
Synthesis of highly-soluble push–pull perylenemonoimide derivatives by regioselective peri-functionalization for switchable memory applications†
Dhananjaya
Sahoo‡
 *a,
Vikas
Sharma‡
a,
Rupam
Roy
a,
Nonu
Varghese
b,
Kallol
Mohanta
b and
Apurba Lal
Koner
*a
*a,
Vikas
Sharma‡
a,
Rupam
Roy
a,
Nonu
Varghese
b,
Kallol
Mohanta
b and
Apurba Lal
Koner
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By pass Road, Bhauri, Bhopal-462066, Madhya Pradesh, India. E-mail: chem.dsahoo@gmail.com; akoner@iiserb.ac.in
bDepartment of Physics, PSG College of Technology & PSG Institute of Advanced Studies, Peelamedu, Avinashi Road, Coimbatore – 641004, Tamilnadu, India
First published on 23rd November 2018
Abstract
A regioselective synthetic protocol is developed via tetrabromination of perylenemonoimide (PMI) which leads to a series of PMI derivatives. The push–pull characteristics of these derivatives are established by spectroscopic and theoretical investigations. Finally, the semiconducting properties of the PMI dyes are utilized for the development of a switchable memory device.
Perylene dyes, with their unique properties such as high fluorescence quantum yield,1–5 outstanding thermal and photochemical stability, and the possibility of chemical modification6–9 to tune their self-assembling behavior, have attracted commendable attention for a wide range of applications in energy transfer,7–9 light harvesting arrays,10–13 organic solar cells,14–16 optical sensors,17 and bio-imaging.18,19 Perylene bisimide derivatives have been extensively explored for optoelectronic applications including solar cells, OFET, and n-type memory applications.20,21 However, perylenemonoimide (PMI) dyes are still underexplored for such applications.
A primary challenge in working with PMI is to overcome their intrinsically-poor solubility. The solubility is improved by introducing bulky/sterically demanding alkyl or aryl groups at the imide position of PMI without affecting the optical properties. The solubility of PMI is further enhanced with fine tuning of the optical properties by functionalization at bay and peri positions.11,12,19,22–25 Therefore, PMI dyes with strong push–pull characteristics, improved solubility in common organic solvents and optically tunable low band gap with suitable regioselective substitution are highly desirable and can serve as promising candidates for optoelectronic applications.26–28 The major contribution towards the push–pull effect in PMI dyes is due to the functionalization at the peri-position with an electron-donating moiety rather than at the bay position.11,17,29 Generally, functionalization at one of the peri-positions of PMIs is carried out either by direct mono-bromination or tri-bromination of PMI followed by further substitution.9–12,17–19,22,23,30 Valiyaveettil et al.23 functionalized both of the peri positions of PMI by tertrabromination and used it for further functionalization. Later, Müllen et al.24 functionalized both of the peri positions of PMI by using a different protocol; 9,10-dibromo-1,6,7,12-tetrachloro PMI was synthesized utilizing the Hunsdiecker reaction followed by further modification at peri positions. In the former work, tetrabrominated PMI (2) was isolated by repetitive precipitation for the purpose of characterization only and a mixture of PMI-Br3 and 2 was used for successive functionalization. Limitations of the latter method are the reluctant substitution of less reactive chlorine atoms at the bay-position and the need for de-chlorination at the end.
Herein, the synthesis of PMI-Br4 is reported in 46% yield with a relatively simple workup procedure. Tetra-substituted PMI derivatives are synthesized and isolated in good yield. Furthermore, the regioselectively substituted products 4a and 4b are synthesized. Later, the (9,10) peri-position of 4b is annulated with S or Se to obtain the push–pull dye 5 (see Scheme 1). The properties of PMI derivatives are investigated using optical spectroscopy, DFT calculations and electrochemistry. Apart from optoelectronic properties, we have also explored the electrical bistability of the synthesized PMIs under visible light and their on/off ratio for switchable memory devices.
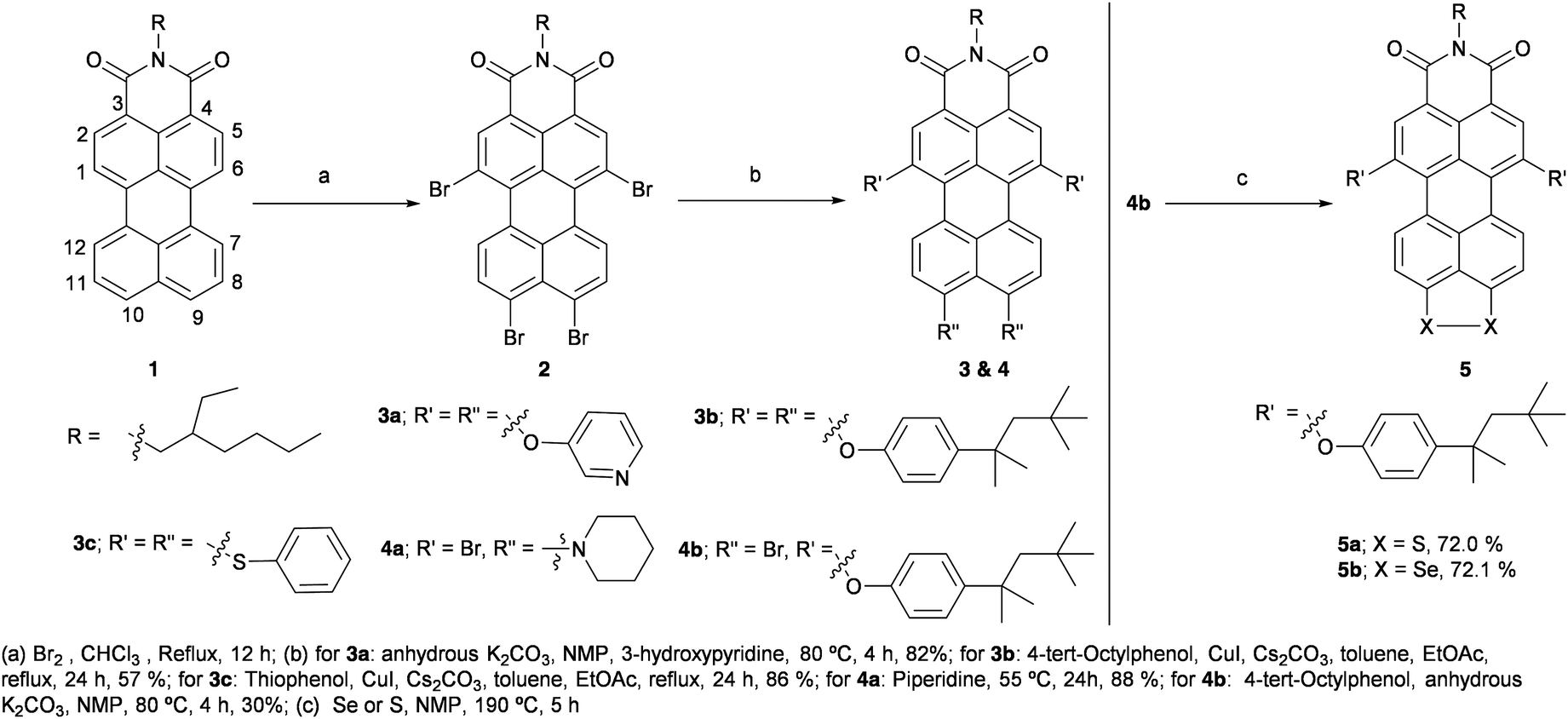 | ||
| Scheme 1 Synthesis of tetra-substituted push–pull PMI derivatives and their regioselective modification. | ||
The synthesis of mono-brominated PMI is achieved either by heating a solution of PMI 1 in chlorobenzene with Br2 at 55 °C for 7 h10,14 or by refluxing a solution of PMI in DCM for 2 h.31 Tri-brominated PMI is obtained by refluxing a solution of PMI in chloroform with a gradual addition of 20 equiv. of Br2 at intervals of 2 h over a period of 7 h. Whereas, bromination at both of the peri-positions of PMI is achieved by tetrabromination of PMI; by refluxing a solution of PMI 1 in chloroform with 50 equiv. of Br2 for 10 h.23 However, in the latter case compound 2 was obtained only in 13% yield after purification and repetitive precipitations. In this report, we have optimized the challenging protocol by using 200 equiv. of Br2 to isolate compound 2 in 46% yield (Scheme 1).23 Note that a mixture of tri- and tetra-brominated products is obtained after precipitation when 50 equiv. and 100 equiv. of Br2 are used for the reaction. For 150 equiv. of Br2, compound 2 is isolated in 25% yield. However, with more than 200 equiv. of Br2 there is no significant improvement in the yield.
Afterwards, the tetra-substituted PMI 3a is synthesised by nucleophilic substitution of all the bromo groups of 2 with 3-hydroxypyridine by using the protocol mentioned by Li et al.32 with minor modifications; compound 2, 3-hydroxypyridine and anhydrous K2CO3 are heated in NMP at 80 °C for 4 h to obtain 3a in 82% yield (Scheme 1, see ESI†). However, by using the modified protocol with 4-tert-octylphenol (TOP), 3b is isolated only in 46% yield. The optimization is carried out by refluxing a solution of compound 2 in DMF along with TOP and K2CO3 for 2 h to provide a complex reaction mixture, but it is difficult to isolate 3b.11 A further modification is carried out by refluxing compound 2, CuI, Cs2CO3 and TOP in a mixture of toluene and EtOAc for 16 h affording compound 3b in 57% yield (Scheme 1).33 By following the latter optimized protocol, 3c is synthesized and isolated in 86% yield (Scheme 1). A regioselective peri-substituted product 4a is isolated with 88% yield instead of the desired tetra-substituted PMI, when compound 2 is heated with piperidine at 60 °C for 36 h.34 Longer reaction time up to 72 h either at room temperature or on heating at 60 °C provided a complex reaction mixture.
The various reaction conditions to annulate at the peri-position of 2 with electron-rich S or Se failed possibly due to the poor solubility of 2 in NMP, which is resolved by regioselective nucleophilic substitution at the bay region by TOP groups. The regioselective product 4b is isolated along with 15–20% of 3b with a reaction time of 4 h, when 2, TOP and anhydrous K2CO3 are heated in NMP at 80 °C (Scheme 1). However, for the shorter reaction time of 3 h, the reaction is incomplete and for a longer reaction time of 5 h, the formation of 3b increased. The S/Se-annulated compound 5a/5b is synthesized in good yield by treating the compound 4b with S or Se powder in NMP at 190 °C for 5 h (Scheme 1).35
After the successful synthesis, we investigated the effect of substitution on the PMI core along the long molecular axis (specifically at 9- and 10-positions), which enabled a systematic comparison of their optical properties. All UV-Vis absorption, steady-state (SS) fluorescence and time-resolved (TR) fluorescence measurements were performed at 25 °C in toluene (Fig. S1 and S2, ESI†). The optical properties of the PMI derivatives are compared with those of PMI 1, indicating a strong push–pull characteristic. The λmaxabs of PMI derivative 3a showed a bathochromic shift of 20 nm in comparison to PMI 1, whereas in the case of 3b and 3c the λmaxabs shows a bathochromic shift of 34 nm and 62 nm respectively (Fig. 1a, Fig. S1, Table 1 and Table S1, ESI†). Interestingly, for the peri-substituted PMIs 4a, and 5a–b the absorption spectra are broad and span the Vis-NIR region with a large bathochromic shift of ∼110 nm in comparison to that of 1. This large shift is due to the push–pull character originating from the intramolecular charge transfer (ICT) process from the electron-rich moiety to the electron-deficient perylene core (vide infra). Moreover, during the steady-state fluorescence measurement in toluene, compounds 3a, 3b, and 3c show λmaxEm values that are significantly red-shifted by 48 nm, 63 nm, and 138 nm, respectively in comparison to 1 (see Fig. 1b and Table 1). The red-shifting of λmaxEm of 4a to the NIR region by 170 nm as compared to that of PMI 1 and the weak fluorescent properties of the annulated PMI derivatives i.e., 5a and 5b, are due to the ICT effect along the long-molecular axis.16,24 The fluorescence quantum yields (ϕf) of 4b, 3a and 3b in toluene are quite high, but those of 3c (0.02) and 4a (0.34) are low (Table 1 and Table S1, ESI†), due to the quenching via PET from the substituents to the PMI core. TR measurements of 2, 3a, 3b, 4a and 4b show that the lifetime spans from 2.3 to 5.7 ns. Similar to ϕf, the lifetime of 3c is only 0.2 ns due to ICT (Fig. S2, ESI†). The redox properties of the PMI derivatives were investigated using cyclic voltammetry (CV) in dry dichloromethane versus Fc+/Fc in 0.1 M Bu4NPF6 (see Table 1, Table S1 and Fig. S3, ESI†). The studied PMI derivatives show quasi-reversible behavior except for 3a and 4a. The electrochemical band gap (Eg) for compound 5a and 5b is the lowest indicating an ICT behavior. The dipole moment and band gap energy were calculated from the onset potential value and validated with DFT calculations (vide infra).
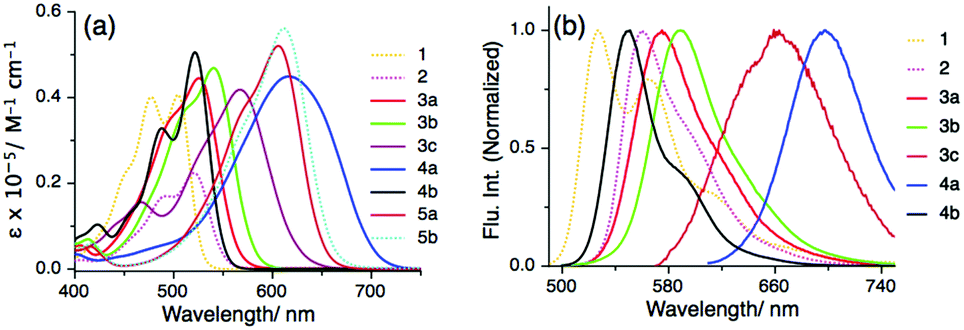 | ||
| Fig. 1 (a) Absorption spectra of PMI derivatives 1–5 in toluene, and (b) normalized emission spectra of PMI 1 and PMI derivatives 2–4 in toluene. | ||
| Entry | λ maxabs (ε × 104/M−1 cm−1) | λ maxEm/nm | ϕ f | τ/ns | HOMOb/eV | LUMOb/eV | E g /eV |
|---|---|---|---|---|---|---|---|
| a Measured in toluene using 2-(2,6-di-tert-butylphenyl)-1H-benzol[10,5]anthrax[2,1,9 def]isoquinone-1,3(2H)-dione as the standard for ϕf.11 b Versus Fc+/Fc in 0.1 M Bu4NPF6 in dry DCM, with a platinum disk as the working electrode with a scan rate of 100 mV s−1, HOMO LUMO gaps are calculated using the relationship EHOMO = −(Eonsetox + 4.7) and ELUMO = −(Eonsetred + 4.7); for CV see Fig. S3. c Electrochemical bandgap. d Not determined due to poor solubility. e E g not determined due to quasi-reversible Eox/Ered. f Weakly fluorescent with ϕf < 0.00,1 due to heavy atom effect. | |||||||
| 1 | 506 (4.0) | 527 | 0.48 | 4.7 | −5.78 | −3.50 | 2.28 |
| 2 | 523 (2.2) | 560 | 0.77 | 5.7 | —d | —d | —d |
| 3a | 526 (4.4) | 575 | 0.85 | 4.7 | —e | —e | —e |
| 3b | 540 (4.7) | 590 | 0.86 | 4.5 | −5.30 | −3.35 | 1.95 |
| 3c | 568 (4.1) | 665 | 0.02 | <0.2 | −5.75 | −3.80 | 1.95 |
| 4a | 616 (4.4) | 697 | 0.34 | 2.8 | −5.30 | —e | —e |
| 4b | 521 (5.0) | 551 | 0.99 | 4.5 | −6.05 | −3.75 | 2.30 |
| 5a | 606 (5.2) | —f | —f | — | −5.45 | −3.75 | 1.70 |
| 5b | 611 (5.6) | —f | —f | — | −5.40 | −3.75 | 1.65 |
To understand the push–pull effect, we performed geometry optimization utilizing DFT using Gaussian 09 at the B3LYP/6-311G level. It is evident from the electron density map and representation of the frontier molecular orbitals that the HOMO and LUMO are localized in the lower and upper naphthalene ring (see ESI,†), respectively. Moreover, a large twisting angle of 14.37° (Fig. 2) in the PMI core for 3b is associated with a moderate dipole moment of 6.75 D, which also indicates a substitution effect of donating as well as bulky TOP groups. A comparison of the twist angle for all compounds is provided in the ESI† (see Fig. S4–S22 and Tables S2–S8). The band gap values obtained from the CV measurements provide good correlation with DFT calculated values (see Table 1, Tables S1, S2 and Fig. S3, ESI†). Among the investigated PMI derivatives, compound 3b shows good electrical bistability in the presence of visible light, which motivated us to perform a detailed investigation (see Fig. 3). This special electrical bistable behavior shown by 3b may arise from the electric field-induced conformational changes in the excited-state of the molecule. Presumably, this is because of the twisting of the PMI core, dipole moment and favorable packing due to the pivotal role of bulky and flexible TOP groups in comparison to the other groups.36 Electrical bistability is demonstrated for compound 3b in a memory device which shows that the OFF or low-conducting state could be switched to an ON or high-conducting state by applying a −1.5 V pulse of width 5 s, while the ON state could be converted to the OFF state by applying a 1.5 V pulse of width 5 s (Fig. 3b–d and Fig. S23, ESI†).
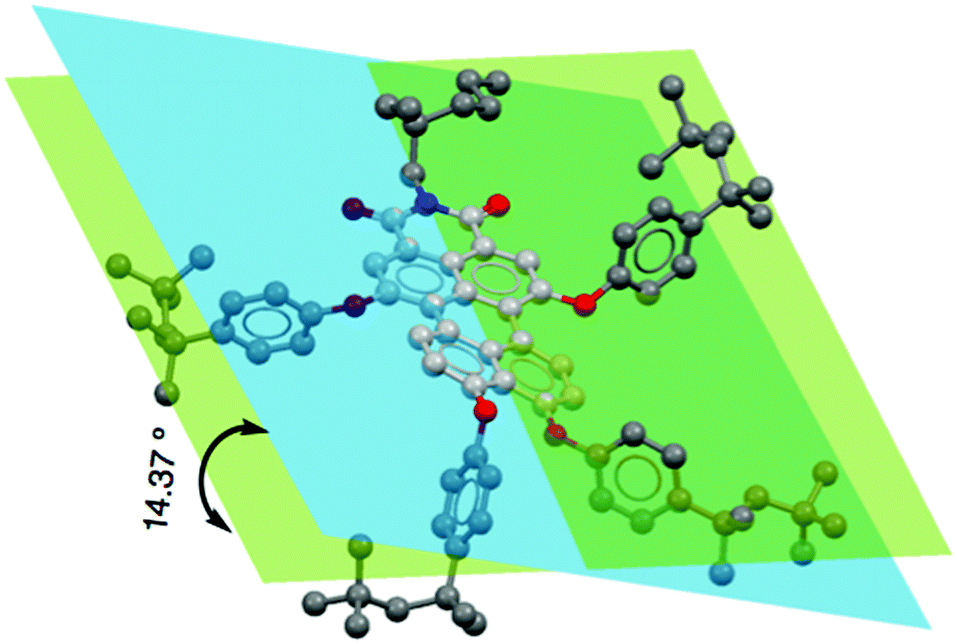 | ||
| Fig. 2 The effect of tetra-substitution on twisting of the perylene core from the energy-optimized structure of 3b calculated by DFT at the B3LYP/6-311G level. | ||
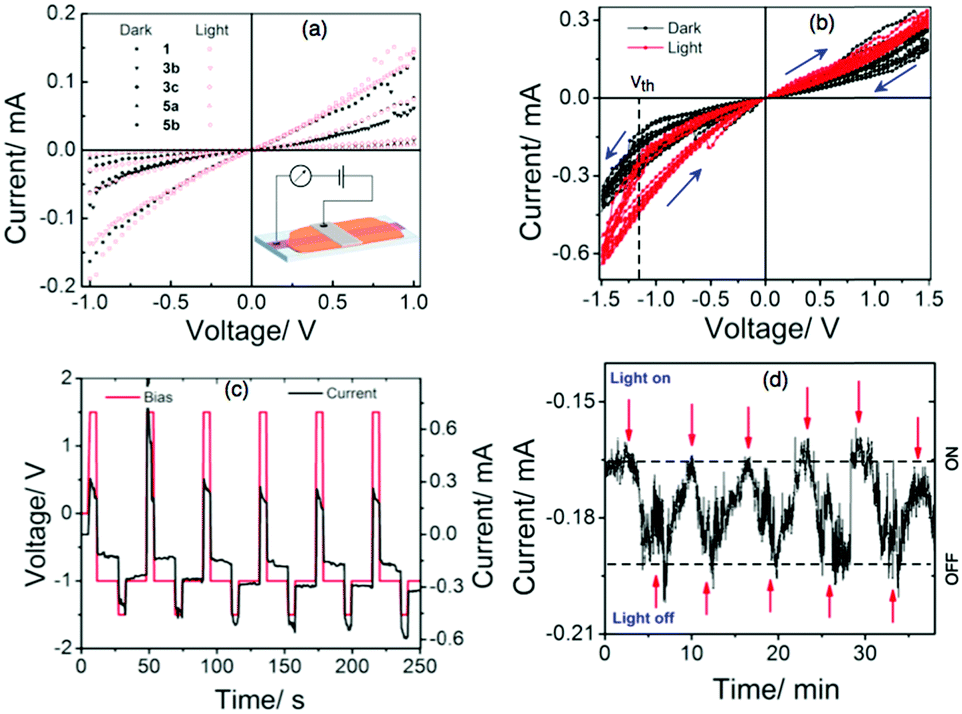 | ||
| Fig. 3 (a) Dark and light I–V characteristics of 1, 3b–c and 5a–b; the inset shows a scheme of the memory device. (b) Electrical bistability in the dark and light for 3b, (c) WRER cycles in the case of 3b under light, and (d) light ON/OFF cycles for 3b. | ||
The ON/OFF current ratio is not high enough for compound 3b to reach the state of the art for memory technologies, but interestingly, it shows a very low switching threshold voltage (−1.13 V), high retention time (>600 s), and reversible switching behavior which are positive indicators for its possible application in memory devices. Additionally, the ON/OFF ratio in the presence and absence of light is also demonstrated which indicates the effect of light on the performance of the designed device. The effect of light and low ON/OFF ratio shown by 3b is not due to any metallic filament formation, but rather due to the molecular property.
In conclusion, tetra-brominated PMI 2 has been synthesized as a promising building block for functional organic materials and switchable memory applications. Subsequently, highly-soluble PMI derivatives 3a–c, and different regioselective electron rich peri-substituted PMI derivatives 4a, and 5a–b have also been synthesized displaying high brightness with a strong push–pull effect. The synthesized PMI derivatives are highly fluorescent except for 3c and the annulated PMI derivatives. Compound 3b shows electrical bistability under visible light. Upon irradiation with visible light, the ON/OFF ratio increases, and a high retention time of the different conducting states promises its application as a memory device. We believe such suitably functionalized PMI derivatives will be highly advantageous for numerous applications including memory devices.
ALK thanks IISERB and CSIR India (Grant no. INGRANT/CHM/2012017 and 02(0134)/17/EMR-II). KM acknowledges PSG for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Chen, C. Li and K. Mullen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS.
- V. Sharma, F. Chandra, D. Sahoo and A. L. Koner, Eur. J. Org. Chem., 2017, 6901–6905 CrossRef CAS.
- Z. Youdi, G. Xia, G. Bing, S. Wenyan, Z. Maojie and L. Yongfang, Adv. Funct. Mater., 2017, 27, 1603892 CrossRef.
- K. Pal, V. Sharma and A. L. Koner, Chem. Commun., 2017, 53, 7909–7912 RSC.
- D. Gosztola, M. P. Niemczyk and M. R. Wasielewski, J. Am. Chem. Soc., 1998, 120, 5118–5119 CrossRef CAS.
- M. A. Miller, R. K. Lammi, S. Prathapan, D. Holten and J. S. Lindsey, J. Org. Chem., 2000, 65, 6634–6649 CrossRef CAS.
- S. I. Yang, S. Prathapan, M. A. Miller, J. Seth, D. F. Bocian, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2001, 105, 8249–8258 CrossRef CAS.
- S. I. Yang, R. K. Lammi, S. Prathapan, M. A. Miller, J. Seth, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, J. Mater. Chem., 2001, 11, 2420–2430 RSC.
- R. S. Loewe, K.-y. Tomizaki, F. Chevalier and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2002, 06, 626–642 CrossRef CAS.
- K.-y. Tomizaki, R. S. Loewe, C. Kirmaier, J. K. Schwartz, J. L. Retsek, D. F. Bocian, D. Holten and J. S. Lindsey, J. Org. Chem., 2002, 67, 6519–6534 CrossRef CAS.
- K.-y. Tomizaki, P. Thamyongkit, R. S. Loewe and J. S. Lindsey, Tetrahedron, 2003, 59, 1191–1207 CrossRef CAS.
- V. Sharma, U. Puthumana, P. Karak and A. L. Koner, J. Org. Chem., 2018, 83, 11458–11462 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CrossRef CAS.
- S. Mathew and H. Imahori, J. Mater. Chem., 2011, 21, 7166 RSC.
- Y. Hu, S. Chen, L. Zhang, Y. Zhang, Z. Yuan, X. Zhao and Y. Chen, J. Org. Chem., 2017, 82, 5926–5931 CrossRef CAS.
- V. Sharma, D. Sahoo, F. Chandra and A. L. Koner, ChemistrySelect, 2017, 2, 11747–11754 CrossRef CAS.
- J. Qu, C. Kohl, M. Pottek and K. Mullen, Angew. Chem., Int. Ed., 2004, 43, 1528–1531 CrossRef CAS.
- K. Pal, V. Sharma, D. Sahoo, N. Kapuria and A. L. Koner, Chem. Commun., 2018, 54, 523–526 RSC.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Adv. Funct. Mater., 2008, 18, 1329–1339 CrossRef CAS.
- R. Centore, L. Ricciotti, A. Carella, A. Roviello, M. Causà, M. Barra, F. Ciccullo and A. Cassinese, Org. Electron., 2012, 13, 2083–2093 CrossRef CAS.
- M. J. Ahrens, M. J. Fuller and M. R. Wasielewski, Chem. Mater., 2003, 15, 2684–2686 CrossRef CAS.
- A. Keerthi, Y. Liu, Q. Wang and S. Valiyaveettil, Chem. – Eur. J., 2012, 18, 11669–11676 CrossRef CAS PubMed.
- Y. Zagranyarski, L. Chen, Y. Zhao, H. Wonneberger, C. Li and K. Müllen, Org. Lett., 2012, 14, 5444–5447 CrossRef CAS.
- N. Kapuria, V. Sharma, P. Kumar and A. L. Koner, J. Mater. Chem. C, 2018, 6, 11328–11335 RSC.
- D. Inan, R. K. Dubey, N. Westerveld, J. Bleeker, W. F. Jager and F. C. Grozema, J. Phys. Chem. A, 2017, 121, 4633–4644 CrossRef CAS.
- T. Kurosawa, Y.-C. Lai, T. Higashihara, M. Ueda, C.-L. Liu and W.-C. Chen, Macromolecules, 2012, 45, 4556–4563 CrossRef CAS.
- Z. Li, M. Jingyu, R. Yi, H. Su-Ting, V. A. L. Roy and Z. Ye, Small, 2018, 14, 1703126 CrossRef.
- C. Li, Z. Liu, J. Schöneboom, F. Eickemeyer, N. G. Pschirer, P. Erk, A. Herrmann and K. Müllen, J. Mater. Chem., 2009, 19, 5405 RSC.
- H. Langhals and A. Hofer, J. Org. Chem., 2012, 77, 9585–9592 CrossRef CAS.
- R. J. Lindquist, K. M. Lefler, K. E. Brown, S. M. Dyar, E. A. Margulies, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2014, 136, 14912–14923 CrossRef CAS.
- C. Li, Z. Liu, J. Schoneboom, F. Eickemeyer, N. G. Pschirer, P. Erk, A. Herrmann and K. Mullen, J. Mater. Chem., 2009, 19, 5405–5415 RSC.
- K. M. Lefler, D. T. Co and M. R. Wasielewski, J. Phys. Chem. Lett., 2012, 3, 3798–3805 CrossRef CAS.
- J. Ma, L. Yin, G. Zou and Q. Zhang, Eur. J. Org. Chem., 2015, 3296–3302 CrossRef CAS.
- Y. Zagranyarski, L. Chen, D. Jänsch, T. Gessner, C. Li and K. Müllen, Org. Lett., 2014, 16, 2814–2817 CrossRef CAS.
- H.-C. Wu, J. Zhang, Z. Bo and W.-C. Chen, Chem. Commun., 2015, 51, 14179–14182 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, and spectroscopic, electrochemical and theoretical investigation. See DOI: 10.1039/c8cc08662a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |