
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis of MoS2 or MoO3via thermolysis of a dialkyl dithiocarbamato molybdenum(IV) complex†
Niting
Zeng
a,
David G.
Hopkinson
 a,
Ben F.
Spencer
a,
Simon G.
McAdams
a,
Aleksander A.
Tedstone
b,
Sarah J.
Haigh
a and
David J.
Lewis
*a
a,
Ben F.
Spencer
a,
Simon G.
McAdams
a,
Aleksander A.
Tedstone
b,
Sarah J.
Haigh
a and
David J.
Lewis
*a
aSchool of Materials, University of Manchester, Oxford Road, M13 9PL, UK. E-mail: david.lewis-4@manchester.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, M13 9PL, UK
First published on 29th November 2018
Abstract
Direct synthesis of either 2H-MoS2 or α-MoO3 is made possible by thermolysis of the same single source precursor in either argon or air at moderate temperatures.
The inorganic compounds molybdenum disulphide (MoS2) and molybdenum trioxide (MoO3) have attracted significant attention. The 2H-MoS2 polytype is an intrinsic semiconductor with a layered structure and has been extensively studied. Its promising applications include semiconductor technology,1,2 catalysts3,4 and energy storage.5,6 Production of atomically thin MoS2 as a complementary 2D material to graphene is also a widespread area of research. Atomically thin MoS2 can be produced by micromechanical exfoliation,7–9 liquid phase exfoliation,10–13 chemical vapour deposition,14–17 hydrothermal synthesis,18,19 and hybrid chemical methods.20,21 Similarly, α-MoO3 also has a layered structure and exhibits excellent properties for charge-generation22,23 and lithium ion storage24,25 as well as catalytic properties.26,27 For both materials, their properties are highly dependent on crystallite size and morphology, as well as the presence of surfactants. For example, MoO3 thin films are used for charge generation, whilst MoO3 nanorods or nanobelts are preferred for lithium storage. Nanocrystals are employed in catalysis.23–27 Hence, controllable synthesis is of interest for both the facile design of crystalline MoO3 and scalable production of atomically thin MoS2.8,22,28
We have been interested in thin film deposition of MoS2 by use of single source precursors. For example, tetrakis(N,N-diethyldithiocarbamato)molybdenum(IV) (Mo(DTC)4, Scheme 1) can be used in combination with other metal dithiocarbamate complexes to produce doped MoS2via aerosol-assisted chemical vapour deposition (AACVD).29 Molybdenum dithiocarbamate (MoDTC) has also been used as a lubricant additive which has been widely investigated for its decomposition mechanism to molybdenum sulfide species in tribology;30,31 MoS2 and MoO3 are reported as two major products in tribological experiments, whilst another study recently revealed that MoS2, FeMoO4 and MoSx (where x > 2) are the dominant species formed under certain conditions.32
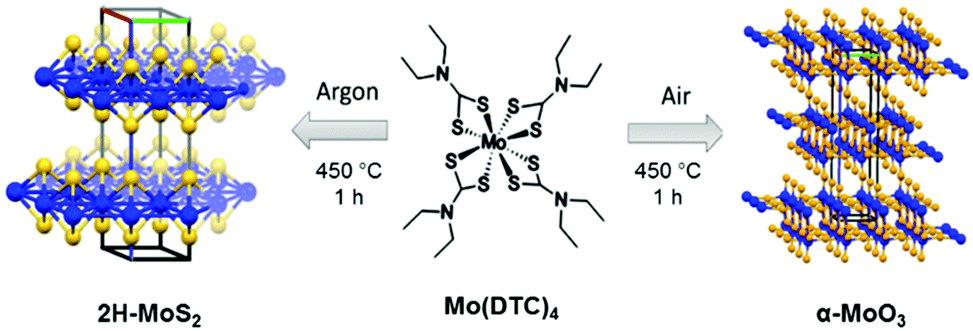 | ||
| Scheme 1 Selective reaction for the solventless synthesis of molybdenum disulphide or molybdenum trioxide. MoS2 was produced under argon while MoO3 was formed in air; both reactions proceed from the same molecular precursor, Mo(DTC)4. The reaction in both cases was carried out in a ceramic crucible placed within a tube furnace maintained at 450 °C for one hour only. | ||
Here we report the direct synthesis of either 2H-MoS2 or α-MoO3 from a single molecular precursor, with the reaction product controlled by choice of processing conditions. Mo(DTC)4 was produced as per Lewis et al.29 The growth of 2H-MoS2 was achieved via thermolysis of Mo(DTC)4 in argon whilst bulk α-MoO3 was formed by the same reaction but in air. The characterisation performed shows that the 2H-MoS2 powder produced is free-standing and nanostructured whilst polycrystalline α-MoO3 is produced in air. The low temperature (450 °C) combined with the short processing time for both routes (1 h) represents a new soft synthetic pathway toward these important functional inorganic materials (Scheme 1).
Raman spectra (Fig. 1) indicate that the reaction under argon produces MoS2 with the in-plane mode E12g observed at 380 cm−1 and the out-of-plane mode A1g located at 403 cm−1 (Δν = 23 cm−1).9 The softening of the two modes together with the broadening of peaks may be caused by phonon confinement,33 which suggested that the material was few-layer. For reaction in air, MoO3 is formed with lattice modes below 200 cm−1, deformation modes between 450 cm−1 and 200 cm−1 and stretching modes above 450 cm−1.34 The peaks at 112 cm−1 (B2g, B3g), 125 cm−1 and 153 cm−1 (Ag, B1g) represent vibrations of the main chains of Mo atoms, and the peaks at 280, 289 cm−1 (B2g, B3g) are assigned to the wagging vibrational mode of pendant oxygens.35 The characteristic peaks at 664 cm−1 (B2g, B3g), 817 cm−1 (Ag, B1g) and 993 cm−1 (Ag, B1g) are associated with the symmetric and asymmetric stretching modes of α-MoO3.34–36
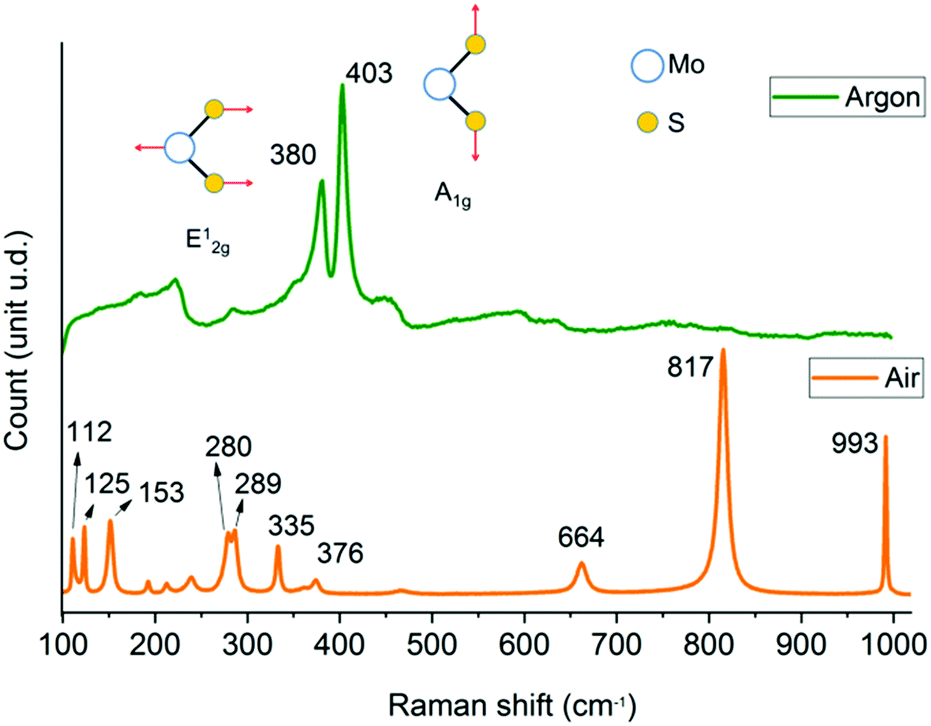 | ||
| Fig. 1 Raman spectra of reaction products of thermolysis after one hour reaction at 450 °C under argon flow at 300 sccm with typical MoS2 peaks at 380 cm−1 and 403 cm−1; air environment with nine peaks that can all be assigned to α-MoO3. | ||
X-ray powder diffraction (XRPD) was used to investigate the crystallographic structure of the materials produced. The diffraction pattern in argon is non-identical compared to standard 2H-MoS2 (JCPDS No. 37-1492). Peaks corresponding to the (101) and (110) planes of MoS2 are relatively intense whereas the peaks corresponding to the (002) and (103) planes are extremely broadened (Fig. 2). A preferred orientation in the (101) and (110) planes is evinced by the pattern, which suggests MoS2 crystallites may grow laterally during the thermolysis process. The reflection of the (002) plane is very weak, indicating a reduced amount of bulk character in the [00l] direction.37 This indicated again that potentially sheets of nanoscale thickness of MoS2 were produced from this reaction pathway.33,38,39 The XRPD diffraction pattern of the product of the decomposition of Mo(DTC)4 in air at 450 °C matches well with orthorhombic α-MoO3 (JCPDS No. 05-0508, Fig. 2). Strong intensities are noticed for the (110), (021) and (002) reflections, which indicates preferred growth orientations along these planes. No reflections assigned to MoO2 or Mo4O11 are observed, which indicates high-purity α-MoO3 is formed in this reaction.
 | ||
| Fig. 2 XRPD patterns of products from reaction in argon (2H-MoS2) and in air (α-MoO3) with peaks indexed to 2H-MoS2 (JCPDS 37-1492) and α-MoO3 (JCPDS 05-0508) respectively. | ||
X-ray photoelectron spectroscopy (XPS) was used to characterise the chemical information and changes occurring at the surface of the materials. A neutraliser was used to remove differential charging occurring to the powders under the X-ray beam, and binding energy scale calibration was performed using the C 1s photoelectron peak at 284.8 eV. The Mo 3d spectrum under argon (Fig. 3a) is fit with three chemical species with binding energies of 228.9, 230.0 and 232.5 eV and are associated with Mo4+ (MoS2), Mo5+ and Mo6+ (MoO3) respectively.41,42 This is in good agreement with Raman results; MoS2 is the dominant material formed from reaction under argon with oxidation occurring at the surface (sampling depth for Al Kα is ca. 6 nm). The Mo 3d spectrum also contains the broad S 2s photoelectron peak at 226.6 eV which is suggestive of the 2H-phase for MoS2 consistent with diffraction results.40 The S 2p spectrum also shows three chemical species, consistent with the Mo spectrum, with S 2p3/2 binding energy positions at 161.6, 162.8 and 168.4 eV (Fig. 3b), associated with S2−, (MoS2), S1− and S6+ (sulfate) respectively, which are again consistent with MoS2 reported in the literature and exhibiting some surface oxidation.33,41,42 The ratio of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S (excluding the oxidized species in the spectra, and adjusted to the relative sensitivity factor each elemental core level) is calculated as 1:2.1, indicating stoichiometric MoS2.
:S (excluding the oxidized species in the spectra, and adjusted to the relative sensitivity factor each elemental core level) is calculated as 1:2.1, indicating stoichiometric MoS2.
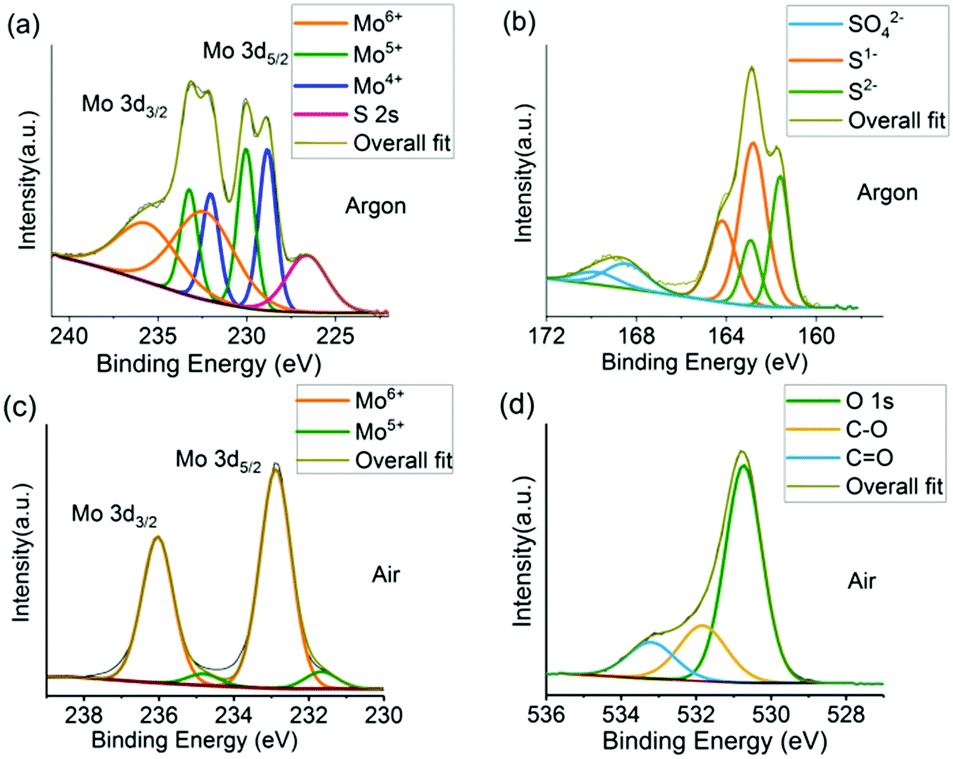 | ||
| Fig. 3 High resolution XPS spectra. Mo 3d spectra for (a) product from reaction in argon and (b) product from reaction in air. (c) S 2p spectrum for product from reaction in argon. (d) O 1s peak for the product from reaction in air. | ||
For the reaction in air, the Mo 3d spectrum is adequately fit with one chemical species with a Mo 3d5/2 binding energy position of 232.9 eV (Fig. 3c), associated with Mo6+ (MoO3).43 This indicates the major oxidation state is (VI). No Mo4+ species are observed, and no signal from S is observed.
The O 1s peak with the largest contribution has a binding energy of 530.7 eV and is associated with metal oxide, with other species at ∼532–534 eV associated with adventitious carbon oxide contamination. This is in agreement with prototypical MoO3 reported in the literature.43 The ratio of Mo:O (adjusted for relative sensitivity factors) is calculated as 1:2.80, indicating a stoichiometric oxide. This is also reflected by the relatively narrow full-width-at-half maximum for the Mo6+, whereas this oxidized species in the MoS2 (argon) sample is much broader indicating an array of partial oxide states (as expected for surface oxidation).
The materials were further investigated using electron microscopy. Scanning electron microscope (SEM) images of the product from reaction in argon shows evenly distributed large crystallites (Fig. 4(b)) of around 200–500 nm in length embedded in a nanostructured surface. Transmission electron microscopy (TEM) reveals that the textured surface is comprised of few-layer nanosheets (Fig. 4c), which must be the dominant species within the sample when taken into consideration together with the SEM images. Atomic force microscopy (AFM) of the powder dispersed by sonication in N-methyl-2-pyrrolidone and drop cast onto silicon substrates for analysis was used to confirm a mean flake thickness of ∼10 nm (N = 83). Selected area electron diffraction (SAED) shows that these sheets give a polycrystalline diffraction pattern which was indexed to 2H-MoS2 (Fig. 4d) in good agreement with XRPD results and as-expected for an ensemble of nanosheets with random orientations. SEM images show that the reaction products from air (Fig. 4a) appear to be tabular nanocrystallites with various sizes and shapes that are noticeably larger compared to the reaction products formed from argon atmospheres.
 | ||
| Fig. 4 Electron microscopy of thermolysis products. (a) SEM imaging after thermolysis at 450 °C in air and (b) in argon. (c & d) TEM image and SAED pattern for reaction products from argon. The SAED pattern is indexed to the diffraction pattern of 2H-MoS2. | ||
In this study, the solventless thermolysis of a single source precursor has been proven to be viable route to produce either 2H-MoS2 or α-MoO3 by judicious choice of reaction conditions. In argon, thin-layer MoS2 nanosheets and nanoparticles are produced, which was confirmed by the broadening of both Raman peaks and XRPD reflections. XRPD further indicates very small polycrystalline sheets formed directly from the reaction which matches with both TEM and SAED results. Based on the thermogravimetric analysis (TGA) data published previously for Mo(DTC)4 under argon the reaction proceeds by loss of the organic parts of the molecule as gaseous species to form amorphous MoSx, followed by formation of crystalline 2H-MoS2.44 In air, these steps are probably followed by a rapid replacement of sulfur by atmospheric oxygen to form MoO3, or by oxidation of the MoSx intermediates to MoOx species followed by formation of MoO3.44
We conclude that the production of high-purity MoS2 or MoO3 is achievable by this method with potential scalability embedded. By using various combinations of precursors (of which for metal dithiocarbamates45 and metal xanthates46 there are many) we should be able to produce large amounts of doped metal oxide and chalcogenide materials for various applications including catalysis, tribology and dilute magnetic semiconductor materials for spintronics. As these are layered structures we might also be able to exfoliate them to the two-dimensional limit to produce bespoke metal oxide and chalcogenide 2D materials. We are currently exploring these possibilities in our laboratory. This approach has already proven to be successful for producing synthetic layered main group sulfides and 2D derivatives thereof.47
DJL and NZ would like to thank the University of Manchester Research Impact Donor Scholarship for funding this study. SGMcA is funded by an EPSRC doctoral prize. DGH is funded by the EPSRC Centre for Doctoral Training in the Science and Applications of Graphene and Related Nanomaterials (GrapheneNOWNANO, EP/L01548X/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Fiori, F. Bonaccorso, G. Iannaccone, T. Palacios, D. Neumaier, A. Seabaugh, S. K. Banerjee and L. Colombo, Nat. Nanotechnol., 2014, 9, 768 CrossRef CAS PubMed.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C.-Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, J. Catal., 2000, 190, 128–143 CrossRef CAS.
- X. Y. Yu, H. Hu, Y. Wang, H. Chen and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 7395–7398 CrossRef CAS.
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839–844 CrossRef CAS.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS.
- J. N. Coleman, M. Lotya, A. O’Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS.
- G. S. Bang, K. W. Nam, J. Y. Kim, J. Shin, J. W. Choi and S.-Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084–7089 CrossRef CAS.
- K. Wang, J. Wang, J. Fan, M. Lotya, A. O’Neill, D. Fox, Y. Feng, X. Zhang, B. Jiang, Q. Zhao, H. Zhang, J. N. Coleman, L. Zhang and W. J. Blau, ACS Nano, 2013, 7, 9260–9267 CrossRef CAS.
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2016, 28, 337–348 CrossRef CAS.
- B. Liu, L. Chen, G. Liu, A. N. Abbas, M. Fathi and C. Zhou, ACS Nano, 2014, 8, 5304–5314 CrossRef CAS.
- M. R. Laskar, L. Ma, S. Kannappan, P. S. Park, S. Krishnamoorthy, D. N. Nath, W. Lu, Y. Wu and S. Rajan, Appl. Phys. Lett., 2013, 102, 252108 CrossRef.
- X. Wang, H. Feng, Y. Wu and L. Jiao, J. Am. Chem. Soc., 2013, 135, 5304–5307 CrossRef CAS.
- Y. Zhan, Z. Liu, S. Najmaei, P. M. Ajayan and J. Lou, Small, 2012, 8, 966–971 CrossRef CAS.
- D. Wang, Z. Pan, Z. Wu, Z. Wang and Z. Liu, J. Power Sources, 2014, 264, 229–234 CrossRef CAS.
- K. Krishnamoorthy, G. K. Veerasubramani, S. Radhakrishnan and S. J. Kim, Mater. Res. Bull., 2014, 50, 499–502 CrossRef CAS.
- H. Samassekou, A. Alkabsh, M. Wasala, M. Eaton, A. Walber, A. Walker, O. Pitkänen, K. Kordas, S. Talapatra, T. Jayasekera and D. Mazumdar, 2D Mater., 2017, 4, 021002 CrossRef.
- T. Sreeprasad, P. Nguyen, N. Kim and V. Berry, Nano Lett., 2013, 13, 4434–4441 CrossRef CAS.
- J. Meyer, S. Hamwi, M. Kröger, W. Kowalsky, T. Riedl and A. Kahn, Adv. Mater., 2012, 24, 5408–5427 CrossRef CAS.
- H. Kanno, R. J. Holmes, Y. Sun, S. Kena-Cohen and S. R. Forrest, Adv. Mater., 2006, 18, 339–342 CrossRef CAS.
- Z. Wang, S. Madhavi and X. W. Lou, J. Phys. Chem. C, 2012, 116, 12508–12513 CrossRef CAS.
- J. S. Chen, Y. L. Cheah, S. Madhavi and X. W. Lou, J. Phys. Chem. C, 2010, 114, 8675–8678 CrossRef CAS.
- M. A. Vuurman and I. E. Wachs, J. Phys. Chem., 1992, 96, 5008–5016 CrossRef CAS.
- A. Manivel, G.-J. Lee, C.-Y. Chen, J.-H. Chen, S.-H. Ma, T.-L. Horng and J. J. Wu, Mater. Res. Bull., 2015, 62, 184–191 CrossRef CAS.
- S. Balendhran, S. Walia, H. Nili, J. Z. Ou, S. Zhuiykov, R. B. Kaner, S. Sriram, M. Bhaskaran and K. Kalantar-zadeh, Adv. Funct. Mater., 2013, 23, 3952–3970 CrossRef CAS.
- D. J. Lewis, A. A. Tedstone, X. L. Zhong, E. A. Lewis, A. Rooney, N. Savjani, J. R. Brent, S. J. Haigh, M. G. Burke, C. A. Muryn, J. M. Raftery, C. Warrens, K. West, S. Gaemers and P. O’Brien, Chem. Mater., 2015, 27, 1367–1374 CrossRef CAS.
- C. Grossiord, K. Varlot, J.-M. Martin, T. Le Mogne, C. Esnouf and K. Inoue, Tribol. Lett., 1998, 31, 737–743 CrossRef CAS.
- M. I. de Barros’ Bouchet, J. M. Martin, T. Le-Mogne and B. Vacher, Tribol. Lett., 2005, 38, 257–264 CrossRef.
- D. N. Khaemba, A. Neville and A. Morina, RSC Adv., 2016, 6, 38637–38646 RSC.
- C. N. R. Rao and A. Nag, Eur. J. Inorg. Chem., 2010, 4244–4250 CrossRef CAS.
- L. Seguin, M. Figlarz, R. Cavagnat and J.-C. Lassègues, Spectrochim. Acta, Part A, 1995, 51, 1323–1344 CrossRef.
- B. C. Windom, W. Sawyer and D. W. Hahn, Tribol. Lett., 2011, 42, 301–310 CrossRef CAS.
- M. A. Py and K. Maschke, Physica B+C, 1981, 105, 370–374 CrossRef CAS.
- T. S. Sahu and S. Mitra, Sci. Rep., 2015, 5, 12571 CrossRef CAS.
- H. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059–4062 CrossRef.
- G. L. Frey, R. Tenne, M. J. Matthews, M. Dresselhaus and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 2883 CrossRef CAS.
- H. W. Wang, P. Skeldon and G. Thompson, Surf. Coat. Technol., 1997, 91, 200–207 CrossRef CAS.
- K.-K. Liu, W. Zhang, Y.-H. Lee, Y.-C. Lin, M.-T. Chang, C.-Y. Su, C.-S. Chang, H. Li, Y. Shi, H. Zhang, C.-S. Lai and L.-J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS.
- M. A. Baker, R. Gilmore, C. Lenardi and W. Gissler, Appl. Surf. Sci., 1999, 150, 255–262 CrossRef CAS.
- D. O. Scanlon, G. W. Watson, D. J. Payne, G. R. Atkinson, R. G. Egdell and D. S. L. Law, J. Phys. Chem. C, 2010, 114, 4636–4645 CrossRef CAS.
- M. N. McCain, B. He, J. Sanati, Q. J. Wang and T. J. Marks, Chem. Mater., 2008, 20, 5438–5443 CrossRef CAS.
- G. Hogarth, Progress in Inorganic Chemistry, John Wiley & Sons, 2005 Search PubMed.
- E. R. T. Tiekink and G. Winter, Rev. Inorg. Chem., 1992, 12, 183 CAS.
- K. Norton, J. Kunstmann, L. Ping, A. Rakowski, C. Wang, A. J. Marsden, G. Murtuza, N. Zeng, S. McAdams, M. A. Bissett, S. J. Haigh, B. Derby, G. Seifert and D. J. Lewis, Chem. Sci., 2019 10.1039/C1038SC04018D.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08932a |
| This journal is © The Royal Society of Chemistry 2019 |