
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of cellular vitamin C dynamics by HPLC-DAD†
Taiki
Miyazawa
 a,
Akira
Matsumoto
*ab and
Yuji
Miyahara
a
a,
Akira
Matsumoto
*ab and
Yuji
Miyahara
a
aInstitute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: matsumoto.bsr@tmd.ac.jp
bKanagawa Institute of Industrial Science and Technology (KISTEC-KAST), Kawasaki 213-0012, Japan
First published on 19th March 2019
Abstract
A redox-sensitive inter-conversion between ascorbic acid (ASC) and its oxidized form dehydroascorbic acid (DHA) in the intracellular environment has been of exceptional interest to recent metabolomics and pharmaceutical research. We developed a chromatographic protocol to instantly determine these vitamers with each identity from cellular extracts, without any labeling and pretreatments. Owing to its simplicity, one can readily continue the assay for hours, an otherwise difficult to cover timescale at which the intracellular DHA–ASC conversion comes into play. The method was validated for the analysis of pancreatic cancer cells, to our knowledge the first-ever study on a nucleated cell type, to trace in detail their kinetics of glucose transporter-dependent DHA uptake and, simultaneously, that for the intracellular ASC conversion. The simplest of all the relevant techniques and yet with the unique ability to provide each vitamer identity on a high-throughput basis, this method should offer the most practical option for VC-involved physiological and pharmaceutical studies including high-dose VC cancer therapy.
Vitamin C (VC) is one of the most fundamental nutrients for sustaining physiology.1 It involves several vitamers that are inter-convertible depending on the redox state, among which ascorbic acid (ASC) and its oxidized form dehydroascorbic acid (DHA) represent two dominant species.2,3 In human plasma, there is a homeostatic mechanism due to the role played by erythrocytes (to reduce DHA to ASC) to maintain the relative abundance of these key vitamers, i.e., 95% ASC and 5% DHA, the disorder of which has been implicated in diabetes and atherosclerosis.4–7 In organs, VC is much more abundant and accumulating evidence shows that the spatiotemporal pattern of this vitamer pair underlies both specificity and kinetic aspects in some important cellular events. For example, the ASC–DHA dynamics between neurons and astrocytes is known to determine the rate of their VC recycling process to create a neuron-specific anti-oxidant microenvironment.8 Another recent study focusing on the ASC–DHA dynamics in human colorectal cancers harboring KRAS or BRAF mutations has provided a mechanistic understanding of the mutation-specific antitumor activity of DHA, reawakening research interest in such VC based cancer therapy.9 Thus, exploring cellular ASC–DHA dynamics has been of exceptional interest at the forefront of metabolomics and pharmaceutical research.
Isotope labeling and mass-spectroscopic techniques are two gold-standard methods to monitor the dynamics of these vitamers.10–12 However, these methods inevitably involve costly instrumentation and complicated parameter optimization. A more cost-effective and readily accessible alternative is based on high performance liquid chromatography (HPLC) coupled with diode array UV detection (HPLC-DAD). Nonetheless, a striking similarity in size and polarity between the pair along with the fact that UV absorption of DHA is much weaker compared to ASC (see the ESI – Fig. S2†) makes this technique poorly reliable, especially for discrimination purposes.12 Alternatively, taking advantage of such an almost “DHA-insensitive” feature, reduction pretreatment using glutathione or dithiothreitol to convert DHA into ASC has been widely attempted; the initial amount of DHA in the sample can then be estimated from an increase in the total VC amount (DHA + ASC) after the treatment.13,14 However, this indirect assay (hereinafter referred to as the “reduction method”) lacking true specificity is prone to interference by other biological compounds and can be feasible only under the premise of quantitative DHA–ASC conversion, which is not always the case; in fact, we found that the presence of a VC stabilizer (metaphosphoric acid: MPA) and alternation in pH can severely interfere with the reductant's efficiency (Fig. 1i). Here we report a modified and remarkably simple chromatographic (HPLC-DAD) protocol which enables, to our knowledge for the first time, simultaneous analysis of the cellular DHA–ASC pair dynamics with each identity in absolute quantity, thus eliminating the abovementioned pre-treatment (“reduction method”). Our protocol (hereinafter referred to as the “direct method”) commences with a cellular VC extraction process using MPA, a commonly used reagent in the food science community. This treatment not only warrants the stability of VC in the sample but also provides ease of hemocyte and protein separations simply by the subsequent centrifugation. Hitherto, however, there is no reported protocol harnessing MPA as the cellular VC extraction reagent because of its severe interference with the “reduction method” (Fig. 1i); instead, existing protocols usually involve extraction using organic solvents such as methanol.9,14 Our MPA-compatible and organic solvent-free protocol, therefore, provides striking advantages over the “reduction method” in terms of the robustness of measurement and sample storage as well as the ease of purification. The simultaneous DHA–ASC measurement was then enabled by tuning the mobile phase in a mixed mode column (Primesep SB, SIELC Technologies, IL, USA) to effectively differentiate the elution time characteristic of each vitamer in a manner not overlapping with those for MPA and any other cell lysed components, and by choosing a single detection wavelength of 240 nm (Fig. 1), because if we use 210 nm as the detection wavelength for DHA, unknown contaminants derived from cells would be detected (see the ESI – Fig. S6†). We show here that such a “direct method” can be readily applied to in vitro assay both on erythrocytes (see the ESI – Fig. S1†), the most extensively studied target, and on a pancreatic cancer cell line (MIA PaCa-2), to our knowledge the first-ever study on nucleated cell types, to trace in detail their GLUT1 (glucose transporter)-dependent or DHA-specific cellular internalization and, concomitantly, time- and dose-dependent intracellular conversion into ASC. The essence of our chromatographic technique (“direct method”) is summarized in Fig. 1. As demonstrated in Fig. 1a and b, two vitamer peaks, i.e., ASC and DHA, become well separated in the elution time under our established mobile phase condition, and neither of these overlaps with those stemming from MPA. These peak identities can be verified from their DAD spectra obtained at each assigned elution time (6.95 min for DHA and 9.11 min for ASC: Fig. 1c and d), which are in agreement with those of the standards (see the ESI – Fig. S2†) and previous reports.12Fig. 1g provides further support for these peak assignments; this analysis on a standard mixture (1 nmol ASC + 1 nmol DHA: red line) reveals that the intensities of both ASC- and DHA-assigned peaks change without tailing in a quantitative fashion as a result of reduction treatment (see also the ESI – Fig. S5†). The linearity of the calibration for each component, i.e., ASC and DHA, is found in the range of 0.001–10 nmol (R = 0.9993) and 0.1–200 nmol (R = 0.9996), with the detection limits being 0.109 pmol and 0.011 nmol, respectively (Fig. 1e and f). Fig. 1h provides a comparison between “direct” and “reduction” methods in the quantification of a standard mixture containing both DHA and ASC at identical 5 mM concentrations, demonstrating their marked relativity (see also the ESI – Tables S1 and S2†). More importantly, this relativity was lost in the presence of MPA (Fig. 1i), which negatively affects the reduction efficiency, leading to substantially underestimated quantification results for the “reduction method” as compared to the “direct method”. The same was true for the erythrocyte DHA-uptake experiment where VC had been extracted using MPA (see the ESI – Fig. S4†). Furthermore, the values of the recovery test on erythrocytes, i.e., spiking and recollecting each VC component, ranged as high as 90% (see the Experimental section), justifying no requirement for the internal reference for the measurement. These observations thus highlight a virtue of our MPA-compatible methodology to secure reliable measurement and sample storage.
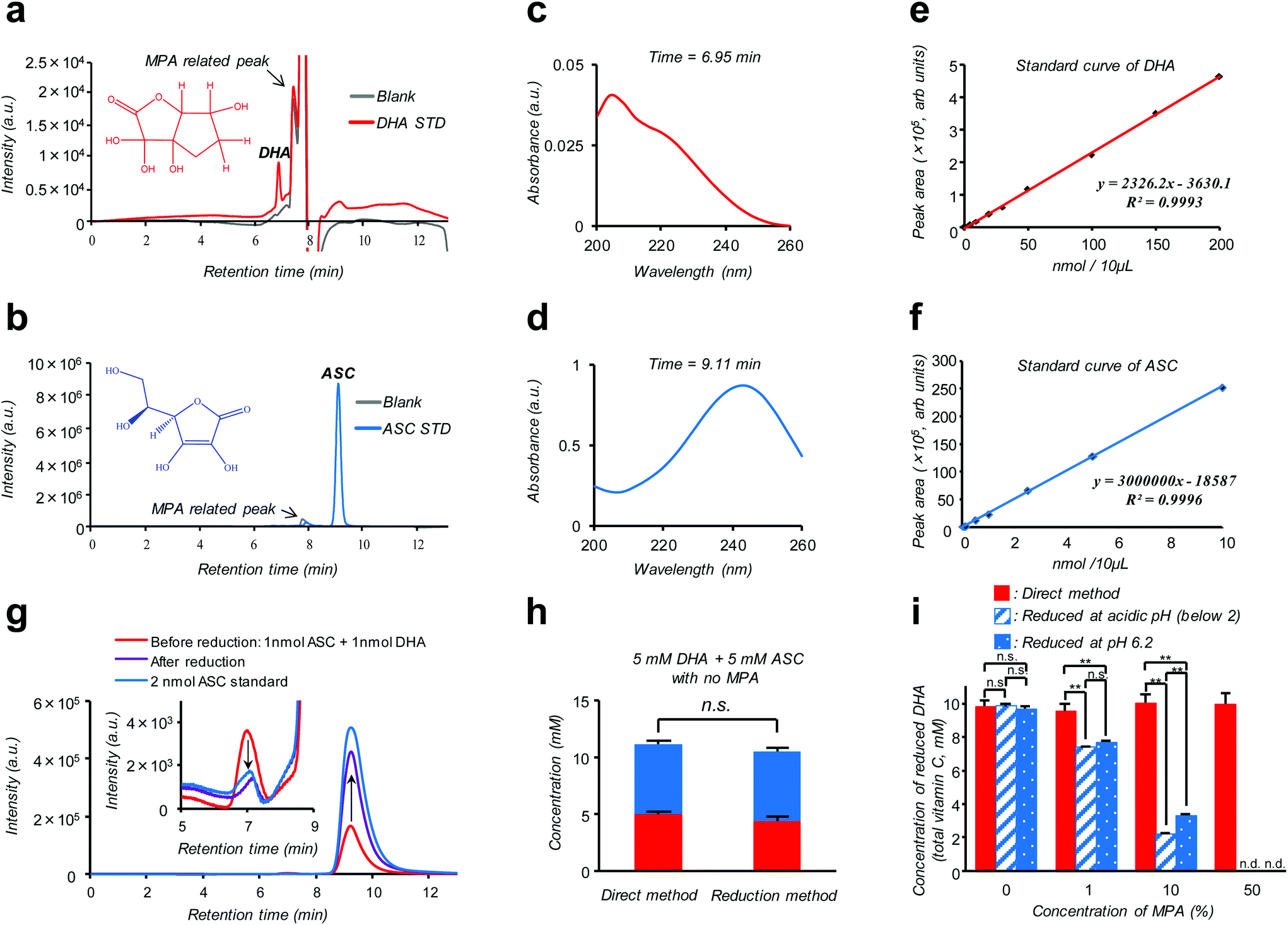 | ||
| Fig. 1 The “direct method” enables simultaneous determination of DHA and ASC using a MPA-compatible HPLC-DAD protocol. a, b; HPLC-DAD chromatograms of 100 nmol DHA (a: red) and 5 nmol ASC (b: blue) standards in aqueous solutions containing 5% MPA (stabilizer), 0.1% formic acid and 2 mM ammonium acetate, analyzed at a wavelength of 240 nm with a flow rate of 0.5 mL min−1. Chromatograms of blank solutions (without DHA or ASC) are shown with gray coloured lines. c, d; DAD spectra obtained for the elutes assigned to DHA (6.95 min in a: red) and ASC (9.11 min in b: blue), respectively. e, f; Standard calibration curves for DHA (e: red) and ASC (f: blue) as determined by their assigned peak area plots as a function of concentration. g; Overlay of HPLC-DAD chromatographs for a standard DHA–ASC mixture (containing 1 nmol of each) before (red) and after (purple) the reduction treatment in the absence of MPA. Chromatograph of 2 nmol ASC standard is shown with a blue coloured line. The inlet focuses on the DHA peak area with an enlarged scale. h; Comparison of the two methods, (left) “direct” and (right) “reduction” methods, in the quantification of a standard DHA–ASC mixture (containing 5 mM of each). Data are presented as means ± SD, n = 3. i; Comparison of protocol reliability; quantification results for 10 mM DHA standard measured by the “direct method” (red), “reduction method” at pH 6.2 (blue stripe) and “reduction method” at acidic pH (below 2) (blue dot). These comparisons reveal the negative effects of MPA and acidic pH on the reduction efficiency. Data are presented as means ± SD, n = 3. Means significantly differed at **P < 0.01. | ||
Amongst many efforts of investigating cellular VC-uptake kinetics primarily focusing on erythrocytes, few of them, despite the importance, have attempted to measure the intracellular DHA–ASC pair dynamics with each identity. To the best of our knowledge, a study by Tu et al. on erythrocytes is so far the only example that achieved this goal, based on a combined technique of isotope-labeling and the “reduction method”.16 Here we demonstrate the first successful experiment on a nucleated cell type, i.e., pancreatic cancer cells (MIA PaCa-2). MIA PaCa-2 cells were obtained from the American Type Culture Collection (VA, USA). Displayed in Fig. 2a–e are the results of our “direct method” analysis on MIA PaCa-2 cells, revealing their kinetics of DHA uptake (red squares) and that for the intracellular conversion into ASC (blue circles) for various DHA-exposure conditions (see also Fig. S6†). It can be observed that MIA PaCa-2 cells initially undergo rapid DHA internalization, maximizing at 10–30 min post-exposure, which then declines over time. In line with this, the intracellular ASC amount increases at a scale that is roughly one order of magnitude smaller than that for DHA. The apparent loss in the total VC amount can be ascribed to those converted into other irreversible DHA metabolites.17 Indeed, we have confirmed that these metabolites can be distinguished from the DHA–ASC pair on the same chromatograph (see the ESI – Fig. S3†), further bolstering the specificity of the method. The observed DHA-specific (see also the Experimental section) and dose-dependent DHA uptake behavior and the fact that the uptake is effectively inhibited by the presence of high glucose concentration (Fig. 2d and e) or GLUT1 blocker cytochalasin B (CytB) (Fig. 2c) collectively support a predominant role of GLUT1 to internalize DHA into the cell, consistent with previous reports.15,18 A recent study by Lu et al. has described the resultant oxidative stress synchronized with the intracellular DHA–ASC conversion, which eventually causes glutathione depletion, as a proposed modality for cell death.11 Accordingly, such an intracellular ASC-induced cytotoxicity is evident in Fig. 2f. Note that although the preceding reports have provided only up to 30 minute kinetic information, the present “direct method”, owing to the simplicity and stability of the measurement, allowed us to readily continue the monitoring for hours, the timescale at which the intracellular ASC conversion takes place in connection with the resultant cytotoxicity, as demonstrated in Fig. 2f.
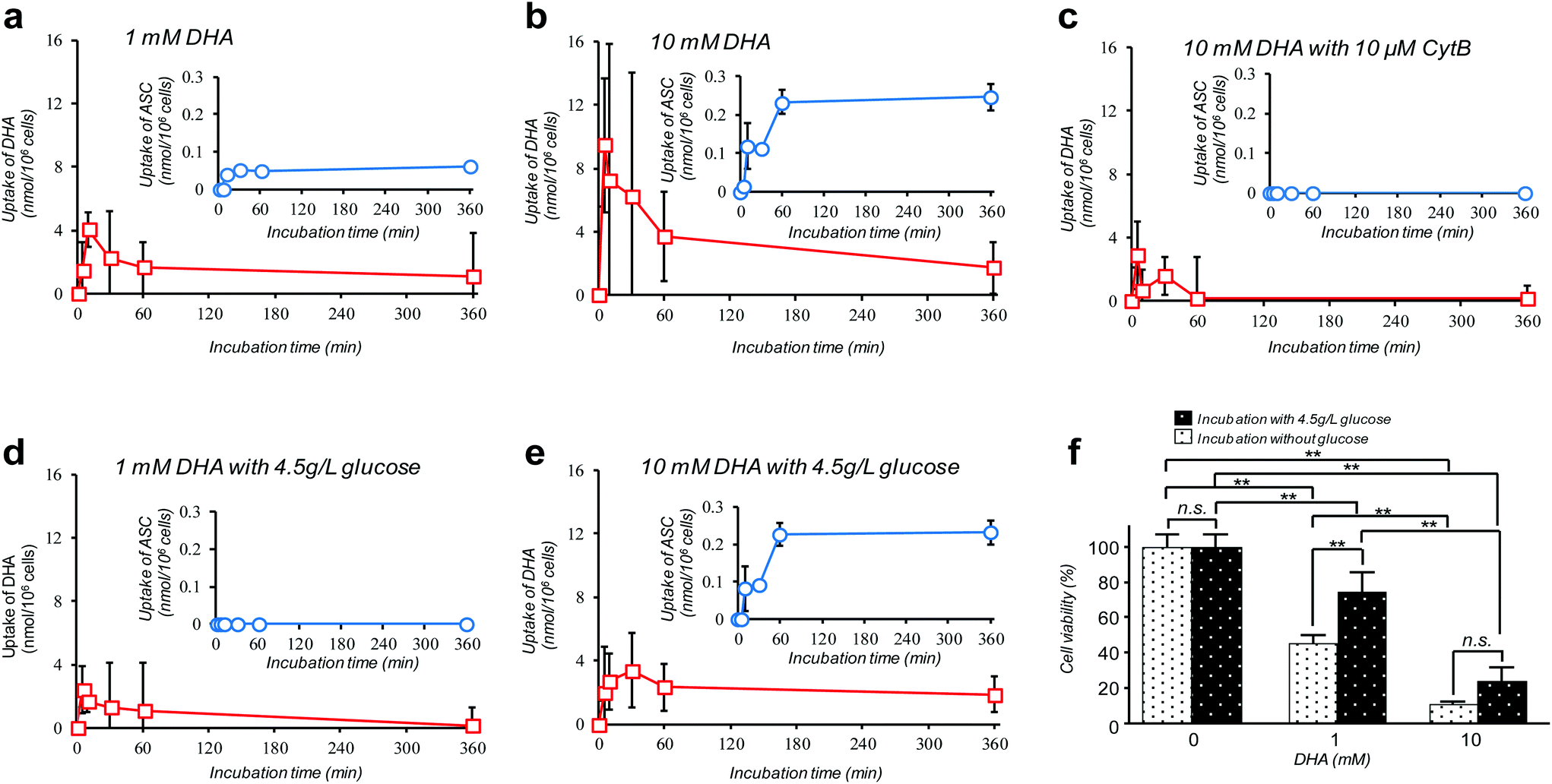 | ||
| Fig. 2 The “direct method” serves as a facile in vitro platform to reveal intracellular ASC–DHA dynamics with each identity and absolute quantity. a–e; Kinetics of GLUT-1-dependent DHA uptake (red squares) and the intracellular conversion into ASC (blue circles) by MIA PaCa-2 cells for various DHA-exposure conditions; a: 1 mM DHA, b: 10 mM DHA, c: 10 mM DHA with 10 μM CyB, d: 1 mM DHA with 4.5 g L−1 glucose, e: 10 mM DHA with 4.5 g L−1 glucose. Data in these figures are presented as means ± SD, n = 3. f: Ensuing viability of MIA PaCa-2 cells after 24 h of DHA exposure for the different doses, with and without glucose (4.5 g L−1), as assessed by a calcein-AM staining assay. Data are presented as means ± SD, n = 3. Means significantly differed at **P < 0.01. | ||
Conclusions
A debate on high-dose (at the millimolar scale) VC cancer therapy may best illustrate the importance of accurate determination of the intracellular DHA–ASC pair dynamics. That is to say, the contradictory clinical data (some studies have indicated anticancer activity of VC while others have shown little effect) have been, at least in part, reasoned by the difference in dose, implying the existence of a threshold VC value at the millimolar concentration range exerting effective cytotoxicity, which is only achievable via intravenous administration, not via oral administration.11,19,20 Indeed, our observation in Fig. 2f discloses that the viability of MIA PaCa-2 cells dramatically changes in a manner that is very sensitive to DHA at such a “millimolar” range of concentration, i.e., 1–10 mM. The cytotoxic threshold VC value may vary depending on the cell type, the metabolic activity and the expression level of GLUT1, the overexpression of which has been implicated in KRAS or BRAF mutations that are prevalent in many types of cancers. It is worth noting again that our HPLC-DAD-based “direct method” represents the simplest technique of all the hitherto known methods and yet offers the unique ability to reveal the DHA–ASC kinetics with each identity, on an inherently robust (stabilizer-compatible) and high-throughput basis requiring no labeling and reduction pre-treatments. Therefore, this technique, by serving as the best-performing screening platform so far, should aid in providing quantitative bases for those therapeutic approaches. It also provides the most practical option for VC involved nutritional science and metabolomics in many scenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowships (17J09342).References
- S. T. Mayne, M. C. Playdon and C. L. Rock, Nat. Rev. Clin. Oncol., 2016, 13, 504 CrossRef PubMed.
- J. Du, J. J. Cullen and G. R. Buettner, Biochim. Biophys. Acta, Rev. Cancer, 2012, 1826, 443 CrossRef CAS PubMed.
- S. E. Bohndiek, M. I. Kettunen, D. E. Hu, B. W. Kennedy, J. Boren, F. A. Gallagher and K. M. Brindle, J. Am. Chem. Soc., 2011, 133, 11795 CrossRef CAS PubMed.
- R. C. Rose, Biochim. Biophys. Acta, 1988, 947, 335–366 CrossRef CAS.
- Y. Kondo, R. Sakuma, M. Ichisawa, K. Ishihara, M. Kubo, S. Handa, H. Mugita, N. Maruyama, H. Koga and A. Ishigami, J. Agric. Food Chem., 2014, 62, 9286 CrossRef CAS PubMed.
- H. Frikke-Schmidt, P. Tveden-Nyborg and J. Lykkesfeldt, Redox Biol., 2016, 7, 8 CrossRef CAS PubMed.
- K. D. Price, C. S. Price and R. D. Reynolds, Atherosclerosis, 2001, 158, 1 CrossRef CAS PubMed.
- M. A. Hediger, Nat. Med., 2002, 8, 445 CrossRef CAS PubMed.
- J. Yun, E. Mullarky, C. Lu, K. N. Bosch, A. Kavalier, K. Rivera, J. Roper, I. I. Chio, E. G. Giannopoulou, C. Rago, A. Muley, J. M. Asara, J. Paik, O. Elemento, Z. Chen, D. J. Pappin, L. E. Dow, N. Papadopoulos, S. S. Gross and L. C. Cantley, Science, 2015, 350, 1391 CrossRef CAS PubMed.
- L. Novakova, P. Solich and D. Solichova, Trends Anal. Chem., 2008, 27, 942 CrossRef CAS.
- Y. X. Lu, Q. N. Wu, D. L. Chen, L. Z. Chen, Z. X. Wang, C. Ren, H. Y. Mo, Y. Chen, H. Sheng, Y. N. Wang, Y. Wang, J. H. Lu, D. S. Wang, Z. L. Zeng, F. Wang, F. H. Wang, Y. H. Li, H. Q. Ju and R. H. Xu, Theranostics, 2018, 8, 1312 CrossRef CAS PubMed.
- L. Wechtersbach, T. Polak, N. P. Ulrih and B. Cigic, Food Chem., 2011, 129, 965 CrossRef CAS PubMed.
- J. Lykkesfeldt, S. Loft and H. E. Poulsen, Anal. Biochem., 1995, 229, 329 CrossRef CAS PubMed.
- H. Y. Li, H. B. Tu, Y. H. Wang and M. Levine, Anal. Biochem., 2012, 426, 109 CrossRef CAS PubMed.
- C. Azzolini, M. Fiorani, A. Guidarelli and O. Cantoni, Br. J. Nutr., 2012, 107, 691 CrossRef CAS PubMed.
- H. B. Tu, H. Y. Li, Y. Wang, M. Niyyati, Y. H. Wang, J. Leshin and M. Levine, EBioMedicine, 2015, 2, 1735 CrossRef PubMed.
- A. Karkonen, R. A. Dewhirst, C. L. Mackay and S. C. Fry, Arch. Biochem. Biophys., 2017, 620, 12 CrossRef CAS PubMed.
- J. C. Vera, C. I. Rivas, R. H. Zhang, C. M. Farber and D. W. Golde, Blood, 1994, 84, 1628 CAS.
- A. Fromberg, D. Gutsch, D. Schulze, C. Vollbracht, G. Weiss, F. Czubayko and A. Aigner, Cancer Chemother. Pharmacol., 2011, 67, 1157 CrossRef PubMed.
- G. Nauman, J. C. Gray, R. Parkinson, M. Levine and C. J. Paller, Antioxidants, 2018, 7, 89 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an02240b |
| This journal is © The Royal Society of Chemistry 2019 |