
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated homogeneous SPARCL™ immunoassay for rapid biomarker detection on a chip†
Natalia
Sandetskaya
 *a,
Nicole
Isserstedt-John
b,
Andreas
Kölsch
a,
Sebastian
Schattschneider
b and
Dirk
Kuhlmeier
a
*a,
Nicole
Isserstedt-John
b,
Andreas
Kölsch
a,
Sebastian
Schattschneider
b and
Dirk
Kuhlmeier
a
aFraunhofer Institute for Cell Therapy and Immunology, Perlickstrasse 1, 04103 Leipzig, Germany. E-mail: natalia.sandetskaya@izi.fraunhofer.de
bMildendo GmbH, Stockholmer Strasse 20, 07747 Jena, Germany
First published on 10th April 2019
Abstract
We developed an integrated version of the homogeneous SPARCL™ (Spatial Proximity Analyte Reagent Capture Luminescence) immunoassay for rapid measurement of biomarkers on a chip. Our development is based on a simple microfluidic design with on-chip preserved dry reagents. The assay requires no washing steps and can be performed in a “mix-and-read” format. A syringe pump and a luminometer supported the detection. Only one manual pipetting step is necessary to load the sample onto the chip, and the entire incubation and measurement can be accomplished in approximately 35 min. We have demonstrated the application of the SPARCL™ on a chip to the quantitative detection of the tissue inhibitor of metalloproteinases 1 (TIMP-1) in spiked mock samples and a limited number of gingival crevicular fluid specimens. The integrated SPARCL™ assay could detect as little as 49.3 pg ml−1 (1.76 pM) TIMP1. To our knowledge, this is the first demonstration of the fully integrated homogeneous SPARCL™ immunoassay in a lab-on-a-chip. Further, we discuss the optimization of the chip and the assay in order to improve its analytical performance.
Introduction
Immunoassay is an essential technique in laboratory diagnostics. Specific detection of antigens or antibodies in liquid samples provides information about a patient's health status, enables assessment of the quality of food and environmental samples, supports identification of biothreats, etc.Yet not only is it the presence or absence of an analyte to be confirmed, but very often also its concentration. Therefore, quantitative immunoassays have become a workhorse for in vitro analysis. The most widely used is the Enzyme Linked Immunosorbent Assay (ELISA).1 Its steps are associated with the sequential addition of reagents and extensive washing of unbound molecules to ensure the specificity of the signal. ELISA is thus quite laborious for manual processing. It is preferred by laboratories with middle to high throughput that perform the assay (semi-)automatically.
However, modern diagnostics often demands rapid tests for individual applications in clinical, field, or home settings, also in the absence of laboratory infrastructure. The transfer of a conventional immunoassay into an integrated and automated format is a challenge, mainly due to the need of washing. An ideal format for an integrated test is “mix-and-read”: a simple introduction of a liquid sample into the reagents and generation of the result in a short time.
Homogeneous immunoassays generally correspond to this concept. They are based on the formation of an antigen–antibody complex in solution, and the signal is generated only in the proximity of the analyte and its specific binding partner. The unbound antigens and antibodies do not lead to signal generation and thus can remain in solution. This principle facilitates the advantageous no-wash protocol of the analysis and makes homogeneous assays very attractive for the rapid detection of biomarkers in lab-on-chip devices.
There are currently two commercially available homogeneous assay technologies on the market: SPARCL™ (Spatial Proximity Analyte Reagent Capture Luminescence) by Lumigen (MI, USA; a Beckman Coulter company) and AlphaLISA® (Amplified Luminescent Proximity Homogeneous Assay) by PerkinElmer (MA, USA). SPARCL™ utilizes an antibody labeled with acridan, a compound whose chemiluminescence is triggered enzymatically via the second horseradish peroxidase (HRP)-labeled antibody. In AlphaLISA®, the two antibodies are conjugated to the small beads (250–350 nm in diameter). The donor beads contain a photosensitizer; upon illumination at 680 nm, they convert ambient oxygen into its singlet form that activates the acceptor beads in the vicinity. The europium chelate in the acceptor beads is activated by the singlet oxygen and emits light at 615 nm.
Both assays are based on the correlation of the optical signal with the concentration of the analyte in the solution. The readout is performed in a microplate reader with the corresponding spectrophotometer functions. Yet the reaction mechanisms of the assays demand specific technical features of the instruments: a jet injection of the reagent substrate with the simultaneous measurement of the luminescence for SPARCL™, and an unconventional filter combination (excitation 680 nm/emission 615 nm) for the down-conversion of light for AlphaLISA®. Moreover, AlphaLISA® needs a sequential addition of the reagents during the incubation time. Although the homogeneous format of the immunoassays would favor their application in point-of-care devices, these technical requirements strongly bind them to the advanced laboratory equipment and infrastructure.
In order to overcome this limitation, we demonstrated the integration of the SPARCL™ immunoassay for the detection of the tissue inhibitor of metalloproteinases 1 (TIMP-1) in a lab-on-a-chip format with a true “mix-and-read” protocol. Our development is based on a simple microfluidic design with the on-chip preserved dry reagents. The manual pipetting step is only necessary to load the sample onto the chip. A syringe pump and a chip luminometer supported the required instrumental steps. To our knowledge, this is the first demonstration of the fully integrated homogeneous SPARCL™ immunoassay in a lab-on-a-chip. Its very simple format requires neither specific user skills nor the bulky lab equipment like the pipetting station, microplate washer or microplate reader. The integrated assay produced quantitative results in the pg ml−1 to ng ml−1 range. The simplicity of the homogeneous immunoassay on a chip facilitates the rapid quantitative biomarker measurement in individual samples that can greatly contribute to the point-of-care diagnostics.
Experimental
Antibody labeling with acridan and peroxidase
A pair of anti-human TIMP1 antibodies from the DuoSet® ELISA Development System (R&D Systems, USA) was used for the development of the SPARCL™ assay. The acridan labeling reagent was a part of the Lumigen SPARCL™ Detection Kit (Beckman Coulter, USA). The labeling reagent (19.8 μl) was mixed in an amber vial with 120 μl of the capture antibody (1 mg ml−1) from the DuoSet® System and 340.2 μl 50 mM sodium borate buffer, pH 8.5. The vial was incubated on a rotating mixer for 30 min at room temperature and overnight at 4 °C, and then aliquoted and stored at −80 °C. A working stock solution was prepared by dilution of the labeled antibodies to 25 μg ml−1 in PBS and stored at −20 °C for up to 1 month.The labeling of the second antibody with peroxidase was accomplished by adding 0.61 ng of Streptavidin-HRP conjugate (Pierce® High Sensitivity Streptavidin-HRP, Thermo Fisher Scientific, USA), per nanogram of the biotinylated human TIMP1 detection antibody; the conjugation occurred directly in the assay reaction.
Samples
The assay development and characterization were performed with the mock samples prepared by spiking PBS + 1% bovine serum albumin (BSA, IgG-free, molecular biology grade; Carl Roth, Germany) with different concentrations of TIMP1. Human TIMP1 was a part of the DuoSet® ELISA Development System.The samples of gingival crevicular fluid (GCF) were taken from clinically healthy volunteers by placing a sterile paper strip (dentognostics, Germany) or a sterile paper point (VDW, Germany) for 30 s into the space between the gingival margin and the tooth pocket until a minimum of resistance was felt. The samples were then eluted in 100 μl PBS at 4 °C for 4 h and spun down at 400 g for 4 min. The absorbing strips or tips were discarded, and the samples were stored at −80 °C until assayed. All specimens were anonymized.
Measurement of TIMP1 in the SPARCL™ immunoassay on a microplate
The working solutions of the antibodies were prepared by diluting the stock solutions straight before the assay. The acridan-labeled antibody was diluted to 2 μg ml−1 in PBS, and the biotinylated antibody was mixed with Streptavidin-HRP conjugate (see above) in PBS + 1% BSA to the final concentration of the antibody 0.1 μg ml−1. The antibodies were then mixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v). Twenty μl of this mixture was pipetted to the wells of a white opaque 384-well OptiPlate (PerkinElmer, USA), and 10 μl of a sample was added. Each reaction contained therefore 20 ng of acridan-labeled antibody and 1 ng of HRP-labeled antibody; this proportion was determined empirically during the assay development and kept constant for all microplate and chip experiments.
:1 (v/v). Twenty μl of this mixture was pipetted to the wells of a white opaque 384-well OptiPlate (PerkinElmer, USA), and 10 μl of a sample was added. Each reaction contained therefore 20 ng of acridan-labeled antibody and 1 ng of HRP-labeled antibody; this proportion was determined empirically during the assay development and kept constant for all microplate and chip experiments.
The plate was covered with aluminum foil to protect the reaction from light and incubated for 30 min at room temperature by continuous agitation at 750 rpm on a Titramax 100 shaker (Heidolph Instruments, Germany). The Background Reducing Agent (BGR) from the Lumigen SPARCL™ Detection Kit was diluted fourfold with 80:1 (v/v) water:ethanol solution. Four μl of BGR was added to each reaction after the incubation, and the plate was transferred to the Mithras LB940 microplate reader (Berthold Technologies, Germany).
The trigger for the chemiluminescence reaction contained 55 mM hydrogen peroxide, 50 mM urea, 8 mM p-coumaric acid, 1 mM EDTA, 0.2% Tween-20, and 3.2% ethanol in 25 mM Tris buffer, pH 8.0. For the measurement, the microplate reader injected 30 μl of trigger to the reaction wells (injection rate “high”), and the luminescence was recorded simultaneously with the injection. Typically, the signal was registered for 5 s with the counting rate 0.05 s.
Measurement of TIMP1 in the SPARCL™ immunoassay on a chip
The microfluidic chips were produced by injection molding of the cycloolefin polymer (COP) and sealed with COP foil (microfluidic ChipShop, Germany). The chips were available from the existing product assortment of the company (fluidic 402).For the lyophilization, 10 μg ml−1 acridan-labeled antibodies in PBS + 6% trehalose (Carl Roth, Germany) and 0.5 μg ml−1 HRP-labeled antibodies in PBS + 1% BSA + 6% trehalose were mixed 1:1 (v:v); four μl of the mixture was spotted on the chip. One μl of BGR was spotted in the adjacent or separate chamber as described in the Results and discussion section. The reagents were freeze-dried in a VaCo 2-II lyophilizer (Zirbus Technology, Germany) for 3 hours (1 h at 10 mbar, 1 h at 8 mbar and 1 h at 6 mbar). Twenty μl of liquid sample was pipetted in the reaction chamber with antibodies and incubated for 30 min at room temperature without agitation. The chips were protected from light during the incubation.
The chip luminometer consisted of a photon counting head H10682-210 (Hamamatsu, Japan) and a counter/timer module DM0101 (Sens-Tech, England) incorporated into a measurement chamber (Fig. S1†). The chip was placed into a fitted slot on the top panel of the instrument and connected to a syringe pump neMESYS 290N (Cetoni, Germany) via a Mini Luer Connector and PTFE tubing (inner diameter 560 μm) (microfluidic ChipShop, Germany) with a 1 ml syringe (Braun, Germany).
The chip luminometer was closed with a lid for the isolation from ambient light during the measurement. The reaction was started by the injection of 20 μl trigger solution at a maximum injection rate. The signal was recorded for 5–10 s using Counter/Timer software supplied with the counter module (Sense Tech, England) with the counting rate 0.02 s.
Evaluation of the SPARCL™ signals
For the quantitative evaluation of the SPARCL™ signals, the absolute maximum (S) of luminescence in the specific peak (straight after the trigger injection) was determined. The software for the Mithras microplate reader (MikroWin 4.41, Miktotek Laborsysteme GmbH) automatically normalized the measurements for each sample to zero baseline luminescence. For the luminescence measurements on the chip, this step was done manually during the analysis of the raw data exported to Microsoft Excel. Namely, the average luminescence for one hundred counts (2 s) before the injection-triggered peak was subtracted from the maximum in the specific peak to derive the absolute maximum S.In each experiment, blank samples (sample volume replaced by PBS) were tested and analyzed in the same manner to determine the blank signal S0. The signal-to-blank ratio S/S0 was calculated for all the samples within the experiment and used for the calibration of the assay and the quantitative assessment of the results.
Results and discussion
Microfluidic structures for the accommodation of the SPARCL™ on a chip
Although SPARCL™ is a mix-and-read assay, certain fluidic parameters are critical for its performance. The main parameter is the jet injection of the trigger solution. SPARCL™ readout is based on the flash luminescence, a reaction that occurs instantly upon the addition of the substrate. Since all reactions contain the same amounts of the enzyme- and acridan-labeled antibodies, they can potentially develop equal signals upon addition of the same amount of the substrate. The specificity and the precision of the assay depend solely on the proximity effect, i.e. the instant flash luminescence occurring only when the labeled antibodies are simultaneously bound to an analyte molecule. This event is registered as a luminescence peak at <1 s after the addition of the trigger. The unbound antibodies will inevitably contribute to the signal; however, since they are further apart from each other, this process requires time for the diffusion of the radicals from the enzymatically converted substrate to the nearest acridan-labeled antibody. The unspecific luminescence in this case gradually develops over a few seconds. To maintain this difference, the substrate must be distributed instantly and evenly over the entire reaction volume to trigger the specific flash luminescence in milliseconds.For the realisation of the assay in a microplate, a laboratory microplate reader with the option for the reagent injection and simultaneous measurement of luminescence is required; moreover, the injection rate must be high enough to enable the instant mixing and should exceed 200 μl s−1 (according to communication with the specialists from Beckman Coulter). The turbulence in the reaction occurring at this injection rate benefits the mixing. In contrast, the intrinsic characteristic of microfluidic systems is laminar flow that naturally occurs in a microscale channel with a pressure-driven fluid actuation. Low flow velocities in microfluidic chips do not favor the integration of the described protocol on the chip. Nevertheless, a combination of a high injection rate and an optimal geometry of the reaction chamber should enable the transfer of the SPARCL™ assay. Therefore, evaluation of fluid mixing in different microfluidic designs was the first step in the development of SPARCL™ on a chip.
Various active and passive mixing strategies are described for lab-on-a-chip systems.2–4 The active mixers such as magnetic stirrers, ultrasound, electrokinetics and others would increase the complexity and the costs for microfluidics and the actuation device that is not rational for a simple assay like SPARCL™. The conventional passive mixers like Y/T, herring bone, zigzag/serpentine or twisted structures2–6 mostly utilize the crossing and recombining laminar liquid flows that are not compatible with the flash luminescence measurement. Embedded barriers can be more favorable for creating local vortices and contributing to the mixing of the liquids. For the instant mixing effect, the flow path must be very short. For this purpose, we have evaluated a mixing chamber chip with two directly interconnected chambers with a ledge barrier between them (Fig. 1).
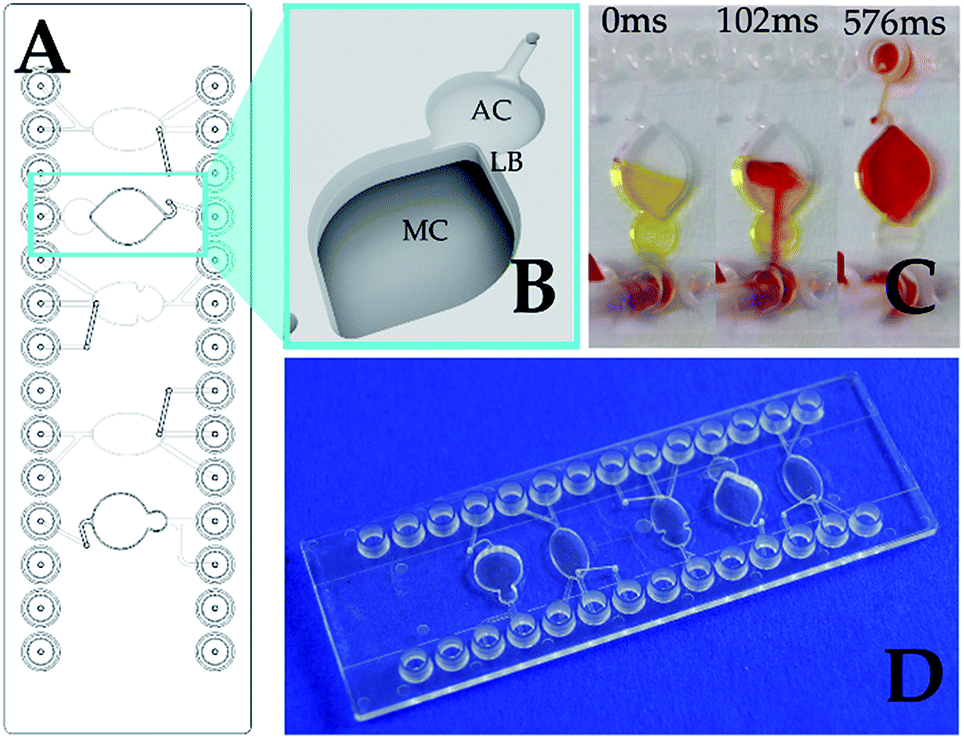 | ||
| Fig. 1 Passive mixing in the microfluidic chip with two interconnected chambers. (A) Macro view of the chip. The chip accommodates five independent fluidic structures; the relevant one is framed. (B) Detailed structure of the interconnected chambers with a ledge barrier. AC, smaller “atrium” chamber; MC, main chamber; LB, ledge barrier. AC/MC volumes: 5/45 μl. (C) Visualization of the mixing for the SPARCL™ assay, manual injection. Time after the injection (ms) is indicated above. (D) The microfluidic chip from the standard product portfolio of microfluidic ChipShop. | ||
The ledge barrier LB is formed due to the different depths (1.5 mm difference) and volumes of the chambers AC and MC shown in Fig. 1. The mixing was evaluated in visualization experiments with colored liquids. Twenty μl of the yellow fluid represented the sample and antibody mix and was loaded on the chip through the AC (Fig. 1C). Then 20 μl of red fluid, that simulated the trigger, was injected manually through the same AC chamber. Since the capacity of the “atrium” AC is significantly smaller than the entire reaction volume, the liquids flow into the adjacent larger chamber MC. The passing through the narrow interconnection area with the ledge supports the vortex mixing. Fig. 1C demonstrates the nearly complete rapid mixing of the yellow and red liquids in approximately 500 ms that fulfils the prerequisites for the effective integration of the SPARCL™ immunoassay on a chip.
Measurement of the flash luminescence on a chip
For the accurate quantitative readout in the SPARCL™ assay on a chip, it is essential to distinguish the specific and unspecific increases of the luminescence after the addition of the trigger. We compared the signal development in the chip luminometer for the chip measurements with the signal in a microplate reader. The specific luminescence peak (0–1 s) on the chip was very well defined, and the flash luminescence developed in the same rate and manner as it was recorded in a conventional assay in a microplate (Fig. 2).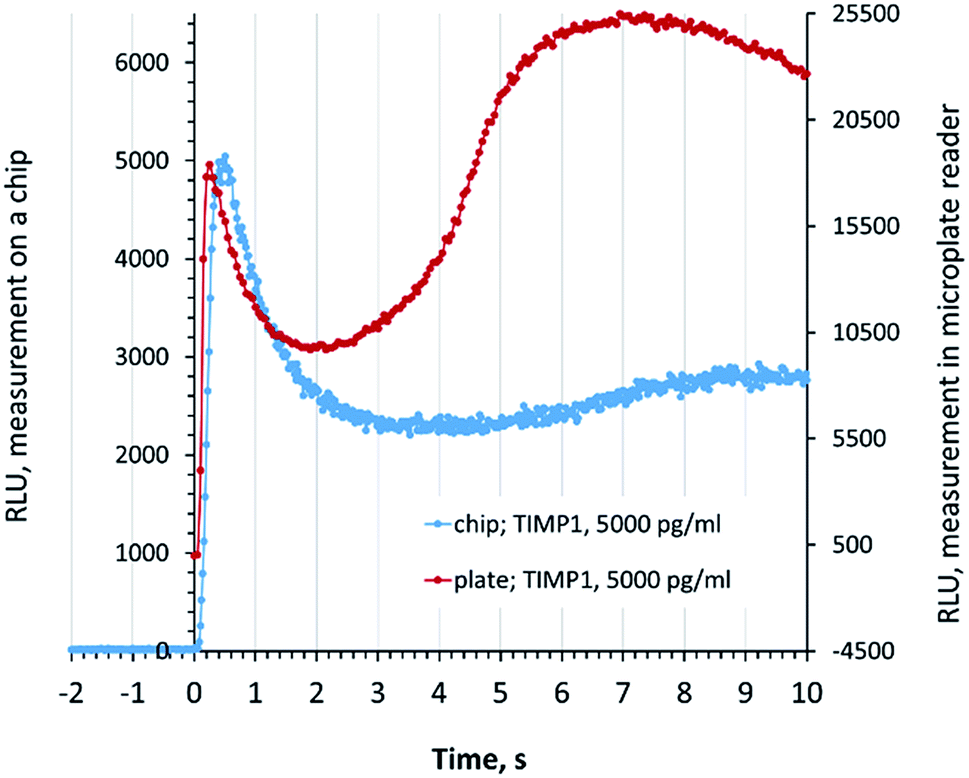 | ||
| Fig. 2 Development of the luminescence in the chip luminometer and the microplate reader. RLU, relative light units. Zero time point corresponds to the injection of the trigger, the first peak (<1 s) indicates the SPARCL signal, and the slowly developing peak indicates the unspecific luminescence. | ||
Fig. 2 also demonstrates the development of the unspecific background signal (1–10 s). These reactions are not promoted by the special proximity of the antibodies and require a longer diffusion of the chemical species. The visually higher background in the microplate-based assay can be explained by the different character of diffusion in a well of a 384-well plate in comparison to the flat reaction chamber of the chip where the development of the background luminescence continues also after the demonstrated 10 s period. These processes do not affect the interpretation of data since only the specific signal is assessed. We demonstrate the exemplary developments of the flash luminescence for various concentrations of analytes in Fig. S2 (ESI†).
In the next step, we evaluated the suitability and the precision of the luminescence measurements for the quantitative assay. The reactions were prepared and incubated off-chip in the vials (the volume of the antibody mix was scaled down from 20 μl to 10 μl while keeping the amount of the antibodies constant). The reactions were transferred to the chips for the measurements where the trigger was injected and the signals were recorded as described in the experimental section. A conventional bench protocol for SPARCL™ in a microplate was used as a reference here.
The on-chip luminescence measurements showed a good correlation with the levels of TIMP1 in the spiked buffer samples (Fig. 3). It was possible to achieve about a two-fold better sensitivity in comparison to the microplate assay. This improvement may be due to a more favorable combination of a lower reaction volume and the chip geometry. Yet the precision of the detection on the plate was obviously higher with the in-run variation not exceeding 10%, while this parameter varied for different biomarker concentrations on a chip but generally was always >10%. Our observations allow suggesting that higher signal deviations on the chip were caused by random, non-standardized distribution of the reaction mix upon its loading into the chamber. Unlike the conventional lab-on-a-chip approach where only reaction volume-fitted microfluidic structures are used, the SPARCL™ chamber is initially filled only to ∼50% of its volume to enable the subsequent injection of the trigger. This led sometimes to suboptimal effects like bubble formations or asymmetric liquid distribution. As an outlook, the optimization of the chip geometry would minimize these negative effects and allow for a better assay reproducibility. The same refers to the linearity range of the assay that was consistent over the tested TIMP1 concentrations (0–5000 pg ml−1) on a microplate but was limited to 2000 pg ml−1 on a chip. It was noticed that the samples with the highest protein concentrations (here: 5000 pg ml−1) were prone to prominent bubble formation or even foaming during the fluidic operations on the chip. This hampered the mixing of the sample with the trigger and resulted in an insufficient recovery of the analyte. The bench assay in a plate was not affected by this phenomenon. We recommend the dilution of the challenging high-protein samples for their analysis on the chip, and we successfully applied this strategy for the investigation of some real GCF samples.
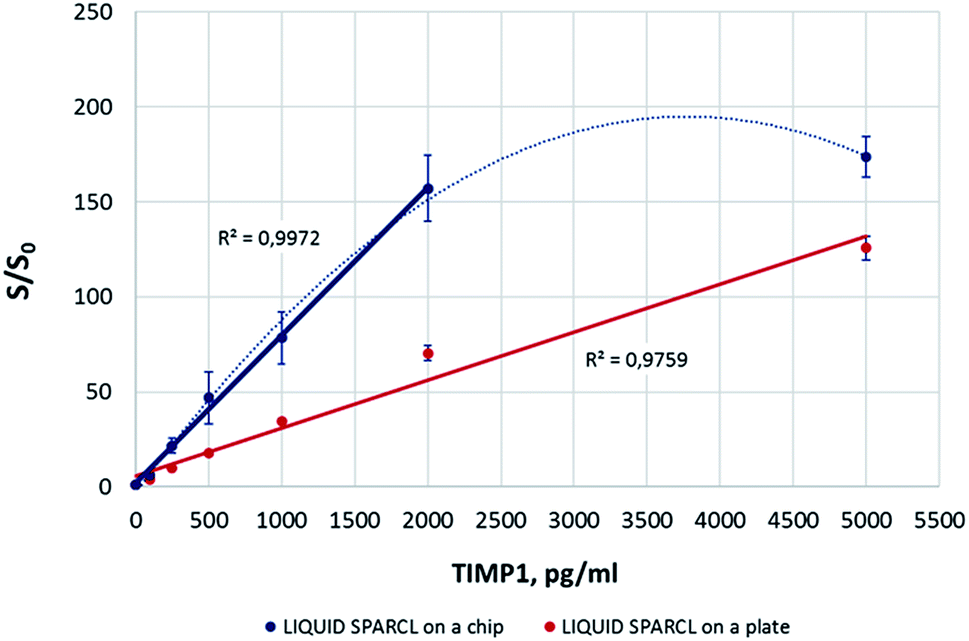 | ||
| Fig. 3 Quantitative measurement of TIMP1 on the chip using liquid reagents and off-chip preparation of reactions. Here and further, the error bars designate the standard deviation; number of replicates n = 3. | ||
Integration of the SPARCL™ assay on the chip
For the complete integration of the assay, the reagents for individual reactions must be readily provided on the chip. The lyophilization of the antibodies and BGR caused a significant drop of the assay sensitivity both in the microplate and chip format. Interestingly, the assay performances in the both formats were almost equal with a slightly better sensitivity on the plate (Fig. 4). This phenomenon might be explained by several consequences of the reagent preservation. Besides the expected reduced efficiency of the freeze-dried enzyme, some assay-specific factors must be taken into consideration.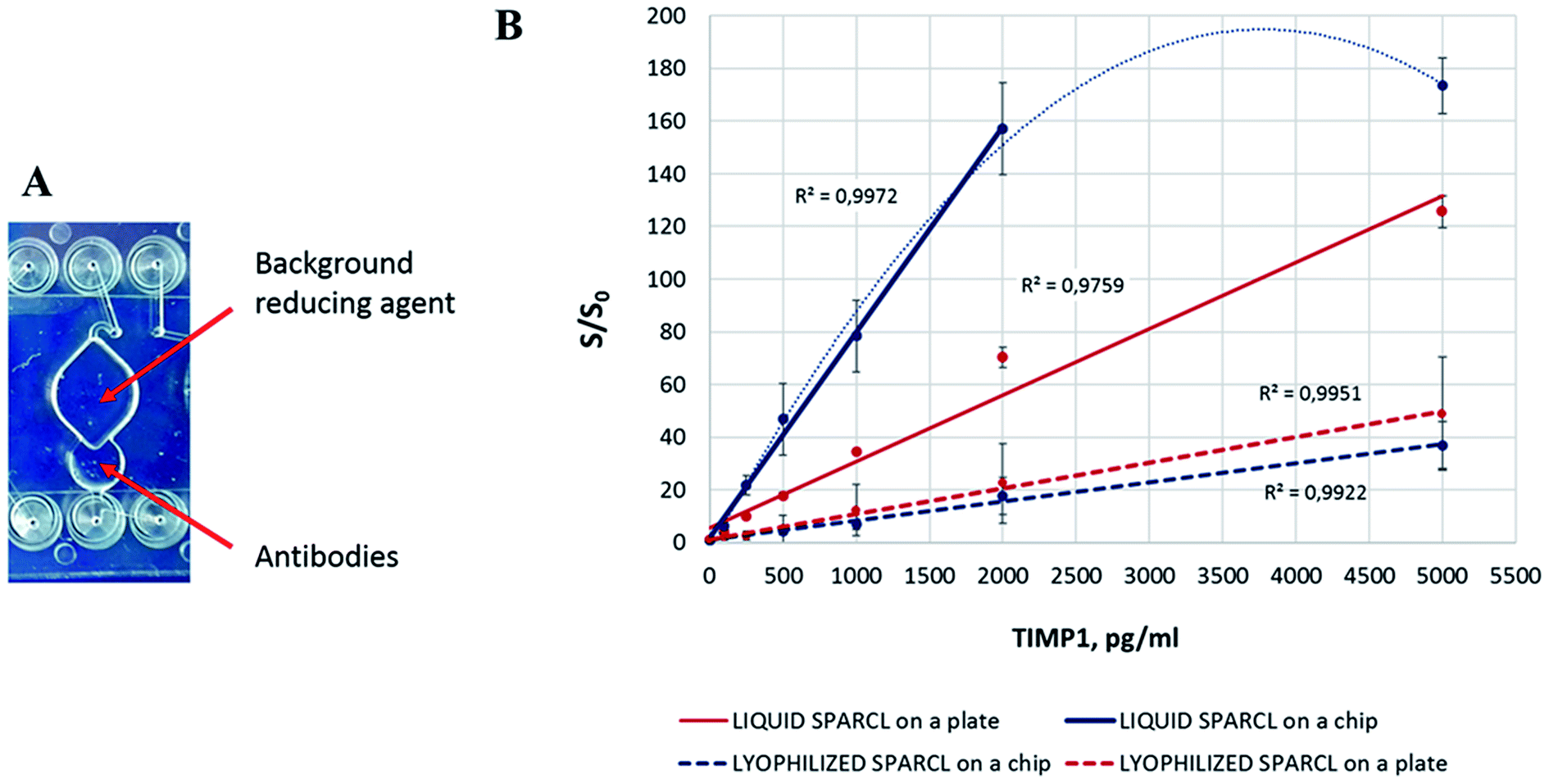 | ||
| Fig. 4 The effect of the reagent lyophilization on the performance of the SPARCL™ assay. (A) Deposition of the lyophilized reagents on the chip. (B) Assay performance with liquid and lyophilized reagents (number of replicates n = 3). | ||
First, little is known about the stability of the freeze-dried acridan label, and its partial inactivation may cause the reduction of the overall assay activity. Thus, Tafti et al. warned about the possible consumption of acridan compounds in side reactions that do not produce light.7 These competing reactions might occur between the chemical species in the buffered antibody formulation or even upon the contact with ambient air before and during the lyophilization. They will decrease the number of the effective interactions and the amount of the produced light in SPARCL.
Second, both labeled antibodies were mixed together for the lyophilization, which exposes the acridan-labeled antibody to HRP prior to the assay. The mechanism of the enzymatic oxidation of acridan is quite complex and needs not only peroxide as the substrate for HRP, but also an obligatory enhancer (e.g. coumaric acid or p-iodophenol, among others) that serves as a mediator for the reactive oxygen species in the reaction.7–9 Both compounds are introduced into the reaction mix only during the injection of the trigger. Although these facts imply that the likelihood of the direct interaction of acridan with the peroxidase is rather low, the enzymatic generation of the reactive oxygen species during the preparation of the chips cannot be excluded. One of the possible mechanisms is the interaction of the chip material (COP) with the enzyme. Some of the manufacturing procedures involve a surface activation of COP, which may lead to the formation of the peroxide groups on the surface.10 This may initiate unspecific reactions in the antibody mix and lead to a preterm inactivation of some acridan labels or cause other types of oxidative damage to the reagents. Under certain conditions, HRP can also generate reactive oxygen species in a peroxide-independent manner;11–14 nevertheless, the contribution of such interactions is unlikely or low in a controlled environment and sufficient reagent purity.
Third, an important change in the assay protocol follows from the complete pre-storage of the reagents on the chip or plate. Namely, BGR, which in the original bench protocol is added after the sample incubation with the antibody mix, now dissolves in the sample-antibody mix during the incubation. BGR is a compound with antioxidant properties; one of the effective BGR agents is for instance ascorbic acid.7 BGR inhibits the background light-producing enzymatic reactions while keeping the specific SPARCL™ luminescence relatively unaffected due to the high rate of proximity reactions. Therefore, it significantly increases the signal-to-blank ratio, i.e. the sensitivity of the assay. At the same time, ascorbate can interfere with the peroxidase-catalysed reactions by different mechanisms including the production of radicals in auto- or enzyme-driven oxidation.15 Hence, both the BGR concentration and the duration of its presence in the reaction must be balanced. The prolonged incubation of the SPARCL™ mix with BGR on a chip can lead to the decreased S/S0 ratio, as we observed. Yet this issue can be resolved by at least three approaches. (a) BGR can be added in liquid form in a separate injection; yet this approach demands more fluidic connections and instrumental manipulations and makes the integrated assay less feasible. (b) A special formulation of lyophilized BGR (e.g. adding a protective polymer layer or carrier that slows down the reconstitution of the dried agent; using a chemically masked BGR compound, etc.) can maintain its controlled release into the reaction during the incubation time. We assume that this or a similar strategy is used for the special SPARCL™ plates provided within the diagnostic kits (Life Diagnostics, PA, USA). (c) Optionally, an alternative design of the microfluidic chip can spatially separate the sample incubation with the antibodies and its subsequent mixing with BGR.
We modelled the latter strategy on the available chip. The antibodies were lyophilized in a spare chamber where the liquid sample was loaded (Fig. 5A). After the incubation, the sample was transferred with a pipette to the measurement chamber with the dried BGR. Thus, the sample was exposed to BGR straight before the measurement as prescribed in the conventional protocol.
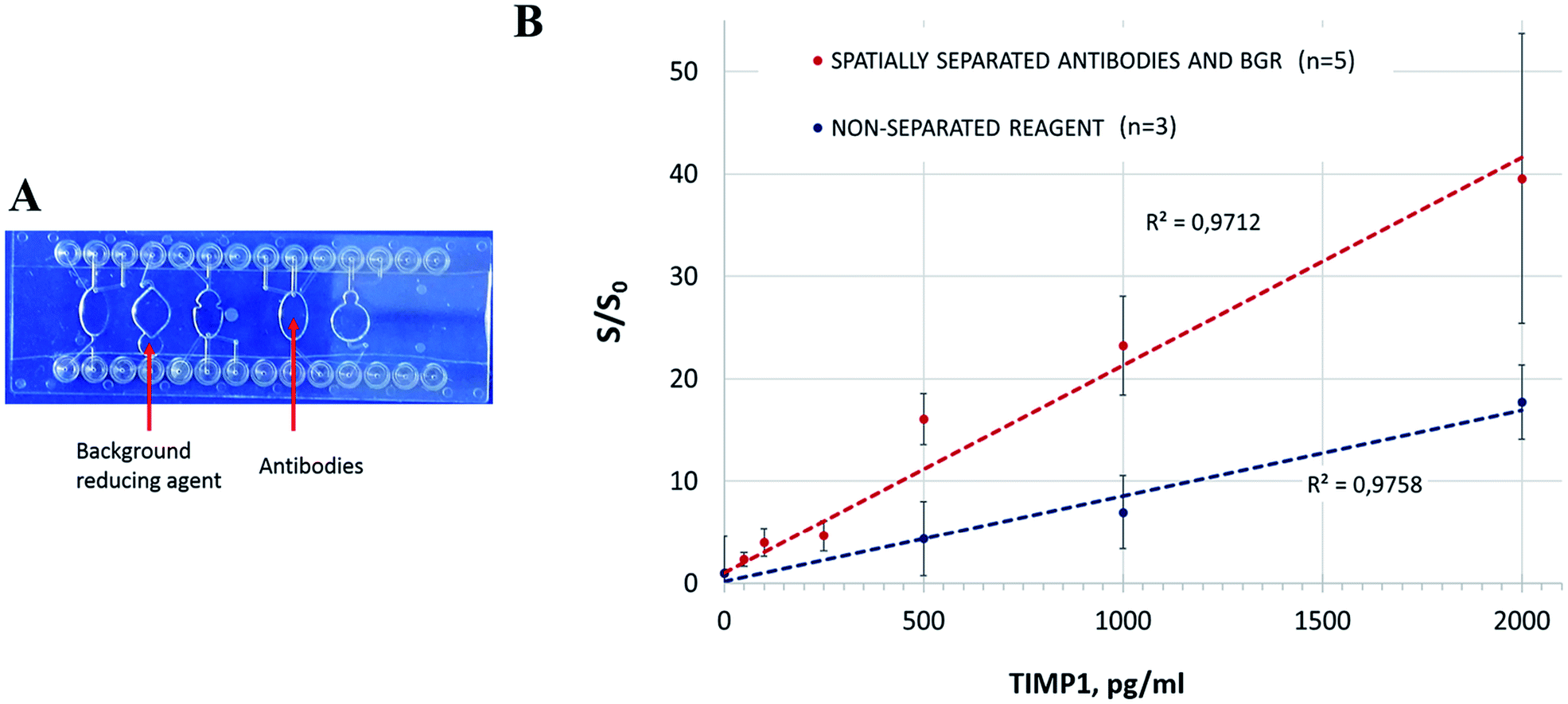 | ||
| Fig. 5 Spatial separation of antibody mix and BGR on a chip. (A) Deposition of the lyophilized reagents on the chip. (B) Assay performance in comparison to non-separated reagent lyophilization (“incubation with BGR”). n – number of replicates. | ||
The spatially separated incubation of the sample with the antibodies and BGR resulted in higher S/S0 values in comparison to the former strategy with the simultaneous reconstitution of the reagents in the reaction mix (Fig. 5B). The manual transfer of the reaction mix between the chambers resulted however in significant deviations. It is apparent that the manual transfer of the sample in a lab-on-a-chip device is not acceptable, and we demonstrated it here solely to confirm the need for the spatial separation of the lyophilized reagents and the corresponding assay steps. The chip design and/or the deposition of the reagents have to be revised accordingly: ideally, the BGR should be provided in the same incubation chamber, but its release into the solution has to be controlled to prevent its preterm effect on the assay. A different reagent formulation (as earlier mentioned here) would be an advantageous option. The spatial separation of the assay steps, while being effective, would require additional chip structures and precise liquid transfer control that would lead to the unnecessary complex concept of integration.
The integrated SPARCL™ assay could detect as little as 49.3 pg ml−1 (1.8 pM) TIMP1 (ESI Table 1, Fig. S3†). The limit of quantification was 157.6 pg ml−1 (5.6 pM) (see Section 2 of ESI†). In order to improve reproducibility, an optimization of the chip design and reagent preparation will be carried out.
We assume that the high standard deviations of the assay are a result of the two factors: (1) incomplete biomarker and/or reagents recovery due to the influence of the chip surface, (2) suboptimal reproducibility of sample distribution on the chip (e.g. foaming of samples with high protein content). Both factors can be compensated in further development of the chip (surface passivation, chamber geometry) and the assay procedure itself (e.g. sample dilution or elution in higher volume to reduce the viscosity of the matrix).
Application of the integrated SPARCL™ for the TIMP1 quantification in gingival crevicular fluid
We evaluated the suitability of the integrated assay for the detection of TIMP1 in biological samples. TIMP1 plays a role in the integrity of the extracellular matrix and is therefore a biomarker for the pathological processes associated with its alteration or degeneration.16,17 Periodontitis, an inflammatory pathology of the tooth supporting tissues, is one of the examples of such diseases. The levels of TIMP1 in GCF together with other biomarkers reflect the severity of the degradation of the connective tissue and can be used for the diagnostics and monitoring of periodontitis.The clinically relevant concentrations of TIMP1 in GCF cannot be specified in absolute values as they strongly depend on the sampling method, elution volume, and measurement procedure itself. However, TIMP1 levels are of clinical significance for the monitoring of periodontitis and its therapy: their measurements before, during and after therapy provide dentists with valuable information on the stage of disease, as the concentrations of this inhibitor of metalloproteinase decrease along with the progression of the tissue degeneration.16,17 Thus, Reddy et al. determined 1.658 ng ml−1 of TIMP1 in periodontitis (mean), while the healthy patients had 8.623 ng ml−1;18 Kumar et al. reported that in the chronic periodontitis group TIMP1 increased from 1.592 ng ml−1 to an after treatment level of 6.408 ng ml−1;19 Ghodpage et al. determined 12.88 ng ml−1, 20.46 ng ml−1 and 25.01 ng ml−1 before, after therapy, and in healthy controls respectively,20 while Popat et al. measured 113.65 ng ml−1 in periodontitis and 351.46 ng ml−1 in the control.17 In our work, we used TIMP1 as a model biomarker aiming additionally at the sub-nanogram range of detection for the evaluation of the SPARCL™ on a chip capabilities, as some other cytokine oral biomarkers like interleukin (IL)-1 beta, IL-8, or monocyte chemoattractant protein (MCP-1) require detection in the pg ml−1 range.21
We measured TIMP1 levels in the limited number of GCF samples of healthy volunteers in the integrated SPARCL™ on a chip in comparison to the bench assay version. The direct measurement resulted first in the high luminescent signal out of the assay linearity range (data not shown). Dilution of the samples 1:10 allowed for the effective detection and quantification of the biomarker (with the remark about the previously discussed insufficient precision) (Fig. 6). The integrated assay generally correlated with its bench version; however, the recovery of the biomarker on a chip was incomplete and resulted in the detection of 2.2 ± 1.6 ng ml−1 less than in a microplate, except for the sample S2. The recovery of the proteins on the chip must be further investigated; a surface passivation strategy e.g. with albumin or skim milk can be beneficial.
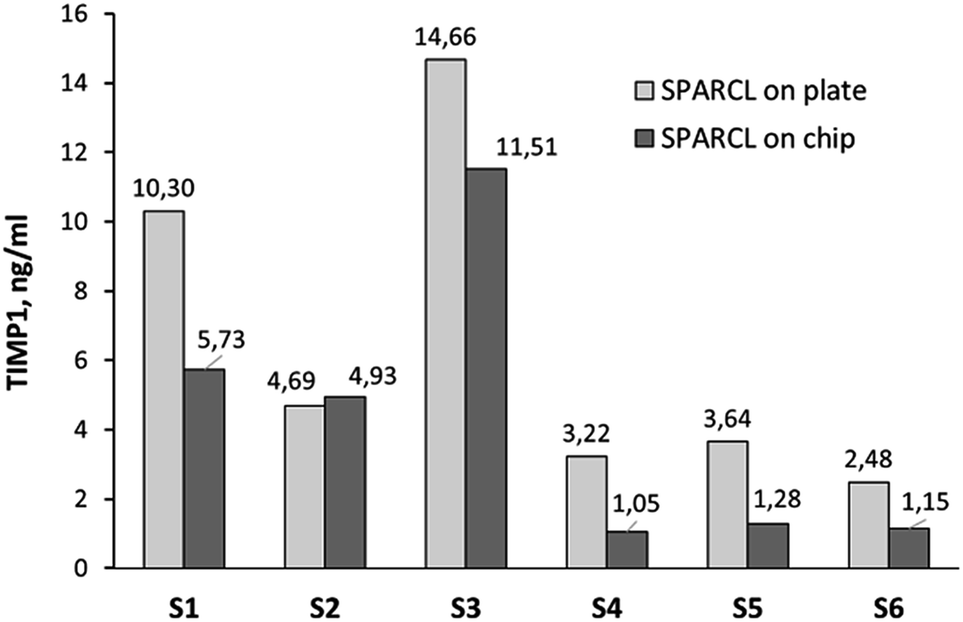 | ||
| Fig. 6 TIMP1 levels in the GCF of healthy volunteers measured by integrated SPARCL (single measurement per sample: n = 1). | ||
Despite the revealed demand for the optimization, the integrated SPARCL™ assay was suitable for the quick, very simple, and sensitive quantitative detection of biomarkers in clinical samples. The integration of the immunoassays into lab-on-a-chip devices has remained challenging over the past years since it was mainly based on the heterogeneous sandwich assays and inevitably required separation and washing steps, storage of multiple reagents on a chip, complex liquid actuation and processing of large reagent volumes.22–25 The homogeneous assays can revolutionize rapid diagnostics, but for the handling feasibility, their integrated format must implement no complex cartridges or instruments and ideally function according to a “mix-and-read” protocol. One of the popular approaches to this is based on fluorescence resonance energy transfer (FRET), yet such assays are generally compatible only with small molecule detection due to the limitation for the FRET distances.26,27 The main advantage of SPARCL over the FRET-based assays is its virtual independence on the size of the analytes. The SPARCL signal is triggered by reactive oxygen species (ROS). They can travel at least 200 nm in aqueous solution before decay.28,29 This distance exceeds the average size of a protein (e.g. 10–15 nm for an antibody), thus allowing proximity interactions within the sandwich (analyte + labeled antibodies), but restricting the unspecific long–distance interaction. With the given travel distance of ROS, it becomes clear that the assay can be applicable to a variety of biomolecules. Nowadays, very few homogeneous quantitative immunoassays on a chip are reported for the protein biomarkers. Tak For Yu et al. demonstrated an integration of another commercial technology, AlphaLISA®, with a limit of detection of 10 pg ml−1.30 However, AlphaLISA® is a fastidious assay in terms of reagent handling (the beads require even distribution; preservation of the dried beads on the chip was not demonstrated; the chip had a complex design with integrated microvalves and micropumps) and detection (a custom optical setup based on a fluorescent microscope was used for the readout). This complexity might preclude AlphaLISA® from the wide implementation in the integrated format.
Liu et al. published a much simpler homogeneous assay based on the analyte concentration-dependent aggregation of the gold nanoparticles (GNPs).31 Yet for the growth of the GNP aggregates, a chemically aggressive reagent, HAuCl4, is necessary, and its deposition on the chip or manual handling by lay users might be unfavorable. The detection limit of the assay for human IgG was 3.74 ng ml−1 and 5.66 ng ml−1 for carcinoembryonic antigen. Many biomarkers require a sub-nanogram sensitivity where this assay will be insufficient. Other examples of GNP-based homogeneous immunoassays could not surmount this sensitivity challenge; e.g. Byun et al. demonstrated a 10 ng ml−1 detection limit for C-reactive protein, and Andresen et al. could determine low nanomolar antibody concentrations in the sample that corresponds to a high ng ml−1 to μg ml−1 range.31–33
Thus, the integrated SPARCL™ assay offers not only simplicity and a high level of integration, but also a remarkably superior sensitivity (pg ml−1, or picomolar range) among the mix-and-read assays.
SPARCL™ can be used for the measurement of biomarkers in other types of samples, yet the impact of the sample matrix must be evaluated in advance. Thus, for the measurements in serum, it is recommended to dilute the specimen at least 8-fold in order to avoid matrix effects.34 A detailed investigation of the assay performance in different body fluids is a subject for future work.
Conclusions
We have developed a fully integrated version of a homogeneous immunoassay based on the SPARCL™ technology. This is the first demonstration of this technology in a lab-on-a-chip format. Due to the short and simple assay protocol, the user only needs to load the sample onto the cartridge and connect it to the injector. Such an advantageous processing enables quantitative detection of various biomarkers without the bulky and expensive equipment and infrastructure for the conventional ELISA; furthermore, the instrumental setup for the luminescence measurement on the chip is also cost-saving in comparison to the conventional laboratory readers. In the outlook, we envisage further optimization of the chip design that would substantially improve the precision of quantitative measurements.Ethical statement
Informed consent was obtained from all subjects who provided GCF samples for the demonstration of the performance of the method in clinical samples.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was funded by the German Federal Ministry for Economic Affairs and Energy (BMWi) within the Central Innovation Programme SME; ZIM-project DentiRisk-Scan, reference number KF 2302710CR4 (Fraunhofer IZI) and KF 2223905 (Mildendo GmbH). We also gratefully appreciate the assistance of Becit GmbH who coordinated the project and kindly provided the paper strips and paper points for the sampling of gingival crevicular fluid.Notes and references
- J. E. Butler, J. Immunoassay, 2000, 21, 165 CrossRef CAS PubMed.
- C.-Y. Lee, C.-L. Chang, Y.-N. Wang and L.-M. Fu, Int. J. Mol. Sci., 2011, 12, 3263 CrossRef CAS PubMed.
- K. Ward and Z. H. Fan, J. Micromech. Microeng., 2015, 25, 094001 CrossRef PubMed.
- L. Capretto, W. Cheng, M. Hill and X. Zhang, Top. Curr. Chem., 2011, 304, 27 CrossRef CAS PubMed.
- P. E. Neerincx, R. P. J. Denteneer, S. Peelen and H. E. H. Meijer, Macromol. Mater. Eng., 2011, 296, 349 CrossRef CAS.
- M. K. Verma, S. R. Ganneboyina, R. Vinayak Rakshith and A. Ghatak, Langmuir, 2008, 24, 2248 CrossRef CAS PubMed.
- H. Akhavan-Tafti, Z. Arghavani, and R. de Silva, US005723295A, 1998.
- A. M. Osman, G. Zomer, C. Laane and R. Hilhorst, Luminescence, 2000, 15, 189 CrossRef CAS PubMed.
- H. Akhavan-Tafti, D. G. Binger, J. J. Blackwood, Y. Chen, R. S. Creager, R. de Silva, R. A. Eickholt, J. E. Gaibor, R. S. Handley, K. P. Kapsner, S. K. Lopac, M. E. Mazelis, T. L. McLernon, J. D. Mendoza, B. H. Odegaard, S. G. Reddy, M. Salvati, B. A. Schoenfelner, N. Shapir, K. R. Shelly, J. C. Todtleben, G. Wang and W. Xie, J. Am. Chem. Soc., 2013, 20, 4191 CrossRef PubMed.
- F. Soberon, Modell. Simul. Mater. Sci. Eng., 2014, 22, 055020 CrossRef.
- H. Mehlhorn, M. Lelandais, H. G. Korth and C. H. Foyer, Ascorbate is the natural substrate for plant peroxidases, FEBS Lett., 1996, 378(3), 203–206 CrossRef CAS PubMed.
- M. Kimura, Y. Umemoto and T. Kawano, Front. Plant Sci., 2014, 5, 285 Search PubMed.
- M. Kimura and T. Kawano, Plant Signaling Behav., 2015, 10, 1000145 CrossRef PubMed.
- S. Palmieri and F. Giovinazzi, Physiol. Plant., 1982, 56, 1 CrossRef CAS.
- R. H. White-Stevens, Clin. Chem., 1982, 28, 578 CAS.
- T. Alpagot, C. Bell, W. Lundergan, D. W. Chambers and R. Rudin, J. Clin. Periodontol., 2001, 28, 353 CrossRef CAS PubMed.
- R. P. Popat, N. V. Bhavsar and P. R. Popat, Singapore Dent. J., 2014, 35, 59–64 CrossRef PubMed.
- N. R. Reddy, A. Deepa, D. S. M. Babu, N. S. Chandra, C. V. S. Reddy and A. K. Kumar, J. Indian Soc. Periodontol., 2014, 18, 301 CrossRef PubMed.
- P. M. Kumar, N. R. Reddy, A. Deepa, D. S. M. Babu, A. K. Kumar and V. Chavan, Dent. Res. J., 2013, 10, 434 Search PubMed.
- P. S. Ghodpage, R. A. Kolte, A. P. Kolte and M. Gupta, Saudi Dent. J., 2014, 26, 171 CrossRef PubMed.
- D. Belstrøm, C. Damgaard, E. Könönen, M. Gürsoy, P. Holmstrup and U. K. Gürsoy, J. Oral Microbiol., 2017, 9, 1364101 CrossRef PubMed.
- A. H. Ng, U. Uddayasankar and A. R. Wheeler, Anal. Bioanal. Chem., 2010, 397, 991 CrossRef CAS PubMed.
- J. Park, V. Sunkara, T. H. Kim, H. Hwang and Y. K. Cho, Anal. Chem., 2012, 84, 2133 CrossRef CAS PubMed.
- G. Wang, C. Das, B. Ledden, Q. Sun and C. Nguyen, SLAS Technol., 2017, 22, 518 Search PubMed.
- K. Cheng, W. Zhao, S. Liu and G. Sui, Biomed. Microdevices, 2013, 15, 949 CrossRef CAS PubMed.
- T. Yokozeki, H. Ueda, R. Arai, W. Mahoney and T. Nagamune, Anal. Chem., 2002, 74, 2500 CrossRef CAS PubMed.
- T. Pulli, M. Höyhtyä, H. Söderlund and K. Takkinen, Anal. Chem., 2005, 77, 2637 CrossRef CAS PubMed.
- R. M. Eglen, T. Reisine, P. Roby, N. Rouleau, C. Illy, R. Bossé and M. Bielefeld, Curr. Chem. Genomics, 2008, 1, 2 CrossRef CAS PubMed.
- F. J. Schmitt, G. Renger, T. Friedrich, V. D. Kreslavski, S. K. Zharmukhamedov, D. A. Los, V. V. Kuznetsov and S. I. Allakhverdiev, Biochim. Biophys. Acta, 2014, 1837, 835 CrossRef CAS PubMed.
- Z. Tak For Yu, H. Guan, M. Ki Cheung, W. M. McHugh, T. T. Cornell, T. P. Shanley, K. Kurabayashi and J. Fu, Sci. Rep., 2015, 5, 11339 CrossRef CAS PubMed.
- H. Liu, P. Rong, H. Jia, J. Yang, B. Dong, Q. Dong, C. Yang, P. Hu, W. Wang, H. Liu and D. Liu, Theranostics, 2016, 6, 54 CrossRef CAS PubMed.
- J. Y. Byun, Y. B. Shin, D. M. Kim and M. G. Kim, Analyst, 2013, 138, 1538 RSC.
- H. Andresen, M. Mager, M. Griessner, P. Charchar, N. Todorova, N. Bell, G. Theocharidis, S. Bertazzo, I. Yarovsky and M. M. Stevens, Chem. Mater., 2014, 26, 4696 CrossRef CAS.
- Skeletal Muscle Troponin-C SPARCL™ Assay, Product information sheet, Life Diagnostics, Inc.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay00198k |
| This journal is © The Royal Society of Chemistry 2019 |