
Molecular and living cell dynamic assays with optical microscopy imaging techniques
Hua
Liu
a,
Zhongju
Ye
a,
Xin
Wang
a,
Lin
Wei
b and
Lehui
Xiao
 *a
*a
aState Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Biosensing and Molecular Recognition, College of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: lehuixiao@nankai.edu.cn; Web: http://www.xiaolhlab.cn
bKey Laboratory of Chemical Biology & Traditional Chinese Medicine Research, Key Laboratory of Phytochemical R&D of Hunan Province, College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha, 410082, China
First published on 31st October 2018
Abstract
Generally, the message elucidated by the conventional analytical methods overlooks the heterogeneity of single objects, where the behavior of individual molecules is shielded. With the advent of optical microscopy imaging techniques, it is possible to identify, visualize and track individual molecules or nanoparticles under a biological environment with high temporal and spatial resolution. In this work, we summarize the commonly adopted optical microscopy techniques for bio-analytical assays in living cells, including total internal reflection fluorescence microscopy (TIRFM), super-resolution optical microscopy (SRM), and dark-field optical microscopy (DFM). The basic principles of these methods and some recent interesting applications in molecular detection and single-particle tracking are introduced. Moreover, the development in high-dimensional optical microscopy to achieve three-dimensional (3-D) as well as sub-diffraction localization and tracking of biomolecules is also highlighted.
Introduction
Exploring the physical and chemical properties of target objects has been the unremitting pursuit of researchers, such as the heterogeneity of individual cells and the kinetics of individual molecules in the process of chemical reactions.1,2 With the development of optical microscopy techniques, research on single-molecule and -cell analysis has received burgeoning attention and rapid advancement. Nowadays, both the reactivity of a single-molecule and the movement of a single-particle within a cell can be revealed.3,4 The conventional ensemble averaged measurements tend to ignore the heterogeneous behavior of many individual objects and even result in false judgment. Different from conventional measurements, microscopy analysis with optical methods can afford statistical and dynamic information in situ. An example is the transcription process of DNA. Studies have shown that polymerases are used to transcribe information stored in DNA and they move along DNA with specific step size (i.e. 3.4 Å).5,6 This continuous and linear process is also associated with random intermittency in the overall process. Quantifying the production rate of proteins within the cell with ensemble measurements (e.g., western blotting) only reflects a continuous process where the step-by-step motion of polymerases cannot be elucidated.7 Masking of individual molecular information in the overall measurement may give us a misunderstanding of the native biological process. Meanwhile, many diseases also occur due to the errors at the single-molecule level, for example, the mutation of a gene. A deep understanding of the molecular feature at the single-molecule level is therefore of great significance.Typically, the merits of single object detection and analysis are covered by the following three points. (1) The ensemble properties can be evaluated from the statistics of enough single objects, while the precise characterization and dynamics of individual objects cannot be captured from ensemble experiments in general.2 (2) Single object analysis with optical microscopy can afford sufficient spatial and temporal resolution which is one of the most important approaches to reveal the precise structure–reactivity relationship of the molecules or nanoparticles. (3) Exploring the essence of reaction dynamics plays crucial roles in manipulating the reaction pathways on the atomic and molecular scale. Basically, our knowledge of molecular interactions and chemical changes comes almost exclusively from experiments on ensembles of molecules. Single object analysis can capture more detailed information from individual behaviors and reveal the structure–activity relationship of a single atom or molecule.
In this review, we firstly introduce the significance and basic principles of TIRFM, SRM, and DFM, which are the three most commonly used optical microscopy techniques for ultra-sensitive detection, such as molecular detection and single-molecule (or -particle) tracking within the cell. Recent achievements of these techniques in molecular detection, single-molecule (or -particle) tracking, and high-dimensional imaging are delineated. The challenges and prospects of optical microscopy methods for bio-analytical oriented applications are also discussed. Finally, perspective regarding the remaining challenges and directions for future advancements are discussed.
Optical microscopy imaging methods for molecular and living cell dynamic assay
The revolution in optical microscopy techniques has spawned a series of robust imaging methods, paving the way for monitoring chemical activity on the atomic or molecular scale and tracking single objects (molecules or particles) inside the living cells. For biological-oriented applications, the commonly adopted optical microscopy techniques can be classified into fluorescence and non-fluorescence modes. For the first mode, fluorescence-based optical microscopy techniques mainly include confocal laser scanning microscopy (CLSM), TIRFM, and SRM. The second mode is based on the scattering or absorption properties of the probes, for example, DFM, differential interference contrast microscopy (DIC) and interferometric scattering microscopy (iSCAT). Previously, the basic principles of these techniques were discussed in detail by many comprehensive reviews.8–10 Photophysical and biological orientated applications of these methods have also been summarized by several excellent studies.11–13 In the following section, we are essentially focusing on the most commonly used optical microscopy techniques for molecular and living cell dynamic assays, including TIRFM, SRM and DFM.Total internal reflection fluorescence microscopy (TIRFM)
Despite the extensive applications of optical microscopy techniques, some challenges still exist. For example, the native fluorescence from the cell sample and the interfering signal from different focal planes make individual events hardly able to be differentiated. The advent of TIRFM makes it possible to solve this problem. Total internal reflection occurs when a propagated wave strikes a medium boundary at an angle larger than a particular critical angle with respect to the normal to the surface. An evanescent wave is then generated when the incident light is totally reflected at the interface (i.e., the glass–water interface) (Fig. 1A). The evanescent electromagnetic field decays exponentially from the interface. The penetration depth can be calculated by using the formula: d = (λ/4π)(n12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin2θ − n22)−1/2, where θ represents the incidence angle of the excitation beam, and n1 and n2 are the refractive index of the cover slip and the specimen, respectively.14 Through controlling the incident angle and laser power, the evanescent wave penetrates to a depth of only approximately 100 nm into the sample medium.15 Thus the TIRFM enables a selective visualization of surface regions such as the basal plasma membrane (which are about 7.5 nm thick) of cells. The fluorescent molecules in the bulk solution or within the cell cannot be excited under this mode, enabling single-molecule sensitivity at the interface.16
sin2θ − n22)−1/2, where θ represents the incidence angle of the excitation beam, and n1 and n2 are the refractive index of the cover slip and the specimen, respectively.14 Through controlling the incident angle and laser power, the evanescent wave penetrates to a depth of only approximately 100 nm into the sample medium.15 Thus the TIRFM enables a selective visualization of surface regions such as the basal plasma membrane (which are about 7.5 nm thick) of cells. The fluorescent molecules in the bulk solution or within the cell cannot be excited under this mode, enabling single-molecule sensitivity at the interface.16
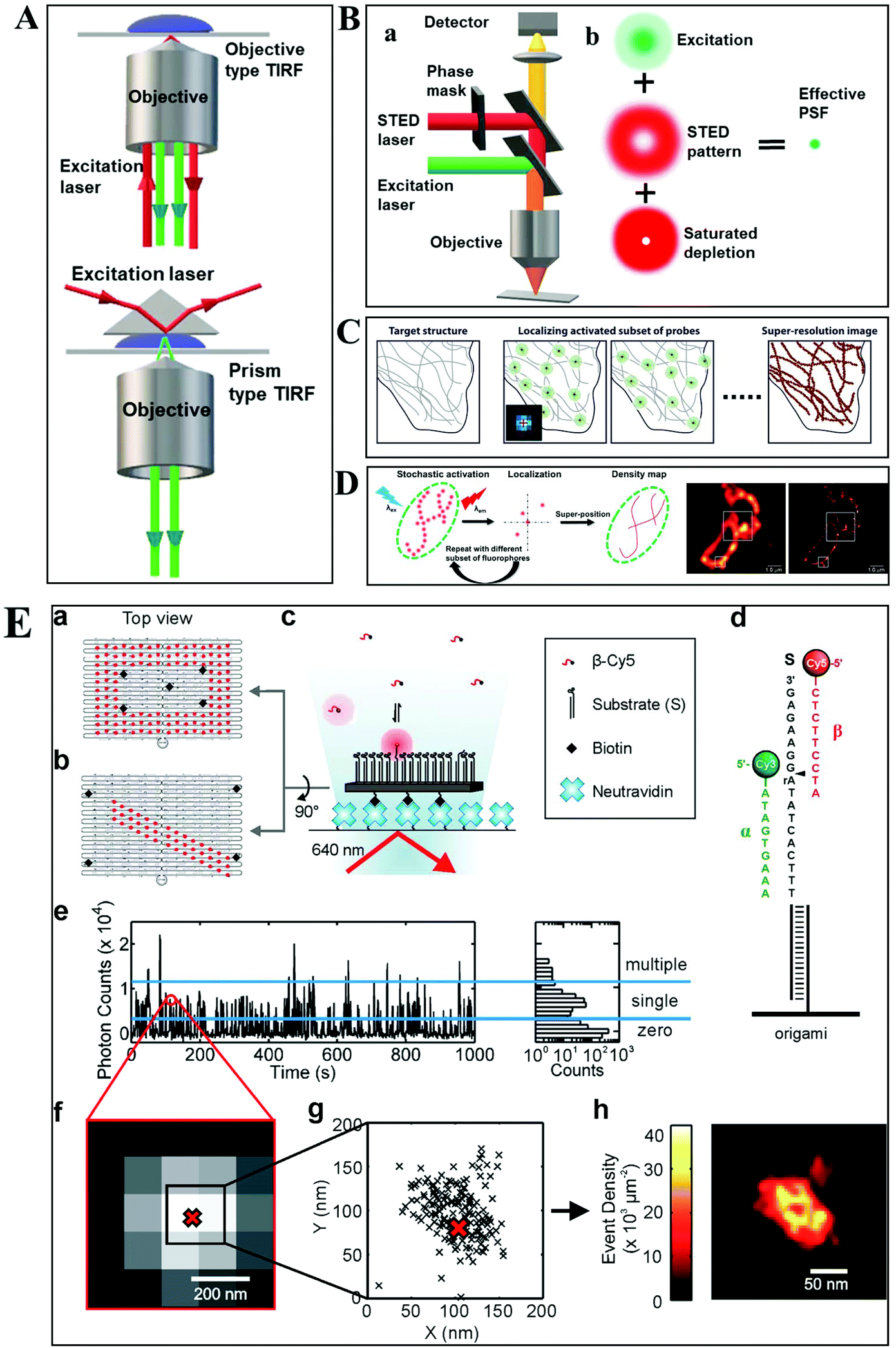 | ||
| Fig. 1 (A) Schematic diagram of objective-type TIRF and prism-type TIRF. (B) Schematics of the light path for a STED microscope.23 (C–D) The principle of STORM and PALM imaging, and the comparison of the TIRF microscopy image (left) and PALM imaging (right) of the thin section from a COS-7 cell (D), scale bar is 1.0 μm.23,25 (E) The principle of the PAINT imaging pattern with origami tile designs (a–d). Selection of data for image reconstruction (e–g). PAINT reconstruction image of the origami shown in g (h).26 | ||
Owing to the greatly improved signal-to-noise ratios (S/N), TIRFM allows the imaging of the movement of single molecules with high spatial resolution, even beyond the optical diffraction limit.17 Yildiz et al. made it possible to observe the movement of motor proteins with 1 nm precision based on Gaussian algorithm fitting of the point spreading function (PSF) from individual molecules.18 Owing to these attractive advantages of TIRFM, many interesting biological processes, particularly membrane events, have been extensively explored.19,20
Super-resolution optical microscopy (SRM)
Typically, lens-based optical microscopy cannot discriminate objects less than 200 nm due to the optical diffraction limit. The recent progress in super-resolution imaging, especially in fluorescence-based optical microscopy, has broken the diffraction limit and achieved tens of nanometers spatial resolution, enabling the observation of biological macromolecules and micro-structures of organelles in living cells with unprecedented clarity.21,22 SRM provides an opportunity to observe protein expression and gene coding processes at the molecular level.Generally, the current SRM techniques can be classified into two groups, which are based on the non-linear effect of fluorophores through saturated depletion/excitation and the accurate localization of individual fluorophores, respectively. Stimulated emission depletion (STED) microscopy is a well-known representative of the first mode, which breaks a diffraction resolution barrier by superimposing excitation and a depletion laser with different phase-spatial distributions. Specifically, as shown in Fig. 1B, there are two beams in STED microscopy.23 The excitation laser stimulates fluorophores, and then the depletion laser drives the excited fluorophores back to the ground state by stimulated emission, suppressing spontaneous fluorescence emission. The emission wavelength of the fluorophore is comparable to that of the depletion laser. The donut-shaped energy distribution of the depletion laser is generated by a phase modulator, which suppresses fluorescence emission in the periphery while the central part is not affected. As a result of the STED pattern, the PSF of individual fluorophores is sharpened, reducing the focal spot area and thus improving the lateral resolution. Willig et al. observed individual synaptic vesicles using a STED microscope with 45 nm spatial resolution in the focal plane.24
Through accurately localizing the spatial position of individual fluorophores, many interesting SRM methods have been developed, including stochastic optical reconstruction microscopy (STORM), photoactivated localization microscopy (PALM), and point accumulation for imaging in nanoscale topography (PAINT) (Fig. 1C–E).23,25,26 Different from the STED strategy, these approaches have overcome the diffraction resolution barrier by iterating images of fluorescent probes at different time points. Provided that the density of probes is low enough, each bright spot in the fluorescence image can be considered as the PSF from individual molecules. Thus, this approach can return the absolute position of the object by replacing the PSF of the object with its centroid point based on a Gaussian function fitting.
In STORM and PALM procedures, organic dyes or fluorescent proteins bound to molecules or particles are successively activated, localized and deactivated. Then the image with high precision can be achieved by overlapping these frames, formed at different time points. A recent study by Soper and colleagues mapped the functional groups present on a surface using STORM, with a fluorophore localization of 17 nm, allowing for an assessment of charge heterogeneity.27 It is reported that the localization precision is positively correlated with the square root of the number of fluorophore position measurements.28 The more the accumulation of frames, the higher the spatial precision that can be achieved. However, the number of measurements is confined by the motion of objects and the photo-stability of the fluorophores.
The PAINT method is considered as a variant of STORM. In the PAINT method, fluorescent probes transiently bind to the target objects through specific or nonspecific interactions. The binding frequency is one of the parameters to control the resolution capability. Since the association of the probe with the sample is reversible, after the initial probe diffuses away, the sample can be labeled by a new coming probe again. The integrated intensity of objects vs. time exhibits intermittency similar to the blinking effect from single quantum dots (QDs). To eliminate the impact of noise and simultaneous multiple bounds, only binding events with a certain range intensity are used in the reconstruction. It is reported that the spatial resolution of this method can achieve 25 nm.29
Dark-field optical microscopy (DFM)
DFM is a simple yet robust technique and well suited for applications in living cells and unstained biological samples. Particularly, with the rapid progress in nanomaterial fabrication (e.g. plasmonic nanoparticles), this technique has been extensively applied for the exploration of molecular–molecular interaction on cell membranes or inside living cells and ultra-sensitive biomolecule detection.30,31 The key point to achieve dark-field mode illumination is the dark-field condenser, which blocks the central part of incident light by the patch stop and allows a portion of the incident light with enough inclination angle to be transported through the sample (Fig. 2A). Different from bright-field illumination, the excitation light cannot enter the detector directly in the dark-field mode, creating a dark background. Only the signals scattered from the samples can be captured by the objective with a numerical value (NA) less than that of the condenser, greatly improving the image contrast and S/N. However, the scattering from the biological sample can interfere with the observation system, which can be reduced by using probes with large scattering cross-section and regulating the refractive index of solution. Plasmonic nanoparticles with large scattering cross-section have been the most commonly used probes in DFM recently.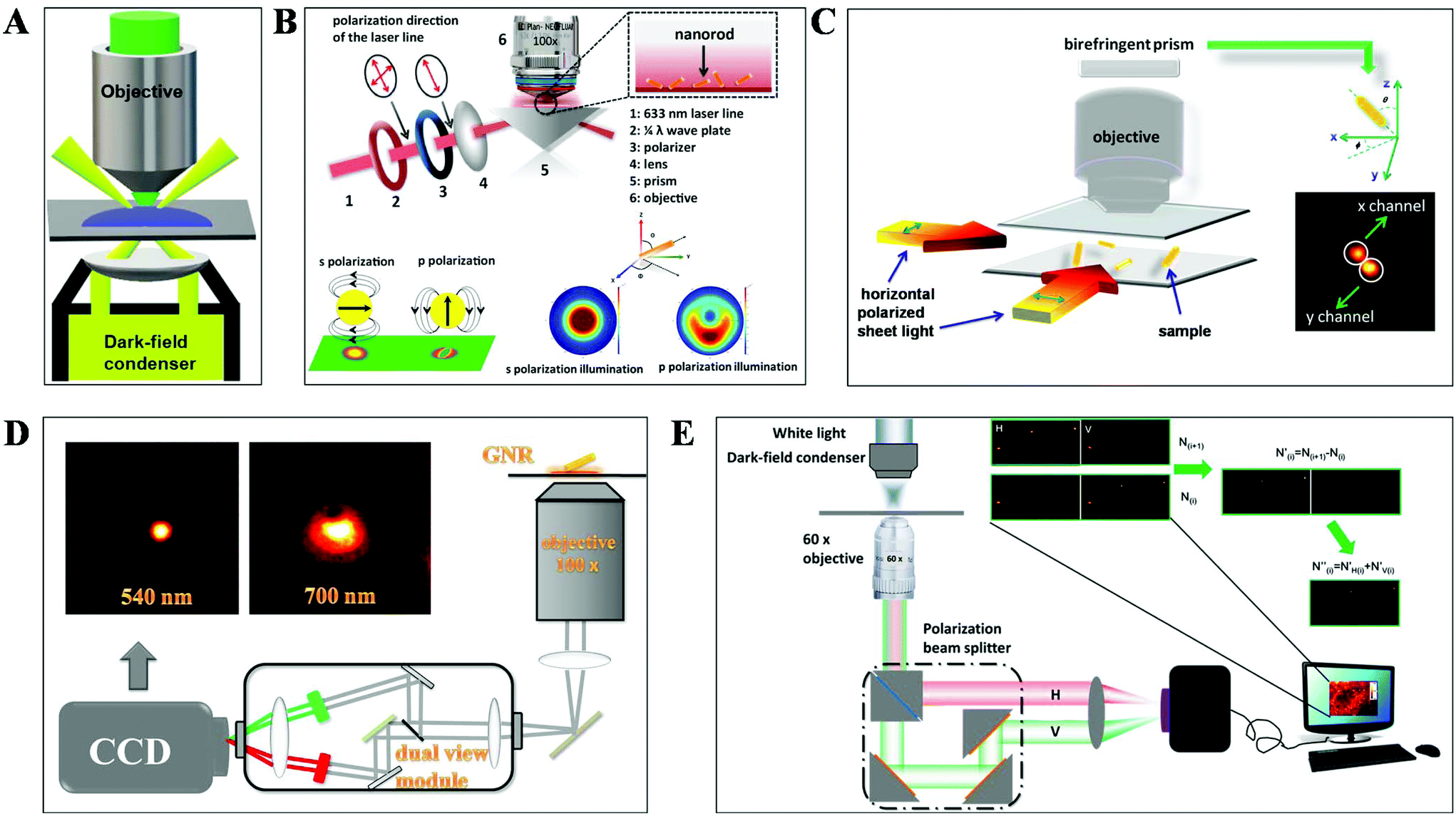 | ||
| Fig. 2 Schematic diagram of (A) DFM, (B) total internal reflection scattering microscopy (TIRSM),32 (C) planar illumination microscopy,33 (D) dual-wavelength upright DFM,34 and (E) DFM with a polarization-resolved dual-channel imaging module.35 | ||
To further improve the sensitivity of DFM for single-particle imaging, several scattering-based detection schemes have been developed recently by our group, such as total internal reflection scattering imaging, sheet light illumination, background-free dual wavelength illumination and so on (Fig. 2B–E).32–35 In contrast to the conventional dark-field illumination mode, greatly improved S/N as well as much more dynamic information can be deduced from these strategies. For example, the 3-D rotational dynamics of the single gold nanorod (GNR) translocating on the microtubule or on the surface trapped by polymers can be readily deduced by these methods (Fig. 2B–D).32–34 Regarding the total internal reflection scattering mode, the S/N can be enhanced through simply adjusting the excitation laser power, which is particularly suitable for high resolution single-particle tracking applications.
The commonly adopted probes for optical microscopy imaging
The detection and tracking of intracellular biomolecules typically require the conjugation of an imaging contrast reagent to the target object. Regarding the fluorescence mode, the most commonly adopted probes mainly include organic fluorophores, QDs and fluorescent proteins. Organic fluorophores have been used for medical diagnosis for a very long time, which are considered as essential reagents in biological research. There are so many organic fluorophores, such as cyanine, oxazine and rhodamine, spanning the entire visible and near infrared wavelength range. The molecular weights of dye molecules are in the range of 200–1000, which are much smaller than that of QDs and fluorescent proteins.2 Fluorescent probes with smaller size dimension exhibit more advantages in single-particle tracking. A less steric hindrance effect from small organic dyes greatly reduces the perturbation of the motion of biomolecules. However, their photo-stability is usually limited, which is detrimental to the long-term observation of the intracellular process.QDs are also known as fluorescent semiconductor nanocrystals. They are typically a few to tens of nanometers in diameter, several orders of magnitude larger than traditional organic fluorophores (Fig. 3A).36 The physicochemical properties, such as absorption and emission, are composition- and size-dependent, controlled by reaction time, ligand molecules and so on.37 QDs are considered as ideal candidates for replacing organic fluorophores, owing to their advantages in tunable photoluminescence, high quantum yield, large Stokes shift and excellent photo-stability. However, the toxicity of semiconductor QDs is still under debate. At present, new kinds of QDs without heavy metal ions synthesized in aqueous solution have sprung up, such as silicon QDs, graphene QDs and so on.
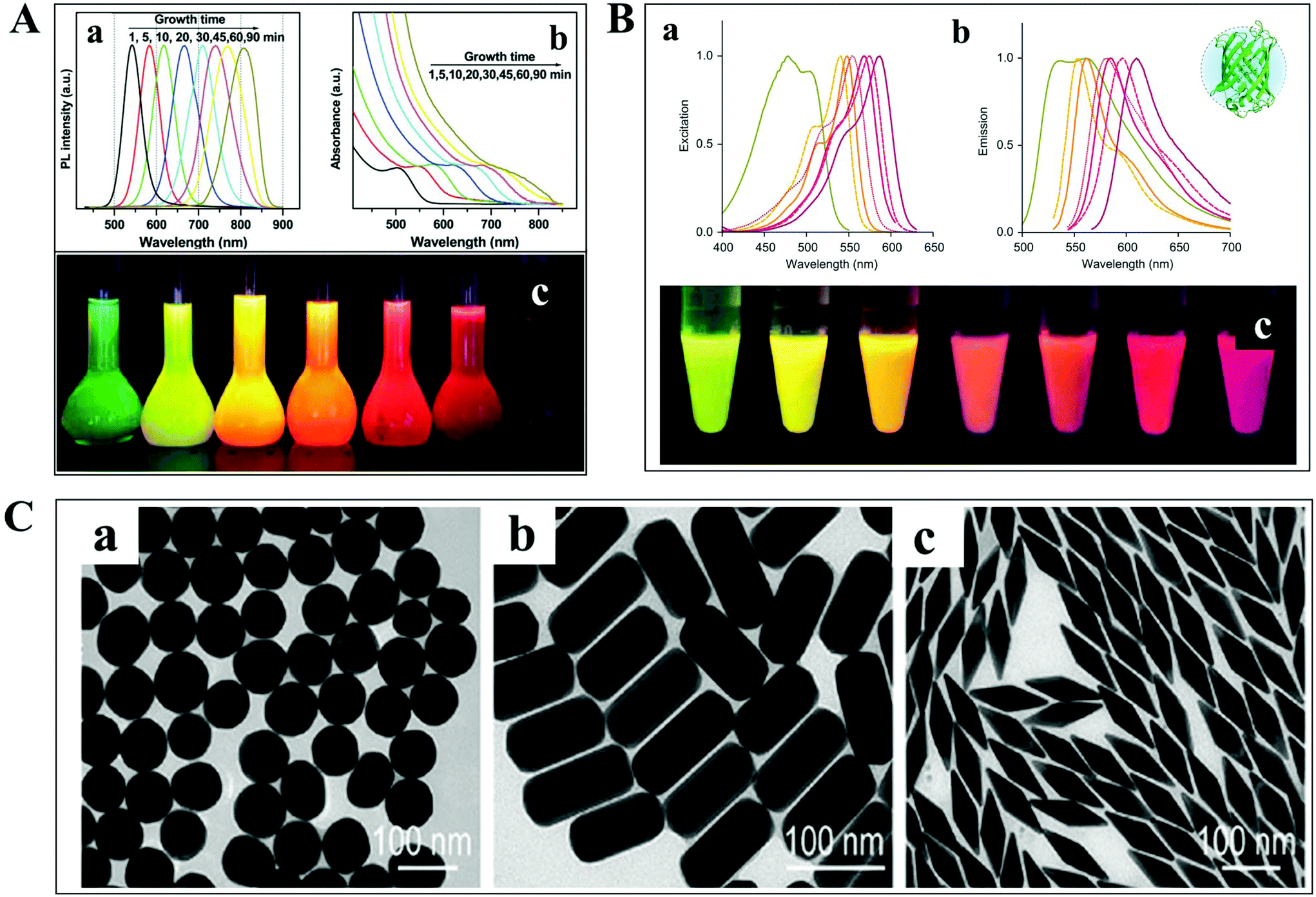 | ||
| Fig. 3 (A) Fluorescence emission spectra (a), UV-vis absorption spectra (b) and photographs (c) under ultraviolet light of cadmium telluride QDs at different reaction times.36 (B) Excitation (a), emission spectra (b), and photographs (c) of different fluorescent proteins under ultraviolet light excitation.38 (C) TEM images of GNPs with different shapes.41 | ||
For living cell labeling, fluorescent proteins are promising candidates for optical imaging applications (Fig. 3B).38 They can easily label intracellular molecules through genetically encoding the target object. Nevertheless, the inherent properties of low quantum yield and reduced photo-stability might affect their biological applications. Great efforts have been paid to the fabrication of fluorescent proteins with greatly improved optical features, such as excellent studies from the Qian group.38–40
In addition to the fluorescent probes, recently, much attention has been paid to the development of non-fluorescent nanoparticles for absorption- and scattering-based imaging applications. One of the most commonly studied materials is the plasmonic nanoparticle which is typically composed of noble metals (Fig. 3C).41 The localized surface plasmonic resonance (LSPR) effect from these particles can be well modulated by changing the composition, size, and morphology of the nanoparticle, which is also sensitive to the refractive index change of the local environment. Therefore, these nanoparticles are perfectly suitable for bio-analytical applications. Another important feature of these particles is the superior photo-stability. In contrast to fluorescent probes, plasmonic nanoparticles never suffer from photobleaching and blinking in scattering mode detection.
Molecular and living cell dynamic assay with optical microscopy methods
Small and macro-molecule detection
The aim of chemical assays is to detect the target object in time with high sensitivity and selectivity. Although amplification techniques, for example, reverse-transcriptase PCR, have been widely used for ultra-sensitive detection of biomolecules, the inherent disadvantages limit their further applications, including poor reproducibility, long reaction time, high background and a false positive output.42,43 Optical microscopy imaging techniques can identify target objects at the single-molecule level without any sample amplification. More importantly, they are capable of quantitatively mapping molecular profiles in single living cells, which are considered as new opportunities in molecular diagnostics.Scattering-based DFM makes it possible to observe scattered light from individual nanometer-sized plasmonic nanoparticles. The LSPR from these particles can be regulated by the composition of the material, and dielectric properties of the medium.44,45 Recently, many sensing principles based on the changes of the nanoparticle size and the nearby environment have been used in the mapping of trace biomolecules.45–47 The binding of biomolecules on the surface of plasma nanoparticles can cause a red-shift in the plasmon resonance of the nanoparticle, which is the result of the changed refractive index of the environment close to the nanoparticle. On this basis, Raschke et al. demonstrated a real-time streptavidin biosensor by observing the resonance shift of biotinylated BSA-functionalized gold nanoparticles (GNPs) after binding streptavidin.45 Moreover, the intracellular NADH can be monitored in real-time based on the red-shift of GNPs caused by the copper deposition on the surface of GNPs (Fig. 4A).47
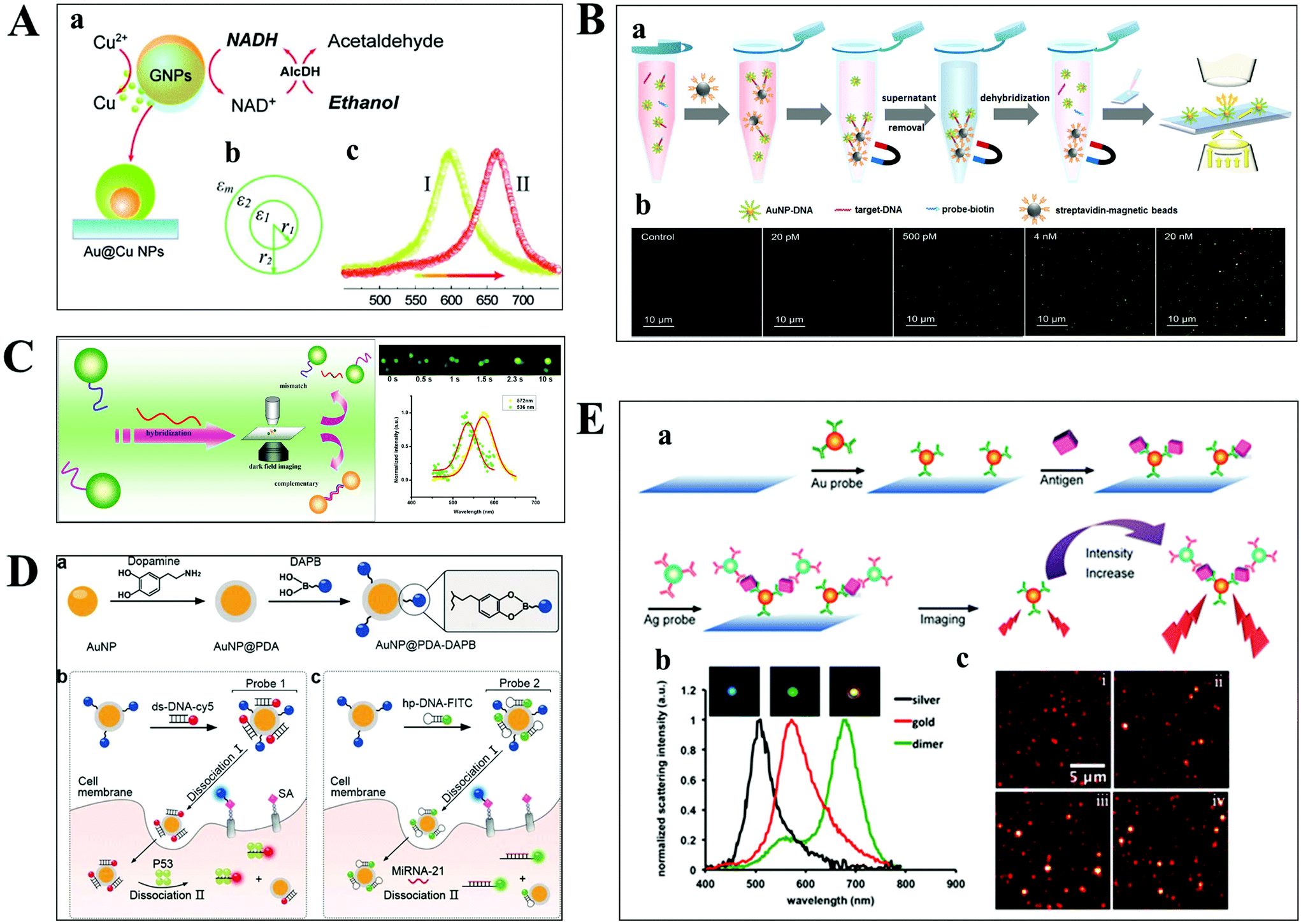 | ||
| Fig. 4 (A) Schematic diagram of copper deposition on the surface of GNPs in the presence of NADH (a), the dielectric constant distribution (b), and the red-shift of scattering spectra of GNPs after copper deposition (c).47 (B) Schematic illustration of the nucleic acid assay based on a nonamplification sandwich method (a), and dark-field images with different concentrations of target DNA (b).48 (C) Schematic diagram of multiplexed nucleic acid detection.46 (D) The multilayer imaging system based on the two-stage disassembly design. In detail, the synthetic route of Au@PDA–DAPB (a), schematic illustration of sensing SAs, p53 protein (b), and miRNA-21 (c).54 (E) Schematic depiction of biomarker detection based on the coupling effect of GNPs and SNPs (a), scattering spectra of probes before and after the immunoassay (b), and representative dark-field scattering images under different carcinoembryonic antigen concentrations (c), where i, ii, iii and iv are 0, 100, 200, and 300 pM, respectively.31 | ||
For genes, the ultrasensitive detection of nucleic acids is fundamentally significant in clinical diagnostics and therapeutics. Li et al. have successfully realized the detection of nucleic acids related to Alzheimer's disease at the femtomolar level based on a nonamplification sandwich assay (Fig. 4B).48 Specifically, target DNA molecules can interact specifically with the DNA sections of GNP–DNA and DNA–biotin to form a sandwich structure. The sandwich structure can be captured by streptavidin-modified magnetic beads owing to the interaction between biotin and streptavidin. Meanwhile, target DNA molecules are proportional to the captured GNPs, and the quantification of target DNA molecules can be achieved by automatically counting GNPs with DFM. Multiplexed nucleic acid detection was also demonstrated by a similar approach (Fig. 4C).46 Moreover, the absolute size and the number of DNA and RNA are important parameters for exploring basic biological processes. Jeynes and co-workers designed a dual GNP–fluorescent probe to measure the length and absolute dimensions of telomeres using SRM.49 Additionally, small molecules and ions in living cells also play important roles in signalling, osmotic regulation, catalysis, and the generation of action potentials, which have been mapped with high sensitivity by various optical microscopes, such as sulphide, Pb2+ and adenosine triphosphate.50–53
Proteins are involved in many cellular processes, such as catalysis, metabolism, neurotransmission, and gene expression. Various important materials in cells consist of proteins, such as insulin, thymus, and enzymes (for example, DNA polymerases), which play critical roles in cell function. Generally, single cell metabolism can be studied by non-invasive and invasive approaches. Invasive methods, for example, mass spectrometry, destroy cellular integrity because of the requirement to extract analyte molecules from cells, whereas non-invasive approaches can monitor biomolecules without disturbing the anatomical and functional integrity of cells. As one of the non-invasive approaches, optical microscopy can continuously monitor target biomolecules and provide molecular profiles in living cells.
Qian designed a multilayer imaging system for mapping molecular profiles of sialic acids (SAs), p53 protein, and microRNA-21 (miRNA-21) in breast cancer cells and explored their expression changes under treatment with various drugs (Fig. 4D).54 Dansylamino phenylboronic acid (DAPB) of Au@PDA–DAPB could be released by reacting with SA on the cell membrane. Because of the specific interaction of ds-DNA and p53 protein as well as hairpin and microRNA-21, Cy5 and FITC were released from the surface of GNPs after entering into the cytoplasm. The imaging of multiple cancer biomarkers in single cells could be achieved by the fluorescence recovery of DAPB, Cy5 and FITC after releasing from the surface of GNPs.
Poon et al. demonstrated the detection of three cancer biomarkers, carcinoembryonic antigen, prostate-specific antigen, and alpha fetoprotein, with picomolar sensitivity in serum based on the plasmonic coupling effect of GNPs and silver nanoparticles (SNPs) using DFM.31 In the presence of target antigens, antibody-modified GNP and SNP probes could be coupled together, enhancing the scattering intensity of the individual one (Fig. 4E). Ideally, multiple antigens could be detected simultaneously by modifying a variety of corresponding antibodies on the probe.
Single-molecule/-particle tracking
Understanding the molecular interaction on the cell membrane is significant for the investigation of reaction mechanisms and the dynamics of a single molecule in living cells (Fig. 5A).2 With the development of optical microscopy, it is possible to observe the movement of biomolecules and molecular motors in living cells. To extract detailed information from huge amounts of data, an effective algorithm is indispensable, which plays a pivotal role in single cell analysis. Mean squared displacement (MSD), dwell time distribution, diffusion coefficient and diffusion types can be extracted from the particle trajectories.55 Stochastic frequency analysis has been used in particle tracking to analyze the transport of biomolecules in biological systems.56 Wei et al. got quantitative information of the particle motion on the lipid membrane by calculating MSD in terms of the formula: 〈Δr(τ)2〉 = 〈|r(t + τ) − r(t)|2〉, where τ is the lag time, r is the nanoparticle coordinate position on the membrane, and the brackets represent the ensemble average of all trajectories (Fig. 5B).30 Moreover, anomalous diffusion can be estimated based on the function of 〈r2(τ)〉 ≅ 2dKατα, where d is the space dimension, Kα is the diffusion coefficient, r(τ) is the physical dimension length, and α is the anomalous diffusion exponent.57,58 | ||
| Fig. 5 (A) Illustration of the content of a highly complex live cell.2 (B) Single-particle tracking the diffusion dynamics of peptide-functionalized GNPs on the lipid membrane. Transient hopping diffusion was observed by analyzing the step size, MSD, and 2-D diffusion coefficient information of GNPs.30 (C) Representative types of diffusion in 2-D (a and b).59 Time-averaged MSD for 20 granules in cells, where blue, magenta, and green represent the different locations of the granule in the cell (c).61 Time-averaged MSD for the motion of potassium channels in plasma membranes at different lag times (d).57 (D) Motor proteins guide vesicles fusing (a) and splitting (b) on cell cytoskeletons observed by DFM.68 (E) Real-time imaging of HIV-1-QD particles entering TZM-bl cells (a and b) and macrophages (f and g) by endocytosis. Analysis of mean velocities (c and h), and MSD plots (d and i) of the viral particles in TZM-bl cells (c and d) and macrophages (h and i). Statistical analysis of the number of HIV-1-QD viral particles per TZM-bl cell (e) and macrophage (j) from 0.5 to 4 h. The scale bars in (a) and (f) are 3 and 10 μm, respectively.70 | ||
The movability of biomolecules in living cells based on MSD can be divided into Brownian diffusion, anomalous diffusion, directed motion, and confined motion (Fig. 5C).59,60 Brownian diffusion is a linear behavior with a diffusion exponent of α = 1. When the motion of biomolecules deviates from Brownian motion, the diffusion exponent will change, and the motion is called anomalous diffusion, which can be further classified into superdiffusion (α > 1) and subdiffusion (0 < α < 1). Directed motion can be considered as the superposition of Brownian and ballistic motion. Confined motion shows MSD saturation after an equilibration time. However, there are some unusual phenomena in living cells, such as the opposition of α calculated from the time-averaged MSD and the ensemble averaged MSD trajectories, and the decreasing exponent over time (Fig. 5C), which is referred to as nonergodic diffusion and aging, respectively.57,61
Most of the substances within the cell are transported by diffusion, while molecular motors, such as kinesin and dynein, are also important transport pathways for certain substances, such as lipid droplets and polymeric particles.62,63 The cells contain a series of microtubules, which connect the cell surface and the nuclei. Kinesin is used to transport macromolecules along microtubules from the surface of the cell membrane, while the vesicles carried by dynein move in the opposite direction. Kinesin and dynein act synergistically to achieve the intracellular bidirectional transport of the substance.64,65 The motion of the substance carried by motor proteins is super-diffusion, which has been demonstrated in living eukaryotic cells (Fig. 5D).62,66–68
Understanding the mechanisms of viral invasion can assist in finding ways to suppress viral infections and offer possibilities for the treatment of diseases. To study the infection pathway of viruses, Liu et al. tracked the infection behaviors of influenza viruses in single cells based on the specific interaction between streptavidin-modified QDs and influenza viruses.69 They monitored individual influenza virus in real time and observed the five-stage infection process of the influenza virus. Additionally, the dynamics of influenza virus transports in the perinuclear region was analyzed by MSD from the trajectories of a single influenza virus. It was found that the intermittent movement of the influenza virus in the perinuclear region was microtubule-dependent.
Li et al. have successfully tracked the viral entry pathway by observing the trajectory and dynamics of QDs encapsulated in the HIV-1 virus (Fig. 5E).70 It was found that there were three requirements to release the viral core into the cytosol, including clathrin-mediated endocytosis, actin-associated virus transport, and the membrane fusion of the viral envelope and the endosomal membrane. They demonstrated that the different stages of HIV-1 entry in macrophages could be inhibited by the corresponding inhibitors, such as clathrin, actin, and endosome fusion inhibitors.
Besides the diffusion kinetics, metabolic pathways in living cells can be unravelled by tracking individual biomolecules in situ, which is crucial to understand the physiological and pathological processes.37,71 Optical microscopy techniques are important tools for mapping the distribution of metabolites in cells, following intracellular metabolic pathways, and observing the effect of drugs during cell metabolism. However, considering the limitations of spatial and temporal resolution from the diffraction limit and the camera frame rate, optical microscopy techniques are mainly suitable for dynamic events with limited reaction rates.72 Currently, optical techniques have been extensively used in biological systems, such as the study of intracellular protein dynamics on the cell membrane, neurotransmitter receptors in synapses, and transcription factors.73–75 Gene synthesis can also be monitored by visualizing the specific site of transcription.76 Levsky achieved simultaneous visualizing of 11 gene expressions in human colon adenocarcinoma cells by combining fluorescence microscopy.77
Progress in high-dimensional optical microscopy
Basically, the motion of molecules is not confined to the 2-D space in biological samples. Much more information can therefore be extracted from the 3-D trajectories (Fig. 6A), such as the depth information and the polar angle of objects, which are vital parameters to elucidate the kinetic mechanisms.78 Although there are some challenges in 3-D imaging with a conventional optical microscope, such as difficulties in collecting data quickly and complicated data processing, recent developments offer possibilities for visualizing molecular dynamics in high dimension.55,79,80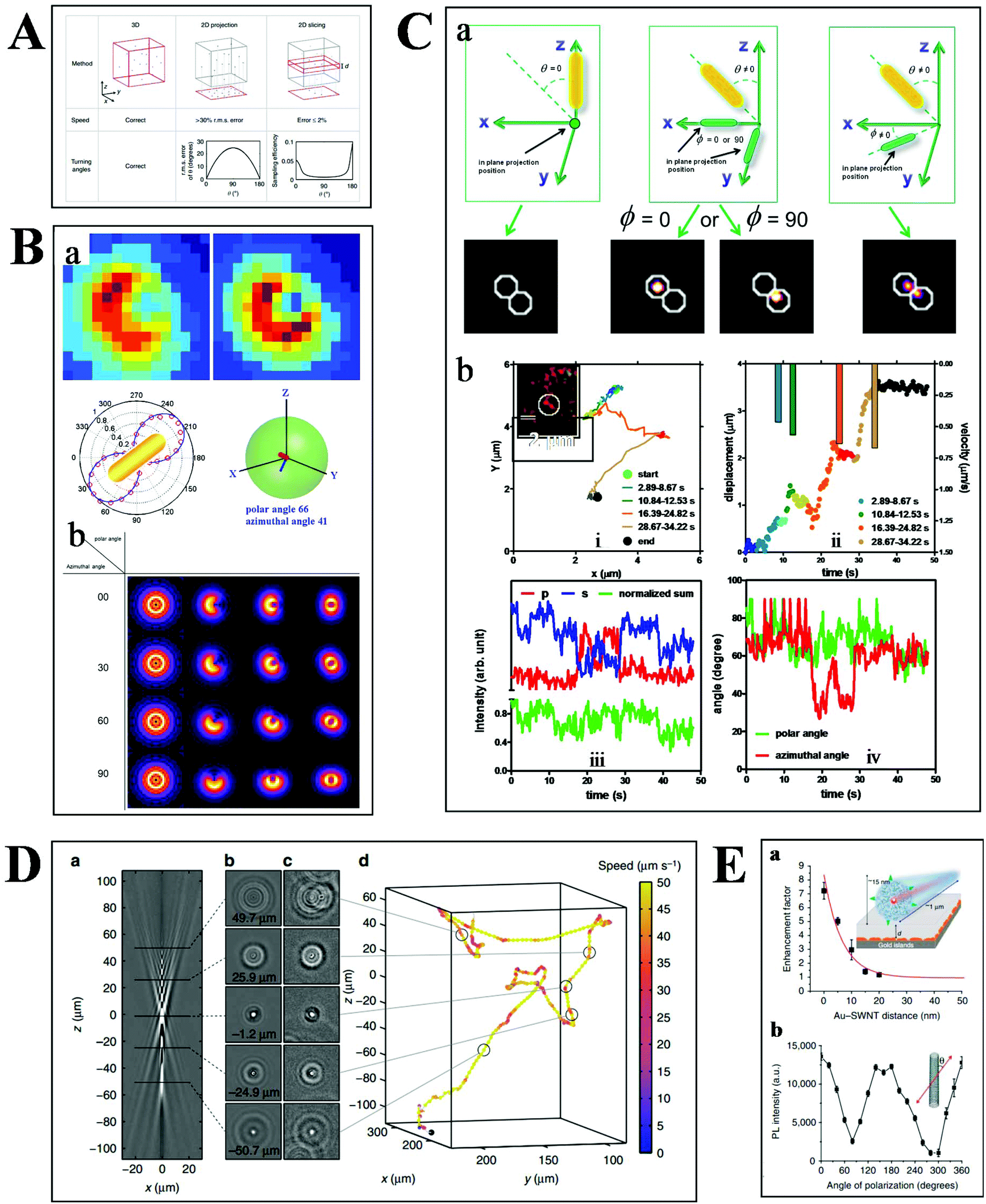 | ||
| Fig. 6 (A) Comparison of 2- and 3-D tracking methods in position, speed and angle information.78 (B) Schematic illustration of 3-D geometric imaging of individual GNRs with defocused DFM.81 Dark-field images of GNRs measured and simulated (a). Defocus imaging of GNRs corresponding to different 3-D angles (b). (C) Scattering images of individual GNRs in different 3-D coordinates (a). Trajectory, relative displacement, velocities, and scattering intensity in two separate and sum channels, the measured polar and azimuthal angle of a GNR stepping on the 2 microtubule inside a living cell as a function of time (b).33 (D) Tracking bacteria in 3-D by comparing their defocused diffraction patterns to a reference library.78 A vertical slice (a) and horizontal slices at different depths (b) of the created reference library. 3-D images of bacteria at different positions (c). Reconstructed 3-D trajectory of the bacterium (d). (E) Distance (a) and polarization (b) dependence of the photoluminescence intensity of SWCNTs.84 | ||
Our previous studies have demonstrated the 3-D geometric imaging of individual GNRs based on the specific relevance of its field distribution pattern and the spatial angle by using defocused DFM (Fig. 6B).81 However, this approach is limited with low S/N, which is not suitable for dynamic tracking in a strong scattering medium. To address this issue, another method for the 3-D orientation imaging of individual GNRs in solution was presented by using sheet light illumination (Fig. 6C).33 This approach is based on the divergence of transverse and longitudinal oscillations of individual GNRs. The 3-D rotational dynamics of individual GNRs in solution and transported by kinesin in living cells were directly imaged by this method.
Besides the orientation imaging, until now, various single-particle tracking methods in the 3-D space were reported, paving the way for a comprehensive understanding of molecular movement in living cells.82,83 Taute et al. observed bacterial motility in a 3-D environment by creating a reference between the intensity patterns and bacterial depth position based on defocused phase contrast microscopy (Fig. 6D).78 Hong et al. successfully demonstrated the 3-D tracking of single nanotube endocytosis within the depth of 10 nm based on the highly sensitive distance dependence of fluorescence enhancement between single-walled carbon nanotubes (SWCNTs) and gold substrates (Fig. 6E).84 Overall, the 3-D tracking technology offers a platform to obtain full positional information about biological pathways, including molecular distance information, translocation motions of molecules, and protein conformational changes.85
Challenges and prospects of optical microscopy technologies in the single cell assay
Recent advancements in theoretical foundations and the design of multiplex probes further expand their applications in biological processes. However, some challenges still exist.32,86–88 Considering the invisibility of most target molecules, it is necessary to tag imaging contrast probes onto target molecules.89 However, it is a complicated task to specifically label target molecules efficiently. Ideally, the number of probes and target molecules is 1:1 by stoichiometry, whereas no labeling methods currently can guarantee this accurately.90 Undercounting and overcounting can be caused by imperfect labeling efficiencies, excessive labeling, and the simultaneous blinking of multiple probes within the diffraction limit.91,92
The long-time tracking of the target objects in living cell requires good photo-stability and excellent biocompatibility. Despite the advantages of the low background in contrast to scattering-based imaging, fluorescence microscopy is limited by the photobleaching and blinking of fluorophores.93–95 Scattering-based optical microscopy is not limited by these restrictions, but the size of probes is usually required to be large enough to separate from background scattering.96 However, the introduction of large size probes may perturb the molecular dynamics in living cells. Hence, it is necessary to develop probes with high selectivity, low cytotoxicity, superior photo-stability and small size dimension.
The large-scale quantification of metabolites, lipids, proteins, and genes in biological samples is the main trend in biological research.97 Optical microscopy makes it possible to detect many target molecules simultaneously by using multiplexed probes.43,98 The combination of optical microscopy techniques with other approaches, such as, optical tweezers and microfluidics, may be the direction of future biological research.99–101
In addition, there are other challenges in the development of optical microscopy technologies, including time consumption, localization efficiency, cytoplasmic auto-fluorescence, high-dimensional imaging, huge data analysis and so on.2,81,89 There is a trade-off among them.72 For instance, higher resolution leads to more photodamage and time consumption for photon collection. 3-D imaging can deduce full positional information but increase the difficulties in data analysis.
Conclusion
Optical microscopy with the capability to detect and track single molecules or nanoparticles can be used to explore the interactions between biomolecules inside living cells, which provide the foundation for the understanding of the fundamental mechanisms of physiological and pathological processes and the design of novel biosensing systems. However, there are still some challenges in the further extension of new optical microscopy techniques, such as efficient localization, spatial resolution, difficulties in 3-D imaging, photodamage, time consumption, and so on. Great efforts have been paid to address these challenges recently. For instance, new labeling strategies, automatic algorithms for digital data analysis and innovative fluorescent probes with higher specificity and multiplex capability have been developed to meet the requirement for efficient localization and high-throughput detection.102–104 Overall, because of the non-invasive and high throughput single object discrimination capability, a new wave of progress in optical microscopy particularly for bio-analytical applications can be envisioned in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, Project no. 21522502), the Excellent Youth Scholars of Hunan Provincial Education Department (17B155) and the Opening Fund of Key Laboratory of Chemical Biology and Tradition Chinese Medicine Research (Ministry of Education of China), Hunan Normal University.Notes and references
- D. Wang and S. Bodovitz, Trends Biotechnol., 2010, 28, 281–290 CrossRef CAS PubMed.
- K. Norregaard, R. Metzler, C. M. Ritter, K. Berg-Sorensen and L. B. Oddershede, Chem. Rev., 2017, 117, 4342–4375 CrossRef CAS PubMed.
- L. Cognet, D. A. Tsyboulski, J.-D. R. Rocha, C. D. Doyle, J. M. Tour and R. B. Weisman, Science, 2007, 316, 1465–1468 CrossRef CAS PubMed.
- P. Zijlstra and M. Orrit, Rep. Prog. Phys., 2011, 74, 106401 CrossRef.
- M. D. Wang, M. J. Schnitzer, H. Yin, R. Landick, J. Gelles and S. M. Block, Science, 1998, 283, 902–907 CrossRef.
- E. A. Abbondanzieri, W. J. Greenleaf, J. W. Shaevitz, R. Landick and S. M. Block, Nature, 2005, 438, 460–465 CrossRef CAS PubMed.
- J.-D. Wen, L. Lancaster, C. Hodges, A.-C. Zeri, S. H. Yoshimura, H. F. Noller, C. Bustamante and I. Tinoco Jr., Nature, 2008, 452, 598–603 CrossRef CAS PubMed.
- L. Xiao and E. S. Yeung, Annu. Rev. Anal. Chem., 2014, 7, 89–111 CrossRef CAS PubMed.
- D. Toomre and J. Bewersdorf, Annu. Rev. Cell Dev. Biol., 2010, 26, 285–314 CrossRef CAS PubMed.
- A. S. Stender, K. Marchuk, C. Liu, S. Sander, M. W. Meyer, E. A. Smith, B. Neupane, G. Wang, J. Li, J.-X. Cheng, B. Huang and N. Fang, Chem. Rev., 2013, 113, 2469–2527 CrossRef CAS PubMed.
- S. Coda, P. D. Siersema, G. W. H. Stamp and A. V. Thillainayagam, Endosc. Int. Open, 2015, 3, E380–E392 CrossRef PubMed.
- L. Wei, D. Zhang, X. Zheng, X. Zeng, Y. Zeng, X. Shi, X. Su and L. Xiao, Nanotheranostics, 2018, 2, 157–167 CrossRef PubMed.
- L. Wei, Y. Ma, X. Zhu, J. Xu, Y. Wang, H. Duan and L. Xiao, Nanoscale, 2017, 9, 8747–8755 RSC.
- V. Beaumont, Biochem. Soc. Trans., 2003, 31, 819–823 CrossRef CAS PubMed.
- S. L. Reck-Peterson, N. D. Derr and N. Stuurman, Cold Spring Harb. Protoc., 2010, 2010, pdb-top73 CrossRef PubMed.
- E. M. Kudalkar, T. N. Davis and C. L. Asbury, Cold Spring Harb. Protoc., 2016, 2016, pdb-top077800 CrossRef PubMed.
- M. Beck, M. Aschwanden and A. Stemmer, J. Microsc., 2008, 232, 99–105 CrossRef CAS PubMed.
- A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman and P. R. Selvin, Science, 2003, 300, 2061–2065 CrossRef CAS PubMed.
- N. Peerboom, E. Schmidt, E. Trybala, S. Block, T. Bergstrom, H. P. Pace and M. Bally, ACS Infect. Dis., 2018, 4, 944–953 CrossRef CAS PubMed.
- M. Stoeber, D. Jullie, B. T. Lobingier, T. Laeremans, J. Steyaert, P. W. Schiller, A. Manglik and M. von Zastrow, Neuron, 2018, 98, 963–976 CrossRef CAS PubMed.
- R. Wombacher, M. Heidbreder, S. van de Linde, M. P. Sheetz, M. Heilemann, V. W. Cornish and M. Sauer, Nat. Methods, 2010, 7, 717–719 CrossRef CAS PubMed.
- B. Huang, Curr. Opin. Chem. Biol., 2010, 14, 10–14 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- K. I. Willig, S. O. Rizzoli, V. Westphal, R. Jahn and S. W. Hell, Nature, 2006, 440, 935–939 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- A. Johnson-Buck, J. Nangreave, D.-N. Kim, M. Bathe, H. Yan and N. G. Walter, Nano Lett., 2013, 13, 728–733 CrossRef CAS PubMed.
- C. E. Oneil, J. M. Jackson, S.-H. Shim and S. A. Soper, Anal. Chem., 2016, 88, 3686–3696 CrossRef CAS PubMed.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- L. Wei, Z. Ye, Y. Xu, B. Chen, E. S. Yeung and L. Xiao, Anal. Chem., 2016, 88, 11973–11977 CrossRef CAS PubMed.
- C.-Y. Poon, L. Wei, Y. Xu, B. Chen, L. Xiao and H.-W. Li, Anal. Chem., 2016, 88, 8849–8856 CrossRef CAS PubMed.
- L. Wei, J. Xu, Z. Ye, X. Zhu, M. Zhong, W. Luo, B. Chen, H. Duan, Q. Liu and L. Xiao, Anal. Chem., 2016, 88, 1995–1999 CrossRef CAS PubMed.
- L. Xiao, Y. Qiao, Y. He and E. S. Yeung, J. Am. Chem. Soc., 2011, 133, 10638–10645 CrossRef CAS PubMed.
- L. Xiao, L. Wei, C. Liu, Y. He and E. S. Yeung, Angew. Chem., Int. Ed., 2012, 51, 4181–4184 CrossRef CAS PubMed.
- Z. Ye, L. Wei, X. Zeng, R. Weng, X. Shi, N. Wang, L. Chen and L. Xiao, Anal. Chem., 2018, 90, 1177–1185 CrossRef CAS PubMed.
- L. Zou, Z. Gu, N. Zhang, Y. Zhang, Z. Fang, W. Zhu and X. Zhong, J. Mater. Chem., 2008, 18, 2807–2815 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- N. C. Shaner, R. E. Campbell, P. A. Steinbach, B. N. G. Giepmans, A. E. Palmer and R. Y. Tsien, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS PubMed.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- R. Heim and R. Y. Tsien, Curr. Biol., 1996, 6, 178–182 CrossRef CAS PubMed.
- W. Lu, N. Jiang and J. Wang, Adv. Mater., 2017, 29, 1604862 CrossRef PubMed.
- X. Su, Z. Li, X. Yan, L. Wang, X. Zhou, L. Wei, L. Xiao and C. Yu, Anal. Chem., 2017, 89, 3576–3582 CrossRef CAS PubMed.
- P. Zrazhevskiy and X. Gao, Nat. Commun., 2013, 4, 1619 CrossRef PubMed.
- C. Sonnichsen, T. Franzl, T. Wilk, G. von Plessen, J. Feldmann, O. Wilson and P. Mulvaney, Phys. Rev. Lett., 2002, 88, 077402 CrossRef CAS PubMed.
- G. Raschke, S. Kowarik, T. Franzl, C. Sonnichsen, T. A. Klar, J. Feldmann, A. Nichtl and K. Kurzinger, Nano Lett., 2003, 3, 935–938 CrossRef CAS.
- L. Xiao, L. Wei, Y. He and E. S. Yeung, Anal. Chem., 2010, 82, 6308–6314 CrossRef CAS PubMed.
- L. Zhang, Y. Li, D. W. Li, C. Jing, X. Chen, M. Lv, Q. Huang, Y. T. Long and I. Willner, Angew. Chem., Int. Ed., 2011, 123, 6921–6924 CrossRef.
- T. Li, X. Xu, G. Zhang, R. Lin, Y. Chen, C. Li, F. Liu and N. Li, Anal. Chem., 2016, 88, 4188–4191 CrossRef CAS PubMed.
- J. C. G. Jeynes, K. Geraki, C. Jeynes, M. Zhaohong, A. A. Bettiol, E. Latorre, L. W. Harries and C. Soeller, ACS Nano, 2017, 11, 12632–12640 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- B. Xiong, R. Zhou, J. Hao, Y. Jia, Y. He and E. S. Yeung, Nat. Commun., 2013, 4, 1708 CrossRef PubMed.
- Q. Liu, C. Jing, X. Zheng, Z. Gu, D. Li, D. W. Li, Q. Huang, Y. T. Long and C. Fan, Chem. Commun., 2012, 48, 9574–9576 RSC.
- Q. He, E. W. Miller, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9316–9317 CrossRef CAS PubMed.
- R. C. Qian, Y. Cao, L. J. Zhao, Z. Gu and Y. T. Long, Angew. Chem., Int. Ed., 2017, 56, 4802–4805 CrossRef CAS PubMed.
- H. Shen, L. J. Tauzin, R. Baiyasi, W. Wang, N. Moringo, B. Shuang and C. F. Landes, Chem. Rev., 2017, 117, 7331–7376 CrossRef CAS PubMed.
- A. Plochowietz, I. Farrell, Z. Smilansky, B. S. Cooperman and A. N. Kapanidis, Nucleic Acids Res., 2017, 45, 926–937 CrossRef CAS PubMed.
- R. Metzler, J. H. Jeon, A. G. Cherstvy and E. Barkai, Phys. Chem. Chem. Phys., 2014, 16, 24128–24164 RSC.
- F. Hofling and T. Franosch, Rep. Prog. Phys., 2013, 76, 046602 CrossRef PubMed.
- C. Manzo and M. F. Garcia-Parajo, Rep. Prog. Phys., 2015, 78, 124601 CrossRef PubMed.
- M. J. Saxton and K. Jacobson, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 373–399 CrossRef CAS PubMed.
- N. Leijnse, J. H. Jeon, S. Loft, R. Metzler and L. B. Oddershede, Eur. Phys. J.: Spec. Top., 2012, 204, 75–84 CAS.
- D. Robert, T.-H. Nguyen, F. Gallet and C. Wilhelm, PLoS One, 2010, 5, e10046 CrossRef PubMed.
- S. P. Gross, M. A. Welte, S. M. Block and E. F. Wieschaus, J. Cell Biol., 2000, 148, 945–956 CrossRef CAS PubMed.
- B. H. Blehm and P. R. Selvin, Chem. Rev., 2014, 114, 3335–3352 CrossRef CAS PubMed.
- C. Kural, H. Kim, S. Syed, G. Goshima, V. I. Gelfand and P. R. Selvin, Science, 2005, 308, 1469–1472 CrossRef CAS PubMed.
- I. M. Tolic-Norrelykke, E. L. Munteanu, G. Thon, L. Oddershede and K. Berg-Sorensen, Phys. Rev. Lett., 2004, 93, 078102 CrossRef PubMed.
- A. Caspi, R. Granek and M. Elbaum, Phys. Rev. Lett., 2000, 85, 5655–5658 CrossRef CAS PubMed.
- L. Wei, Q. Yang and L. Xiao, Nanoscale, 2014, 6, 10207–10215 RSC.
- S.-L. Liu, Z.-L. Zhang, Z.-Q. Tian, H.-S. Zhao, H. Liu, E.-Z. Sun, G. F. Xiao, W. Zhang, H.-Z. Wang and D.-W. Pang, ACS Nano, 2012, 6, 141–150 CrossRef CAS PubMed.
- Q. Li, W. Li, W. Yin, J. Guo, Z. P. Zhang, D. Zeng, X. Zhang, Y. Wu, X. E. Zhang and Z. Cui, ACS Nano, 2017, 11, 3890–3903 CrossRef CAS PubMed.
- L. Liotta and E. Petricoin, Nat. Rev. Genet., 2000, 1, 48–56 CrossRef CAS PubMed.
- Z. Liu, L. D. Lavis and E. Betzig, Mol. Cell, 2015, 58, 644–659 CrossRef CAS PubMed.
- N. Li, Y. Yang, K. He, F. Zhang, L. Zhao, W. Zhou, J. Yuan, W. Liang and X. Fang, Sci. Rep., 2016, 6, 33469 CrossRef CAS PubMed.
- D. Nair, E. Hosy, J. D. Petersen, A. Constals, G. Giannone, D. Choquet and J.-B. Sibarita, J. Neurosci., 2013, 33, 13204–13224 CrossRef CAS PubMed.
- I. Izeddin, V. Recamier, L. Bosanac, I. I. Cisse, L. Boudarene, C. Dugast-Darzacq, F. Proux, O. Benichou, R. Voituriez, O. Bensaude, M. Dahan and X. Darzacq, eLife, 2014, 3, e02230 CrossRef PubMed.
- J. Elf, G.-W. Li and X. S. Xie, Science, 2007, 316, 1191–1194 CrossRef CAS PubMed.
- J. M. Levsky, S. M. Shenoy, R. C. Pezo and R. H. Singer, Science, 2002, 297, 836–840 CrossRef CAS PubMed.
- K. M. Taute, S. Gude, S. J. Tans and T. S. Shimizu, Nat. Commun., 2015, 6, 8776 CrossRef CAS PubMed.
- Y. Shechtman, A.-K. Gustavssonn, P. N. Petrov, E. Dultz, M. Y. Lee, K. Weis and W. E. Moerner, Biomed. Opt. Express, 2017, 8, 5735–5748 CrossRef PubMed.
- P. Memmolo, L. Miccio, M. Paturzo, G. D. Caprio, G. Coppola, P. A. Netti and P. Ferraro, Adv. Opt. Photonics, 2015, 7, 713–755 CrossRef.
- L. Xiao, Y. Qiao, Y. He and E. S. Yeung, Anal. Chem., 2010, 82, 5268–5274 CrossRef CAS PubMed.
- M. A. Thompson, J. M. Casolari, M. Badieirostami, P. O. Brown and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17864–17871 CrossRef CAS PubMed.
- F. Long, J. Zhou and H. Peng, PLoS Comput. Biol., 2012, 8, e1002519 CrossRef CAS PubMed.
- G. Hong, J. Z. Wu, J. T. Robinson, H. Wang, B. Zhang and H. Dai, Nat. Commun., 2012, 3, 700 CrossRef PubMed.
- K. Kim, J. Yoon and Y. Park, Optica, 2015, 2, 343–346 CrossRef CAS.
- M. S. Michie, R. Goetz, C. Franke, M. Bowler, N. Kumari, V. Magidson, M. Levitus, J. Loncarek, M. Sauer and M. J. Schnermann, J. Am. Chem. Soc., 2017, 139, 12406–12409 CrossRef CAS PubMed.
- I.-H. Lee, M. T. Mahmood, S.-O. Shim and T.-S. Choi, Multimed. Tools Appl., 2014, 71, 247–262 CrossRef.
- X. Bai, Y. Zhang, F. Zhou and B. Xue, Inf. Fusion, 2015, 22, 105–118 CrossRef.
- M. Liebel, J. T. Hugall and N. F. van Hulst, Nano Lett., 2017, 17, 1277–1281 CrossRef CAS PubMed.
- R. F. Laine, G. S. Kaminski Schierle, S. van de Linde and C. F. Kaminski, Methods Appl. Fluoresc., 2016, 4, 022004 CrossRef PubMed.
- C. Coltharp, X. Yang and J. Xiao, Curr. Opin. Struct. Biol., 2014, 28, 112–121 CrossRef CAS PubMed.
- N. Durisic, L. L. Cuervo and M. Lakadamyali, Curr. Opin. Chem. Biol., 2014, 20, 22–28 CrossRef CAS PubMed.
- W. E. Moerner and M. Orrit, Science, 1999, 283, 1670–1676 CrossRef CAS PubMed.
- R. M. Dickson, A. B. Cubitt, R. Y. Tsien and W. E. Moerner, Nature, 1997, 388, 355–358 CrossRef CAS PubMed.
- P. Tinnefeld, D. P. Herten and M. Sauer, J. Phys. Chem. A, 2001, 105, 7989–8003 CrossRef CAS.
- A. Weigel, A. Sebesta and P. Kukura, ACS Photonics, 2014, 1, 848–856 CrossRef CAS.
- J. M. Burkhart, M. Vaudel, R. P. Zahedi, L. Martens and A. Sickmann, Proteomics, 2011, 11, 1125–1134 CrossRef CAS PubMed.
- F. S. Fritzsch, C. Dusny, O. Frick and A. Schmid, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 129–155 CrossRef CAS PubMed.
- P. Gross, G. Farge, E. J. G. Peterman and G. J. L. Wuite, Methods Enzymol., 2010, 475, 427–453 CAS.
- J. Mameren, M. Modesti, R. Kanaar, C. Wyman, G. J. Wuite and E. J. Peterman, Biophys. J., 2006, 91, L78–L80 CrossRef PubMed.
- A. Meister, M. Gabi, P. Behr, P. Studer, J. Voeroes, P. Niedermann, J. Bitterli, J. Polesel-Maris, M. Liley, H. Heinzelmann and T. Zambelli, Nano Lett., 2009, 9, 2501–2507 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohane, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- G. C. Rollins, J. Y. Shin, C. Bustamante and S. Presse, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E110–E118 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |