out in an asymmetric fashion.
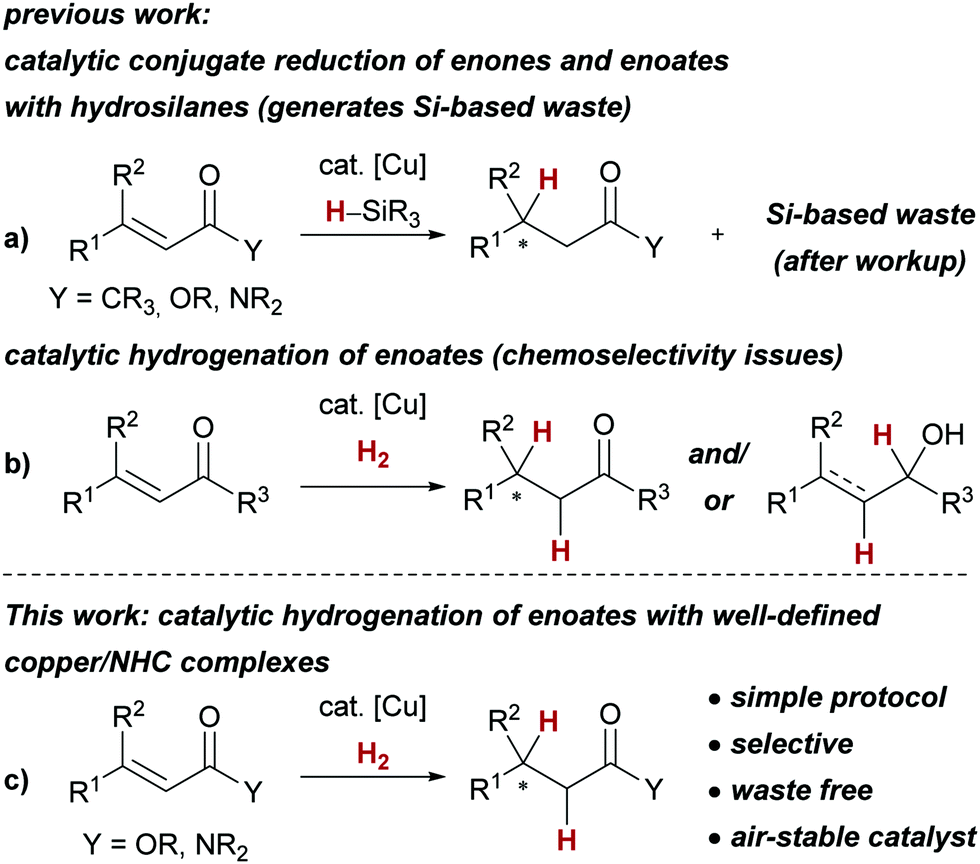
One of the main challenges of contemporary method development for synthetic chemistry is the development of atom economic and sustainable transformations.1,2 In this vein, catalytic hydrogenations are much desired reactions, as they serve to replace complex and waste-generating reducing agents such as borohydrides, aluminium hydrides or hydrosilanes.3,4 Catalytic reactions involving copper hydride intermediates5 serve as a prime example for this challenge: commonly, hydrosilanes are used as stoichiometric reducing agents, whereas the use of dihydrogen (H2) for the – considerably more atom economic – generation of the desired copper hydride complexes has been much less explored.6,7 The hallmark reaction of the so-called copper hydride catalysis5 is arguably the conjugate reduction of α,β-unsaturated carbonyl or carboxyl compounds employing hydrosilanes (Scheme 1a). Even though the replacement of hydrosilanes with H2 has been attempted for the reduction of enones, the chemoselectivity of the 1,2- vs. the 1,4-reduction turned out to be challenging to control (Scheme 1b).6 Furthermore, the corresponding enoates emerged as too unreactive in combination with H2.8,9 Enoates are common substrates for the catalytic hydrogenation with precious metals such as rhodium and ruthenium,3,10 and replacement with base metal catalysts such as copper complexes is highly desirable.
 | ||
| Scheme 1 Challenges in atom-economic copper-catalysed conjugate reductions. | ||
We herein report the first copper-catalysed conjugate reduction of previously unreactive α,β-unsaturated esters and amides with H2 employing well-defined and air-stable copper(I)/N-heterocyclic carbene complexes11 (Scheme 1c). Additionally, this approach circumvents the use of waste-generating hydrosilanes, resulting in a simple and sustainable protocol for the catalytic hydrogenation of α,β-unsaturated esters and amides.
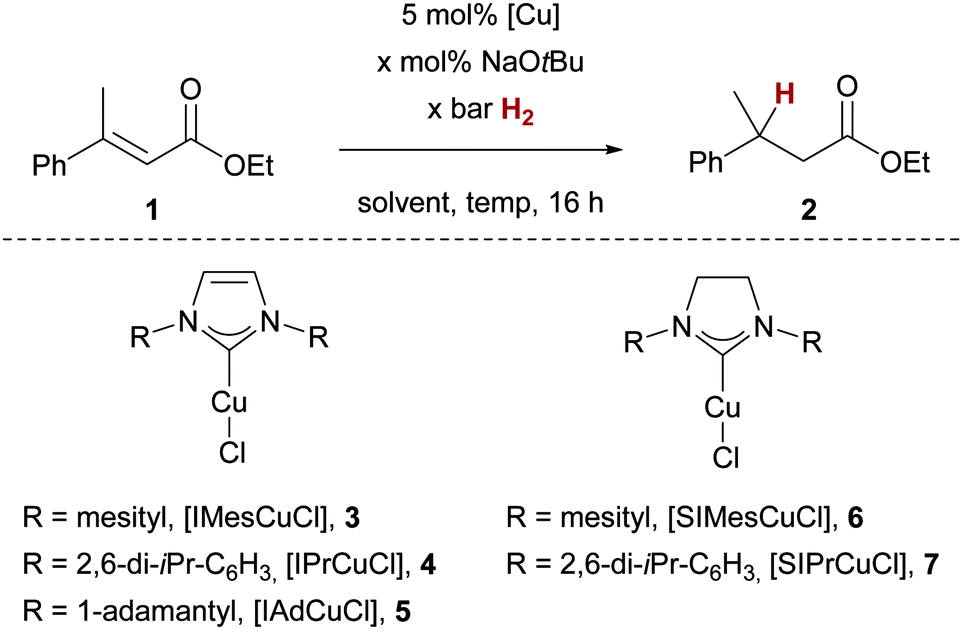
The copper-catalysed hydrogenation of enoates was optimised employing ethyl β-methyl cinnamate (1, Table 1). Using sodium tert-butanolate (NaOtBu) as additive for the generation of the key Cu–O-bond for H2 activation,12 we investigated common copper(I)/NHC complexes 3–7 at 90 bar H2 and 60 °C in THF (Table 1, entries 1–5). From these experiments, mesityl-derived complexes 3 and 6 emerged as most active, as judged by the conversion of 1 to 2. Notably, no other side-products were observed. We found that the amount of NaOtBu could be lowered to 30 mol% while maintaining full conversion, however, even smaller amounts of the additive (10 mol%) led to an almost complete halt of the reaction (Table 1, entries 6 and 7). Lowering the H2 pressure to 50 bar at these limiting conditions led to diminished conversion of 32% (Table 1, entry 8). A subsequent solvent optimisation revealed 1,4-dioxane as optimal,13 with full conversion of 1 reached at lowered H2 pressure of 50 bar (Table 1, entry 9). When comparing catalysts 3 and 6 at 10 bar H2 pressure, imidazolinium-based copper complex 6 turned out to be more active (Table 1, entries 10 vs. 12), even though no reactivity difference could be detected at higher temperature of 100 °C. The investigation of the substrate scope was therefore carried out with catalyst 6. Notably, lowering the H2 pressure with catalyst 6 to 1 bar still led to a detectable conversion of 1 (32% conv. Table 1, entry 14).
|
|
|||
|---|---|---|---|
| Entry | [Cu] | Conditions | Conv.b |
| a Reactions were carried out on a 0.25 mmol scale. b Determined by 1H NMR spectroscopy. c 71% isolated yield. | |||
| 1 | 3 | 110 mol% NaOtBu, 90 bar H2, THF, 60 °C | >95% |
| 2 | 4 | As entry 1 | 67% |
| 3 | 5 | As entry 1 | 17% |
| 4 | 6 | As entry 1 | >95% |
| 5 | 7 | As entry 1 | 41% |
| 6 | 3 | 30 mol% NaOtBu, 90 bar H2, THF, 60 °C | >95% |
| 7 | 3 | 10 mol% NaOtBu, 90 bar H2, THF, 60 °C | 6% |
| 8 | 3 | 30 mol% NaOtBu, 50 bar H2, THF, 60 °C | 32% |
| 9 | 3 | 30 mol% NaOtBu, 50 bar H2, 1,4-dioxane, 60 °C | >95% |
| 10 | 3 | 30 mol% NaOtBu, 10 bar H2, 1,4-dioxane, 60 °C | 5% |
| 11 | 3 | 30 mol% NaOtBu, 10 bar H2, 1,4-dioxane, 100 °C | >95% |
| 12 | 6 | 30 mol% NaOtBu, 10 bar H2, 1,4-dioxane, 60 °C | 79% |
| 13 | 6 | 30 mol% NaOtBu, 10 bar H 2 , 1,4-dioxane, 100 °C | >95% |
| 14 | 6 | 30 mol% NaOtBu, 1 bar H2, 1,4-dioxane, 100 °C | 32% |
With optimised reaction conditions in hand, we set out to investigate the substrate scope of the copper-catalysed conjugate reduction of α,β-unsaturated carboxylic acid derivatives 8 (Scheme 2). We found that the catalytic hydrogenation could be applied to a variety of aryl or alkyl substituted enoates 8. Next to the successful generation of simple naphthyl derivate 9a, also the sterically more demanding tert-butyl ester 9b as well as diphenyl-substituted ester 9c could be furnished in good yields (68–84%). Both electron donating and withdrawing groups were tolerated as substituents of the cinnamic acid derivatives 9d–9h with similar results in terms of yield. Notably, no protodehalogenation was observed with bromide 9f or chloride 9g. As in previous studies,7 the presence of a nitro or a carbonyl group (9i, 9k) led to no or diminished conversion, whereas nitrile derivative 9j led to full conversion. Of note is the fact that protic groups such as a free phenol (9l) were susceptible to the conjugate reduction.14 Also, the silyl-protected variant (9m) could be converted successfully. Dimethylaniline 9n and thiophene-derived 9o as possibly coordinating substrates could be successfully hydrogenated. The generation of cyclopropane-substituted 9p in almost quantitative yield (95%) gives an important indication that no carbon-based radical is involved in the overall process. The clean formation of alkene-substituted ester 9q underscores the chemoselectivity of the present catalyst, as no alkene hydrogenation was observed.15 Next to the successful formation of diester 9r, also 9s, bearing a methyl group in α-position, which generally slows down the conjugate addition in other copper-catalysed processes,16 was turned over by the catalyst. Ester 9s was formed as a 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :41 mixture of diastereomers. Finally, dialkyl-substituted enoates 9t–9v could be converted with similarly good results in terms of yield. We could demonstrate that even enamide 10, which generally is too electron-rich for reactivity in other copper-catalysed conjugate addition reactions,17 displays some reactivity with our catalyst (60% conv., 17% yield for 11). This result underscores the fact that the copper/NHC complexes employed in this transformation serve a key role for the generation of copper hydride intermediates with higher reactivity in comparison to the commonly used phosphine complexes.5 The latter could not realize any conversion of carboxylic acid derivatives.
:41 mixture of diastereomers. Finally, dialkyl-substituted enoates 9t–9v could be converted with similarly good results in terms of yield. We could demonstrate that even enamide 10, which generally is too electron-rich for reactivity in other copper-catalysed conjugate addition reactions,17 displays some reactivity with our catalyst (60% conv., 17% yield for 11). This result underscores the fact that the copper/NHC complexes employed in this transformation serve a key role for the generation of copper hydride intermediates with higher reactivity in comparison to the commonly used phosphine complexes.5 The latter could not realize any conversion of carboxylic acid derivatives.
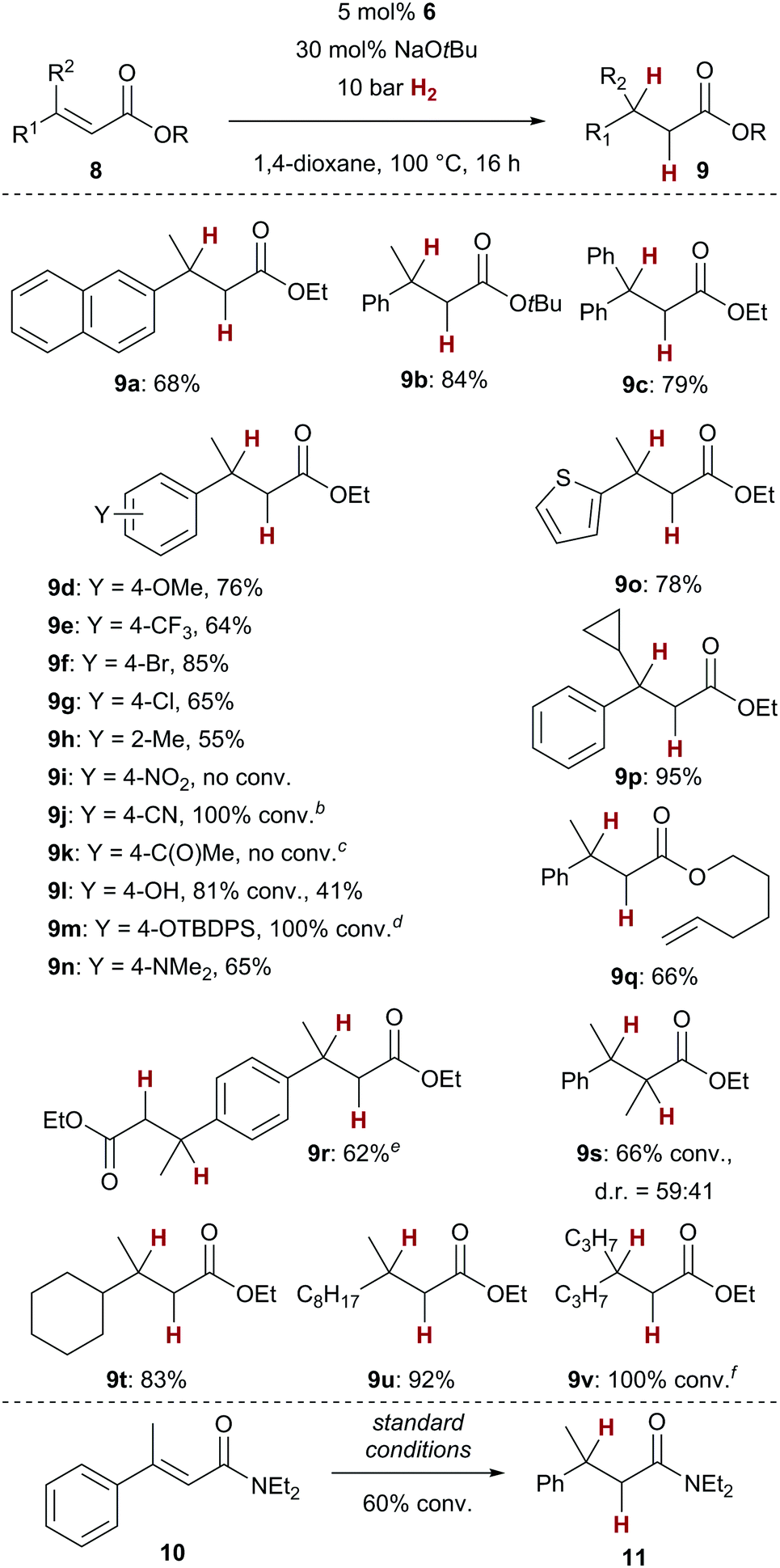 | ||
Scheme 2 Cu-catalysed hydrogenation of enoates and enamides, scopea. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) If not noted otherwise, E-configured enoates 8 have been employed. E/Z ratios: 8f: 90:10, 8i: 88:12, 8k: 87:13, 8m: 93:7, 8u: 77:23. bThe product decomposed during purification, 5% isolated yield. cNo 1,2-reduction of the ketone was observed. dA mixture of the corresponding ethyl and tert-butyl ester were observed (83:17). The separation turned out to be tedious, resulting in lower yields (OEt: 40%, OtBu: 7%). e10 mol% 6, 60 mol% NaOtBu used. fProduct is volatile. If not noted otherwise, E-configured enoates 8 have been employed. E/Z ratios: 8f: 90:10, 8i: 88:12, 8k: 87:13, 8m: 93:7, 8u: 77:23. bThe product decomposed during purification, 5% isolated yield. cNo 1,2-reduction of the ketone was observed. dA mixture of the corresponding ethyl and tert-butyl ester were observed (83:17). The separation turned out to be tedious, resulting in lower yields (OEt: 40%, OtBu: 7%). e10 mol% 6, 60 mol% NaOtBu used. fProduct is volatile. | ||
Sorbic acid derivative 12 gave a mixture of 1,4- and 1,6-addition products, with unsaturated ester 13a (from 1,4-addition of the hydride nucleophile) as the major product (Scheme 3). The formation of 13b and 13c can be explained by prior 1,6-addition of the copper hydride, and, in the case of 13c, subsequent 1,4-addition.
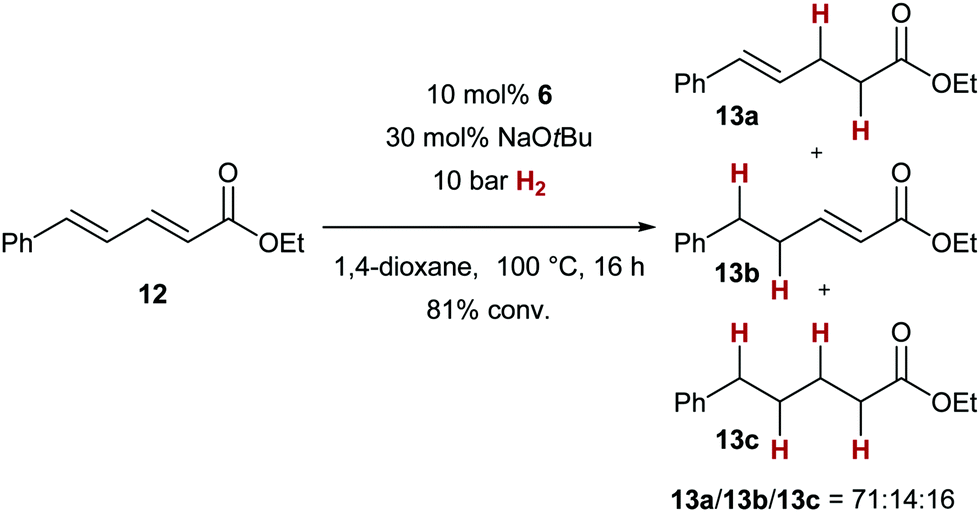 | ||
| Scheme 3 Conjugate reduction of an α,β,γ,δ-unsaturated ester. | ||
To gain some insight into the mechanism of the present protocol, we carried out the conjugate reduction in the presence of deuterium gas (D2, Scheme 4). With ethyl esters 8c and 8d, as expected for a hydride transfer reaction, the deuterium incorporation in the β position was high (≥90% D), but also significant isotope labeling in the α position was found, indicating an enolisation process with t-BuOD after the conjugate addition. Unexpectedly, also deuterium incorporation was observed in the ethyl ester (23–39% D, D1′ and D2′).18
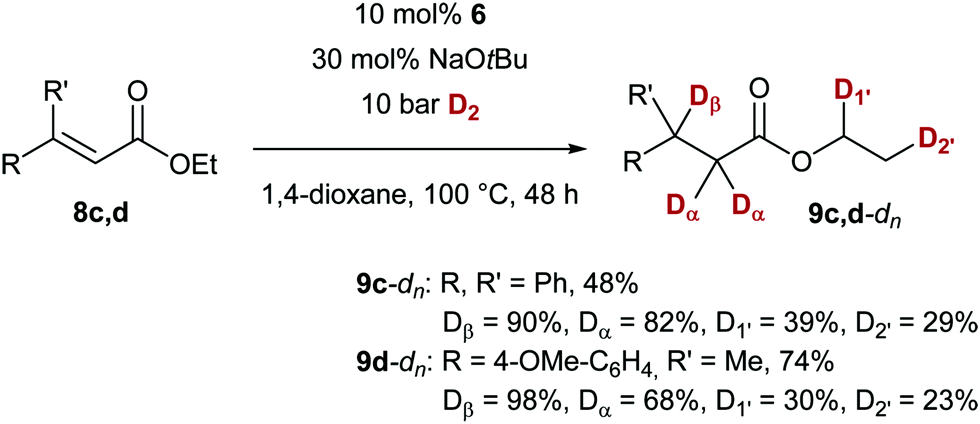 | ||
| Scheme 4 Cu-catalysed conjugate reduction with D2. | ||
As an asymmetric variant of catalytic conjugate reduction with H2 is highly attractive,16 we turned our attention to chiral NHC ligands. In preliminary experiments, we were able to demonstrate that indeed stereoinduction with chiral NHC ligands is possible, even though more drastic reaction conditions (100 bar H2, 48 h reaction time) had to be employed with chiral NHC precursors 1419 and 1520 (Scheme 5). The corresponding reduced ester 2 was obtained with an enantiomeric ratio of up to 82:18 with chiral NHC precursor 14, demonstrating the viability of an asymmetric reaction.
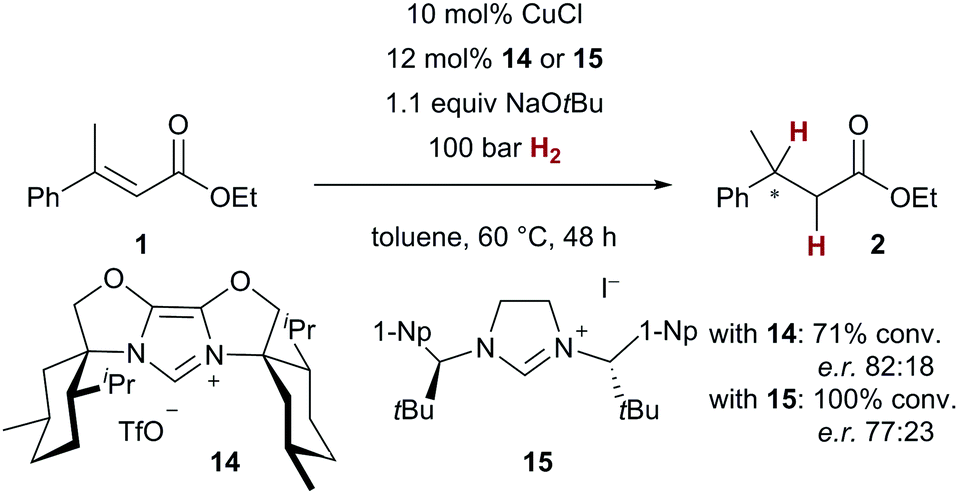 | ||
| Scheme 5 Asymmetric conjugate reduction of cinnamate 1 (1-Np = 1-naphthyl). | ||
In summary, we have developed a simple and efficient copper-catalysed conjugate reduction of enoates and enamides with H2. Well-defined and easily accessible copper(I)/NHC complexes are employed as catalysts, giving the desired products which had so far not been accessible with copper/phosphine complexes due to the lower reactivity of these substrates employing H2. The present catalytic protocol serves as an atom economic alternative to replace the commonly used and waste-generating hydrosilanes as stoichiometric reducing agents with dihydrogen (H2). Isotope labelling studies indicate a polar mechanism involving a copper hydride intermediate. In addition, the viability of an asymmetric reaction has been demonstrated.
This work was supported by the German Research Council (DFG, Emmy Noether Fellowship for J. F. T., TE1101/2-1), by the Fonds der Chemischen Industrie (Liebig-Stipendium for J. F. T.) and by the Daimler and Benz Foundation (postdoctoral fellowship for J. F. T.). Prof. Dr Martin Oestreich (TU Berlin) is kindly thanked for generous support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For a review on green chemistry, see: P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- For a reviews on atom economy and atom efficiency, see: (a) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC; (b) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- The handbook of homogeneous hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
- The use of H2 as reagent has been put forward, see for example: (a) R. Noyorii, Chem. Commun., 2005, 1807–1811 RSC; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- For reviews, see: (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (b) B. H. Lipshutz, in Copper-catalysed asymmetric synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 2014, vol. 110, pp. 179–202 Search PubMed; (c) C. Deutsch, B. H. Liphsutz and N. Krause, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS; (d) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed.
- For the catalytic hydrogenation of enones, enals and carbonyl compounds, see: (a) W. S. Mahoney and J. M. Stryker, J. Am. Chem. Soc., 1989, 111, 8818–8823 CrossRef CAS; (b) J.-X. Chen, J. F. Daeuble, D. M. Brestensky and J. M. Stryker, Tetrahedron, 2000, 56, 2153–2166 CrossRef CAS; (c) J.-X. Chen, J. F. Daeuble and J. M. Stryker, Tetrahedron, 2000, 56, 2789–2798 CrossRef CAS; (d) H. Shimizu, D. Igarashi, W. Kuriyama, Y. Yusa, N. Sayo and T. Saito, Org. Lett., 2007, 9, 1655–1657 CrossRef CAS PubMed; (e) H. Shimizu, N. Sayo and T. Saito, Synlett, 2009, 1295–1298 CrossRef CAS; (f) H. Shimizu, T. Nagano, N. Sayo, T. Saito, T. Ohshima and K. Mashima, Synlett, 2009, 3143–3146 CrossRef CAS; (g) K. Junge, B. Wendt, D. Addis, S. Zhou, S. Das, S. Fleischer and M. Beller, Chem. – Eur. J., 2011, 17, 101–105 CrossRef CAS PubMed.
- For copper-catalysed semihydrogenations of alkynes, see: (a) K. Semba, R. Kameyama and Y. Nakao, Synlett, 2015, 318–322 CrossRef CAS; (b) F. Pape, N. O. Thiel and J. F. Teichert, Chem. – Eur. J., 2015, 21, 15934–15938 CrossRef CAS PubMed; (c) N. O. Thiel and J. F. Teichert, Org. Biomol. Chem., 2016, 14, 10660–10666 RSC; (d) T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Organometallics, 2016, 35, 1354–1357 CrossRef CAS; (e) F. Pape and J. F. Teichert, Synthesis, 2017, 2470–2482 CAS; (f) N. O. Thiel, S. Kemper and J. F. Teichert, Tetrahedron, 2017, 73, 5023–5028 CrossRef CAS.
- This reactivity trend was also observed employing hydrosilanes, see: V. Jurkauskas, J. P. Sadighi and S. L. Buchwald, Org. Lett., 2003, 5, 2417–2420 CrossRef CAS PubMed.
- For a recently published report on a related heterogeneous hydrogenation of α,β-unsaturated carbonyl compounds, see: J. Mendes-Burak, B. Ghaffari and C. Copéret, Chem. Commun., 2019, 55, 179–181 RSC.
- J. F. Hartwig, Organotransition metal chemistry, From bonding to catalysis, University Science Books, Mill Valley, 2010 Search PubMed.
- For reviews, see: (a) F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293-294, 48–79 CrossRef CAS; (b) F. Lazreg and C. S. J. Cazin, in N-Heterocyclic carbenes. Effective tools for organometallic synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014, vol. 113, pp. 199–242 Search PubMed.
- (a) A. J. Chalk and J. Halpern, J. Am. Chem. Soc., 1959, 81, 5852–5854 CrossRef; (b) J. Halpern, J. Phys. Chem., 1959, 63, 398–403 CrossRef CAS; (c) G. V. Goeden and K. G. Caulton, J. Am. Chem. Soc., 1981, 103, 7354–7355 CrossRef CAS.
- See the ESI† for details.
- For investigation of further protic additives, see the ESI†.
- The copper-catalyzed alkyne semihydrogenation (compare ref. 7) is competitive, see the ESI†.
- For reviews, see: (a) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC; (b) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; (c) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed.
- For a notable exception, see: M. Rodríguez-Fernández, X. Yan, J. F. Collados, P. B. White and S. R. Harutyunyan, J. Am. Chem. Soc., 2017, 139, 14224–14231 CrossRef PubMed.
- The mechanism of the D incorporation of the ester is unclear at present. Experiments also show D incorporation in the ester moeity when the reduced ester 2 is submitted to the standard conditions employing D2. Further studies in shedding light on this process are currently underway. At present, a second, heterogenous process cannot be excluded. For the distinction between homogeneous and heterogenous catalysis, see: (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed; (b) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS; (c) ref. 9 .
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed.
- D. Katayev, Y.-X. Jia, A. K. Sharma, D. Banerjee, C. Besnard, R. B. Sunoj and E. P. Kündig, Chem. – Eur. J., 2013, 19, 11916–11927 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation and NMR spectra. See DOI: 10.1039/c8cc09853k |
| This journal is © The Royal Society of Chemistry 2019 |