
In situ paper-based 3D cell culture for rapid screening of the anti-melanogenic activity†
Naricha
Pupinyo
 a,
Moragot
Chatatikun
a,
Anchalee
Chiabchalard
b and
Wanida
Laiwattanapaisal
*b
a,
Moragot
Chatatikun
a,
Anchalee
Chiabchalard
b and
Wanida
Laiwattanapaisal
*b
aGraduate Program in Clinical Biochemistry and Molecular Medicine, Department of Clinical Chemistry, Faculty of Allied Health Sciences, Chulalongkorn University, Bangkok 10330, Thailand
bDepartment of Clinical Chemistry, Faculty of Allied Health Sciences, Chulalongkorn University, Bangkok, 10330, Thailand. E-mail: Wanida.L@chula.ac.th
First published on 15th October 2018
Abstract
Recently, paper has gained traction in the biotechnology research field due to its ability to be a substrate for 3D cell culture. In this work, we demonstrate the application of paper-based 3D cell culture for rapid and easy screening of the effect of natural compounds on melanin production. Whatman No. 1 filter paper was used as the substrate for B16F10 melanoma cell culture. The use of paper is beneficial for supporting the 3D structure of cells, which makes the result more reliable due to the similarity to in vivo conditions. Furthermore, paper is beneficial for melanin observation due to melanin's black color, which is easily in situ visualized after it is cultured on white paper. Matrigel was used to encapsulate cells before being pipetted onto the paper to prevent the passing of cells through paper pores. The intensity of melanin can then be observed with the naked eye and analyzed by scanning the paper. The analysis process took only 20 minutes, which is faster than that of the conventional absorbance spectroscopy, owing to the elimination of centrifugation, melanin solubilization, and the absorbance measurement step. The color intensity on the paper showed a direct proportion with increased α-MSH concentrations, confirming that the color on the paper was melanin. The 3D structure of cells was confirmed by using a scanning electron microscope. To demonstrate the application of the paper-based scaffold, paper-based 3D cell culture was used for screening the effects of Kojic acid and Arbutin on melanin production, which showed increased anti-melanogenesis effects with increased concentrations of natural compounds. High cell viability was observed over 120 hours. In conclusion, the developed paper-based scaffold can be used for screening the effect of natural compounds on melanin production, as a rapid and simple method with low cost.
1. Introduction
In human skin, melanocytes reside in the basal layer of the epidermis, where they form the epidermal melanin units.1 The most common pigment disorders are not disorders of melanin quality, but rather of the quantity of pigment-producing cells, which may be reduced in number, absent, or hyperactive.2 Hence, the study of melanin-producing melanocytes under different conditions is the key to finding substances that enhance or inhibit melanin production. Tyrosinase is the key enzyme of the melanogenesis pathway. In malignant melanoma, patients struggle with hypermelanogenesis, which results from the heightened function of the tyrosinase enzyme.3 Thus, finding inhibitors for tyrosinase is of great interest in melanoma treatment. Arbutin, a glycosylated hydroquinone, is a well-known tyrosinase inhibitor and has a dominant effect on depigmentation.4 Another tyrosinase inhibitor is Kojic acid, an organic acid produced by different types of fungi during aerobic fermentation.5 Both Kojic acid and Arbutin have been studied for the mechanism of melanongenesis inhibition. Furthermore, these natural compounds have been tested as cosmetics where they are used as whitening agents.6 As a consequence, screening the effect of natural compounds does not only provide information on the study of melanoma, but is also beneficial for the cosmetic and pharmaceutical fields.3To date, several methods have been developed for melanin quantification. For instance, the most extensively used method nowadays is the absorbance measurement method.7 In short, melanocytes are collected after seeding and treatment. Next, the produced melanin is solubilized in 1 N NaOH. The total amount of melanin is measured through absorbance measurement and compared to the standard curve of synthetic melanin or melanin isolated from Sepia officinalis. However, the absorbance measurement method is a labor intensive process, has low sensitivity and specificity, and is time-consuming due to the incubation step of melanin solubilization.8 Thus, conventional melanin quantification takes at least 1 hour for the incubation step of melanin solubilization and takes around 2 hours to complete every step. Another method used for melanin quantification is electron spin resonance spectrometry (ESR) which allows the measurement of an electron spin resonance signal based on free radicals derived from melanin. ESR is specific to melanin but lacks sensitivity.9 High performance liquid chromatography (HPLC) is another method used for melanin quantification and can differentiate between eumelanin and pheomelanin. However, HPLC requires specific equipment and expertise.10 Rosenthal et al. originally developed fluorescent quantification of melanin by subjecting melanin to hydrogen peroxide solution. The advantage of fluorescent quantification of melanin is that the fluorescent signal from melanin is not affected by proteinaceous or lipid contamination. However, the fluorescent quantification of melanin still requires multiple steps which take around 4–5 hours to complete.1,7 Recently, near-infrared (NIR) fluorescence was applied for non-invasive melanin content analysis both in vitro and in vivo. However, this method was only tested in normal skin and a specific instrument, a spectrometer, was required.11 Moreover, most of the conventional melanin quantification methods still require an expensive instrument and special skill to perform. Besides, the conventional methods are still based on 2D cell culture in which cells cannot behave properly causing significantly different biological responses.12–14
Recently, in vitro 3D culture has become an important model for studying cell change. Cells in the 3D culture environment can develop morphology and physiology similar to those of analogous cell types in vivo. Moreover, the extracellular matrix that surrounds the cells influences the distribution of cell–cell and cell–matrix contacts on the surface of the cells which affects the cell polarity and cell signaling.15–17 Therefore, 3D cell culture can reflect the in vivo condition better than the rigid 2D culture. Currently, paper is very attractive in the biotechnology field due to its ability to be a substrate for 3D cell culture.18 Researchers have demonstrated the “Cell-in-Gel-in-Paper or CiGiP” technique, which is the use of hydrogels to encapsulate cancer cells which are placed in paper.18–20 Moreover, Derda et al. confirmed that the geometry of cells cultured in the paper using Matrigel encapsulation shows cellular behavior similar to that of in vivo.19 This technique has also been adapted for various cell culture applications, such as paper-based cell culture platforms for the construction of in vitro disease models,18,21–26 drug screening,27,28 coculture systems,29–31 and cell cryopreservation applications.32,33 Paper is made of a bundle of cellulose microfibers and contains micro-fibrous pores which are suitable for constructing the 3D scaffold.33,34 In particular, paper is biocompatible and can easily be established with extremely low costs.34–38 Thus, paper is now an interesting alternative substrate for 3D cell culture.
To simplify the method for screening the amount of melanin production, we developed a rapid and easy method for screening the effects of natural compounds on melanin production by constructing a paper-based 3D cell culture. Due to its black color, the melanin produced by the melanocytes can be easily observed on the white paper-based scaffold after a few days of culturing. In this work, we initially applied a paper-based scaffold for melanoma cell culture and then demonstrated the application for screening the effect of Kojic acid and Arbutin on melanin production. Paper-based scaffolding can be beneficial for 3D cell culture, allowing melanoma cells to behave similar to the in vivo structure. Furthermore, the melanin amount can be easily screened by scanning the paper and analyzing the melanin intensity with ImageJ software, thus decreasing the duration of conventional melanin content analysis (absorbance spectroscopy). Moreover, the color on the paper was confirmed as melanin by the treatment of α-MSH at various concentrations. The amount of melanin production in every experiment was confirmed by absorbance spectroscopy.
2. Materials and methods
2.1. Cells and culture reagents
Mouse melanoma cells (B16F10) were purchased from the American Type Culture Collection (ATCC, USA). B16F10 cells were cultured in standard T25 cm2 flasks and incubated in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin (Hyclone). Cells were incubated at 37 °C in a humidified incubator with a 5% CO2 atmosphere. Prior to seeding cells on the paper, cell suspensions were prepared by standard trypsinization using a trypsin-EDTA solution (Hyclone). Cells were centrifuged at 1500 rpm, 25 °C for 5 minutes. The cell number was determined by using a standard hemocytometer. The desired cell densities were prepared by diluting the initial cell suspension with the culture medium.Kojic acid was purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). Arbutin was purchased from TCI America (Portland, USA).
2.2. Design and fabrication of the paper-based scaffold for melanoma cell culture
The pattern of the paper-based scaffold for melanin quantification was designed using Photoshop CS4 software (Fig. 1). A Xerox ColorQube 8870 printer was used to print wax patterns onto one side of a sheet of cellulose filter paper. The papers were baked on a hot plate at 150 °C for 2 minutes to melt the wax through the thickness of papers. After cooling, the wax solidified in the paper and became a hydrophobic wall.35 The paper consisted of 3 test zones and 1 blank zone. Each zone has 5 mm diameter. Before cell seeding, the paper was sterilized using UV radiation on both the front and back sides of the paper for 15 minutes each. | ||
| Fig. 1 The experimental setup of paper-based 3D cell culture for screening melanin quantification. | ||
2.3. Conventional melanin content analysis
B16F10 cells were seeded into the 6-well plate. Next, cells were stimulated with α-melanocyte stimulating hormone or α-MSH (Abcam) and treated with natural compounds for 48 hours. α-MSH was used for confirming the melanin color on the paper. The suspension of cells and culture medium served as the negative control. After treatment, cells were washed twice with phosphate-buffered saline or PBS (Hyclone) and trypsinized in 0.25 (% v/v) trypsin EDTA solutions. The collected cells were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 minutes. Cell pellets were dissolved in 1 N NaOH at 80 °C for 1 h. The relative melanin content was determined by measuring the absorbance at 475 nm on an ELISA reader.9 The percentage of produced melanin was determined using the following formula (a).
000g for 15 minutes. Cell pellets were dissolved in 1 N NaOH at 80 °C for 1 h. The relative melanin content was determined by measuring the absorbance at 475 nm on an ELISA reader.9 The percentage of produced melanin was determined using the following formula (a). | (a) |
ODTest represented the absorbance of produced melanin under the treatment of α-MSH (positive control) or natural compounds. ODBlank represented the absorbance of produced melanin from untreated cells.
2.4. Melanin content determination on the paper-based scaffold
B16F10 cells were suspended in concentrated phenol red-free Matrigel (8.5 mg ml−1 protein concentration) with a ratio of 1:1. Then, 5 μl of cell suspension was pipetted onto the paper. The paper-based scaffold was then placed in 2 ml of phenol red-free DMEM (Corning cellgro) with 10% fetal calf serum, 4 mM L-glutamine (Corning cellgro) 100 U mL−1 penicillin, or 100 U mL−1 streptomycin with or without α-MSH, Kojic acid, or Arbutin. After that, paper was incubated at 37 °C in a humidified incubator with a 5% CO2 atmosphere at the desired time. Next, the paper was removed from the cell culture medium and air-dried. The dry paper was scanned by using a HP Deskjet F300 series scanner at a resolution of 600 dpi and the intensity was analyzed by using ImageJ software. After importing to ImageJ, the color RGB images were split and the blue channel image was used for analyzing the intensity of melanin on the paper.
2.5. Optimization of cell culture conditions on the paper-based scaffold
To find the most suitable type of paper for paper-based cell culture, 200000 cells of the B16F10 cell line were suspended in Matrigel (Corning Life Sciences) and seeded on the Whatman No. 1, Whatman No. 2, Whatman No. 4, CF4, LF1, and MF1 (GE healthcare life sciences). Next, cell-containing papers were incubated at 37 °C with 5% CO2. After 48 hours of culturing, papers were air-dried. The intensity of melanin on the paper was analyzed using ImageJ software. The same process was also performed without the use of Matrigel encapsulation.
After that, the various cell concentrations were cultured on the paper to find the best concentration that fit with the test zone diameter. In brief, 100000, 200000, 400000, and 800000 cells of B16F10 in Matrigel were seeded on the paper with a test zone of 5 and 10 mm in diameter. Cell-containing papers were incubated at 37 °C with 5% CO2. After 48 hours of culturing, papers were air-dried. Next, the intensity of melanin on the paper was analyzed using ImageJ software.
To find the least incubation time which is suitable for analyzing the melanin amount using ImageJ software, 200000 cells of B16F10 in Matrigel were seeded on the paper. The cell-containing paper was incubated at 37 °C with 5% CO2. Next, the intensity on the paper was analyzed after 24, 48, 72, 96, and 120 hours of incubation using ImageJ software.
2.6. Confirmation of the cell morphology and cell viability on the paper
200000 cells of B16F10 in Matrigel were seeded on the Whatman No. 1 filter paper. The cell-containing paper was incubated at 37 °C with 5% CO2. After 48 hours of culturing, the paper was air-dried. To confirm 3D cell morphology, after sputter-coating with gold, the surface morphology of all samples was monitored with a scanning electron microscope (SEM).
To measure cell viability, after 48 hours of culturing, the paper was washed twice using a phosphate buffer solution or PBS, and then fluorescently stained with a solution of calcein-AM (Invitrogen; 4 μg mL−1 in PBS) and propidium iodide (BioLegend; 4 μg mL−1 in PBS) for 20 min at 37 °C in an environment of 5% CO2. After that, the paper was washed with PBS twice before imaging with a confocal microscope (100-micron resolution images, Zeiss LSM 700 confocal microscope). The fluorescent images captured 5 fields before analyzing the intensity of calcein-AM and propidium iodide of each picture using ImageJ software. The average intensity of calcein-AM and propidium iodide of the 5 captured images was calculated as the mean intensity and converted to the percentage of cell viability from the ratio of living cells to total cells (living and dead cells).
2.7. Detection of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) of B16F10 cells on the paper
200000 cells of B16F10 in Matrigel were seeded on the paper. The cell-containing paper was incubated at 37 °C with 5% CO2. After 48 hours of culturing, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich®) and permeated with 0.5% Tritonx-100 (PanReac AppliChem). After that, cells were captured with anti-GAPDH antibody as a primary antibody (Ambion) and anti-rabbit IgG Alexa flour (R) 488 as a secondary antibody (Cell Signaling Technology), respectively. The nucleus was stained with DAPI (Cell Signaling Technology). The fluorescent image was then captured by using a confocal microscope (Zeiss LSM 700 confocal microscope).
3. Results and discussion
3.1. Culture conditions of the B16F10 cell line on the paper-based scaffold
To find the most suitable type of paper for culturing the B16F10 cell line, which provides the best melanin intensity on paper under 3D cell culture conditions, B16F10 cells were cultured in 6 types of paper, which were available in our laboratory, including MF1, LF1, CF4, Whatman No. 1, Whatman No. 2, and Whatman No. 4. These papers were selected due to their difference in the pore size, thickness, and fiber type. To prevent cells passing through the paper pores, cells were encapsulated in Matrigel. The result was compared with paper-based cell culture without gel encapsulation. To eliminate errors resulting from culture medium color, we used phenol red free DMEM medium when culturing on paper. The result in Fig. 2 shows that when cells were cultured without gel encapsulation, the intensity of the melanin on the paper was lower than the culture with Matrigel encapsulation because some cells passing through the pores of the paper caused low and unstable intensity of melanin on the paper. The result of melanin intensity in gel encapsulation shows high dispersion among each type of paper. The differences in the mean values among the Matrigel encapsulation in each type of paper are greater than those would be expected by chance, which shows a statistically significant difference (P = 0.002) after analysis by one-way ANOVA, due to the various physiological properties of the paper, such as the thickness and pore size, which is explained in brief in the next paragraph.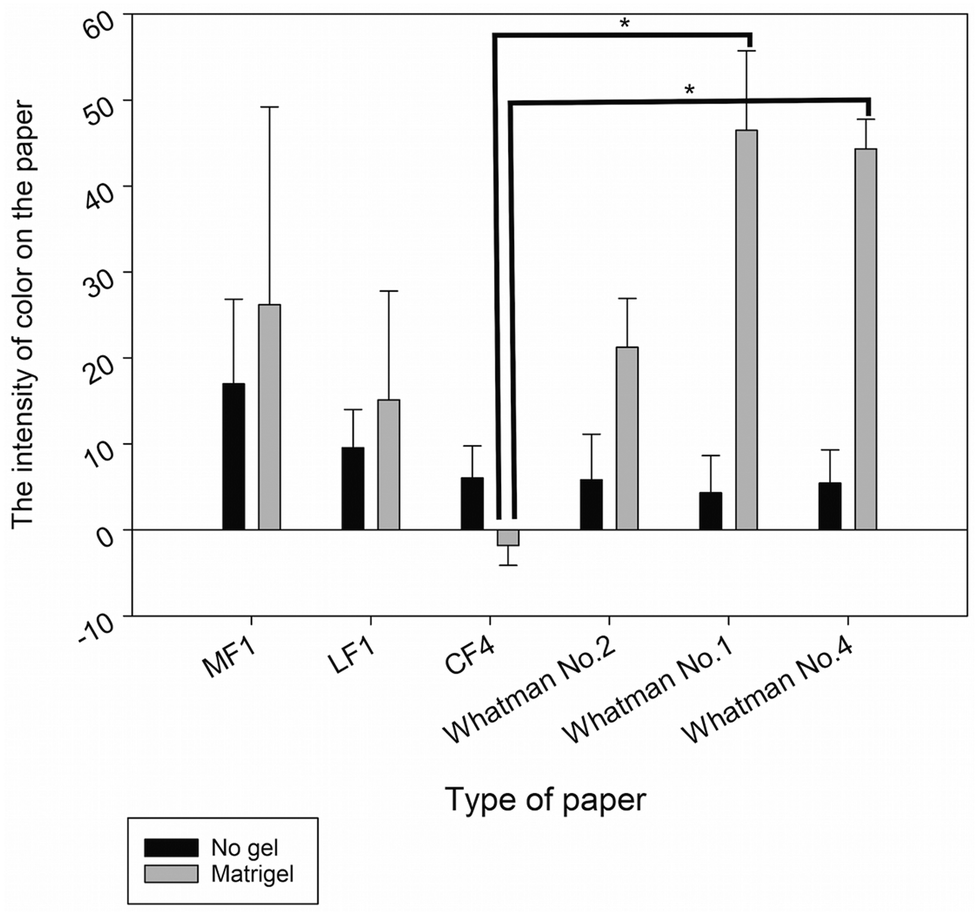 | ||
| Fig. 2 Optimization of paper for paper-based cell culture. Data were analyzed by one-way ANOVA, followed by the Holm-Sidak test. Data are presented as the mean ± SD, n = 3 (*p < 0.05). | ||
The culture of cells on MF1, LF1, and Whatman No. 2 paper with or without gel encapsulation can cause high intensity of melanin on the paper because of the small pore size (MF1 and LF1 ≤2 μm; Whatman No. 2–8 μm),39 which is smaller than the cell size (∼10 μm; Fig. 8). Thus, cells cannot pass through the pore of the paper and behave as a 2D structure above the surface of the paper. Even though MF1, LF1, and Whatman No. 2 promoted high intensity of melanin on the papers, they are not suitable for 3D cell culture.26 Besides, the intensities of melanin on MF1 and LF1 paper with gel encapsulation both show a high standard deviation value due to the difficulty in ensuring exact cell numbers on the small pore size paper, thus causing variation of melanin color on the paper. CF4 was stable when placed in the culture medium for a long time, but it has a high wettability property;40 thus the paper took a very long time to dry, so it cannot decrease the duration of the conventional melanin content analysis. Moreover, the intensity of melanin on the CF4 paper was not seen when cultured with Matrigel encapsulation resulting in a minus value of melanin intensity after being analyzed with ImageJ software (Fig. 2). This is due to the higher level of thickness of the CF4 paper.40 As a result, a high cell number was required for the consistent distribution of cells under the culture area. In contrast, the culture of B16F10 cells on Whatman No. 1 and Whatman No. 4 filter paper with Matrigel encapsulation provided the best intensity of melanin and showed a statistically significant difference with CF4 paper (Fig. 2), due to their suitable pore sizes, which are ∼11 and ∼25 μm in Whatman No. 1 and Whatman No. 4, respectively.39 Moreover, the use of Matrigel is easy to handle due to its viscous transitional property. In brief, Matrigel can be liquefied at 4 °C, which is appropriate for mixing cells and Matrigel together, and pipetted onto the paper. After placing the paper in the 37 °C incubator, Matrigel was polymerized and solidified,41 which means that the total cells were encapsulated within the paper without passing through the pores of the paper. This result also supported that the use of paper can improve the handle and manipulation of the hydrogel for 3D cell support because the hydrogel alone has weak mechanical properties.42,43
Next, Whatman No. 1 and Whatman No. 4 were used for culturing B16F10 cells in various concentrations which are 100000, 200000, 400000, and 800000 cells per test zone in designed paper with 5 mm and 10 mm test zone diameters. The result shows that the intensity of melanin has a direct variation with the cell number in both Whatman No. 1 and Whatman No. 4 (Fig. S1†). However, 10 mm diameter of the designed paper was excessive in every cell concentration and caused inconsistent distribution of melanin on the paper, whereas 5 mm diameter paper showed consistent distribution of melanin throughout the paper surface in cell concentrations of 200000, 400000, and 800000 cells per test zone (Fig. 3). However, the melanin intensity on the paper was higher in Whatman No. 1, due to the 11 μm pore size,39 which better fits the B16F10 cell size (∼10 μm; Fig. 8). Therefore, Whatman No. 1 was the most suitable paper for this work. Nonetheless, the cell concentration of 100000 cells per test zone is too low for screening the effect of a whitening substance on melanin production (Fig. 3). Thus, the lowest amount of cells which can be used for screening the effects of natural compounds on melanin production is 200000 cells per test zone with the encapsulation of Matrigel. Although the required cell amount was around 2 times higher than conventional melanin content analysis, which normally requires 100000 cells per well of the 6-well plate, the total volume of cells suspended in Matrigel (5 μl) was very low compared to conventional melanin content analysis.
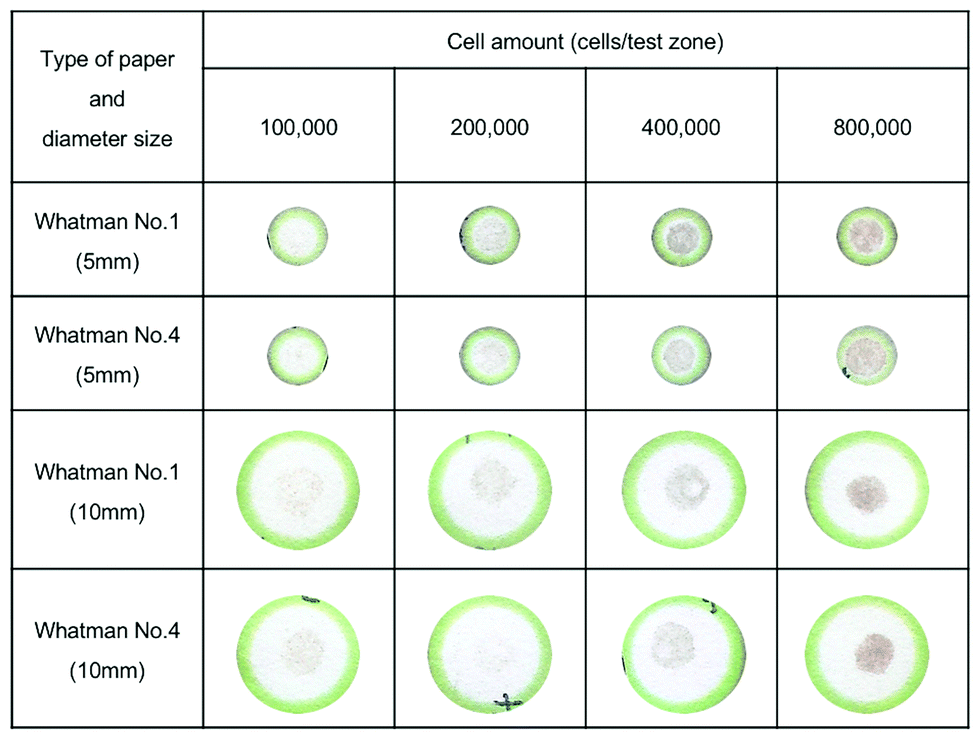 | ||
| Fig. 3 Example of paper after seeding cells in various concentrations on Whatman No. 1 and Whatman No. 4 with a test zone diameter of 5 and 10 mm. | ||
Next, we optimized the most suitable incubation time for screening the melanin intensity on the paper. B16F10 cells were encapsulated with Matrigel and cultured in Whatman No. 1 with 5 mm test zone diameter for 24, 48, 72, 96, and 120 hours. The result showed that after culturing for 48 hours, B16F10 cells produced suitable amounts of melanin that can be observed with the naked eye. Moreover, the intensity of melanin on the paper increased with incubation time, showing the ability of cells to produce melanin for at least 120 hours during the culturing on the paper (Fig. 4). By using paper for melanoma cell culture, the color of melanin can be easily observed with the naked eye due to the contrast of the white color of the paper and the dark color of melanin. Moreover, the total analysis time was just 20 minutes for scanning the paper and analyzing the intensity with ImageJ software, which is very quick compared to conventional melanin content analysis, which takes about 1 hour and 30 minutes (15 minutes to centrifuge melanoma cells, 1 hour to solubilize melanin in NaOH, and another 10 minutes to perform the absorbance measurement).9
 | ||
| Fig. 4 Optimization of the incubation time. Data are presented as the mean ± SD (n = 3). | ||
3.2. Identification of B16F10 cell morphology on the paper-based scaffold
To confirm that cells are able to form a 3D structure during the culturing in Whatman No. 1 paper, B16F10 cells were encapsulated with Matrigel, pipetted onto Whatman No. 1 paper and incubated at 37 °C with 5% CO2 for 48 hours. The morphology of the paper was then analyzed by using a scanning electron microscope (SEM). Fig. 5a shows the structure of naked Whatman No. 1 paper which contains fibrous pores which are suitable to be a scaffold for cell support. Fig. 5b shows the 3D structure of B16F10 cells that tended to form clusters. Fig. 5c shows B16F10 cells in a 3D shape surrounded by produced melanin on the paper fiber. Thus, the 3D architecture of Whatman No. 1 paper was able to provide the 3D cell culture condition. In addition, the use of Matrigel encapsulation can provide the native extracellular matrix which enables cells to behave similar to those seen in vivo.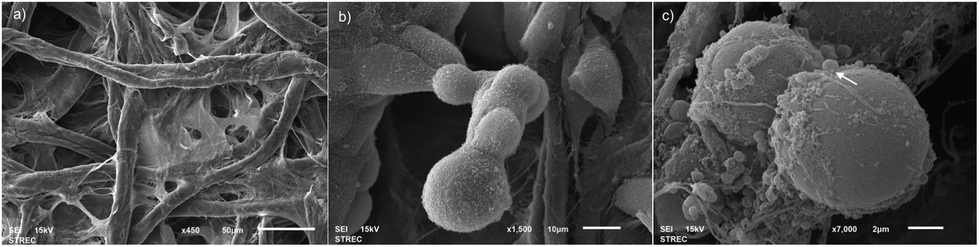 | ||
| Fig. 5 SEM images of microarchitecture of (a) the paper-based scaffold of Whatman No. 1 filter paper characteristic (b) B16F10 morphology after culturing under the paper-based scaffold for 2 days representing a cluster of cells (c) B16F10 morphology after culturing under the paper-based scaffold for 2 days representing the produced melanin (arrow) on and near the cell surface. | ||
To confirm the existence of B16F10 cells in the paper, we also performed immunofluorescent staining of Glyceraldehyde 3-phosphate dehydrogenase or GAPDH which served as the internal control of the experiments due to the constant expression of the GAPDH gene, which can be found in the cells44 (represented in green color in Fig. 6). Also, nuclei were stained by DAPI, a DNA-specific probe, allowing the fluorescent imaging of nuclei (represented in blue color of Fig. 9).45Fig. 6 shows the immunofluorescent image of GADH of B16F10 on the paper-based scaffold. Some stained cells were unclear due to the obstruction of fibers in the paper. Therefore, this image can confirm that the paper-based scaffold contains B16F10 cells.
 | ||
| Fig. 6 B16F10 cells were stained for GAPDH (green – GAPDH; blue – DAPI; 1000-micron resolution images). | ||
3.3. Identification of B16F10 cell viability under paper-based cell culture
To measure cell viability, B16F10 cells were cultured on paper for 24, 48, 72, 96, and 144 hours without changing the culture medium. After that, viable cells were stained with calcein-AM and dead cells were stained with propidium iodide (PI). In brief, calcein-AM can be converted to green fluorescence after acetoxymethyl ester hydrolysis by esterase in living cells.46 Propidium iodide (PI) binds to the double strand DNA of dead cells with the intact plasma membrane.47Fig. 7 shows high cell viability during culturing for the first 72 hours. At 96 and 120 hours, percent cell viability decreased due to the high cell number and the limited culture medium. However, cells were able to proliferate under the paper-based cell culture. The result shows single viable cells in the paper after 24 hours of culturing. After 48 hours of culturing, cells started proliferating and forming clusters of cells. After 72 and 96 hours, cell numbers were higher than after 24 and 48 hours of incubation with some dead cells shown in red color. After 120 hours, the cells continued forming clusters, covering almost the entire area of paper thus indicating cell proliferation (Fig. S2†).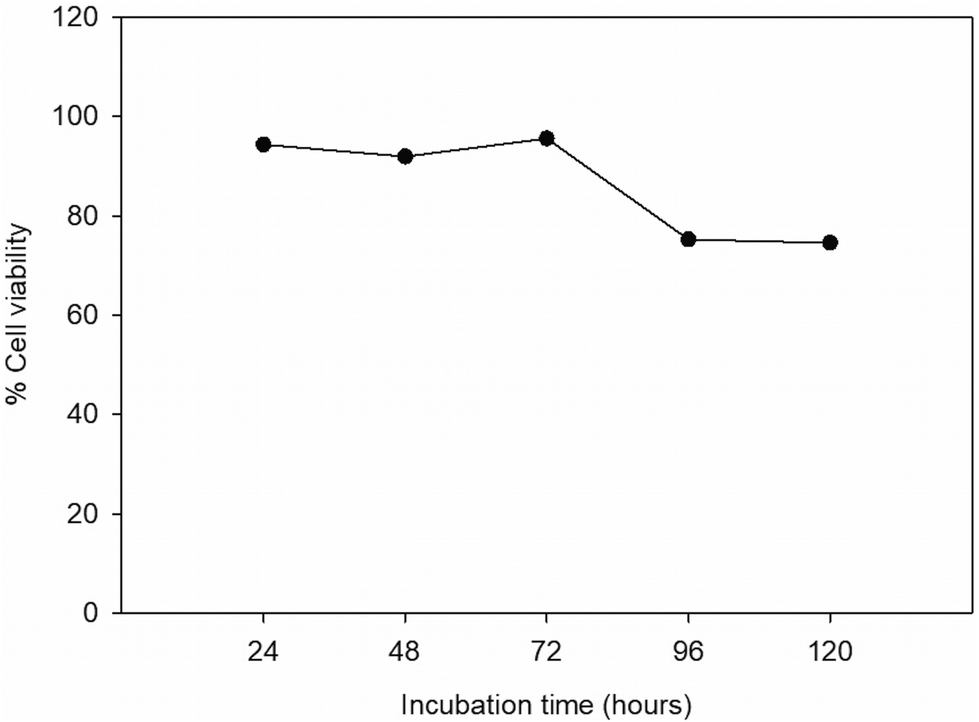 | ||
| Fig. 7 Calcein-AM staining showed consistent cell viability over 120 hours of culture. | ||
3.4. Confirmation of melanin color on the paper-based scaffold
α-Melanocyte stimulating hormone or α-MSH is known as a melanotropic hormone and a major physiologic stimulus for murine pigmentation.48–50 To confirm that the color in the paper was from the produced melanin, B16F10 cells on the paper were treated with α-MSH at a concentration of 0, 0.1, 1, and 10 μM. The conventional absorbance measurement was performed to confirm the percentage of melanin production under α-MSH treatment. Fig. 8 shows the intensity of color on the paper which increased with higher concentrations of α-MSH. Intensity of melanin at 0 μM α-MSH can be observed because initial seeded melanoma cells naturally produce melanin without α-MSH stimulation. Above each bar of Fig. 8 is shown an example of increased color on the paper with higher concentrations of α-MSH, confirming that the color on the paper was a produced melanin from B16F10 cells. | ||
| Fig. 8 The intensity of melanin on the paper under α-MSH treatment at various concentrations. Data are presented as the mean ± SD (n = 3). | ||
3.5. Comparison between the percentage of melanin production with the conventional method (absorbance measurement) and melanin intensity on the paper-based scaffold
To compare the melanin production between the conventional assay and paper-based scaffold, 200000 cells of B16F10 were seeded in a 6-well plate and paper-based scaffold and treated with α-MSH at a concentration of 0.1, 1, and 10 μM. Fig. 9 shows the percentage of melanin production in the conventional method, which increased with higher concentrations of α-MSH. Moreover, percentage of melanin production showed a direct proportion with the intensity of melanin on the paper when treated with the same concentration of α-MSH with the conventional method (Fig. 9). This result confirmed that the intensity of melanin on the paper was consistent with the percentage of melanin production in the conventional assay.
 | ||
| Fig. 9 Bar graph shows the relationship between % melanin production in the conventional method and the intensity of melanin on the paper with α-MSH at a concentration of 0.1, 1, and 10 μM. Data are presented as the mean ± SD (n = 3). | ||
Several studies have been used a paper as a scaffold for cell culture. Yet, there was no evidence on paper-based melanoma cell culture for the melanin study. In this work, we firstly applied a paper-based scaffold for melanoma cell culture for screening the effect of natural compounds on melanin production. We confirmed that the melanin color on the paper can reflect the effect of treated substances on melanin production by treating with α-MSH which is generally known as a stimulant for melanin production. The color of melanin on the paper was consistent with the amount of melanin in conventional assay after the treatment of α-MSH which can also confirm that the visible color on the paper was melanin.
3.6. The use of the paper-based scaffold for screening the effects of natural compounds on melanin production
To validate the ability of the paper-based scaffold to detect the effect of natural compounds on melanin production, cells on the paper-based scaffold were treated with Kojic acid and Arbutin, which are known as tyrosinase inhibitors, a crucial enzyme in synthesizing melanin through melanogenesis.51–53 In brief, the suspension of 200000 cells of B16F10 and Matrigel was treated with 2 natural compounds, which are Kojic acid with a concentration of 0, 0.2, 0.4, 0.8, and 1.6 mM, and Arbutin with a concentration of 0, 0.1, 0.2, 0.4, and 0.8 mM. The suspension of cells was then seeded on the paper and incubated at 37 °C with 5% CO2. The suspension of the cells and hydrogel alone served as the negative control. The intensity on the paper was then analyzed using ImageJ software. Furthermore, the conventional melanin content analysis was performed on both non-treatment and natural compound treatments to confirm the effect on melanin production.
Fig. 10 shows the intensity of color on the paper which decreased with increased concentrations of Kojic acid and Arbutin in a dose dependent manner, confirming the whitening effect of Kojic acid and Arbutin on melanin production. This result confirmed the ability of paper-based melanoma cell culture to detect the effects of whitening substances on melanin production. Nevertheless, the paper-based scaffold was only used with low concentrations of whitening substances because of the limitation of melanin color on the paper which required a longer incubation time to perceive the effect of high concentration of whitening substances on melanin production.
 | ||
| Fig. 10 The intensity of melanin on the paper under (a) Kojic acid treatment with concentrations of 0, 0.2, 0.4, 0.8, and 1.6 mM. (b) Arbutin treatment with concentrations of 0, 0.1, 0.2, 0.4, and 0.8 mM. Data are presented as the mean ± SD (n = 3). | ||
Thereby, the paper-based scaffold can be quickly used for screening substances which affect both the increase and decrease of melanin production. Moreover, scanning and analyzing the paper-based scaffold with ImageJ software can reduce the cell seeding step before the treatment of compounds, and the cell centrifugation and melanin solubilization steps of the conventional method before further analysis in the research field. Moreover, our paper-based scaffold can eliminate the use of specific equipment such as spectrophotometers and HPLC. Thus, the specific expertise was not necessary (Table 1).
| Paper-based 3D cell culture | Conventional absorbance measurement | |
|---|---|---|
|
a Fernandes et al. have reported that the detection limit of absorbance measurement was 310000 cells per ml in the SK-MEL-1 cell line and 630000 cells per ml in the SK-MEL-23 cell line.7
|
||
| Limit of detection | 10183 cells per mm2 of paper culture area |
Not performed in the B16F10 cell line so fara |
| Preparation step | Cells can be treated with anti-melanogenic substance directly after seeding on the paper | Treatment can be performed only after cells attach to the bottom of well (which requires at least 4 hours) |
| Analysis time | 20 minutes | 120 minutes (ref. 9) |
However, the melanin color on the paper was not stable enough to measure the melanin amount in a semi-quantitative manner due to the various effects of natural compounds on melanin production. Currently, our paper-based scaffold is suitable for screening for the effect of natural compounds on melanin production which can shorten the primary step of choosing whether the compound has anti-melanogenic effect or not. Thus, we aim to improve our paper-based scaffold to have the ability to screen the melanin amount in various crude extracts in both semi-quantitative and quantitative manner in the future.
Since our work aims to demonstrate the semi-quantitative screening effect of anti-melanogenic substances, the comparison between the melanogenesis of B16F10 in paper-based 3D cell culture and conventional 3D cell culture was not performed due to the lack of standard methods for the murine 3D cell culture model. However, the general method for analyzing the melanin synthesis was performed in a 3D reconstructed human skin model and animal model.53–56 Matsui et al. have demonstrated that the color of a reconstructed 3D human skin model after treatment with kojic acid showed lower intensity of melanin color compared to the vehicle.57 This result confirms that the anti-melanogenic effect of Kojic acid found in the paper-based 3D cell culture yields a similar result to the conventional 3D skin model.
4. Conclusions
In this work, a paper-based scaffold has been proposed for rapid screening anti-melanogenic activity of natural compounds, in which Kojic acid and Arbutin were demonstrated as examples. By using the advantage of existing white color of the paper substrate, the black color of melanin complexion can be easily observed after 48 hours of culture. The paper-based scaffold was easy to fabricate by using the wax printing technique to create the culture zone. Furthermore, the treatment of natural compounds can be performed with the seeding step of cells in the paper, which is faster than the conventional 2D cell culture that needs at least 4 hours for cell attachment before natural compound treatment. The analysis of the effects of natural compounds can be easily analyzed by scanning the paper and measuring the intensity of melanin with ImageJ software and comparing with the untreated paper. In addition, the paper-based scaffold was able to culture B16F10 cells in 3D conditions where cells can behave similar to in vivo structures. Besides, fluorescent staining can be performed directly on the paper. Thus, cell viability and cell biomarkers can be immediately detected after the desired incubation time on the paper. The paper-based melanoma cell culture served as the platform for in vitro 3D cell culture for quick and in situ screening of the effects of compounds of interest on melanin production and provides great opportunities for developing further high-throughput platforms that could be used for quantitative melanin content analysis with more efficacy and accuracy.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
N. P. gratefully acknowledges the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (under grant no. PHD/0012/2558) and the 90th Year Anniversary Ratchadaphiseksomphot Endowment Fund, Chulalongkorn University. Ms Sakawrat Janpaijit and Ms Surangrat Thongkorn are acknowledged for providing technical assistance with fluorescent image acquisition.References
- M. H. Rosenthal, J. W. Kreider and R. Shiman, Anal. Biochem., 1973, 56, 91–99 CrossRef CAS PubMed.
- J. P. Ebanks, R. R. Wickett and R. E. Boissy, Int. J. Mol. Sci., 2009, 10, 4066–4087 CrossRef CAS PubMed.
- N. Baurin, E. Arnoult, T. Scior, Q. Do and P. Bernard, J. Ethnopharmacol., 2002, 82, 155–158 CrossRef CAS PubMed.
- K. Maeda and M. Fukuda, J. Pharmacol. Exp. Ther., 1996, 276, 765–769 CAS.
- H. Kaatz, K. Streffer, U. Wollenberger and M. G. Peter, Z. Naturforsch., C: J. Biosci., 1999, 54, 70–74 CAS.
- N. Smit, J. Vicanova and S. Pavel, Int. J. Mol. Sci., 2009, 10, 5326–5349 CrossRef CAS PubMed.
- B. Fernandes, T. Matamá, D. Guimarães, A. Gomes and A. Cavaco-Paulo, Pigm. Cell Melanoma Res., 2016, 29, 707–712 CrossRef CAS PubMed.
- K. Wakamatsu and S. Ito, Pigm. Cell Melanoma Res., 2002, 15, 174–183 CrossRef CAS.
- T. Watanabe, A. Tamura, Y. Yoshimura and H. Nakazawa, Anal. Biochem., 1997, 254, 267–271 CrossRef CAS PubMed.
- S. Ito, A. Pilat, W. Gerwat, C. Skumatz, M. Ito, A. Kiyono, A. Zadlo, Y. Nakanishi, L. Kolbe and J. M. Burke, Pigm. Cell Melanoma Res., 2013, 26, 357–366 CrossRef CAS PubMed.
- S. Kalia, J. Zhao, H. Zeng, D. McLean, N. Kollias and H. Lui, Pigm. Cell Melanoma Res., 2018, 31, 31–38 CrossRef CAS PubMed.
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15(5), 405–412 CrossRef PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839 CrossRef CAS PubMed.
- Y. Zhang, Y.-W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- D. Gao, H. Liu, Y. Jiang and J.-M. Lin, TrAC, Trends Anal. Chem., 2012, 35, 150–164 CrossRef CAS.
- M. Mehling and S. Tay, Curr. Opin. Biotechnol., 2014, 25, 95–102 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2004, 22, 151 CrossRef CAS PubMed.
- B. Mosadegh, B. E. Dabiri, M. R. Lockett, R. Derda, P. Campbell, K. K. Parker and G. M. Whitesides, Adv. Healthcare Mater., 2014, 3, 1036–1043 CrossRef CAS PubMed.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef CAS PubMed.
- M. C. Sapp, H. J. Fares, A. C. Estrada and K. J. Grande-Allen, Acta Biomater., 2015, 13, 199–206 CrossRef CAS PubMed.
- M. W. Boyce, R. M. Kenney, A. S. Truong and M. R. Lockett, Anal. Bioanal. Chem., 2016, 408, 2985–2992 CrossRef CAS PubMed.
- M. W. Boyce, G. J. LaBonia, A. B. Hummon and M. R. Lockett, Analyst, 2017, 142, 2819–2827 RSC.
- G. Huang, X. Zhang, Z. Xiao, Q. Zhang, J. Zhou, F. Xu and T. J. Lu, Soft Matter, 2012, 8, 10687–10694 RSC.
- K. A. Simon, B. Mosadegh, K. T. Minn, M. R. Lockett, M. R. Mohammady, D. M. Boucher, A. B. Hall, S. M. Hillier, T. Udagawa and B. K. Eustace, Biomaterials, 2016, 95, 47–59 CrossRef CAS PubMed.
- L. Wang, C. Xu, Y. Zhu, Y. Yu, N. Sun, X. Zhang, K. Feng and J. Qin, Lab Chip, 2015, 15, 4283–4290 RSC.
- Y. Wei, Q. Zhang, C. Bin, G.-T. Liang, L. Wei-Xuan, Z. Xiao-Mian and L. Da-Yu, Chin. J. Anal. Chem., 2013, 41, 822–827 CrossRef.
- B. Hong, P. Xue, Y. Wu, J. Bao, Y. J. Chuah and Y. Kang, Biomed. Microdevices, 2016, 18, 21 CrossRef PubMed.
- S. K. Min, C. R. Kim, S. H. Kim and H. S. Shin, J. Nanomater., 2014, 2014, 1 CrossRef.
- G. Camci-Unal, D. Newsome, B. K. Eustace and G. M. Whitesides, Adv. Healthcare Mater., 2016, 5, 641–647 CrossRef CAS PubMed.
- Y. Wang, W. Su, L. Wang, L. Jiang, Y. Liu, L. Hui and J. Qin, Toxicol. Res., 2018, 7, 13–21 RSC.
- M. J. Aebersold, G. Thompson-Steckel, A. Joutang, M. Schneider, C. Burchert, C. Forró, S. Weydert, H. Han and J. Vörös, Front. Neurosci., 2018, 12, 94 CrossRef PubMed.
- K. Ng, B. Gao, K. W. Yong, Y. Li, M. Shi, X. Zhao, Z. Li, X. Zhang, B. Pingguan-Murphy and H. Yang, Mater. Today, 2017, 20, 32–44 CrossRef CAS.
- G. F. Petersen, B. J. Hilbert, G. D. Trope, W. H. Kalle and P. M. Strappe, Biotechnol. Lett., 2015, 37, 2321–2331 CrossRef CAS PubMed.
- E. A. Peraza-Hernandez, D. J. Hartl, R. J. Malak Jr. and D. C. Lagoudas, Smart Mater. Struct., 2014, 23, 094001 CrossRef.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS PubMed.
- J. Lee, M. J. Cuddihy and N. A. Kotov, Tissue Eng., Part B, 2008, 14, 61–86 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2009, 81, 5990–5998 CrossRef PubMed.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- Whatman Price Catalog, http://www.iitk.ac.in/centralstores/data/price/15%20Whatman_Pricelist_2014_INR.pdf, (accessed March 20, 2018).
- CF4, https://www.gelifesciences.com/en/bd/shop/protein-analysis/immunoassays-biochemical-assays-accessories-and-reagents/components-for-lateral-flow-and-flow-through-immunoassays/cf4-p-00786?current=8114–2250, (accessed March 20, 2018).
- Frequently Asked Questions of Matrigel, https://www.corning.com/media/worldwide/cls/documents/CLS-DL-CC-026%20DL.pdf, (accessed March 20, 2018).
- D. Lantigua, Y. N. Kelly, B. Unal and G. Camci-Unal, Adv. Healthcare Mater., 2017, 6, 1700619 CrossRef PubMed.
- X. Wu, S. Suvarnapathaki, K. Walsh and G. Camci-Unal, MRS Commun., 2018, 8, 1–14 CrossRef.
- R. D. Barber, D. W. Harmer, R. A. Coleman and B. J. Clark, Physiol. Genomics, 2005, 21, 389–395 CrossRef CAS PubMed.
- J. Kapuscinski, Biotech. Histochem., 1995, 70, 220–233 CrossRef CAS PubMed.
- Calcein, AM, cell-permeant dye, https://www.thermofisher.com/order/catalog/product/C3100MP, (accessed June 28, 2018).
- eBioscience™ Propidium Iodide Staining Solution, https://www.thermofisher.com/order/catalog/product/00–6990–50?gclid=Cj0KCQjwjtLZBRDLARIsAKT6fXxNTDQQvgGYC6DXeIitdkTerFnNIo_PXJuSSVzTFSfwNp5ao5LxnmcaAhK4EALw_wcB&s_kwcid=AL!3652!3!256657343038!b!!g!!%2Bpropidium%20%2Biodide%20%2Bstaining&ef_id=WcsodwAAAIQYShDK:20180628140913:s, (accessed June 28, 2018).
- S. Ito, Pigm. Cell Melanoma Res., 2003, 16, 230–236 CrossRef CAS.
- E. R. Price, M. A. Horstmann, A. G. Wells, K. N. Weilbaecher, C. M. Takemoto, M. W. Landis and D. E. Fisher, J. Biol. Chem., 1998, 273, 33042–33047 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Brenner and V. J. Hearing, J. Biol. Chem., 2007, 282, 27557–27561 CrossRef CAS PubMed.
- M. Gonçalez, M. A. Correa and M. Chorilli, BioMed Res. Int., 2013, 2013 Search PubMed.
- D.-H. Ki, H.-C. Jung, Y.-W. Noh, P. Thanigaimalai, B.-H. Kim, S.-C. Shin, S.-H. Jung and C.-W. Cho, Drug Dev. Ind. Pharm., 2013, 39, 526–533 CrossRef CAS PubMed.
- K. Kumar, M. G. Vani, S. Y. Wang, J. W. Liao, L. S. Hsu, H. L. Yang and Y. C. Hseu, BioFactors, 2013, 39, 259–270 CrossRef CAS PubMed.
- H. Ando, A. Ryu, A. Hashimoto, M. Oka and M. Ichihashi, Arch. Dermatol. Res., 1998, 290, 375–381 CrossRef CAS PubMed.
- M.-S. Jang, H.-Y. Park and K.-H. Nam, Food Sci. Biotechnol., 2014, 23, 555–560 CrossRef CAS.
- M. Kim, S. Shin, J.-A. Lee, D. Park, J. Lee and E. Jung, BMC Complementary Altern. Med., 2015, 15, 449 CrossRef PubMed.
- M. S. Matsui, M. J. Petris, Y. Niki, N. Karaman-Jurukovska, N. Muizzuddin, M. Ichihashi and D. B. Yarosh, J. Invest. Dermatol., 2015, 135, 834–841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c8an01725e |
| This journal is © The Royal Society of Chemistry 2019 |