
Integrated microfluidic device for the separation, decomposition and detection of low molecular weight S-nitrosothiols†
Gerson F.
Duarte-Junior
 ab,
Abdulghani
Ismail
a,
Sophie
Griveau
a,
Fanny
d'Orlyé
a,
José Alberto
Fracassi da Silva
c,
Wendell K. T.
Coltro
b,
Fethi
Bedioui
a and
Anne
Varenne
*a
ab,
Abdulghani
Ismail
a,
Sophie
Griveau
a,
Fanny
d'Orlyé
a,
José Alberto
Fracassi da Silva
c,
Wendell K. T.
Coltro
b,
Fethi
Bedioui
a and
Anne
Varenne
*a
aChimie ParisTech, PSL Research University, INSERM 1022, CNRS 8258, Paris Descartes, Unité de Technologies Chimiques et Biologiques pour la Santé, 75005 Paris, France. E-mail: anne.varenne@chimieparistech.psl.eu
bInstituto de Química, Universidade Federal de Goiás, Campus Samambaia, Goiânia, GO 74690-900, Brazil
cInstituto de Química, Universidade Estadual de Campinas, UNICAMP, Campinas, SP 13083-970, Brazil
First published on 17th October 2018
Abstract
S-nitrosothiols (RSNOs) are very important biomolecules that play crucial roles in many physiological and physiopathological processes. They act as NO-donors and are candidates for future medicines. Their identification and quantitation are therefore important for biomedical applications. One, two or more RSNOs can then be combined to design a drug and therefore, the quantification of each is important to establish an acceptable quality control process. Till date, miniaturized devices have been used to detect RSNOs based on their total quantitation without a preceding separation step. This study reports on an original and integrated microdevice allowing for the successive electrokinetic separation of low molecular weight RSNOs, their decomposition under metal catalysis, and their quantitation by amperometric detection of the produced nitrite in the end-channel arrangement, leading to their quantitation in a single run. For this purpose, a commercial SU-8/Pyrex microfluidic system was coupled to a portable and wireless potentiostat. Different operating and running parameters were optimized to achieve the best analytical data, allowing for an LOD equal to 20 μM. The simultaneous separation of S-nitrosoglutathione and S-nitrosocysteine was successfully obtained within 75 s. The proposed methodology using SU-8/Pyrex microfluidic devices opens new possibilities to investigate future drug candidates for NO-donors.
Introduction
S-Nitrosothiols (RSNOs) are nitric oxide (NO) carrier molecules that play important roles in several physiological functions (vasodilatation and relaxation,1,2 antiplatelet aggregation,3,4 antimicrobial function,5 regulation and signaling protein function,6etc.) and pathological events (neurodegenerative diseases such as Parkinson's and Alzheimer's,7 apoptosis,8 chronic obstructive pulmonary disease,9 preeclampsia,10 and diabetes11). RSNOs can be divided into low molecular weight (LMW) and high molecular weight (HMW) RSNOs. Although there is no defined border in terms of molecular mass, it is common to use the term “low molecular weight” for peptides and amino acid-S-nitrosothiols (such as S-nitrosoglutathione (GSNO) and S-nitrosocysteine (CySNO)) and “high molecular weight” for S-nitrosylated proteins (such as S-nitrosoalbumin (AlbSNO) and S-nitrosohemoglobin (HbSNO)). RSNOs store, transport and release NO. They can also inter-exchange NO through a transnitrosation reaction.12 The variation of RSNO concentrations has been shown to occur in many diseases.13 For all these reasons, the development of RSNO-based drug candidates is essential to regulate physiological dysfunctions.14 One, two or more RSNOs can then be combined to design a drug and therefore, the quantification of each is important to establish an acceptable quality control process. Thus, the development of powerful methodologies for the simultaneous quantitation of mixtures of RSNOs in standard samples is crucial.Numerous methods have been developed for RSNO analysis based on direct or indirect detection. Examples of direct detection consist of the separation of RSNOs from other species by capillary electrophoresis (CE) or liquid chromatography, followed by mass spectrometric or spectrophotometric detection.15 For example, the simultaneous separation of S-nitrosoglutathione (GSNO) and S-nitrosocysteine (CySNO) was performed by our group using conventional CE equipped with capacitively coupled contactless conductivity detection, but in a conventional system.16 In another study, the simultaneous separation of GSNO, GSH, glutathione sulfonic and sulfinic acid by CE coupled to mass spectrometry was obtained.15 Most standard methods developed for RSNO quantitation reported in the literature remain indirect. They are based on the detection of their decomposition products,17 which are obtained through homolytic or heterolytic cleavage of the S–NO bond, generating NO or NO+ and finally leading to NO2−. These decomposition products are then detected by spectrophotometry, fluorimetry, electrochemistry or chemiluminescence.17,18 Various reagents have been used to decompose RSNOs, such as metal cations19 (Hg2+, Cu+), light20 and heat,21 leading to different decomposition products: NO is generated19 if Cu+, light or heat are employed, whereas nitrite is directly generated19,22 when Hg2+ or Ag+ are used.
Nowadays, miniaturization in chemical analysis has become a powerful tool contributing to the reduction of the amount of samples/reagents, analysis time and waste generation. Such an approach can be beneficial for the quantitation of RSNOs. Indeed, our group has recently reported the colorimetric analysis of RSNOs in a microfluidic paper-based analytical device.23 This system allowed for the analysis of total RSNOs in plasma samples without any separation step. Other approaches were proposed by Hunter et al. for NO24 and total RSNO detection25 (after light decomposition) using a single PDMS microfluidic channel with amperometric detection. In all cases, no separation of RSNOs occurred before detection in these miniaturized devices. Moreover, Gunasekara et al.26 used microchip capillary electrophoresis (MCE) with amperometric detection to separate an NO donor (DEA-NONOate or Proli-NONOate) from NO and nitrite in less than one minute. Tu et al.27 used MCE with fluorescence detection to separate and detect NO, reduced glutathione (GSH) and cysteine (Cys). Herein, we report the design and optimization of a single-run MCE analytical strategy allowing for the first time the simultaneous quantitation of two low molecular weight RSNOs (S-nitrosoglutathione GSNO and S-nitrosocysteine Cys-NO) owing to the integration of successive electrokinetic separation, RSNO decomposition by Hg2+ to nitrite and nitrite quantitation by amperometry.
Experimental
Chemicals
All reagents were of analytical grade and used as received. L-Arginine (Arg), 2-(N-morpholino) ethanesulfonic acid (MES), acetic acid (HAc), L-histidine (His), sodium tetraborate, sodium nitrite, N-acetyl-p-aminophenol (Paracetamol), mercury(II) chloride, EDTA, hydrochloric acid, sodium phosphate monobasic, sodium phosphate dibasic, L-cysteine (Cys) and reduced glutathione (GSH) were purchased from Sigma Aldrich (St Louis, MO, USA). All aqueous solutions were made using ultra-pure water with a resistivity of 18.2 MΩ cm from a Pure Lab Flex system (ELGA Labwater, France).Synthesis of S-nitrosothiols
GSNO was synthesized as described elsewhere.28 Briefly, an equimolar amount of nitrite was added to an equimolar amount of GSH and HCl. The resulting pure solid was rinsed once with 80% acetone, twice with 100% acetone and three times with diethyl ether and then stocked in the dark at 20 °C.S-Nitrosocysteine (CySNO) was synthesized daily using the method described by Peterson et al.29 Briefly, solutions of 5 mM CysNO were prepared by reacting cysteine with an equimolar concentration of nitrite in acidic medium (0.1 M HCl) in a dark flask to avoid light decomposition. After 5 min, more than 90% of cysteine was converted into CysNO. The solution was neutralized by 0.1 M PBS buffer (pH 7.4) containing 0.5 mM EDTA to prevent decomposition by trace metal cation contaminants.
The final concentrations of RSNOs were determined spectrophotometrically in aqueous solution at 335 nm (ε = 586 and 503 M−1 cm−1 for GSNO and CysNO, respectively).30
Instrumentation
Electrophoretic experiments were performed using SU-8/Pyrex microchips with integrated microband platinum electrodes (MCE-SU8-Pt001T, Micrux Technologies, Oviedo, Spain) at the outlet end of the separation channel (see Fig. S1 in ESI†). Only working (WE) and reference (RE) electrodes were used, with widths of 50 μm and 250 μm, respectively. The separation and injection channel lengths were 35 mm and 10 mm, respectively. The width and depth of the microchannels were 50 μm and 20 μm, respectively. A microfluidic platform (Oviedo, Spain, MCE-HOLDER-DC02) was used to interface the microchip with the amperometric detector and the high-voltage source. The high-voltage source was a programmable HVS448-3000V 8-channel high-voltage supply (LabSmith Inc., CA, USA) controlled by Sequence software v.1.165. Amperometric detection was performed by a modified model 9051, 2-channel, wireless, portable and electrically isolated potentiostat (Pinnacle Technology, Lawrence, KS, USA) operating in a two-electrode format at a 5 Hz sampling rate (gain = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 V A−1, resolution = 30 fA). The potentiostat is isolated, due to which interferences from the high-voltage power supply system used for the separation are eliminated. This potentiostat was controlled by Sirenia Acquisition Software v.1.7.6. The WE and RE were connected to the corresponding electrodes using a commercial chip holder.
000000 V A−1, resolution = 30 fA). The potentiostat is isolated, due to which interferences from the high-voltage power supply system used for the separation are eliminated. This potentiostat was controlled by Sirenia Acquisition Software v.1.7.6. The WE and RE were connected to the corresponding electrodes using a commercial chip holder.
C4D detection was performed using a commercial detector model ER815 acquired from eDAQ Pty (Denistone East, Australia). A microfluidic platform EDAQ ET121 containing external electrodes was used to interface a commercial PMMA microchip (model 02-0750-0082-01, ChipShop, Jena, Netherlands) with the detection system. The microchip layout comprised separation and injection channels (50 μm wide/deep) that are 87 and 10 mm in length, respectively.
Electrophoresis and decomposition procedure
Prior to analysis, microchannels were conditioned with 0.1 M NaOH, deionized water and running buffer. Samples of RSNOs and paracetamol (1 mM each) were electrokinetically injected via gated mode31 by applying potentials of 800 V and 1000 V to sample and buffer reservoirs, respectively, while both waste reservoirs were grounded for the loading step. Sample injection was performed by floating the potential at the buffer reservoir for 3 s to initialize the separation step. The same procedure but under reversed polarity was performed for nitrite (1 mM) quantitation. For the decomposition step, HgCl2 (10 mM) was added in the detection reservoir. Before RSNOs reached the buffer waste reservoir, the polarity was inverted, allowing the detection of nitrite generated from decomposition. For the amperometric detection of nitrite and paracetamol, potentials from 0.7 to 1.2 V vs. Pt were applied. Analysis using C4D detection was realized under the same electrophoretic conditions, and detection was performed by applying a sinusoidal signal of 600 kHz and 90 Vpeak-to-peak.Results and discussion
Herein, a method for the successive separation, decomposition, and detection of RSNOs is proposed by integrating all three steps using a microfluidic device. In order to achieve this goal, (i) the decomposition should be much faster than the separation process (otherwise the peaks originating from different RSNOs will overlap), (ii) the decomposition should be quantitable, and (iii) the decomposition product should be stable through the analysis time and operating conditions. It is well known that the decomposition product of RSNOs depends on the decomposition agent. As indicated above, the use of Cu+ or light leads to homolytic cleavage and the formation of NO, while the use of Hg2+ leads to heterolytic cleavage and the production of NO+ that transforms immediately to NO2−. Decomposition of RSNOs by light is slow (tens of minutes are needed to decompose the sample16) and only partial decomposition of RSNOs was obtained by Hunter et al.25 in a microfluidic device using 530 nm LED. Cu+ decomposition is faster than light decomposition, but still insufficient (>2 min (ref. 32)) in comparison with the separation time scale. Moreover, Cu+ is poorly soluble and stable in aqueous solution and is usually produced by the reduction of Cu2+ with a reducing agent such as GSH. Decomposition using Cu2+ is affected by the variation of GSH concentration in the sample, which is difficult to control.32 Decomposition using mercuric(II) is instantaneous, leading to NO2− which is stable and electroacive.33 Consequently, Hg2+ was chosen as the decomposition agent.BGE plays an important role in the migration and electrochemical detection steps in microchip electrophoresis (MCE).34,35 As the objective was the detection of nitrite generated from RSNO decomposition, BGE optimization was focused on the nitrite signal-to-noise (S/N) ratio during detection. Several BGEs usually used for biological samples during MCE were tested: 20 mM MES/His (pH 6.0), 20 mM MES/Arg (pH 7.5), and 20 mM Arg/acetic acid (pH 5.8). Nitrite (1 mM) was injected in the gated mode (see Experimental section), separated and then detected by amperometry using these various BGEs. For each BGE, the detection potential applied between WE and RE was varied from +0.5 V to +1.5 V, maintaining constant electrophoretic injection and separation conditions. The optimal potential for nitrite detection in all BGEs was 0.7 V. Fig. 1 shows the electropherograms for the separation of nitrite in various BGEs. It can be seen that 20 mM MES/His (pH. 6.0) provided the highest amperometric detection signal. However, this BGE was not selected for our design as Hg2+, which will be used as the decomposition reagent for RSNO, reacts with histidine to form a precipitate. Although MES/Arg leads to the highest signal intensity, it results in the lowest signal-to-noise ratio. Therefore, the selected BGE, leading to the highest S/N ratio without interference with other molecules in the solution, was 20 mM Arg adjusted to pH 5.8 with HAc.
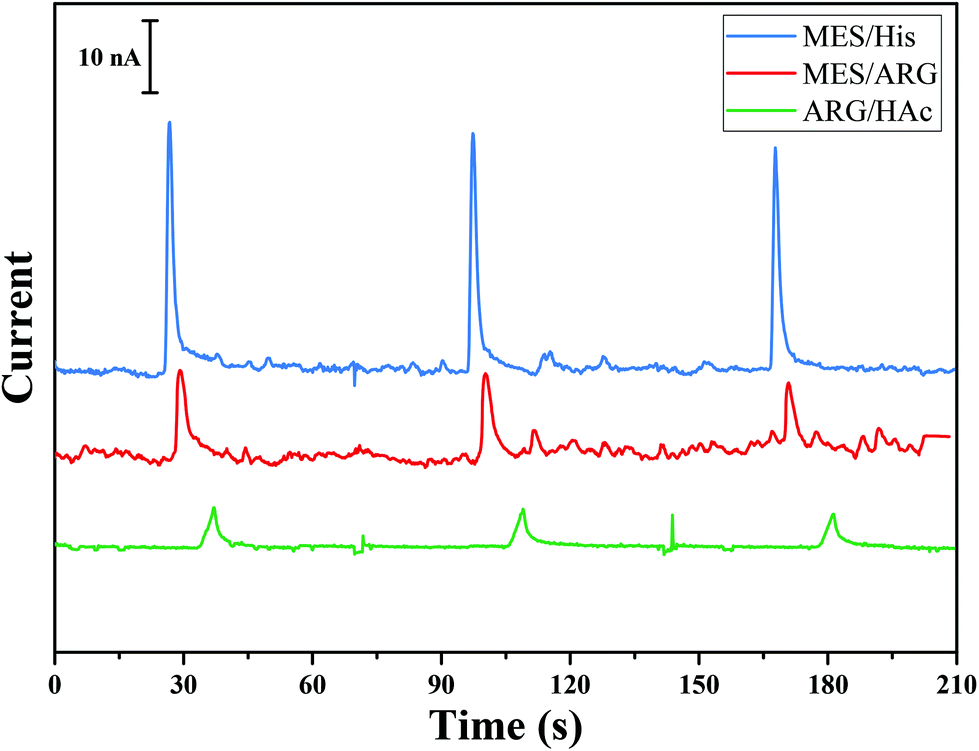 | ||
| Fig. 1 Electrophoretic separation of 1 mM nitrite in the SU-8/Pyrex microchip. BGE: MES (20 mM)/His (20 mM) pH 6.0 in blue, MES (20 mM)/Arg (20 mM) pH 7.5 in red, Arg (20 mM) pH 5.8 adjusted with acetic acid in green. Gated injection V1 = −800 V, V2 = −1000 V; injection time = 3 s, successive injections at t = 70 s; detection at 0.7 V vs. Pt. | ||
For the optimized on-chip integration of the three steps involved in RSNOs characterization (separation, decomposition and detection) in the microchip, the apparent mobility of each of the compounds (different RSNOs, nitrite, mercury) were determined. The overall procedure was first optimized for GSNO, as it is the most abundant low molecular weight RSNO. Control experiments by direct addition of nitrite into the BW reservoir with or without the application of electrophoretic voltage resulted in an amperometric signal, proving the efficiency of the electrochemical detection step. A neutral electroactive marker (paracetamol) allowed the determination of the electroosmotic mobility as 1.85 ± 0.07 × 10−4 cm2 V−1 s−1, and GSNO electrophoretic mobility was determined as −0.64 ± 0.06 × 10−4 cm2 V−1 s−1 by employing a C4D detector as GSNO is not electroactive (results not shown).
Under these experimental conditions, GSNO migrates towards the detector under positive polarity. The device was primarily developed as follows: GSNO was electrokinetically injected from the sample reservoir (S) in the gated mode under positive polarity (see Experimental section). Hg2+ was introduced into the waste reservoir (connected to the cathode) where it should decompose GSNO into nitrite upon reaching the buffer waste reservoir (BW). However, no amperometric signal was observed (results not shown). One hypothesis is based on the fact that Hg2+ undergoes diffusion from BW within the separation channel, inducing GSNO decomposition within the separation channel. As nitrite electrophoretic mobility under these experimental conditions (−4.25 × 10−4 cm2 V−1 s−1) is higher in absolute value than the electroosmotic mobility, nitrite moves back to the sample reservoir (S) instead of the BW reservoir where it should be detected.
A new design was then developed, including an additional step allowing voltage inversion just before GSNO decomposition (Fig. 2A). In this new design, the loading, injection and separation steps were performed under positive polarity (step I, II and III, respectively), Hg2+ was added before GSNO reaches the channel end in step III, and the polarity was inverted (step IV). This inversion of polarity led to the migration of Hg2+ and GSNO to the sample reservoir (S). As Hg2+ migrates faster than GSNO, the migration zone of Hg2+ enters that of GSNO, allowing for GSNO decomposition. The produced nitrite migrates towards the BW reservoir and is detected (see details in step IV, Fig. 2A). A typical electropherogram obtained characterizing all the analytical steps is presented in Fig. 2B. Control experiments (without Hg2+ or without GSNO) did not show any signals (data not shown).
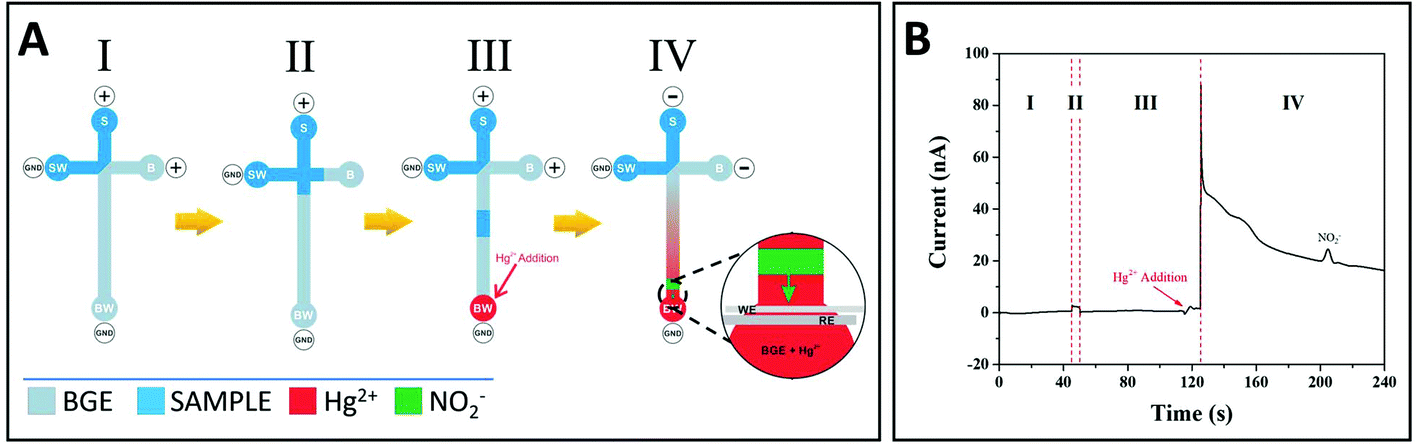 | ||
| Fig. 2 (A) Schematic of the main steps for RSNOs quantitation. Loading step (I): voltages of 800 and 1000 V were applied for 45 s to the sample (S) and buffer (B) reservoirs, respectively, grounding both waste reservoirs (SW and BW). Injection step (II): samples were injected into the separation channel by floating the voltage applied to the B reservoir for 3 s. Migration step (III): the potentials were then re-established to the conditions of step I, allowing the migration of the RSNO sample towards the separation channel. Then, 15 s before the end of this step, Hg2+ was added to the BW reservoir. Inversion and detection step (IV): in this step, the potential polarity was inverted, which led to the migration of Hg2+ into the separation channel faster than RSNO, leading to RSNO decomposition. This was followed by the opposite migration of nascent nitrite (in green) towards the electrodes, and detection was performed by applying a potential of 1.2 V vs. Pt. (B) Typical electropherogram obtained for GSNO (1 mmol L−1) analysis characterising all the steps of the process. | ||
Therefore, the overall integrated protocol includes (1) separation of the RSNOs under positive polarity, (2) inversion of the separation polarity, (3) decomposition of RSNO owing to the on-line crossing and mixing of RSNO and Hg2+ zones, respectively, due to different migration velocities, and (4) the migration of the produced nitrite to the detector. To further optimize the experimental conditions to improve the limit of detection for nitrite, three parameters were studied: the BGE ionic strength (from 10 to 50 mM), the detection voltage (from 0.5 to 1.5 V vs. Pt) and the time of polarity inversion. Considering the first two parameters, the best signal intensities and S/N ratios for nitrite detection were obtained for a detection potential of 1.2 V (Fig. 3).
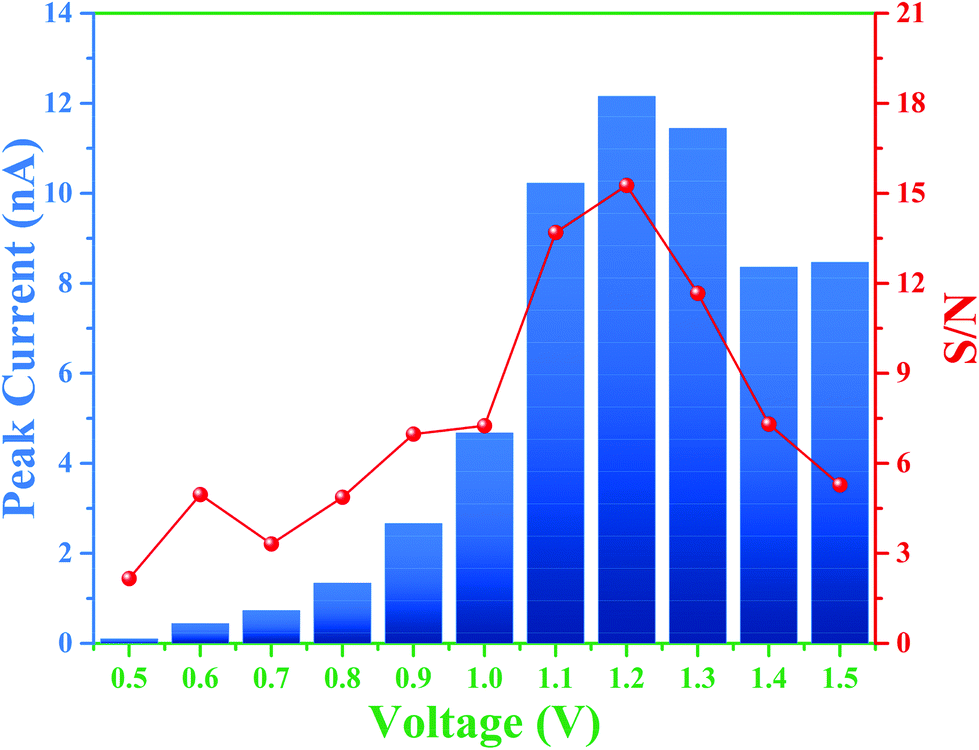 | ||
| Fig. 3 Histogram of peak current (in nA) and signal-to-noise ratio (S/N) of nitrite (1 mM) injection in a SU-8/Pyrex microchip obtained via variation of the detection potential from 0.5 to 1.5 V vs. Pt. BGE: 20 mM arginine solution adjusted at pH 5.6 with acetic acid. | ||
The time of polarity inversion is a crucial parameter as it should allow for the best separation of the RSNOs, their total decomposition and their optimal detection. For each studied compound, the electroosmotic and apparent mobility must therefore be determined. For RSNOs of positive apparent mobility, the time of inversion should be chosen between the migration time of the neutral marker and that of the RNSOs. This parameter was optimized for GSNO and CysNO. CysNO, another important nitrosothiol, as it is smaller than GSNO and has a similar charge at this pH, presents a higher apparent mobility than GSNO. The best signal intensities were obtained at the inversion times of 90 s and 75 s, for GSNO and CysNO, respectively (Fig. 4). These results indicate the versatility of the procedure for all types of RSNOs.
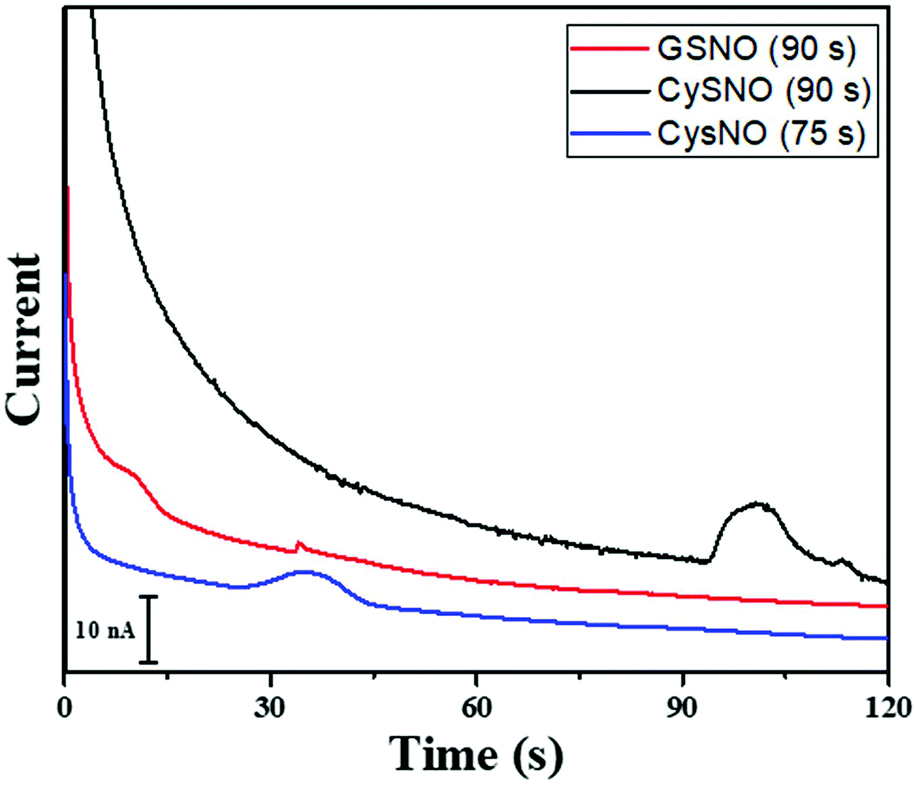 | ||
| Fig. 4 Electropherograms for the detection of 1 mM GSNO (black) and 1 mM CySNO (red and blue) to determine the time of polarity inversion. In black and red 90 s was used as the inversion time, while in blue 75 s was used as the inversion time. BGE: 20 mM arginine solution adjusted at pH 5.6 with acetic acid. | ||
In these experimental conditions, the analytical performance of this methodology was determined for GSNO. The linearity was verified in the 130 to 675 μM concentration range (y = 0.0485x − 5.0485, R2 = 0.9936) with an LOD equal to 20 μM. The run-to-run repeatability was investigated for each tested concentration using three injections. It is important to note that between each run, the microchannels were washed with BGE to remove the Hg2+ used for the decomposition of GSNO. Considering this procedure, the relative standard deviation (RSD) values ranged from 0.4 to 6.0%.
The final objective of such a micro-total analysis system is to allow for the simultaneous quantitation of various RSNOs. Therefore, three main challenges have to be addressed: (i) the efficient separation of the different RSNOs, (ii) the choice of a unique time of polarity inversion in the process, and (iii) the efficient detection of the successive nitrite zones produced from each RSNO decomposition.
The simultaneous separation of GSNO and CysNO was performed so as to prove the versatility of the system. For this purpose, GSNO and CysNO were first separated and detected individually. The crucial parameter being the inversion time, different values were applied from 75 to 90 s. In the second step, an equimolar mixture of CySNO and GSNO (1 mM each) was separated and detected. Fig. 5 presents the resulting electropherograms for an optimized inversion time of 75 s that corresponds to the migration time of CysNO. As expected, the signal arising from GSNO appears after that of CysNO as CysNO has higher apparent mobility. These results indicate a powerful simultaneous separation and indirect detection of GSNO and CysNO. The similar intensities for GSNO and CySNO, injected at the same concentration, indicate that the decomposition efficiency is similar in both cases. This method is therefore applicable to the quantitation of pharmaceutical RSNOs for future drug candidates. Some work is in progress for decreasing the LODs to reach biological concentrations (less than 16 μM (ref. 23)).
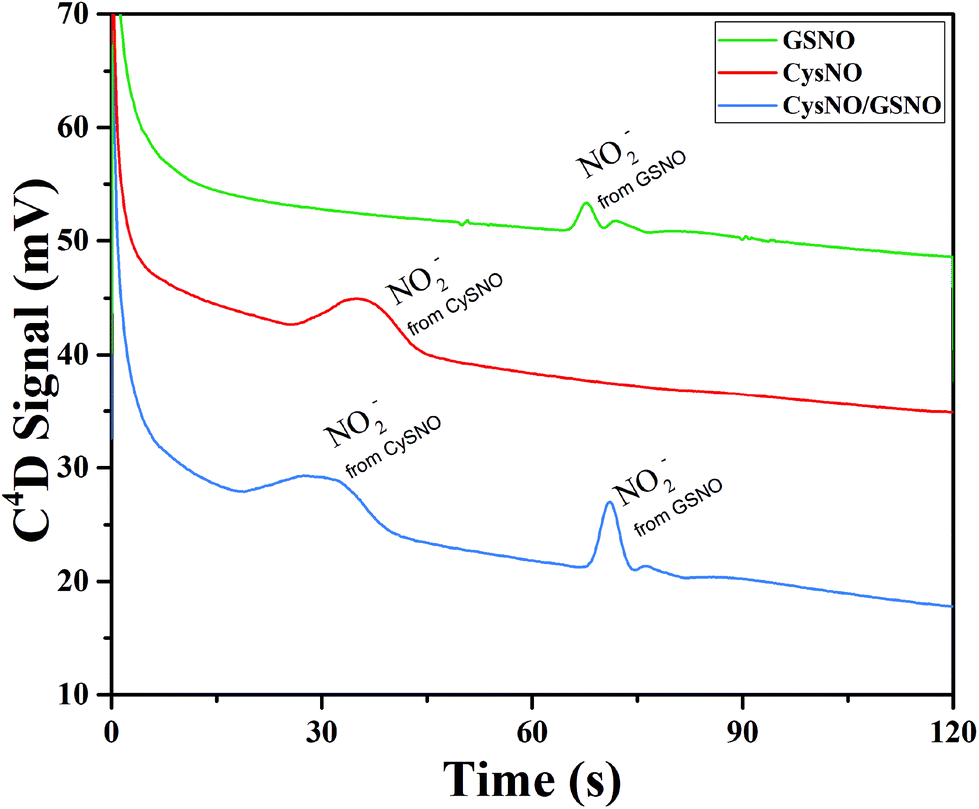 | ||
| Fig. 5 Electropherograms corresponding to the electrophoretic profile of CySNO (1 mM), GSNO (1 mM) and the GSNO mixture (1 mM)/CySNO (1 mM) in blue, red and black, respectively. Experiments were performed in a Su-8/Pyrex microchip with gated injection. Procedure: (1) 3 s injection, (2) application of V1 = 800 V and V2 = 1000 V for 3 minutes (not visible on the graph as it occurs before running the electropherogram), (3) addition of Hg2+ at t = 0 s of the electropherogram, and inversion of polarity V1 = −800 V and V2 = −1000 V for t = 100 s. Detection 1 V vs. Pt. BGE: ARG (20 mM) adjusted to pH 5.8 with HAc. | ||
Conclusions
An original method to simultaneously quantify two low molecular RSNOs in a mixture using MCE was developed. A commercial SU-8/Pyrex microchip and a wireless isolated potentiostat were used. After the electrokinetic separation step, inversion of electrokinetic polarity was necessary to mix the RSNOs with the decomposition agent within the separation channel and detect the produced nitrite by amperometric detection at the buffer waste reservoir. Optimization of BGE composition and detection potential were performed in order to obtain the best signal intensity and S/N ratio. The LOD was 20 μM for both GSNO and CysNO. This methodology can be applied to the quantitation of pharmaceutical RSNOs for future drug candidates.Conflicts of interest
The author declares no conflicts of interest.Acknowledgements
Financial support from “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)” and “French Committee for the Evaluation of Academic and Scientific Cooperation with Brazil (COFECUB)” (grant no. 802-14) is acknowledged.References
- B. T. Mellion, L. J. Ignarro, C. B. Myers, E. H. Ohlstein, B. A. Ballot, A. L. Hyman and P. J. Kadowitz, Mol. Pharmacol., 1983, 23, 653–664 CAS.
- J. S. Stamler, D. I. Simon, J. A. Osborne, M. E. Mullins, O. Jaraki, T. Michel, D. J. Singel and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 444–448 CrossRef CAS.
- E. J. Langford, A. S. Brown, R. J. Wainwright, A. J. Debelder, M. R. Thomas, R. E. A. Smith, M. W. Radomski, J. F. Martin and S. Moncada, Lancet, 1994, 344, 1458–1460 CrossRef CAS.
- M. W. Radomski, D. D. Rees, A. Dutra and S. Moncada, Br. J. Pharmacol., 1992, 107, 745–749 CrossRef CAS PubMed.
- G. F. P. de Souza, J. K. U. Yokoyama-Yasunaka, A. B. Seabra, D. C. Miguel, M. G. de Oliveira and S. R. B. Uliana, Nitric Oxide, 2006, 15, 209–216 CrossRef PubMed.
- C. G. Kevil and R. P. Patel, Curr. Opin. Invest. Drugs, 2010, 11, 1127–1134 CAS.
- M. Ito, Annu. Rev. Neurosci., 1989, 12, 85–102 CrossRef CAS PubMed.
- A. R. Butler and P. Rhodes, Anal. Biochem., 1997, 249, 1–9 CrossRef CAS PubMed.
- K. M. Beeh, J. Beier, N. Koppenhoefer and R. Buhl, Chest, 2004, 126, 1116–1122 CrossRef CAS PubMed.
- V. A. Tyurin, S. X. Liu, Y. Y. Tyurina, N. B. Sussman, C. A. Hubel, J. M. Roberts, R. Taylor and V. E. Kagan, Circ. Res., 2001, 88, 1210–1215 CrossRef CAS PubMed.
- A. B. Milsom, C. J. H. Jones, J. Goodfellow, M. P. Frenneaux, J. R. Peters and P. E. James, Diabetologia, 2002, 45, 1515–1522 CrossRef CAS PubMed.
- B. C. Smith and M. A. Marletta, Curr. Opin. Chem. Biol., 2012, 16, 498–506 CrossRef CAS PubMed.
- M. W. Foster, T. J. McMahon and J. S. Stamler, Trends Mol. Med., 2003, 9, 160 CrossRef CAS PubMed.
- H. H. Al-Sa'doni and A. Ferro, Mini-Rev. Med. Chem., 2005, 5, 247–254 CrossRef.
- A. Ismail, F. d'Orlyé, S. Griveau, J. A. F. Da Silva, F. Bedioui and A. Varenne, Anal. Bioanal. Chem., 2015, 407, 6221–6226 CrossRef CAS PubMed.
- A. Ismail, F. d'Orlye, S. Griveau, F. Bedioui, A. Varenne and J. A. F. da Silva, Electrophoresis, 2015, 36, 1982–1988 CrossRef CAS PubMed.
- S. Griveau and F. Bedioui, Analyst, 2013, 138, 5173–5181 RSC.
- D. Giustarini, A. Milzani, I. Dalle-Donne and R. Rossi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 851, 124–139 CrossRef CAS PubMed.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- M. M. Veleeparampil, U. K. Aravind and C. T. Aravindakumar, Adv. Phys. Chem., 2009, 2009 Search PubMed , 890346.
- M. G. de Oliveira, S. M. Shishido, A. B. Seabra and N. H. Morgon, J. Phys. Chem. A, 2002, 106, 8963–8970 CrossRef CAS.
- H. R. Swift and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1997, 1933–1935 RSC.
- A. Ismail, M. O. Araujo, C. L. S. Chagas, S. Griveau, F. D'Orlye, A. Varenne, F. Bedioui and W. K. T. Coltro, Analyst, 2016, 141, 6314–6320 RSC.
- R. A. Hunter, B. J. Privett, W. H. Henley, E. R. Breed, Z. Liang, R. Mittal, B. P. Yoseph, J. E. McDunn, E. M. Burd, C. M. Coopersmith, J. M. Ramsey and M. H. Schoenfisch, Anal. Chem., 2013, 85, 6066–6072 CrossRef CAS PubMed.
- R. A. Hunter and M. H. Schoenfisch, Anal. Chem., 2015, 87, 3171–3176 CrossRef CAS PubMed.
- D. B. Gunasekara, M. K. Hulvey, S. M. Lunte and J. A. F. da Silva, Anal. Bioanal. Chem., 2012, 403, 2377–2384 CrossRef CAS PubMed.
- F. Q. Tu, L. Y. Zhang, X. F. Guo, Z. X. Zhang, H. Wang and H. S. Zhang, J. Chromatogr. A, 2014, 1359, 309–316 CrossRef CAS PubMed.
- J.-W. Yoo, G. Acharya and C. H. Lee, Biomaterials, 2009, 30, 3978–3985 CrossRef CAS PubMed.
- L. A. Peterson, T. Wagener, H. Sies and W. Stahl, Chem. Res. Toxicol., 2007, 20, 721–723 Search PubMed.
- R. A. Hunter and M. H. Schoenfisch, Anal. Chem., 2015, 87, 3171–3176 CrossRef CAS PubMed.
- S. C. Jacobson, S. V. Ermakov and J. M. Ramsey, Anal. Chem., 1999, 71, 3273–3276 CrossRef CAS PubMed.
- A. Ismail, S. Griveau, F. d'Orlyé, A. Varenne and F. Bedioui, Electroanalysis, 2015, 27, 2857–2863 CrossRef CAS.
- B. Thirumalraj, S. Palanisamy, S.-M. Chen and D.-H. Zhao, J. Colloid Interface Sci., 2016, 478, 413–420 CrossRef CAS PubMed.
- P. Kubáň and P. C. Hauser, Electrophoresis, 2009, 30, 3305–3314 CrossRef PubMed.
- J. L. Beckers and P. Boček, Electrophoresis, 2003, 24, 518–535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an00757h |
| This journal is © The Royal Society of Chemistry 2019 |