
Development of a method for multiple vitamin D metabolite measurements by liquid chromatography coupled with tandem mass spectrometry in dried blood spots†
R.
Rola
 *ab,
K.
Kowalski
b,
T.
Bieńkowski
b,
A.
Kołodyńska-Goworek
b and
S.
Studzińska
a
*ab,
K.
Kowalski
b,
T.
Bieńkowski
b,
A.
Kołodyńska-Goworek
b and
S.
Studzińska
a
aChair of Environmental Chemistry and Bioanalytics, Faculty of Chemistry, Nicolaus Copernicus University in Toruń, 7 Gagarin St., PL – 87-100 Toruń, Poland. E-mail: r.rola@doktorant.umk.pl; Fax: +48-56-6114837; Tel: +48-56-6114308
bMasdiag – Diagnostic Mass Spectrometry Laboratory, 33 Stefana Żeromskiego St., PL – 01-882 Warsaw, Poland. Tel: +48-602-228-824
First published on 18th October 2018
Abstract
There are two forms of vitamin D which are essential to the human body, i.e. vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol). The inactive metabolites of vitamin D are commonly used for quantitative analysis because of their longer half-life, stability, and relatively high blood concentrations. This paper presents the development of a high-throughput and sensitive method for determining four vitamin D metabolites in dried blood spots using liquid chromatography coupled with tandem mass spectrometry. This method allows for the determination of 25(OH)D2 and 25(OH)D3 concentrations, as well as the epimeric form 3-epi-25(OH)D3 and 24,25(OH)2D3. The analyzed material is capillary blood taken from the fingertip, deposited on filter paper. Four different chromatographic columns were tested to separate all compounds, in particular, the epimeric form. The column of choice was F5 (Phenomenex, Torrance, CA, USA). In order to prove the consistency between the results for DBS, used as an alternative biological matrix, and serum, comparative studies of these two materials were carried out in nearly 100 individuals. The results indicated their positive correlation. The evaluation of short-term stability of metabolites in DBS within the month showed no change in metabolite concentration. During the validation, the impact of the matrix on the ionization of the tested compounds was evaluated. Capillary blood and venous blood collected for different anticoagulants were also compared. The smallest differences in the results were obtained for citrate. In order to achieve a limit of quantitation of 0.2 ng ml−1, sample preparation involved derivatization using a Cookson-type reagent, 4-(4′-dimethylaminophenyl)-1,2,4-triazoline-3,5-dione (DAPTAD).
1. Introduction
Vitamin D belongs to secosteroids, a group of steroid organic compounds. It has two main forms: vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol).1 The most important source of vitamin D3 is the process of skin biosynthesis, which occurs under the influence of sunlight. Vitamin D3 precursor is 7-dehydrocholesterol, which, due to its absorption of UVB radiation (285–320 nm), is ultimately converted to vitamin D3.1 In turn, vitamin D2 is found in plant products, mainly in shiitake mushrooms and yeast.1 Vitamin D primarily affects the skeletal system, but also the nervous, muscular, immunological, circulatory, and reproductive systems and many other tissues and organs.2The complex metabolism of vitamin D results in a relatively large number of structurally diversified metabolites, a few of which are monitored during routine assays. Although more than 50 metabolites of vitamin D are known to date, the most common method is the measurement of the total calcidiol concentration, i.e. the sum of 25(OH)D3 and 25(OH)D2 metabolites and, to a lesser extent, calcitriol – the active form that is present in serum at 1000-fold lower concentrations.3 It is considered that measurement of the 25(OH)D concentration makes it more clinically useful, especially to assess the body's supply of vitamin D.3,4 There are several techniques used to determine the concentration of vitamin D metabolites, and they differ primarily in their accuracy. These include radioimmunoassays (RIA), immunoenzymatic assays (ELISA), chemiluminescent assays (CLIA) and direct techniques: high-performance liquid chromatography with UV detection (HPLC-UV) or tandem mass spectrometry (LC-MS/MS) and gas chromatography coupled with mass spectrometry (GC-MS).5–7 The results obtained by RIA, ELISA, and CLIA, depending on the manufacturer, are not consistent, largely due to the cross-reactivity to the various vitamin D isoforms.7 The disadvantages of these tests are the lower affinity for 25(OH)D2 and the impossibility to determine the epimeric form. What is more, the obtained result is the sum of the metabolites of vitamin D2 and D3, without distinguishing the individual components (25(OH)D2 and 25(OH)D3).8,9
The LC-MS/MS technique allows for a highly specific and selective analysis of all the relevant metabolites, including compounds with lower biological activity.10–14 The advantage of MS is the ability to determine multiple metabolites simultaneously. Analysis of vitamin D using MS most often utilizes two modes of ionization in a positive polarization: electrospray (ESI) and atmospheric pressure chemical ionization (APCI), although there are single cases using fast atom bombardment (FAB) or atmospheric pressure photoionization (APPI).12 The lipophilic nature of the metabolites of vitamin D, the lack of functional groups that facilitate ionization and the relatively low concentration in the biological material make their analysis using MS a challenge, especially in terms of sensitivity. However, the most common solution to the above problems is a derivatization process, which not only facilitates the ionization process in MS, but also improves the specificity.12,15 In spite of all the advantages of mass spectrometry, attention should be paid to the following issues: ion suppression induced by matrix components, selectivity and specificity for both vitamin D and potential interferents (isobaric and isomeric compounds).16 Except for chromatographic separation, a possible solution to the last problem can be the utilization of high-resolution MS or ion mobility spectrometry.15,16
An important step in the determination of vitamin D using LC-MS/MS is the preparation of the sample. The serum, plasma, dried blood spots (DBS), milk, tissue, urine, cerebrospinal fluid and even saliva can be analyzed.13,17–30 Depending on the matrix, different methods of sample preparation are used, where, in addition to the efficient isolation of the analytes, the effective release of the studied compounds from the VDBP complex (Vitamin D Binding Protein) is crucial. The most common methods are protein precipitation (PP), liquid–liquid extraction (LLE) and solid phase extraction (SPE).15,31 The common solution is to combine several methods, such as PP with LLE or LLE with SPE, which improves the purification efficiency of the sample, but it is also laborious and time-consuming. Kassim et al.32 compared two methods of sample preparation: saponification and PP. The results showed that PP more effectively extracts vitamin D compounds from the blood.32 Recently, a method has been developed that uses the pentafluorophenyl (PFP) guard column as online SPE in the determination of 12 vitamin D compounds in serum, both hydrophobic and hydrophilic.33 For serum/plasma measurements, usually, 50 to 1000 μl of sample is used. In turn, the use of DBS in the analysis of vitamin D metabolites significantly reduces the volume of biological material (up to approximately 6 μl). Usually, discs with diameters of 3.2 mm or 6 mm are punched.20,34 Such a material also facilitates sample preparation, and results in lower reagent consumption, and high throughput analysis. However, the utilization of DBS requires a very sensitive detection method and derivatization.18,20,34,35
The aim of this work was to develop a high-throughput method for profiling vitamin D metabolites using LC-MS/MS in DBS, including 24,25(OH)2D3, which is an important metabolite formed as a result of an alternative pathway of vitamin D hydroxylation, and the epimeric form of 25(OH)D3. The goal was also to reduce the volume of the test material to use DBS for assays, which is why (dimethylaminophenyl)-1,2,4-triazoline-3,5-dione (DAPTAD) was used as a derivatization agent to increase the sensitivity of the method. To the best of our knowledge, to date there are no studies in the literature describing the use of DAPTAD in such a comprehensive analysis of vitamin D metabolites in DBS. In addition, extensive validation of the developed methodology was performed, including the comparison of the results from the analyses of two biological materials: serum and DBS, collected from nearly 100 individuals.
2. Materials and methods
2.1. Materials
Four metabolites of vitamin D (25(OH)D3, 3-epi-25(OH)D3, 25(OH)D2, 24,25(OH)2D3) and their deuterated standards (d6-25(OH)D3, d3-3-epi-25(OH)D3, d3-25(OH)D2, d6-24,25(OH)2D3) were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). Dried blood spots were collected using custom prepared Munktell TFN-Specimen Collection Cards, measuring 76 × 108 mm (Bärenstein, Germany). Calibration cards were prepared using a blood substitute prepared from red blood cell concentrate that was taken from the Regional Blood Donation Center and Blood Treatment (Warsaw, Poland) and human albumin, recombinant, expressed in rice, which was purchased from Sigma-Aldrich (Gillingham, Dorset, UK). During the validation process, blood collection tubes with different anticoagulants and clot activators were used (Sarstedt Ag&Co., Numbrecht, Germany). All experiments were performed in accordance with the Guidelines for Good Clinical Practice, and approved by the ethics committee at Medical University of Bialystok (consent of the bioethics commission no. R-I-002/143/2017). Informed consent was obtained from the human participants of this study.Various reagents were used in the sample preparation procedure. 4-(4′-Dimethylaminophenyl)-1,2,4-triazoline-3,5-dione (DAPTAD) was used as a derivatization agent. It was synthesized by Masdiag Laboratory (Warsaw, Poland). Additionally, solvents, such as water, ethyl acetate (POCh S.A., Gliwice, Poland) and methanol (Honeywell, Sigma-Aldrich, Gillingham, Dorset, UK), were used.
Mobile phases were prepared using acetonitrile (ACN) (Honeywell, Sigma-Aldrich, Gillingham, Dorset, UK), water (POCh S.A., Gliwice, Poland) and formic acid (FA) (Merck KGaA, Darmstadt, Germany). All solvents were of LC-MS grade.
Four different chromatographic columns were tested during retention studies, i.e. COSMOSIL Cholester (2.6 μm; 100 × 2.1 mm; Nacalai Tesque, Kyoto, Japan), COSMOSIL PBr (2.6 μm; 100 × 2.1 mm; Nacalai Tesque, Kyoto, Japan), Kinetex F5 (1.7 μm; 50 × 2.1 mm; Phenomenex, Torrance, CA, USA) and Kinetex Biphenyl (1.7 μm; 100 × 2.1 mm; Phenomenex, Torrance, CA, USA).
2.2 Apparatus and chromatographic conditions
A Shimadzu Nexera high-performance liquid chromatograph (Kyoto, Japan) equipped with a CTC PAL autosampler (Zwinger, Switzerland) coupled with a Shimadzu MS-8050 Triple Quadrupole MS/MS system (Kyoto, Japan) was used in the main part of the research. In addition, two other mass spectrometers were used in some parts of the experiments, i.e. QTRAP® 3200 and QTRAP® 4500 MS/MS systems (Sciex, Framingham, MA, USA). The liquid chromatograph was equipped with a degasser, two pumps, and a column oven. Analyses were performed in the positive mode using electrospray ionization (ESI). The product ion scan for each parent ion of the studied compounds was recorded within the mass range of m/z 50–650 and with a collision energy ramp of 0 to 180 V. For quantitative analysis, multiple reaction monitoring (MRM) was used. The ion source parameters were optimized by the infusion and series of multiple injections of the standard mixture. Then, the following operating parameters of the MS/MS system were applied: nebulizing gas 3 L min−1, heating gas 10 L min−1, interface temperature 300 °C, DL temperature 250 °C, heat block temperature 400 °C, and drying gas 10 L min−1.The raw data were collected using LabSolutions LCGC. To process and quantify the collected data, LabSolutions LCGC was also used.
The final chromatographic analysis was performed using Kinetex F5 1.7 μm (50 × 2.1 mm) (Phenomenex, Torrance, CA, USA) at a flow rate of 0.45 ml min−1. The temperature of the column oven was 40 °C. The mobile phase consisted of water and acetonitrile with 0.1% formic acid as an additive.
2.3 Preparation of the calibration curves using DBS
The first step was to prepare artificial human serum with the addition of a standard solution containing vitamin D metabolites in order to obtain fortified samples at eight different concentration levels. Artificial human serum was a 2% solution of human serum albumin in phosphate buffer saline. The standard solution was a mixture of four metabolites in methanol. The final serum concentrations for 24,25(OH)2D3, 25(OH)D2 and 3-epi-25(OH)D3 were 0.2, 0.5, 1.0, 3.0, 5.0, 10.0, 15.0, and 20.0 ng ml−1 and it was 2.0, 5.0, 10.0, 30.0, 50.0, 100.0, 150.0, and 200.0 ng ml−1 for 25(OH)D3. Then, the prepared material was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with red blood cell concentrate (hematocrit at 50%) and applied onto filter paper (50 μl on each spot). The prepared DBS were allowed to dry overnight at room temperature and then stored in a refrigerator at +4 °C in a string bag containing silica gel as a desiccant.
:1 ratio with red blood cell concentrate (hematocrit at 50%) and applied onto filter paper (50 μl on each spot). The prepared DBS were allowed to dry overnight at room temperature and then stored in a refrigerator at +4 °C in a string bag containing silica gel as a desiccant.
2.4 DBS sample preparation
The analyzed material was capillary blood taken from the fingertip, deposited on filter paper. An automatic DBS puncher was used to cut out a disc of 3 mm diameter from the paper cards into the polystyrene 96-well plate. During the validation process, various conditions have been investigated, such as the number of discs, type and volume of extractant, time of extraction, and the number of extractions. The developed sample preparation procedure is based on the extraction of two 3 mm discs (∼6.2 μl of blood) using a methanol solution of an isotopic standard (containing 4 deuterated standards). 150 μl of such a solution is added to each well and then, the plate is incubated for 30 minutes at room temperature on a shaker (450 rpm). After this time, 110 μl of supernatant is collected from each well and transferred to a polypropylene 96-well plate, which is dried at 50 °C under the stream of nitrogen for about 15 minutes. Subsequently, 60 μl of derivatization agent is added to each well. The solution used for derivatization is a five-fold diluted DAPTAD solution with ethyl acetate (the final concentration is 200 μg ml−1). The mixture is kept at room temperature for 30 minutes on a shaker (450 rpm). To deactivate DAPTAD, 30 μl of methanol is added. After that, the 96-well plate is dried at 50 °C under a stream of nitrogen for about 15 minutes. The residue is dissolved in a methanol/water (1:1) mixture and a 20 μl aliquot is subjected to LC-MS/MS analysis.
2.5 Assay validation
Validation of the developed method was performed according to the guidelines for Bioanalytical Method Validation of FDA Center for Drug Evaluation and Research.36 It included the limit of detection (LOD), limit of quantitation (LOQ), accuracy, and intra-day and inter-day assay precision. Also, sample stability testing under seven different conditions was performed. To examine the impact of DBS in relation to the serum results, several experiments were used, including the change in the volume of the blood spot or punching at different positions. In addition, comparative studies were conducted to investigate the differences between capillary blood and venous blood collected in test tubes with various anticoagulants, both deposited on paper cards. All validation studies were performed using both capillary and venous blood collected from healthy adult volunteers. Since there are currently no commercially available vitamin D reference materials in the form of dried blood spots, the accuracy was determined by comparing the dried blood spots to serum, both collected from nearly 100 individuals. Serum sample preparation was conducted according to the method presented by Lee et al.37 An additional step was the derivatization reaction.2.6 Quantitation of vitamin D metabolites in DBS
Multiple reaction monitoring was used to determine the concentration of vitamin D metabolites in DBS samples. Table 1 presents precursor/product ion transitions. It has to be pointed out that precursor ions were selected on the basis of signals obtained after the derivatization process. Consequently, Table 1 also presents changes in molecular weights of derivatized compounds. Quantitative analysis was based on the ratio of the area of a given metabolite peak to the area of the internal standard peak. The obtained values were compared with those from calibration curves. Since vitamin D is bound to serum proteins, the obtained concentrations have been recalculated considering the mean hematocrit for females (0.42) and males (0.47), using the following formula: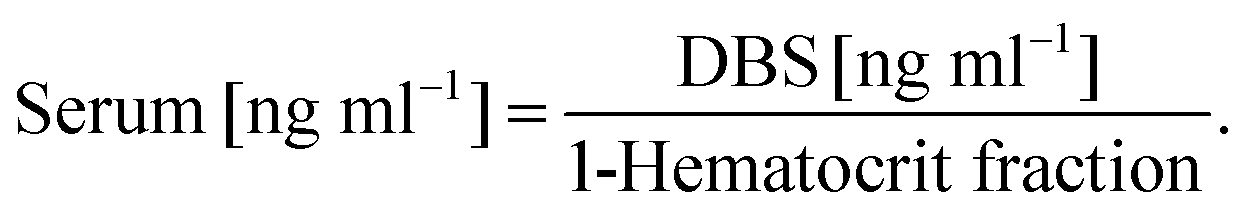
| Compound | Monoisotopic mass of intact metabolite [g] | Monoisotopic mass of DAPTAD derivative [g] | Q 1 [m/z] | Q 3 [m/z] |
|---|---|---|---|---|
| 25(OH)D3 | 400.3 | 618.4 | 619.5 | 341.1 |
| d 6-25(OH)D3 | 406.4 | 624.5 | 625.5 | 341.1 |
| 3-epi-25(OH)D3 | 400.3 | 618.4 | 619.5 | 341.1 |
| d 3-3-epi-25(OH)D3 | 403.4 | 621.4 | 622.5 | 344.1 |
| 25(OH)D2 | 412.3 | 630.4 | 631.5 | 341.1 |
| d 3-25(OH)D2 | 415.4 | 633.4 | 634.5 | 344.1 |
| 24,25(OH)2D3 | 416.3 | 634.4 | 635.5 | 341.1 |
| d 6-24,25(OH)2D3 | 422.4 | 640.5 | 641.5 | 341.1 |
3. Results and discussion
3.1 Mass spectrometry of vitamin D metabolites
The Product Ion Scan was utilized to obtain fragment spectra of four vitamin D metabolites and their corresponding isotopically labeled standards. The purpose of this stage was to determine whether the fragmentation of vitamin D derivatives is specific and will allow distinguishing similar metabolites. As a result of derivatization, the characteristic fragment is observed, which contains in its structure a moiety derived from the metabolite and DAPTAD, respectively. This is an advantage as opposed to the non-specific loss of water molecule observed in the analysis of unmodified vitamin D. This fragment is derived from the cleavage of the vitamin D skeleton and is of the highest intensity. All fragmentation spectra for standard substances are presented in Fig. 1 and Fig. 1S (ESI†), along with an additional identification of fragment ions.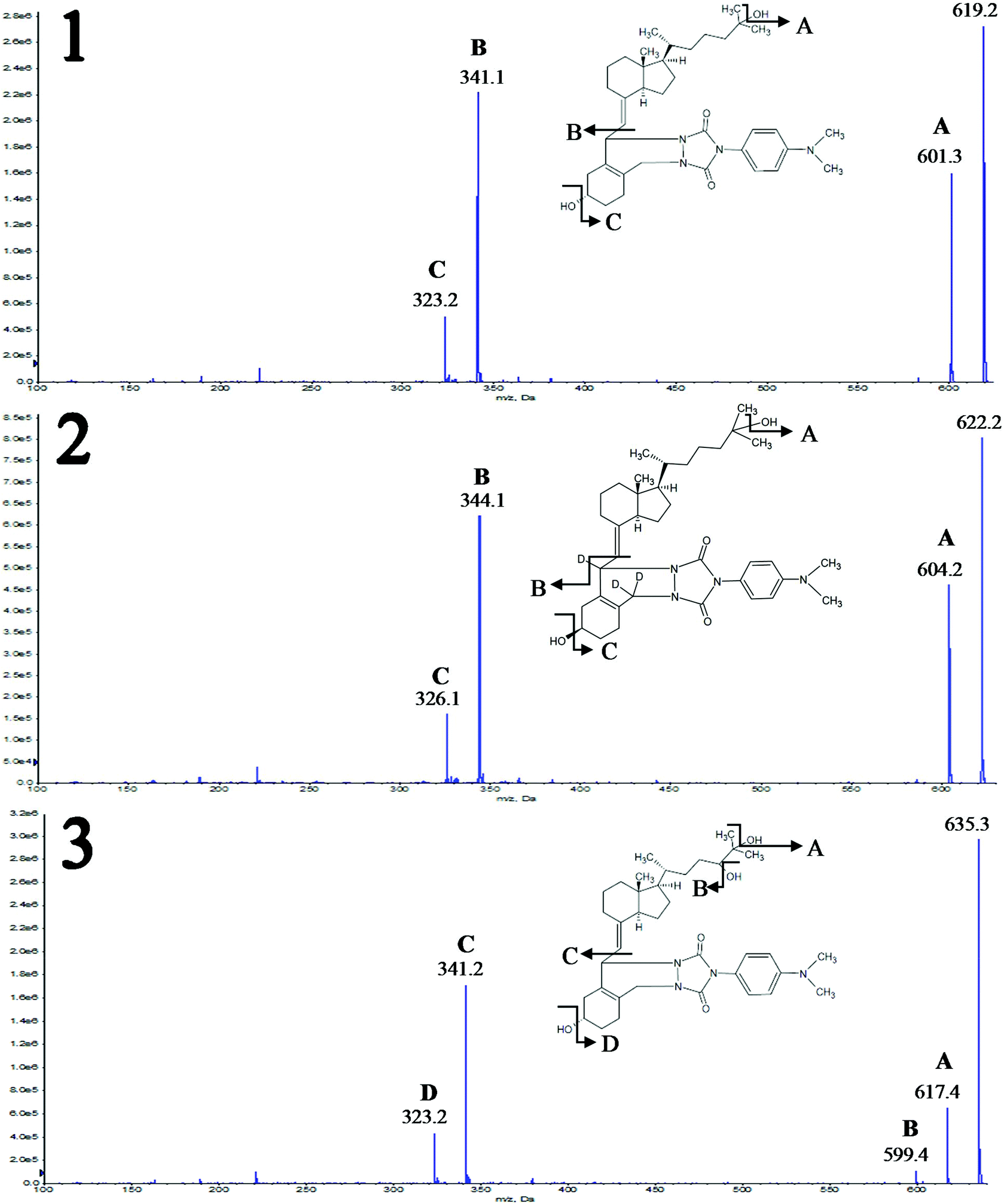 | ||
| Fig. 1 Fragment spectra of the studied compounds with the identification of daughter ions. Operation parameters of the MS/MS system: curtain gas (CUR) 30, collision gas (CAD) 7, ion spray voltage (IS) 3500 V, temperature of drying gas (TEM) 500 °C, ion source gas 1 (GS1) 40, ion source 2 (GS2) 50, declustering potential (DP) 40 V, entrance potential (EP) 10 V, collision energy (CE) 30 V, collision cell exit potential (CXP) 13 V. Notation: 1 – 25(OH)D3, 2 – d3-3-epi-25(OH)D3, and 3 – 24,25(OH)2D3. | ||
Regardless of the number of hydroxyl groups present in the side chain, the characteristic fragment is the ion with the m/z value of 341.1 Da (Fig. 1 and 1S†). This is due to the fact that the fragmentation reaction occurs between C6 and C7 of the vitamin D molecule, so any differences in the structure of the alkyl chain do not affect the mass of the resulting fragment. Different observations were made as a result of substitution on the cyclohexane moiety. Hence the fragmentation of two deuterated standards – d3-3-epi-25(OH)D3, d3-25(OH)D2 – gives the characteristic fragment at m/z = 344.1 Da. This is caused by the substitution of hydrogen atoms at positions 6, 19, and 19 with deuterium atoms, which is shown in Fig. 2S (ESI†). Apart from fragments at m/z = 341.1 and 344.1 Da, other signals can be observed in the spectra corresponding to fragments resulting from the loss of a water molecule [M + H − H2O]+ (Fig. 1 and 1S†).
The obtained results indicate that the derivatization reaction increases the specificity of the fragmentation reaction, as compared to non-derivative compounds. However, any modifications within the side chain will not give different fragment spectra, so there is no possibility to differentiate isomeric metabolites of vitamin D. Therefore, proper attention should be paid when developing a chromatographic method in order to separate these metabolites.
3.2 Attempts at chromatographic separation of vitamin D metabolites
As already mentioned, due to the derivatization of vitamin D metabolites, a mixture of two stereoisomers (6S and 6R) is formed, in a reagent-dependent constant ratio, which for DAPTAD is 5:1.38 This is an additional difficulty when developing a method for their separation. Furthermore, the addition of these dienophiles to the vitamin D molecule increases its polarity. When optimizing the chromatographic conditions, the presence of epimeric forms was also taken into account, since they have the same molecular mass and consequently the same MRM transitions as their non-epimeric forms. This results in additional systematic errors if they are not efficiently separated. Epimeric forms are created as a result of the C-3 epimerization process, where the hydroxyl group located at the 3rd asymmetric carbon is converted from alpha to beta orientation (a change in spatial configuration occurs).
Two different mobile phases were tested during this stage of studies. The first was the mixture of methanol and water, and the second was the mixture of acetonitrile and water, both with the addition of 0.1% formic acid. Different types of stationary phases were tested, including cholesteryl (Cholester), pentafluorophenyl (F5), pentabromophenyl (PBr) and biphenyl (Biphenyl) groups. The selection was based on a literature review;13,15 however, PBr was applied for the first time for the separation of the studied compounds. Due to the presence of epimers and stereoisomers, octadecyl-phase columns were not tested, because they do not have enough resolving power to separate all the studied compounds. Analyses were performed using gradient elution. Each time, the composition of the mobile phase was changed for a given column. Representative chromatograms are shown in Fig. 2.
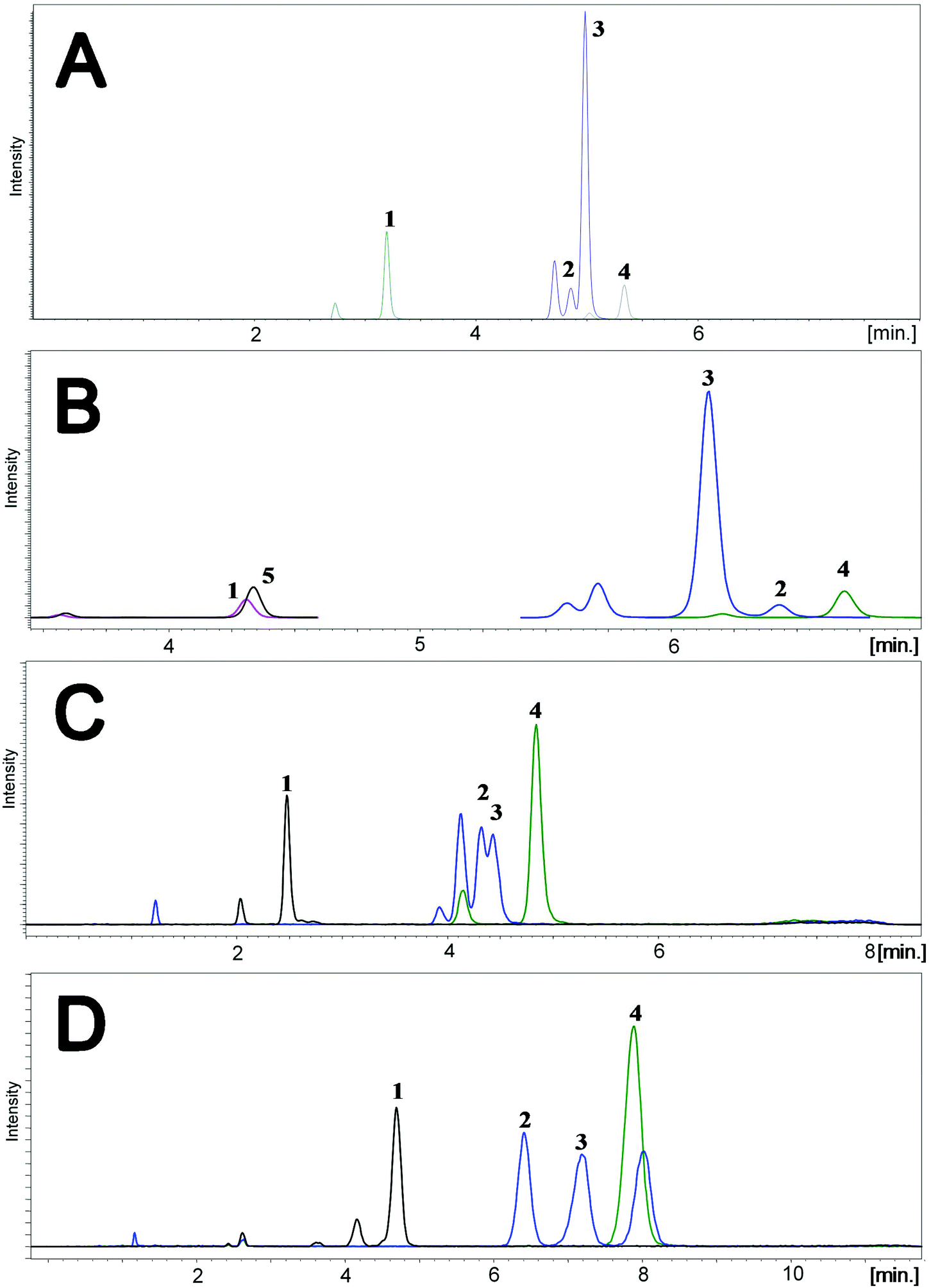 | ||
| Fig. 2 Representative chromatograms for the four tested stationary phases. Conditions: F5: Kinetex F5 (1.7 μm; 50 × 2.1 mm); mobile phase: water (A) and acetonitrile (B) with 0.1% FA; flow rate: 0.45 ml min−1; gradient elution program: 0 min – 30%B, 5.5 min – 50%B, 5.7 min – 95%B, 6.6 min – 95%B, 6.8 min – 30%B, 8 min – 30%B; column oven temperature: 40 °C; Biphenyl: Kinetex Biphenyl (1.7 μm; 100 × 2.1 mm); mobile phase: water (A) and acetonitrile (B) with 0.1% FA; flow rate: 0.45 ml min−1; gradient elution program: 0 min – 40%B, 6.5 min – 65%B, 6.7 min – 95%B, 7.6 min – 95%B, 7.8 min – 40%B, 9 min – 40%B; column oven temperature: 40 °C; Cholester: COSMOSIL Cholester (2.6 μm; 100 × 2.1 mm); mobile phase: water (A) and acetonitrile (B) with 0.1% FA; flow rate: 0.45 ml min−1; gradient elution program: 0 min – 35%B, 5.5 min – 55%B, 5.7 min – 95%B, 6.6 min – 95%B, 6.8 min – 35%B, 8 min – 35%B; column oven temperature: 40 °C; PBr: COSMOSIL PBr (2.6 μm; 100 × 2.1 mm); mobile phase: water (A) and methanol (B) with 0.1% FA; flow rate: 0.60 ml min−1; gradient elution program: 0 min – 70%B, 0.3 min – 89%B, 7 min – 89%B, 7.2 min – 95%B, 8 min – 95%B, 8.2 min – 70%B, 10 min – 70%B; column oven temperature: 45 °C. Notation: A – F5, B – Biphenyl, C – Cholester, D – PBr, 1 – 24,25(OH)2D3, 2 – 3-epi-25(OH)D3, 3 – 25(OH)D3, 4 – 25(OH)D2, 5 – d6-24,25(OH)2D3. | ||
Comparison of the results obtained for F5 and Biphenyl columns revealed that the main disadvantage of the second stationary phase was a shift in the retention time of the analyzed compounds in relation to their internal standards (Fig. 2A and B). In such a situation there is no certainty regarding the compensation of ion suppression resulting from the matrix effect. Both columns enabled almost complete separation of 3-epi-25(OH)D3 and 25(OH)D3, wherein the 3-epi-25(OH)D3 isomers were eluted at the same time, resulting in a single peak (Fig. 2A and B). The analysis time for F5 was 1 minute shorter than for Biphenyl and it was equal to 8 minutes. Both the stationary phases were used for two different particle sizes: 1.7 μm and 2.6 μm. The advantage of 1.7 μm over 2.6 μm particle size was obvious and typical of ultra-high performance liquid chromatography, namely better resolution and narrower peaks due to greater column efficiency.
The Cholester column did not have adequate resolving power to separate 3-epi-25(OH)D3 and 25(OH)D3 metabolites; therefore it was not possible to quantify them separately (Fig. 2C). This effect may be a consequence of the greater size of cholesterol molecules bonded to the silica surface, compared to pentafluorophenyl or biphenyl molecules. Consequently, the steric effects can reduce the interactions (between the studied metabolites and stationary phase) and resolution. Moreover, the particle size of Cholester (2.6 μm) will have also a significant impact on the resolution.
An interesting separation was obtained using F5-like stationary phase, where the fluorine atoms are replaced with bromine atoms – PBr. Such a column has a different selectivity, resulting in changed elution order of 3-epi-25(OH)D3 and 25(OH)D3 (Fig. 2D). For this reason, it can be used as an alternative stationary phase, especially because of the complete separation of 3-epi-25(OH)D3 and 25(OH)D3 (Fig. 2D). To date, there are no literature reports on the use of this kind of stationary phase in the analysis of vitamin D metabolites. In this case, the retention time has been extended to 10 minutes, which results from a compromise between the resolution and run time. Despite its resolving power, this column was not used in further studies due to the relatively wide peaks, which would make it impossible to achieve the desired sensitivity regarding the signal-to-noise ratio. However, the results for PBr may be significantly improved in the future by changing the particle size from 2.6 μm to 1.7 μm. It will allow both reduction of time and improvement of the peak shape and consequently also resolution and sensitivity. The final choice was a F5 column with a particle size of 1.7 μm, due to the separation of metabolites allowing individual determination during one analysis, while maintaining narrow peaks with a symmetrical shape.
The use of methanol as an organic modifier in the case of tested chromatographic columns gave wider peaks compared to acetonitrile. What is more, the analytes eluted at the higher percentage of the organic phase, which is associated with a lower elution strength of methanol compared to acetonitrile. In the case of Biphenyl and PBr phases, the substitution of methanol with acetonitrile caused a change in the elution order, which resulted from the altered selectivity of the stationary phase with respect to the analytes depending on the mobile phase being used.
3.3 DBS sample preparation
Sample preparation of the DBS is based on the extraction process with the chosen solvent. The extraction efficiency was determined on the basis of the MRM peak intensity. During method development, different types of solvents in different combinations were tested, including methanol, acetonitrile, isopropanol, a mixture of methanol and ethyl acetate, hexane and isopropanol. Additionally, various mixtures of organic solvent and water were examined. The addition of water was supposed to moisten the surface of the filter paper and facilitate extraction. Different incubation times were also tested and it was checked whether the re-extraction process would increase the overall efficiency by means of peak intensity. The last examined parameter was the temperature applied during the derivatization process.Depending on the solvent used, the extraction proceeded with a different efficiency, which could be related to the affinity of the analytes to the solvent, as well as its ability to wet the surface of the paper. Both the volume of the extractant and the number of discs to be extracted have been chosen to reach the quantification limit at the level of 0.2 ng ml−1. The best results were obtained using methanol as an extractant, as well as a mixture of methanol and ethyl acetate (70:30% v/v). Due to the rapid evaporation of ethyl acetate during extraction, methanol was finally selected because of the less problematic sample preparation. The addition of water lowered the efficiency of the extraction (based on the peak intensity at the MRM chromatogram). Moreover, the samples were not clear, which would require an additional centrifugation step.
Regarding the re-extraction impact on the extraction efficiency, no significant changes were observed after the double extraction (the difference in peak intensity was less than 10%). The optimal extraction time was 30 minutes. Above this limit, no significant increase in the peak intensity was observed.
Another parameter investigated during the study was the application of ultrasonication and checking, if it would accelerate or increase the extraction efficiency. However, no significant changes were observed. Increasing the temperature during derivatization up to 50 °C gradually changed the quantitative ratio of the resulting isomers. For this reason, the reaction was finally carried out at room temperature.
The purpose of the developed sample preparation method was primarily high throughput, which is why all operations were limited to the minimum while maintaining the sensitivity of 0.2 ng ml−1 with such a low volume of biological material (∼6.2 μl).
3.5 Assay validation
Calibration curves retain the required linearity (R > 0.995) in the given concentration ranges (Table 2). With a probability of 95%, the correlations between the peak area ratio of the analyte to the internal standard and the concentration of a given vitamin D metabolite are statistically significant. LOD and LOQ of the method were calculated based on the equations of the calibration curves (Table 2). The dynamic range for 24,25(OH)2D3, 25(OH)D2 and 3-epi-25(OH)D3 was 0.2–10 ng ml−1, and for 25(OH)D3 it was 2–100 ng ml−1. These values result from the levels at which these metabolites are found in the body.15,39| Metabolite | 25(OH)D3 | 25(OH)D2 | 3-epi-25(OH)D3 | 24,25(OH)2D3 |
|---|---|---|---|---|
| LOD [ng ml−1] | 0.16 | 0.02 | 0.01 | 0.03 |
| LOQ [ng ml−1] | 0.48 | 0.06 | 0.04 | 0.10 |
| Equation | y = 0.0342x − 0.0114 | y = 0.05x − 0.0008 | y = 0.0144x − 0.0018 | y = 0.0346x − 0.0037 |
| R 2 value | 0.9992 | 0.9989 | 0.9981 | 0.9981 |
The intra-day and inter-day assay precision was calculated on the basis of sample measurements in five replicates for five consecutive days and is expressed by the coefficient of variation. Intra-day precision for 25(OH)D3 was 3% and for the rest of the metabolites it was 2–6%. Inter-day precision was 6% for 25(OH)D3 and 7–10% for the rest of the metabolites. These results are presented in Table 3. They are consistent with the guidelines for Bioanalytical Method Validation of the FDA Center for Drug Evaluation and Research. Based on the obtained values, the normality of the data distribution was also tested in accordance with the Shapiro–Wilk test. The value W determined on the basis of the test statistic is compared with the tabulated values W(n,α) for the appropriate confidence levels and sample size. The obtained values were W = 0.949 and W(n = 24, α = 0.05) = 0.916. So there is an inequality W > W(n,α), which means that there is no reason to reject the hypothesis about the normality of the distribution of the analyzed data.
| Metabolite | 25(OH)D3 | 25(OH)D2 | 3-epi-25(OH)D3 | 24,25(OH)2D3 |
|---|---|---|---|---|
| Intra-day precision | 3% | 5% | 6% | 2% |
| Inter-day precision | 6% | 10% | 7% | 7% |
During method development, the matrix effect on ionization suppression or enhancement of tested compounds was examined. This impact was determined on the basis of two experiments. The first involved the monitoring of different classes of phospholipids using the positive precursor ion scan of m/z = 184 Da, negative precursor ion scan of m/z = 153 Da, positive neutral loss of m/z = 141 Da and single reaction monitoring of m/z 184→184 Da.40 The second experiment included permanent post-column infusion of deuterated vitamin D standards while injecting the matrix into the chromatographic system.41,42 Due to the fact that artificial blank serum based on the expressed albumin does not reflect the actual “real” sample, deuterated standards were used, since they are not present in the human body. Both experiments were carried out with pre-set chromatographic conditions. Qualitative assessment of plasma phospholipids in the case of the developed methodology is particularly important, because the studied material is a whole blood, and phospholipids present in the plasma are considered to be the main contributors to the matrix effect. The results of the first experiment showed that the phospholipid-rich fractions were eluted mainly in the 6–7.5 min time range, which results from their hydrophobic nature. A high percentage of acetonitrile in the mobile phase increases its elution strength so that the non-polar matrix components can be effectively eluted, which positively affects the lifetime of the chromatographic column. Interpretation of the obtained mass spectra revealed the presence of lysophosphatidylethanolamine, lysophosphatidylcholine, lysophosphatidic acid, lysophosphatidylserine, and plasmalogen.
The second experiment was used to evaluate the matrix effect throughout the entire chromatographic run time. For this purpose, a mixture of water and methanol was injected, followed by a matrix (real sample). During both analyses, four deuterated vitamin D metabolites were post-column infused via a T-piece. Fig. 3 presents the Total Ion Current chromatogram (TIC) intensity during the analysis time. The results indicate that matrix components altogether exhibit ion suppression under the given chromatographic conditions of about 10%. The greatest matrix effect occurs in the 6–7 min time range and it is equal to about 35%. These observations are consistent with the results of the first experiment, where the most phospholipid-rich fractions eluted at the same time. A slight increase in intensity results from the fact that the analyzed compounds ionize more efficiently with a higher percentage of the organic solvent. The results of both experiments indicated that the components of the matrix that affect the electrospray ionization of the analyzed compounds elute at a different retention time than the metabolites of vitamin D (Fig. 3). Therefore, there was no need to re-optimize the chromatographic conditions. Considering the relatively simple preparation of the sample, without the extract purification, it can be concluded that the matrix effect does not have a significant effect on the ionization and chromatographic behavior of the tested compounds.
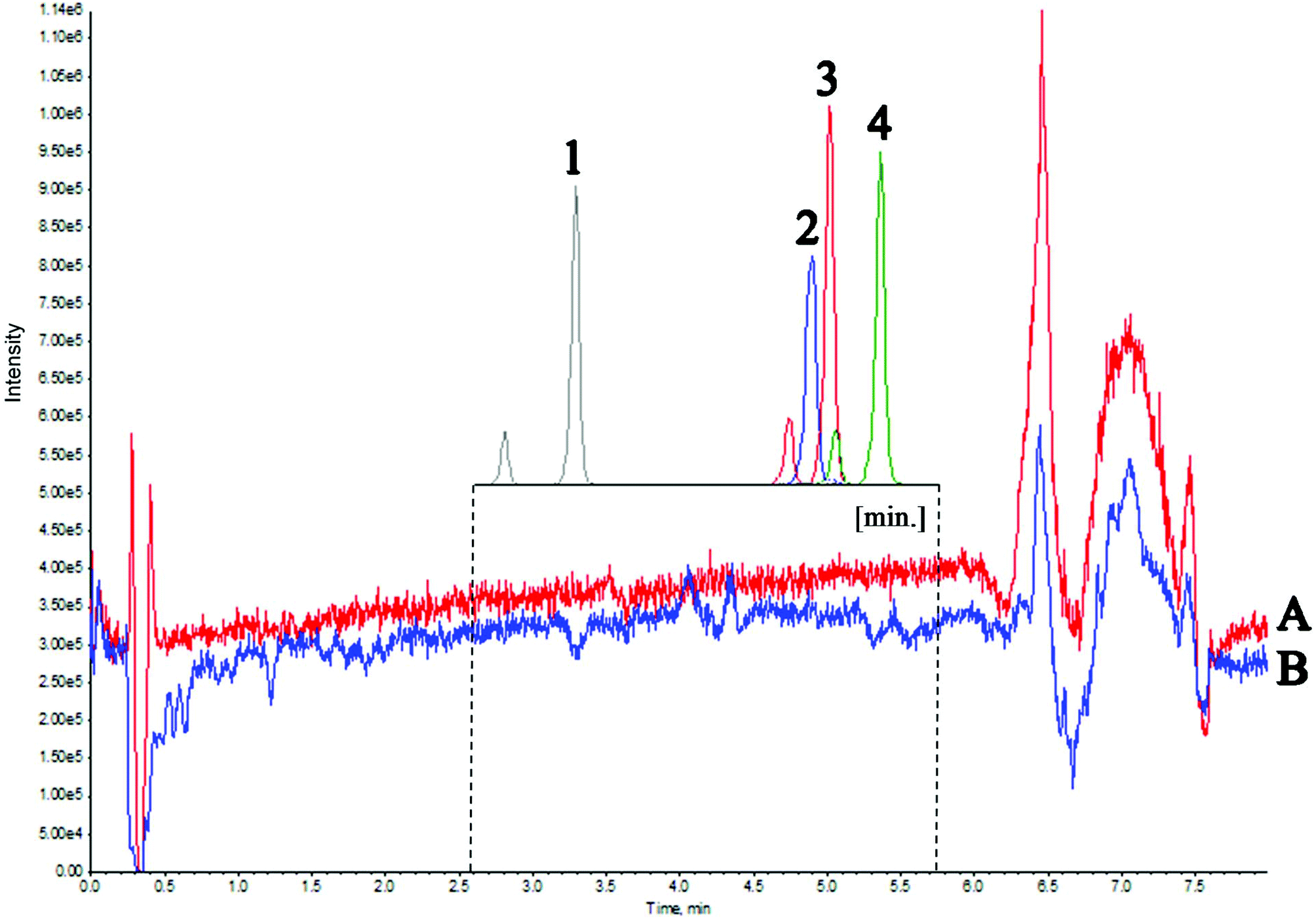 | ||
| Fig. 3 Total ion chromatogram of the four deuterated vitamin D metabolites after the injection of pure solvent and DBS sample extract. Conditions: Kinetex F5 (1.7 μm; 50 × 2.1 mm); mobile phase: water (A) and acetonitrile (B) with 0.1% FA; flow rate: 0.45 ml min−1; gradient elution program: 0 min – 30%B, 5.5 min – 50%B, 5.7 min – 95%B, 6.6 min – 95%B, 6.8 min – 30%B, 8 min – 30%B; column oven temperature: 40 °C. Notation: A – pure solvent; B – DBS sample extract; 1 – d6-24,25(OH)2D3; 2 – d3-3-epi-25(OH)D3; 3 – d6-25(OH)D3; 4 – d3-25(OH)D2. | ||
During the validation of the analytical method, the influence of both the size of the spot of blood and the place of cutting out the disc on the metabolite concentration was examined. Fig. 4S (ESI†) shows the exemplary filter paper, where blood drops of different volumes were applied and the possibilities of cutting the discs were center and edge (A), center twice (B), edge twice (C), respectively. A drop of blood deposited on filter paper isn't homogeneous over the entire surface. For this reason, compounds have lower concentration in the center of the spot than on the peripheries. This is caused by the concentration effect as the drops spread. These conclusions are consistent with those obtained by Kvaskoff et al.20 The chromatographic effect seen is due to the fact that the morphotic elements of the blood, including the red blood cells, migrate more slowly as the blood spreads on the filter paper compared to the serum. This results in higher concentration of serum on the periphery, and because vitamin D is largely bound to proteins in the serum, cutting the discs at the periphery is associated with inflated concentrations of the determined metabolites. The differences in the obtained 25(OH)D concentration values at various punching locations (center and edge, center twice, and edge twice) reach about 10–15% (between 44.5 and 51.6 ng ml−1). The solution to this problem is to punch one disc from the center and one disc from the edge of the spot. This will ensure averaging the results.
To determine the relative accuracy and to examine the effect of blood volume on the final result, an experiment was conducted, where vitamin D metabolites were analyzed in nearly 100 individuals using two different materials – serum and DBS. Serum preparation involved liquid–liquid extraction with hexane and its analysis was done identically to the DBS samples.37 The mean of all 25(OH)D results for DBS was 16.5 ng ml−1, while for the serum it was19.0 ng ml−1. The average difference between the DBS and serum results was 11.4%. Fig. 4 presents graphs comparing the results obtained for the serum and DBS samples for the corresponding compounds. The results for these two biological materials are positively correlated, with the determination coefficients of 0.9 for all the examined metabolites. Additionally, the Bland–Altman plot is shown in Fig. 5 in order to prove the good agreement between these two methods. The mean and the standard deviation of the differences between two measurements are 2.53 ng ml−1 and 2.59 ng ml−1, respectively. All of the data points lie within ±2 s of the mean difference. Hence, it may be concluded that both methods may be used interchangeably since the results are highly correlated. It should also be taken into account that the quantitative results for DBS were obtained using the average hematocrit for a given sex. It has to be pointed out that the obtained results strictly depend on the size of the blood drop. This effect is related to the degree of saturation of the sorption material from which the paper is made. The smallest differences, between DBS and serum, were obtained for a well-filtered blood drop of about 50 μl (diameter of 12 mm). Then, these differences were approximately 2–5%. When the drop of blood had a volume of approx. 25 μl, the differences in the obtained results were up to 15%. In turn, when a drop of blood was about 10 μl, the differences reached even 25%. These observations indicate that the material collection stage is crucial for the quality of the results.
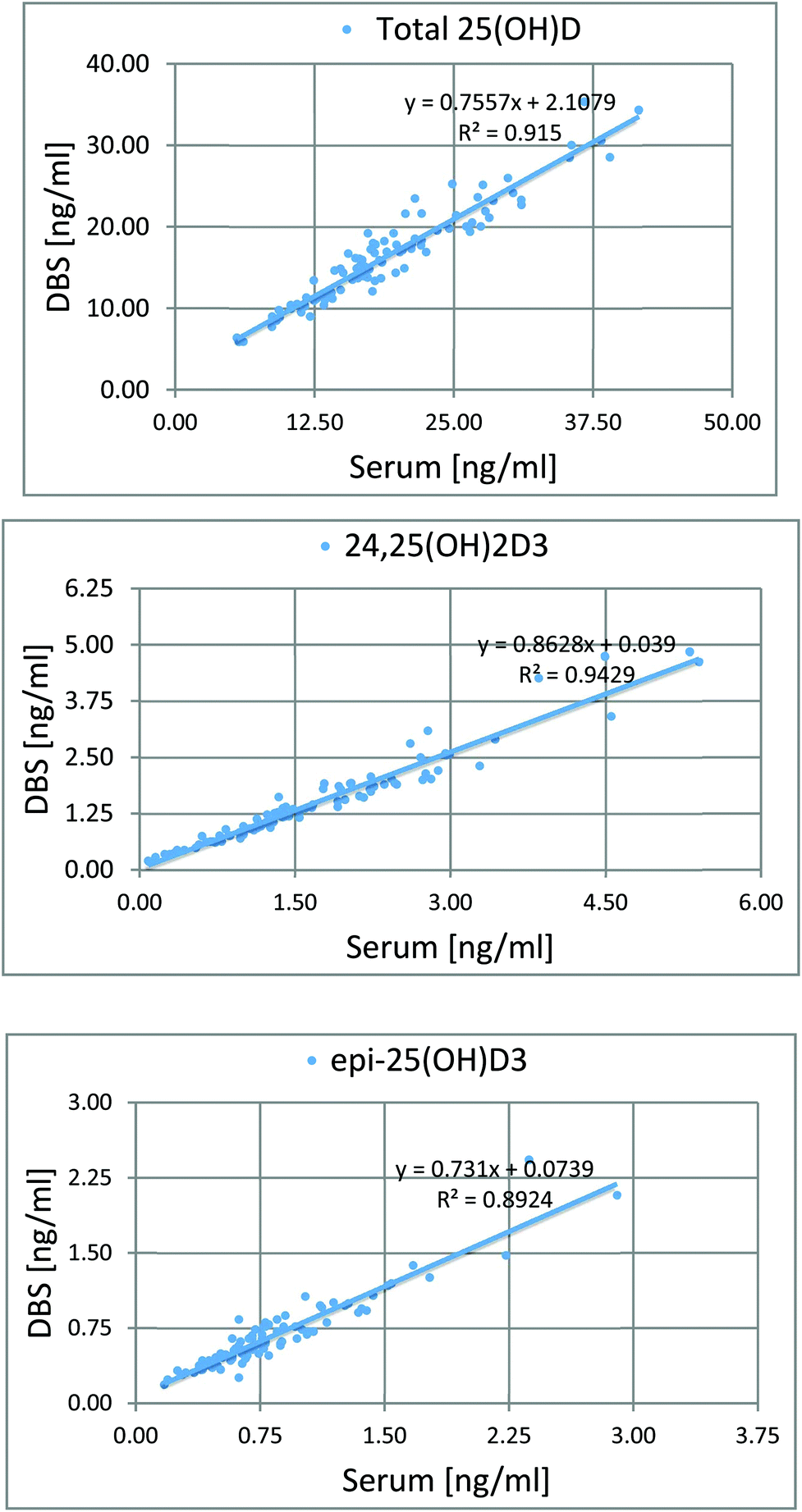 | ||
| Fig. 4 Graphs showing the good correlation of the results obtained from the analysis of serum and DBS. | ||
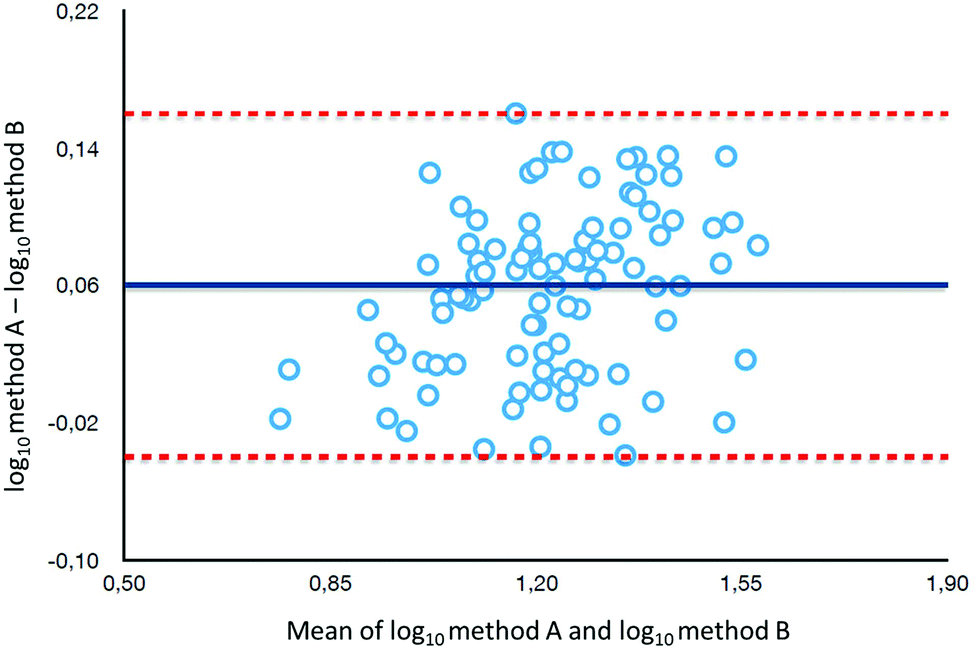 | ||
| Fig. 5 A Bland–Altman plot of log-transformed data. | ||
Another parameter verified during the validation was the impact of the anticoagulant on the total 25(OH)D concentration with respect to the capillary blood, since there are no data in the literature on this subject. The goal of this step was to study whether the blood collected from the peripheral vein can be deposited on the paper and subjected to identical determination as capillary blood. For this purpose, blood from the vein was collected into test tubes with three different anticoagulants, i.e. sodium citrate, EDTA and lithium heparin. Differences in the total concentration of 25(OH)D for citrate, heparin, and EDTA with respect to capillary blood were 6.7%, 13.6%, and 7.4%, respectively. The results indicate a significant difference in the determined concentrations when using heparin as an anticoagulant (about two times greater error compared to the other ones).
Method validation included sample stability testing under certain conditions such as exposure to light, high/low humidity, different temperatures (room temperature, refrigerator, and freezer), in different combinations. Table 1S (ESI†) presents between-assay imprecision calculated from DBS samples. Within a month the concentration of individual metabolites did not change, regardless of the condition, and the differences in results arose only from the size of the blood drop or the place of cutting out the disc (center or edge of the spot).
4. Conclusion
This study provided a method for simultaneous determination of four vitamin D metabolites, including 24,25(OH)2D3 and the epimeric form of 25(OH)D3, using a DBS-based methodology. Such a comprehensive analysis of vitamin D metabolites in DBS using DAPTAD as a derivatizing agent was performed for the first time. Chromatographic separation was carried out using an F5 column. Also, the PBr column was used for the first time, which ensured the complete separation of 3-epi-25(OH)D3 and 25(OH)D3. Therefore, it can be successfully used as an alternative stationary phase. An important step was to establish whether DBS concentrations reflect those in serum. The obtained results show good agreement between these two materials, with the determination coefficients of 0.9 for all the examined metabolites. The stage that significantly influenced the accuracy of the obtained results was the collection of materials. Due to the lack of literature data on the anticoagulant effect on the concentration of vitamin D metabolites, three commonly used vitamin D metabolites were examined. The smallest differences in the results between capillary and venous blood were obtained for citrate. The storage conditions for the stability of the tested compounds were evaluated during the month. The results showed that the concentrations of the analyzed metabolites did not change, and the differences were only due to the different size of the blood spot and the place of cutting out the disc. The use of DBS for the determination of vitamin D metabolites is a big step towards personalized medical diagnostics, due to the possibility of self-collection of blood through the prick of a fingertip. DBS is a material that requires a very small volume of blood (∼50 μl), it is easy to ship and store, and it significantly simplifies the process of sample preparation, including its collection. The developed method allows for the simultaneous measurement of multiple vitamin D metabolites. In this way, the study of metabolite distribution, rather than the monitoring of a single compound, can have high diagnostic potential.Conflicts of interest
There are no conflicts to declare.References
- S. S. Gropper and J. L. Smith, J. Adv. Nutr. Hum. Metab., 2013, 390–400 Search PubMed.
- M. F. Holick, N. Engl. J. Med., 2007, 357, 266–281 CrossRef CAS PubMed.
- J. E. Zerwekh, Am. J. Clin. Nutr., 2008, 87, 1087S–1091S CrossRef CAS PubMed.
- C. T. Sempos, A. C. Heijboer, D. D. Bikle, J. Bollerslev, R. Bouillon, P. M. Brannon, H. F. DeLuca, G. Jones, C. F. Munns, J. P. Bilezikian, A. Giustina and N. Binkley, Br. J. Clin. Pharmacol., 2018, 84, 2194–2207 CrossRef CAS PubMed.
- M. Wójcik, E. Czekuć-Kryśkiewicz, B. Parafiniuk, E. Skorupa, A. Kępka and P. Płudowski, Post. Nauk. Med., 2016, 10, 726–733 Search PubMed.
- B. W. Hollis, Curr. Opin. Endocrinol., Diabetes Obes., 2008, 15, 489–494 CrossRef CAS PubMed.
- J. H. Lee, J. Choi, O. J. Kweon and A. J. Park, J. Bone Metab., 2015, 22, 107–112 CrossRef PubMed.
- D. Bailey, K. Veljkovic, M. Yazdanpanah and K. Adeli, Clin. Biochem., 2013, 46(3), 190–196 CrossRef CAS PubMed.
- L. Li, Q. Zeng, J. Yuan and Z. Xie, J. Clin. Biochem. Nutr., 2016, 58(3), 186–192 CrossRef CAS PubMed.
- C. L. Farrell, S. Martin, B. McWhinney, I. Straub, P. Williams and M. Herrmann, Clin. Chem., 2012, 58(3), 531–542 CrossRef CAS PubMed.
- Z. Bartoszewicz, A. Kondracka, R. Jaźwiec, M. Popow, M. Dadlez and T. Bednarczuk, Endokrynol. Pol., 2013, 22–30 Search PubMed.
- D. A. Volmer, L. R. B. C. Mendes and C. S. Stokes, Mass Spectrom. Rev., 2015, 34, 2–23 CrossRef CAS PubMed.
- M. J. Müller and D. A. Volmer, Clin. Chem., 2015, 61(8), 1033–1048 CrossRef PubMed.
- G. Jones and M. Kaufmann, J. Steroid Biochem. Mol. Biol., 2016, 164, 110–114 CrossRef CAS PubMed.
- J. M. W. van den Ouweland, Trends Anal. Chem., 2016, 84, 117–130 CrossRef CAS.
- Y. Qi, T. Geib, P. Schorr, F. Meier and D. A. Volmer, Rapid Commun. Mass Spectrom., 2015, 29, 1–9 CrossRef CAS PubMed.
- M. S. Newman, T. R. Brandon, M. N. Groves, W. L. Gregory, S. Kapur and D. T. Zava, J. Diabetes Sci. Technol., 2009, 3(1), 156–162 CrossRef PubMed.
- D. Eyles, C. Anderson, P. Ko, A. Jones, A. Thomas, T. Burne, P. B. Mortensen, B. Norgaard-Pedersen, D. M. Hougaard and J. McGrath, Clin. Chim. Acta, 2009, 403, 145–151 CrossRef CAS PubMed.
- T. Higashi, M. Suzuki, J. Hanai, S. Inagaki, J. Z. Min, K. Shimada and T. Toto'oka, J. Sep. Sci., 2011, 34, 725–732 CrossRef CAS PubMed.
- D. Kvaskoff, P. Ko, H. A. Simila and D. W. Eyles, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 901, 47–52 CrossRef CAS PubMed.
- U. Hoeller, Br. J. Nutr., 2016, 115, 202–211 CrossRef CAS PubMed.
- S. Baecher, A. Leinenbach, J. A. Wright, S. Pongratz, U. Kobold and R. Thiele, Clin. Biochem., 2012, 45, 1491–1496 CrossRef CAS PubMed.
- C. J. Hedman, D. A. Wiebe, S. Dey, J. Plath, J. W. Kemnitz and T. E. Ziegler, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 62–67 CrossRef CAS PubMed.
- S. Zhang, W. Jian, S. Sullivan, B. Sankaran, R. W. Edom, N. Weng and D. Sharkey, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 961, 62–70 CrossRef CAS PubMed.
- H. Ketha, R. Kumar and R. J. Singh, Clin. Chem., 2016, 61(1), 236–242 CrossRef PubMed.
- T. Ishige, M. Satoh, S. Ogawa, M. Nishimura, K. Matsushita, T. Higashi and F. Nomura, Clin. Chim. Acta, 2017, 473, 173–179 CrossRef CAS PubMed.
- E. Kasalova, J. Aufartova, L. Kujovska Krcmova, D. Solichova and P. Solich, Food Chem., 2015, 171, 177–190 CrossRef CAS PubMed.
- T. Higashi, Y. Shibayama, M. Fuji and K. Shimada, Anal. Bioanal. Chem., 2008, 391, 229–238 CrossRef CAS PubMed.
- W. Li and F. L. S. Tse, Biomed. Chromatogr., 2010, 24, 49–65 CrossRef CAS PubMed.
- T. Holmoy, S. M. Moen, T. A. Gundersen, M. F. Holick, E. Fainardi and M. Castellazzi, Mult. Scler., 2009, 15, 1280–1285 CrossRef PubMed.
- M. L. Musteata and F. M. Musteata, Future Sci., 2011, 1987–2002 CAS.
- N. S. A. Kassim, F. G. Gomes, P. N. Shaw and A. K. Hewavitharana, Bioanalysis, 2016, 8(5), 397–411 CrossRef PubMed.
- N. S. A. Kassim, P. N. Shaw and A. K. Hewavitharana, J. Chromatogr. A, 2018, 1533, 57–65 CrossRef PubMed.
- A. J. Makowski, J. A. Rathmacher, R. L. Horst and C. T. Sempos, J. AOAC Int., 2017, 100(5), 1328–1336 CrossRef CAS PubMed.
- M. J. Müller, C. S. Stokes and D. A. Volmer, Talanta, 2017, 165, 398–404 CrossRef PubMed.
- Bioanalytical Method Validation – Guidance for Industry https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf (accessed February 2018).
- D. Lee, T. J. Garrett, B. A. Goldberger and L. A. Bazydlo, Bioanalysis, 2015, 7(2), 167–178 CrossRef CAS PubMed.
- S. Ogawa, S. Ooki, M. Morohashi, K. Yamagata and T. Higashi, Rapid Commun. Mass Spectrom., 2013, 27, 2453–2460 CrossRef CAS PubMed.
- D. Ferrari, G. Lombardi and G. Banfi, Biochem. Med., 2017, 27(3), 1–14 Search PubMed.
- Y. Xia and M. Jemal, Rapid Commun. Mass Spectrom., 2009, 23, 2125–2138 CrossRef CAS PubMed.
- H. Stahnke, T. Reemtsma and L. Alder, Anal. Chem., 2009, 81, 2185–2192 CrossRef CAS PubMed.
- R. Bonfiglio, R. C. King, T. V. Olah and K. Merkle, Rapid Commun. Mass Spectrom., 1999, 13, 1175–1185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01422a |
| This journal is © The Royal Society of Chemistry 2019 |