
A size-tunable and multi-responsive nanoplatform for deep tumor penetration and targeted combinatorial radio-/chemotherapy†
Fatima Zohra
Dahmani
ab,
Danni
Zhong
b,
Yuchen
Qi
b,
Aboubaker El G.
Dahmani
c,
Tingting
Xie
b,
Bo
Zhou
 b,
Wanli
Li
b,
Ke
Yao
ad,
Lei
Li
*e and
Min
Zhou
*abfg
b,
Wanli
Li
b,
Ke
Yao
ad,
Lei
Li
*e and
Min
Zhou
*abfg
aEye Center, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, China. E-mail: zhoum@zju.edu.cn
bInstitute of Translational Medicine, Zhejiang University, Hangzhou 310009, China
cJiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, China
dZhejiang Provincial Key Lab of Ophthalmology, Hangzhou, China
eShanghai Key Laboratory of Regulatory Biology, Institute of Biomedical Sciences, School of Life Sciences, East China Normal University, Shanghai 200241, China. E-mail: lli@bio.ecnu.edu.cn
fKey Laboratory of Cancer Prevention and Intervention, National Ministry of Education, Zhejiang University, Hangzhou 310009, China
gState Key Laboratory of Modern Optical Instrumentations, Zhejiang University, China
First published on 26th June 2019
Abstract
Nowadays, the design of multistimuli-responsive and deep penetrating nanotherapeutics remains an auspicious means for improving the efficiency of anticancer therapeutics. Herein, we develop a targeted size-tunable nanosystem that would concurrently and uniformly release its therapeutic cargo into tumor cells by exploiting both extracellular and intracellular signals. This nanosystem (HPDAu) is composed of self-assembled pH-responsive DOX–PAMAM (PD) conjugates and ultrasmall PAMAM-stabilized gold nanoparticles (AuNPs), incorporated into a hyaluronidase-responsive hyaluronic acid nanoshell. HPDAu nanoparticles with an initial particle size of ∼100 nm could disassemble into tiny cationic nanostructures (∼5 nm, PD and AuNPs) upon incubation with HAase, and showed a burst drug-release under tumor microenvironment-mimicking conditions (HAase and acidic conditions). Such characteristics resulted in significantly improved tumor penetration both in vitro and in vivo, along with a higher cellular uptake efficiency and tumor accumulation as compared to free drug solutions. Taking advantage of these features along with the chemotherapeutic effect (DOX) and AuNP/DOX-induced radiosensitization, HPDAu plus radiotherapy (RT) treatment resulted in a substantial increase of apoptotic and cytotoxic effects against 4T1 cells. More importantly, the radio-/chemotherapeutic feasibility of HPDAu was further validated in a 4T1 orthotropic model, revealing a prominent antitumor efficacy and reduced side-toxicity compared to monotherapy and free drug solutions. Therefore, this versatile nanoplatform with active targeting, size tunability and radio-/chemotherapeutic features could be a promising tool for tumor combinatorial therapy.
Introduction
With the ever-growing application of nanomedicine in cancer therapy, the development of versatile nanosystems with multiple intrinsic features has become an obvious target for efficient therapeutic outcomes.1 Such delivery systems would preferably be safe and efficiently target the tumor site, while providing an effective drug accumulation in tumor cells without disrupting healthy organ function. Nevertheless, various biological barriers substantially affect the nanoparticles’ fate in vivo, such as bloodstream circulation, non-specific accumulation in normal organs, impaired diffusion into the tumor interstitium, inefficient uptake by the tumor cells and intracellular delivery of loaded therapeutics.1,2 Numerous nanotherapeutics have been developed in order to bypass such barriers; yet achieving a uniform nanoparticle penetration into the tumor tissue is still challenging.2–4 This issue is related to the impaired delivery of therapeutic molecules into the tumor's deeper parts due to the compact extracellular matrix (ECM), aberrant intratumoral vasculature and elevated interstitial fluid pressure (IFP) in the tumor microenvironment.1,3,5 Convincing evidence from previous reports showed that nanoparticles with a larger particle size are more likely to be propitious for improved pharmacokinetic behavior and tumor accumulation; however, their restricted penetration into the tumor interstitium remains unfavorable.6–8 In addition, the application of smaller nanoparticles is greatly hindered by their short circulation time and deficient tumor accumulation despite their prominent penetration ability.2,9,10 To address this dilemma, designing a nanoplatform with size shrinking characteristics upon reaching the tumor microenvironment seems an ideal approach.4,9,11 Recent reports were devoted to designing stimuli-responsive nanosystems, i.e., enzyme, UV, light, and pH/redox-responsive, with the aim to promote their intratumoral penetration efficiency.6,9,12–15 For instance, Ruan et al. developed a shrinkable gelatin nanoparticle with an initial particle size of ∼190 nm that could disassemble into tiny nanoparticles (60 nm) as a response to MMP-2 degradation, a highly expressed enzyme in the tumor microenvironment.15 Importantly, such a characteristic resulted in substantially enhanced tumor penetration and antitumor efficacy in a B16F10 tumor model. Besides the particle size, the surface charge may also affect the nanomedicine's penetrating ability.3 Cationic nanoparticles were reported to display better tumor penetration compared to their neutral and slightly anionic counterparts, while these latter nanoparticles remain favorable for long circulation times.3,16,17 Therefore, surface modification of positively charged nanosystems with biodegradable materials becomes essential to bypass their rapid clearance by the mononuclear phagocytic system (MPS) and extend their circulation time.Radio-/chemotherapy, a widely used combinative therapeutic platform for most solid tumors, holds considerable appeal in improving the therapeutic efficacy of antitumor medicines, which is likely related to the sensitizing effect of certain chemodrugs to radiotherapy.18–21 However, the non-specific action and associated side-toxicity of such therapies is still hindering their clinical application.22,23 Of note, gold nanoparticles (AuNPs) have been lately explored as radiosensitizing agents owing to their amplifying capability of ionizing radiation-induced damaging effects.24–26 Their use has also expanded to optical sensing, imaging, drug delivery, and therapeutic applications owing to their size- and shape-dependent physical properties, good biocompatibility and ease of preparation.27–29 Recent reports have shown that the radiosensitizing level of AuNPs increased as the particle size decreased and their cell uptake efficiency augmented.30–32 Poly(amidoamine) (PAMAM) dendrimers are well-defined and highly branched polymeric nanostructures with numerous surface amino groups, narrow polydispersity and nanoscaled size.33 Recently, these cationic dendrimers have been implicated for therapeutic and biomedical applications due to their conjugating and/or entrapping ability of small drugs and imaging moieties along with their stabilizing ability of metal nanoparticles. Such nanostructures can be easily internalized by the tumor cells via adsorption-mediated endocytosis after electrostatic binding to the negatively charged cell membrane lipids, while having the ability to rupture the endo-/lysosomal membranes and promote subsequent escape from the vesicle trap.34,35 Owing to these properties, using ultrasmall PAMAM dendrimers (∼5 nm) as a stabilizing agent for AuNPs may improve their internalization efficiency and the resulting radiosensitizing effect.
Over the past few decades, polysaccharide-based nanosystems have arisen as a promising means for targeted delivery of anticancer therapeutics owing to their biodegradability, biocompatibility and low immunogenicity.36–38 These nanosystems would promote a preferential accumulation of delivered therapeutics (passive targeting) via the eminent enhanced permeability and retention (EPR) effect, besides their anchoring capability of numerous molecules and easier chemical modification. Among others, hyaluronic acid (HA), an anionic linear polysaccharide composed of β (1 → 3)-linked disaccharide (N-acetyl-D-glucosamine (GlcNAc) and D-glucuronic acid (GlcA)), has been extensively explored for nanoparticle surface functionalization. The reasons behind this are its capability to improve the nanoparticle stability and circulation time, and its targeting ability towards a variety of tumor cells overexpressing some membrane receptors, such as CD44.39,40 Moreover, this polysaccharide is known to be enzymatically degradable by the tumor extracellular and endo-/lysosomal enzymes, i.e., hyaluronidase (HAase), thereby representing an attractive biomaterial with not only an active targeting capability but also an on-site degradability.39,41
Herein, we focused on combining radio-/chemotherapeutic and active targeting features into a single nanosystem by incorporating ultrasmall PAMAM-stabilized gold nanoparticles (AuNPs) and a pH-responsive PD conjugate (obtained through conjugation of PAMAM's amino group to the carboxylic group of cis-aconityl-doxorubicin) into a HA-based nanoshell (Fig. 1). This latter material was used as a coating material for oppositely charged dendritic nanoparticles in order to shield them from plasma opsonization and immunogenicity, while providing a selective targeting ability towards the tumor cells and an on-site size changeability upon enzymatic degradation by hyaluronidase. The resulting nanosystem would concomitantly take advantage of the attractive biological features of HA biopolymer along with the radio-/chemotherapeutic effects of AuNPs and pH-responsive DOX prodrug (PD), while exploiting both extracellular and intracellular signals of the tumor microenvironment (HAase and mildly acidic pH) to promote drug penetration and internalization efficiency and subsequent drug release within the tumor cells. As shown in Fig. 1, after their passive extravasation through the EPR effect, HPDAu nanoparticles would undergo hyaluronidase-mediated degradation to release ultrasmall cationic nanoparticles (∼5 nm) that are expected to provide a uniform and deep intratumoral penetration of delivered drugs. Meanwhile, such a nanosystem may improve the drug's uptake efficiency by the tumor cells not only via an active targeting mechanism (CD44-mediated internalization) but also a direct binding of the exposed cationic nanoparticles to plasma membranes via electrostatic interactions. Consequently, the mildly acidic pH in endosomal/lysosomal structures would instigate the release of the nanoparticles’ cargo (DOX), followed by an endo–lysosomal escape owing to PAMAM's ability to rupture the endo-/lysosomal membranes.35 Overall, this approach may promote the drug's cytosolic and nuclear accumulation, thereby improving the cytotoxic effect of DOX and AuNP/DOX-induced radiosensitization, and thus the resulting therapeutic outcome.42
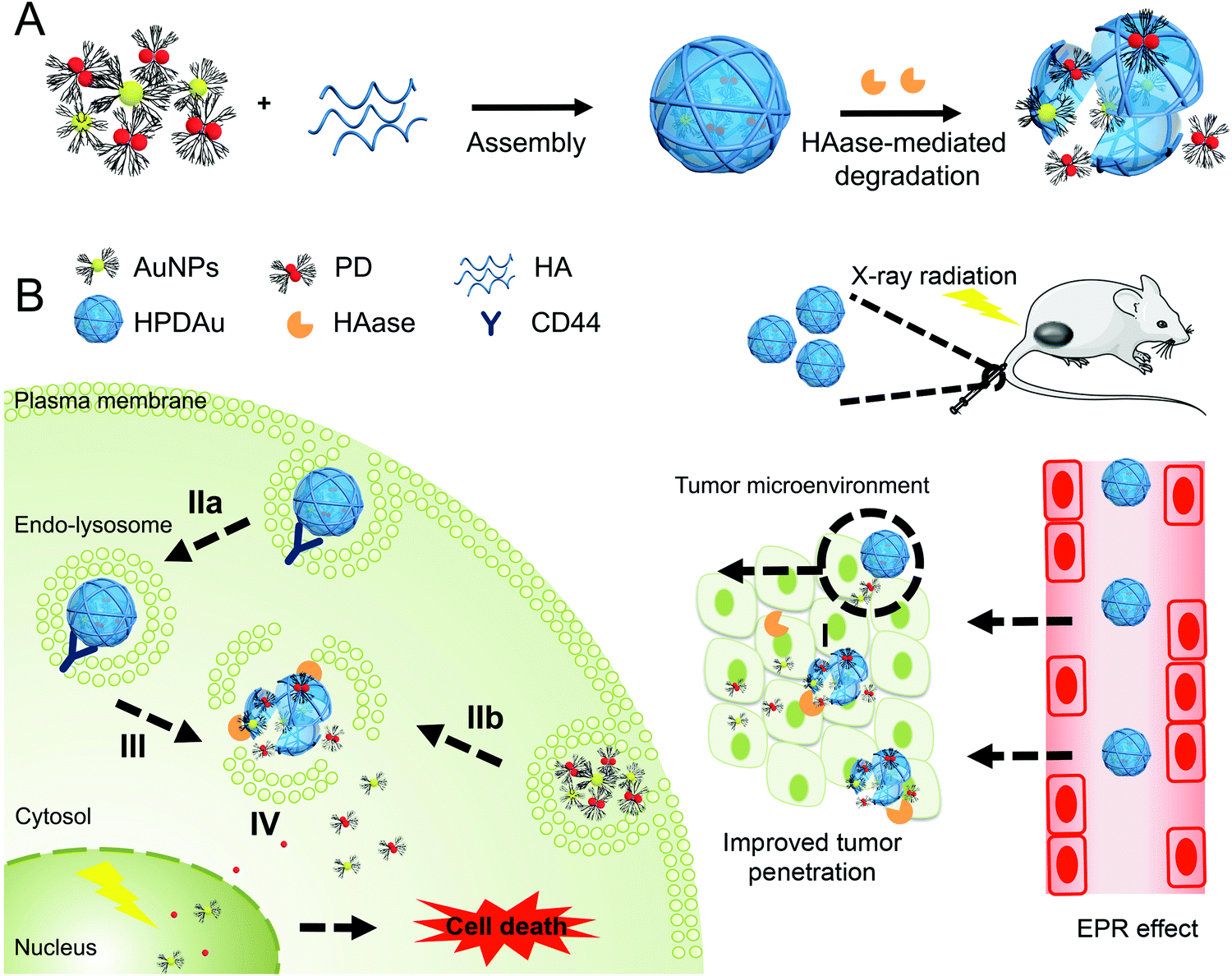 | ||
| Fig. 1 (A) Schematic representation of the assembly and disassembly process of HPDAu. (B) Mechanistic representative diagram of the passive extravasation of HPDAu nanoparticles into the tumor tissue (EPR effect) after IV injection, followed by stimuli-triggered disassembly, cellular uptake and drug release: (I) hyaluronidase (HAase)-mediated degradation of hyaluronic acid (HA) nanoshell to release ultrasmall gold nanoparticles (AuNPs) and DOX–PAMAM conjugates (PD) that can deeply penetrate into the tumor, (IIa) recognition of CD44 receptors by HPDAu followed by receptor-mediated internalization, (IIb) adsorption-mediated endocytosis of the exposed cationic nanoparticles, (III) HAase and acidic pH-triggered disassembly of HPDAu nanoparticles and drug release in endo–lysosomes, and (IV) endo–lysosomal escape leading to enhanced cytosolic and nuclear accumulation of DOX and AuNPs. | ||
Experimental section
Materials
Doxorubicin hydrochloride (DOX·HCl) and cis-aconitic anhydride were purchased from Beijing Huafeng United Technology Co. (Beijing, China) and Alfa Aesar (Lancashire, UK), respectively. PAMAM G5.0 was obtained from Weihai CY Dendrimer Technology Co. Ltd (Shandong, China). N-Hydroxysuccinimide (NHS), 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), adipic acid dihydrazide (ADH), anhydrous dimethylformamide (DMF), triethylamine (TEA) and acetone were purchased from Aladdin (Shanghai, China). Hyaluronic acid (MW ≈ 103 kDa, Pd 1.34) was obtained from Freda Biochemical Co. (Shandong, China). Chloroauric acid (HAuCl4), sodium borohydride (NaBH4), IR780 and FITC-labeled Lycopersicon esculentum lectin were purchased from Sigma Chemical Co. (St. Louis, Mon USA). Immortalized human hepatocytes (HepLL) and neonatal mouse cardiac fibroblasts (NMCF) were generously provided by Prof. Li Lanjuan (The First Affiliated Hospital of Zhejiang University School of Medicine) and Prof. Chen Jinghai (Institute of Translational Medicine, Zhejiang University), respectively.Characterization
The synthesized CAD was analyzed using HPLC (Water Alliance 2690, USA) and a Symmetry ShieldTM RP18 column (4.6 × 250 mm, 5 μm) at a detection wavelength of 490 nm. The mobile phase consisted of ammonium carbonate (3 w%), MeOH and acetonitrile at a volume ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45:5, with a 20 μL injection volume and 1.0 mL min−1 flow rate. 1H NMR and UV spectra were recorded using a Bruker Avance III-HD 500 NMR Spectrometer (Bruker, Germany) and a UV-vis spectrometer (Shimadzu UV-2600, Japan), respectively. The DOX content (DC) of PD conjugate was determined using a UV-vis spectrometer (Shimadzu UV-2600, Japan) at 490 nm. DC = mDOX/mPD × 100, w% (mDOX: mass of detected DOX in PD conjugate; mPD: mass of PD conjugate). DOX and Au contents of HPDAu nanoparticles were measured using a spectrofluorophotometer (Shimadzu RF-6000, Japan, Ex/Em wavelengths = 480/595 nm) and an ICP-MS system (NexION 300×, PerkinElmer, USA), respectively. Loading content (w%) = (m1 − m2)/mHPDAu × 100 (m1 is the total mass of DOX or Au used, m2 is the mass of unloaded DOX or Au, and mHPDAu is the mass of HPDAu). The dynamic light scattering (DLS) and zeta potential measurements of the aqueous solutions of different nanoformulations were obtained using a Malvern Zetasizer (Nano ZS90, Malvern Instruments, Worcestershire, UK) at 25 °C and a scattering angle of 90°. The nanoparticles’ 3D morphology was observed using scanning electron microscopy (SEM, Hitachi SU-70, Japan) operated at 4 kV. Besides, the 2D morphology of HPDAu without and with HAase treatment was characterized using high-resolution transmission electron microscopy (H-7650 HR, Hitachi High Technologies, Tokyo, Japan) operated at 90 kV. Element mapping was obtained using a Hitachi SU8010 FETEM (Hitachi, Tokyo, Japan). Herein, all HAase + HPDAu treatments were carried out after 12 h pre-incubation of HPDAu with HAase (1 mg mL−1) at 37 °C.
:45:5, with a 20 μL injection volume and 1.0 mL min−1 flow rate. 1H NMR and UV spectra were recorded using a Bruker Avance III-HD 500 NMR Spectrometer (Bruker, Germany) and a UV-vis spectrometer (Shimadzu UV-2600, Japan), respectively. The DOX content (DC) of PD conjugate was determined using a UV-vis spectrometer (Shimadzu UV-2600, Japan) at 490 nm. DC = mDOX/mPD × 100, w% (mDOX: mass of detected DOX in PD conjugate; mPD: mass of PD conjugate). DOX and Au contents of HPDAu nanoparticles were measured using a spectrofluorophotometer (Shimadzu RF-6000, Japan, Ex/Em wavelengths = 480/595 nm) and an ICP-MS system (NexION 300×, PerkinElmer, USA), respectively. Loading content (w%) = (m1 − m2)/mHPDAu × 100 (m1 is the total mass of DOX or Au used, m2 is the mass of unloaded DOX or Au, and mHPDAu is the mass of HPDAu). The dynamic light scattering (DLS) and zeta potential measurements of the aqueous solutions of different nanoformulations were obtained using a Malvern Zetasizer (Nano ZS90, Malvern Instruments, Worcestershire, UK) at 25 °C and a scattering angle of 90°. The nanoparticles’ 3D morphology was observed using scanning electron microscopy (SEM, Hitachi SU-70, Japan) operated at 4 kV. Besides, the 2D morphology of HPDAu without and with HAase treatment was characterized using high-resolution transmission electron microscopy (H-7650 HR, Hitachi High Technologies, Tokyo, Japan) operated at 90 kV. Element mapping was obtained using a Hitachi SU8010 FETEM (Hitachi, Tokyo, Japan). Herein, all HAase + HPDAu treatments were carried out after 12 h pre-incubation of HPDAu with HAase (1 mg mL−1) at 37 °C.
Synthesis of PD prodrug
DOX was firstly conjugated to cis-aconitic anhydride to obtain CAD that was subsequently coupled to the amino groups of PAMAM dendrimers. Firstly, DOX·HCl (50 mg) and cis-aconitic anhydride (15 mg) were dissolved in 3 mL of anhydrous DMF, and 13 μL of TEA was added followed by overnight reaction under argon protection and dark conditions. The reaction mixture was extracted with 25 mL of cold ethyl acetate and then sequentially washed twice with brine solution. The organic layer was collected and then evaporated using a rotavap (IKA-RV10, Stanfer, Germany) followed by vacuum drying to get CAD as a red powder. The obtained product was analyzed using a Waters HPLC system.Secondly, the carboxylic groups of CAD were activated with NHS (32.8 mg) and EDC·HCl (54.8 mg) for 30 min using DMSO as solvent. Thereafter, PAMAM (50 mg) solution in DMSO was added followed by an overnight reaction (50:1 CAD:PAMAM molar ratio). The mixture was diluted in deionized water and then purified using Amicon® ultra-centrifugal filters (10 kDa) followed by 48 h dialysis against deionized water and then freeze drying. The synthesized product was then characterized using 1H NMR and UV-vis spectroscopy. 1H NMR of PD (400 MHz, DMSOd6/D2O, δ ppm): 1.18 (3H, d, DOX), 3.93 (3H, d, –O–CH3, DOX), 3.19 (2H, m, cis-aconitic anhydride), 4.59 (2H, d, –C(O)CH2–, DOX), 6.51 (1H, s, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CO–, cis-aconitic anhydride), 7.52–7.85 (1H, m, aromatic protons of DOX).
CH–CO–, cis-aconitic anhydride), 7.52–7.85 (1H, m, aromatic protons of DOX).
Synthesis of AuNPs
AuNPs were prepared using amine-terminated PAMAM dendrimers as a stabilizer. First, 100 μL of HAuCl4 solution (100 mM) was added into 50 mL of dendrimer solution (1 μM) in ultrapure water with vigorous stirring for 2 h. Thereafter, the complexed Au ions were chemically reduced using a five-fold molar excess of 25 mM NaBH4 solution in 0.3 N NaOH and then allowed to react for 6 h. The obtained nanoparticles were concentrated using Amicon® ultra-centrifugal filters (10 kDa) and analyzed using UV-vis spectrometry.Synthesis of HPDAu
PD and AuNPs coated with HA were prepared by a nanoprecipitation method. Briefly, 50 mg of HA (100 kDa) and 10 mg equivalent of PAMAM (PD, AuNPs) were dissolved in 40 mL of ultrapure water; then 50 mL of acetone was added and stirred for 30 min. Thereafter, EDC·HCl and ADH were introduced into the reaction mixture followed by 30 min reaction (EDC:ADH:HA carboxylic group molar ratio 1:1:8, 2:2:8, 4:4:8, and 8:8:8). Another 50 mL of acetone was added and allowed to react overnight. The organic layer was evaporated using an IKA-RV10 rotary evaporator; the obtained solution was then sonicated using an ultrasonicator probe (JY92-II, Scientz Biotechnology Co., Ningbo, China) for 30 min. The unreacted and unloaded reagents were removed using ultrafiltration and dialysis against deionized water (48 h) followed by freeze-drying.
3D spheroid experiment
4T1 cell suspensions (5000 cells per well) were seeded in ultra-low attachment ninety six-well plates (Corning® 7007), then centrifuged at 1000 rpm for 5 min and incubated at 37 °C for 5 days. As the spheroid diameter reached roughly 200 μm, the culture medium (100 μL per well) was carefully replaced with the drug-containing medium (free DOX, HPDAu, or HAase + HPDAu at 1 μg mL−1 DOX concentration) followed by 4-h incubation. The spheroids were neatly washed with PBS, and high-resolution Z-stacks were then acquired using a Nikon A1 confocal laser-scanning microscope (CLSM, Tokyo, Japan) at an excitation wavelength of 488 nm.In vivo tumor penetration and microdistribution of HPDAu in tumor tissue
All animal use protocols were approved by the University Animal Care and Use Committee of Zhejiang University. The orthotropic 4T1 tumor xenograft model was established by inoculating 2 × 106 4T1 cells into the right mammary fat pad of BALB/c nude mice (female, 20 ± 2 g, four-weeks-old, Shanghai Slac Laboratory Animal Company, China). When the tumor size achieved about 10 mm, the mice were randomly assigned to two groups, which were injected with free DOX and HPDAu (5 mg kg−1 DOX), respectively. After 24 hours, and 5 min before being euthanized, the mice were administered with FITC-labeled Lycopersicon esculentum lectin to indicate the tumor vasculature endothelial cells.7 The tumor tissues were then respectively harvested, covered with OCT and sectioned into ten-micrometer frozen sections using a cryostat (Leica CM3050S, Germany). After fixation in cold acetone (15 min) and PBS washing, the tissue slides were mounted using Fluoromount-G with DAPI, then imaged using a CLSM.In vivo tumor targeting capability
4T1-tumor bearing mice were randomly assigned to two groups (tumor diameter around 10 mm) that were intravenously injected with IR780 and IR780-labeled HPDAu (100 μg IR780 per kg, n = 4), respectively. The mice were anesthetized, at certain time points, with isoflurane, and then imaged using an IVIS Lumina LT Series III (PerkinElmer, USA). After 48 h, the mice were sacrificed and dissected to obtain major organs (heart, kidneys, lungs, spleen, and liver) and tumor tissues; then, the ex vivo fluorescence images were acquired using the same imaging system. The semi-quantitative evaluation of the fluorescence signals of the excised tissues was performed through region-of-interest (ROI) analysis using Living Image 4.5 software (PerkinElmer).In vivo antitumor efficacy
As the tumor diameter reached roughly 3 mm, mice were randomly assigned to six groups (n = 6 mice per group): saline (control group), DOX, RT, DOX + RT, HPDAu, and HPDAu + RT, wherein DOX and HPDAu (5 mg kg−1 DOX and 20 mg kg−1 Au) were administered via tail vein injection on day 0 and day 3 then RT was performed on day 1 and day 4 (by in situ X-ray irradiation, 6 Gy). The mice behavioral change, body weight, and tumor size were noted every three days. After 18 days, serum samples were obtained for the quantification of enzymatic markers of heart (CK), liver (AST, ALT) and kidney (BUN, CREA) functions. The mice were then euthanized and dissected to harvest major organs and tumor tissues that were fixed in formalin accordingly. Thereafter, formalin-fixed paraffin-embedded tissues were prepared for histopathological examination.Statistical analysis
Statistical differences between groups were determined using the two-tailed Student's t-test. All data are presented as mean ± SD. P < 0.05 was considered as significant.Results and discussion
Preparation and physicochemical characterization of HPDAu
The PD conjugate was obtained by grafting DOX molecules to PAMAM dendrimers via a pH-sensitive cis-aconityl linkage (Fig. S1, ESI†). Firstly, CAD was synthesized by coupling DOX to cis-aconitic anhydride and then characterized using HPLC (Fig. S2, ESI†). Two isomers were detected, cis-CAD and trans-CAD (Rt 5.91 and 6.67 min), while the free DOX signal (Rt 8.53 min) was almost negligible, indicating the successful synthesis of CAD. Thereafter, CAD was subsequently coupled to the amino groups of PAMAM to obtain the PD conjugate that was characterized using 1H NMR spectra and UV-vis spectroscopy. As displayed in Fig. S3 (ESI†), DOX characteristic peaks appeared at δ 1.18, 3.93 ppm (methyl protons), δ 4.59 ppm (methylene protons) and δ 7.52–5.85 ppm (anthracene protons). Besides, the peak emerging at 6.51 ppm was attributed toCH–CO– in the cis-aconitic anhydride linker. UV-vis spectroscopy showed comparable results, wherein the characteristic absorption peak of DOX emerged at 490 nm (Fig. 2A). The corresponding DOX content in the PD conjugate was calculated as (32.57 ± 2.29%, w%) with an estimated molar grafting ratio (DOX/PAMAM) of 28.5.
 | ||
| Fig. 2 Physicochemical characterization of HPDAu nanoparticles: (A) UV-vis spectra of DOX, PAMAM, DOX–PAMAM conjugate (PD), gold nanoparticles (AuNPs) and HPDAu aqueous solutions. (B) Particle size distribution of different nanoformulation solutions as determined using dynamic light scattering (DLS). (C) Transmission electron microscopy (TEM) image of HPDAu without (a) and with (b) hyaluronidase (HAase) treatment; elemental mapping of HPDAu nanoparticles, electron micrograph region (c), and respective gold element distribution (d). (D) Scanning electron microscopy (SEM) image of HPDAu. (E) Photographic images of DOX, AuNPs, PD, and HPDAu solutions. (F and G) Release rates of DOX from HPDAu upon incubation under different pH conditions with and without HAase treatment. Data are presented as mean ± SD (n = 3). | ||
AuNPs were prepared via electrostatic interaction between AuCl4− and the amino groups of PAMAM that were characterized by the broad band emerging at 216 nm with a shoulder at 261 nm, as displayed in the UV-vis spectra (Fig. 2A). Also, the surface plasmon resonance (SPR) peak of the formed AuNPs was observed at 520 nm after reduction with NaBH4. Subsequently, HPDAu nanoparticles were obtained through a co-assembly process of HA, PD, and AuNPs by nanoprecipitation and electrostatic interactions. Acetone was used as the nanoprecipitation solvent with the aim to break intense inter-chain hydrogen bonds between HA molecules and increase the hydrophobicity, resulting in the formation of globular HA structures. Meanwhile, HA chains were cross-linked via the formation of peptide bonds between the neighboring carboxylic groups of HA and the amino groups of adipic acid dihydrazide. In this respect, the size distribution of HPDAu nanoparticles at different EDC:ADH:HA molar ratios was assessed using DLS. As displayed in Fig. S4 (ESI†), the optimum formulation (4:4:8 molar ratio) showed a relatively uniform size distribution with a narrow PDI (0.12). The hydrodynamic size of this nanoformulation was around 100 nm (Fig. 2B and Table S1, ESI†), whereas the zeta potential switches from positive to negative values owing to the HA-based polyanionic outer shell surrounding the AuNPs and PD. The corresponding loading contents of DOX and Au in HPDAu nanoparticles were 7.06 ± 0.54% and 27.94 ± 1.79%, respectively. Nanoparticle morphology was then observed using HR-TEM and SEM. As shown in Fig. 2C and D, the as-prepared HPDAu nanoparticles displayed a quasi-oval shape that could disassemble into ultrasmall nanoparticles (∼5 nm) after incubation with HAase. The EDS spectra further confirmed the presence of Au element, wherein the Au characteristic peak emerged at 2.3 keV (Fig. 2C and Fig. S5, ESI†).
To estimate the nanoparticles’ responsiveness to tumor microenvironment-simulated conditions (mildly acidic pH and enzymatic degradation by HAase), the change in particle size and zeta potential of HPDAu incubated with HAase at pH 7.4 (plasma pH) and pH 6.5 (tumor extracellular pH) was assessed using DLS. As shown in Fig. S6 (ESI†), the particle size reduced from ∼100 nm to ∼60 nm within the first 4 h, then it gradually decreased to reach ∼7 nm. Moreover, the zeta potential reversed from ∼−20 mV to ∼+15 mV, suggesting that HAase treatment can trigger the degradation of the HA nanoshell to release the loaded cationic nanostructures (AuNPs and PD). The change in DOX release rates of HPDAu was also determined upon incubation under different conditions using fluorescence spectroscopy. The release rate of DOX under neutral conditions (pH 7.4) was trivial (Fig. 2F). In comparison, a prominent increase was observed under acidic conditions (pH 6.5 and pH 5.5), with a more obvious difference at pH 5.5. Additionally, the co-incubation with HAase enzyme further augmented the DOX release rates under acidic conditions (Fig. 2G). Overall, these results indicate that HPDAu nanoparticles would sequentially respond to the endogenous features of tumor tissue to form ultrasmall cationic nanoparticles, which would presumably promote the tumor penetration of the delivered molecules.3,9,16,17,43 In addition, the pH-responsiveness of the PD conjugate is expected to provide a selective drug release after reaching the tumor microenvironment (pH 6.5) and internalization by the tumor cells (endo–lysosomal structures, pH 5.5).
Penetration of HPDAu into multicellular tumor spheroids
The in vitro penetration efficiency of HPDAu nanoparticles was investigated using multicellular spheroids (MCSs) derived from the murine breast cancer cell line 4T1. This latter cell line was suggested as a versatile 3D model to estimate the effectiveness of anticancer therapeutics.9,44 MCSs were incubated with DOX, HPDAu, and HAase + HPDAu for 4 h, then washed with PBS and observed using a CLSM (Fig. 3 and Fig. S7, ESI†). For DOX treatment, the DOX red fluorescence was mainly detected in the MCSs’ periphery, whereas no noticeable signal was observed in the internal area. Likewise, a similar effect was noticed with HPDAu, indicating the limited tumor penetration of larger nanoparticles. In comparison, HAase + HPDAu treatment resulted in a clearly increased signal in the MCSs’ internal area, likely owing to the degradation of the HA-based outer shell by HAase. Previous reports have shown that small and positively charged nanoparticles remain favorable for improving tumor penetration.3,9,16,17 Thence, these data indicate the evident ability of the size-changeable HPDAu nanoparticles to promote the tumor penetration of DOX, owing to the smaller size and cationic charge of the released nanostructures upon HAase degradation.39,41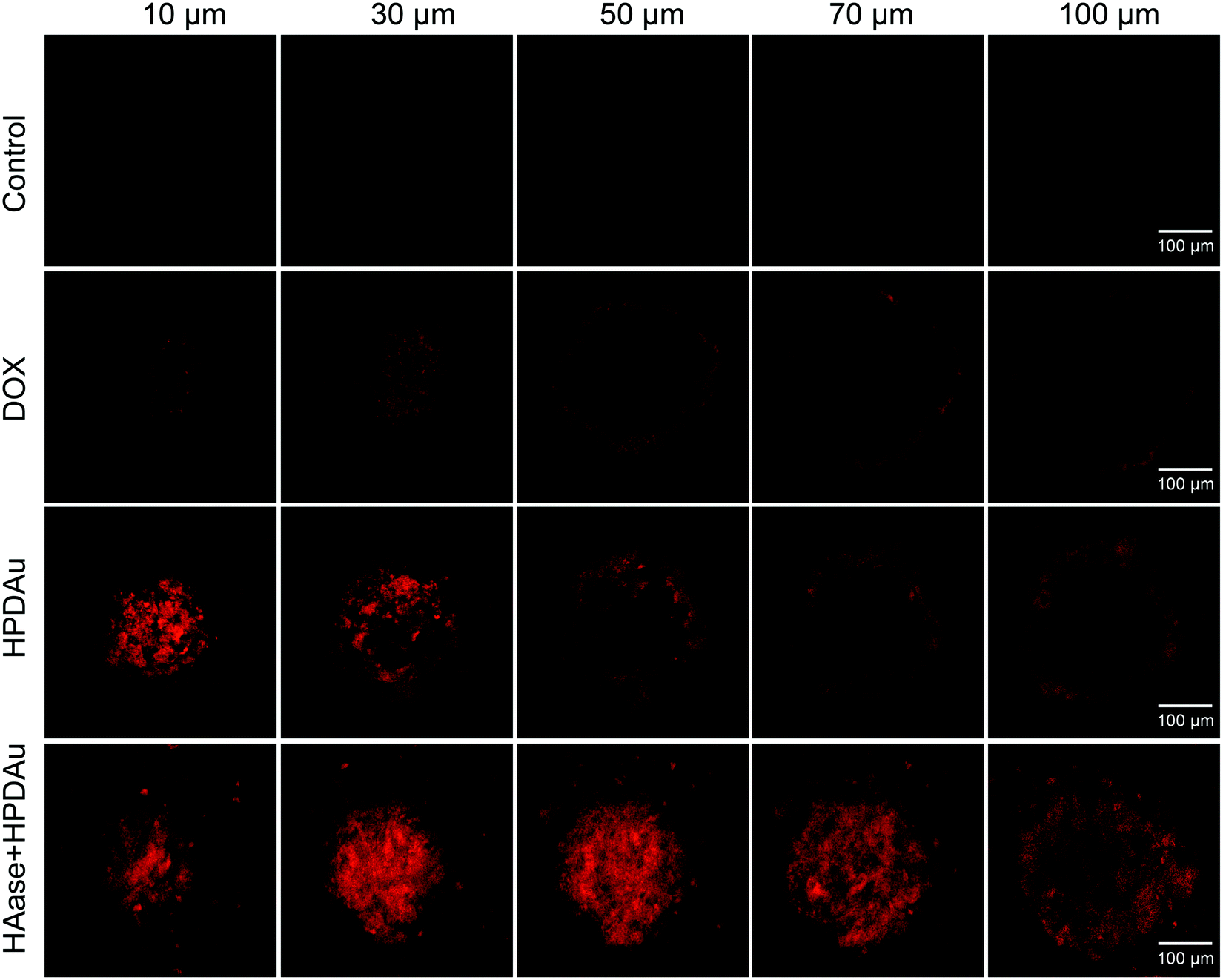 | ||
| Fig. 3 In vitro penetration efficiency of HPDAu nanoparticles; Z-stack confocal images of 4T1 multicellular spheroids, acquired from the surface top towards the equatorial plane at 20 μm intervals, after 4 h incubation with DOX, HPDAu and hyaluronidase plus HPDAu (HAase + HPDAu) at 1 μg mL−1 DOX concentration. The scale bars indicate 100 μm in length. | ||
Cellular uptake study
The in vitro targetability and uptake efficiency of the developed HPDAu nanoparticles were assessed using fluorescence microscopy. In this respect, 4T1 cells were incubated with DOX, HPDAu, and HAase + HPDAu for 6 and 12 h, respectively. Moreover, a blocking experiment was performed after 1 h incubation with an excess of free HA followed by the same treatment with HPDAu. Quantitative estimation of the fluorescence signals in each group was carried out using Harmony software. As shown in Fig. 4A–C, the uptake efficiency of free DOX was markedly lower than HPDAu, whereas the latter's fluorescence signal increased with extended incubation time. In comparison, HAase + HPDAu treatment resulted in comparable signals (P > 0.05 vs. HPDAu), indicating that the released PD prodrugs could also be internalized by the tumor cells via direct binding to cell membranes.42 In addition, the pretreatment with HA duly resulted in a significant reduction of fluorescence signals, suggesting that HPDAu nanoparticles were internalized via a CD44-mediated process.40,45–47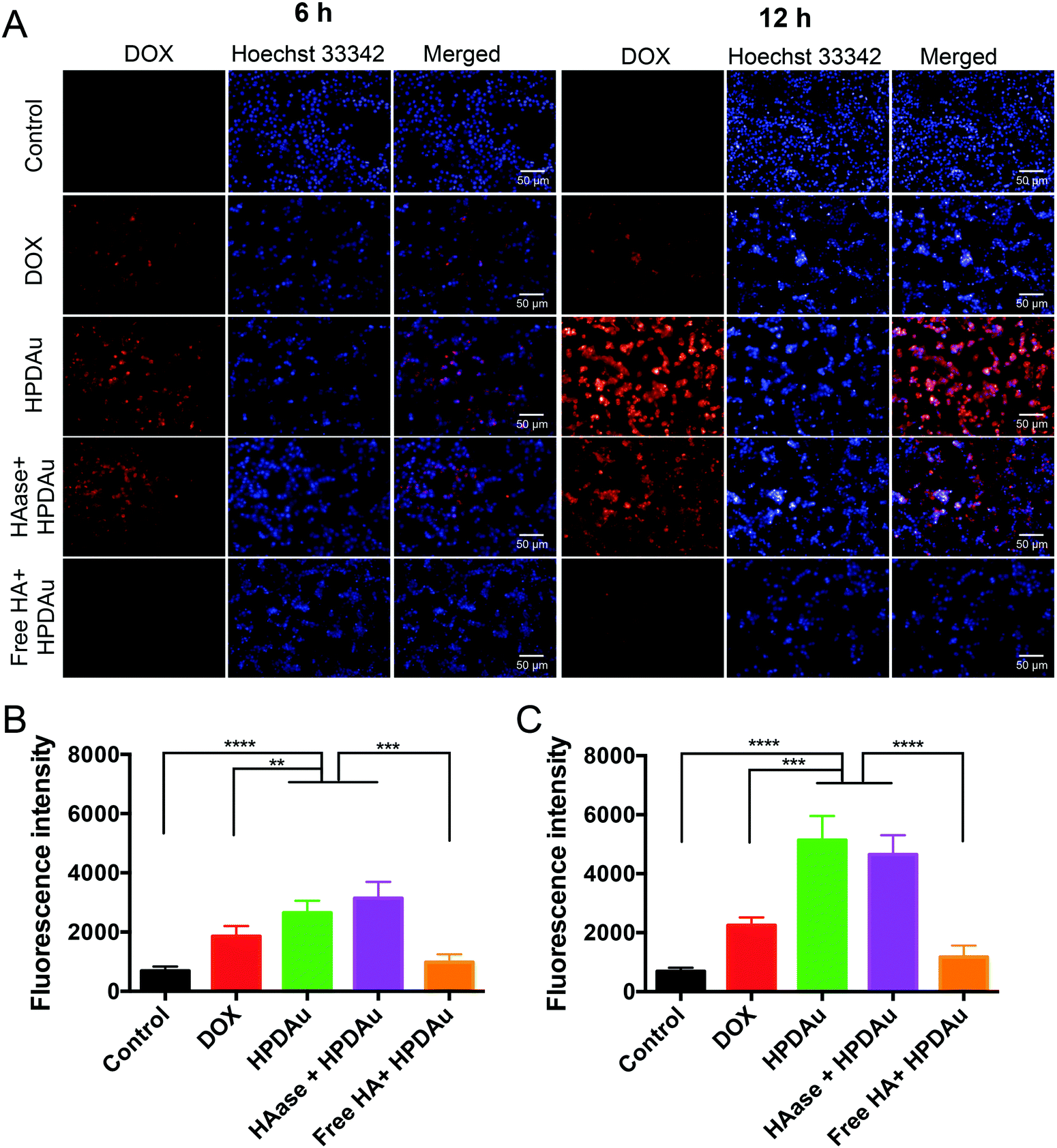 | ||
| Fig. 4 Cellular uptake study: (A) uptake efficiency of DOX, HPDAu, hyaluronidase plus HPDAu (HAase + HPDAu), and free hyaluronic acid pretreatment plus HPDAu (free HA + HPDAu) in 4T1 cells at different time points (6 h and 12 h) as observed using an Operetta high-content imaging system (20× magnification). The cell nuclei were stained using Hoechst 33342 (blue). (B and C) Quantitative evaluation of DOX channel fluorescence intensities in different groups after 6 and 12 h incubation, respectively (n = 6). All data are presented as mean ± SD. **P < 0.01, ***P < 0.001, and ****P < 0.0001. The scale bars indicate 50 μm in length. | ||
In vitro apoptosis detection and cytotoxicity
The in vitro radio-/chemotherapeutic efficacy of HPDAu nanoparticles against 4T1 cells was evaluated using Annexin V-FITC/PI apoptosis assay. As displayed in Fig. 5A and B, RT, DOX, and AuNPs alone resulted in a slight apoptotic effect (∼11–18%), whereas an obvious increase was noted with DOX + RT and AuNPs + RT, indicating the radiosensitizing effect of DOX and AuNPs, as reported previously.18–21,48 In comparison, the apoptotic rate of HPDAu + RT rose by 1.8- to 2-fold as compared to those of DOX + RT, AuNPs + RT, and HPDAu (P < 0.01).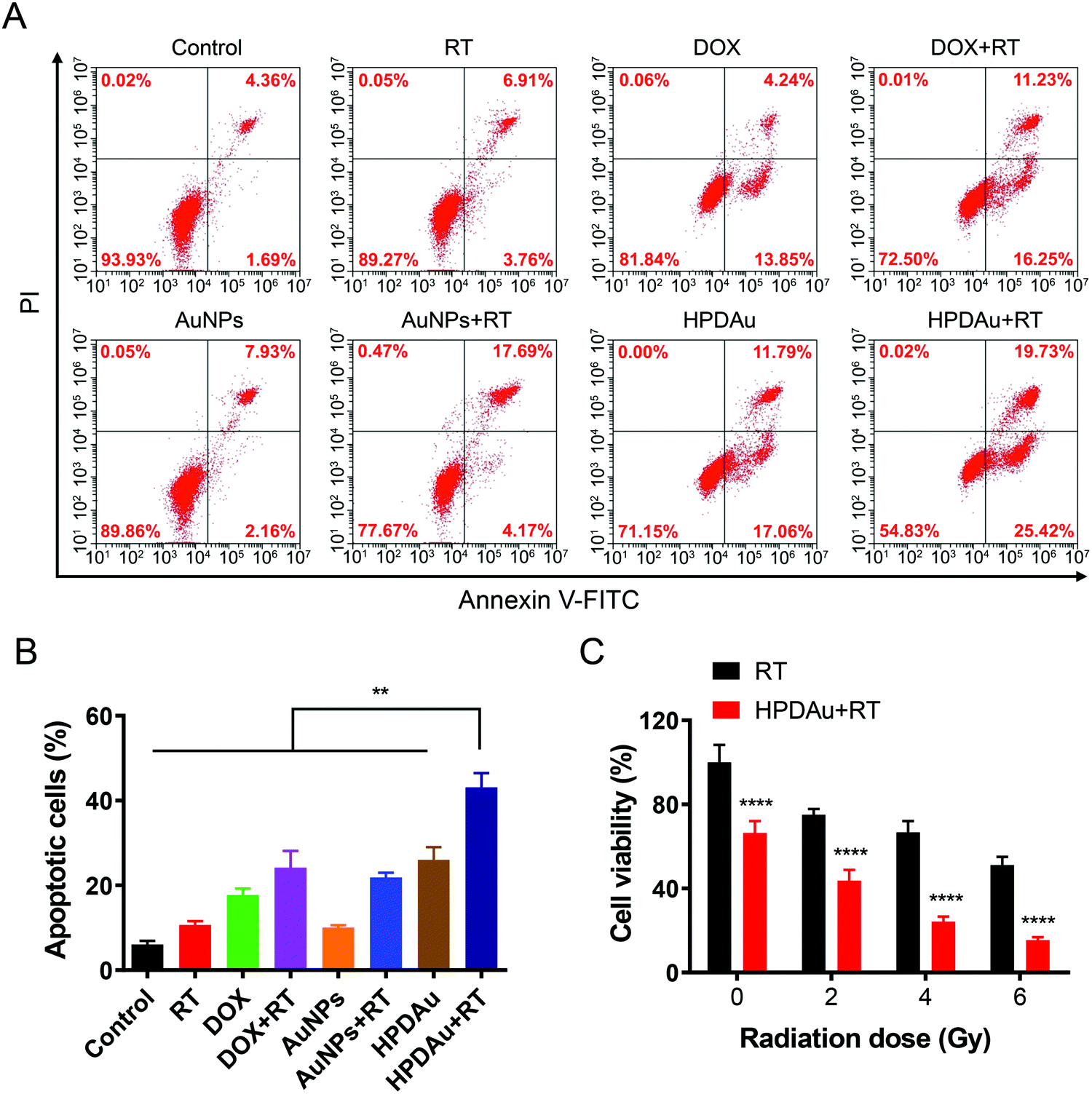 | ||
| Fig. 5 Evaluation of the in vitro apoptotic and cytotoxic effects of HPDAu + RT: (A) flow cytometric analysis of 4T1 cells after 4 h treatment with different formulations (1 μg mL−1 DOX, 4 μg mL−1 Au) using the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit: untreated cells (control), radiotherapy (RT), DOX, DOX plus RT (DOX + RT), AuNPs, AuNPs plus RT (AuNPs + RT), HPDAu, and HPDAu plus RT (HPDAu + RT). The cells in the RT groups were irradiated with a 4 Gy X-ray dose after 4 h treatment with the corresponding formulation followed by 12 h incubation and fluorescence-activated cell sorting (FACS) analysis. (B) Apoptotic cell percentage comprising early (Annexin V+/PI−) and late (Annexin V+/PI+) stage apoptotic cells in different groups (n = 3). (C) In vitro cytotoxic effect of RT and HPDAu + RT (1 μg mL−1 DOX, 4 μg mL−1 Au) at different radiation doses towards 4T1 cells after 48 h incubation (n = 6). All data are presented as mean ± SD. **P < 0.01, and ****P < 0.0001. | ||
Next, the cytotoxic effect of HPDAu nanoparticles was further assessed after irradiation with different X-ray doses (0–6 Gy) and compared to the control group (RT) using MTT assay. As shown in Fig. 5C, compared to the RT group, the viability of 4T1 cells in the HPDAu + RT group reduced with increasing radiation dose, wherein the lowest cell viability rate was noticed with the 6 Gy dose (15.48 ± 1.33%). This treatment resulted in a more obvious decrease of cell viability as compared to the apoptosis assay (Fig. 5A and B), which might be explained by the longer incubation time in the MTT assay (48 h vs. 4 h), along with the higher X-ray radiation dose (6 Gy vs. 4 Gy). In accordance with previous reports, combinatorial radio-/chemotherapy achieved greatly ameliorated oncotherapeutic outcomes.20,21,49 Thence, these results provide conclusive proof of the improved antiproliferative activity of HPDAu + RT in vitro, which is likely due to DOX- and AuNP-induced radiosensitization along with the apoptotic effect of DOX.50
In order to assess potential toxicity of the developed nanoparticles towards normal cells, the cell viability of primary human hepatocytes (HepLL) and neonatal mouse cardiac fibroblasts (NMCF) was estimated after treatment with DOX and HPDAu using MTT assay (Fig. S8, ESI†). Interestingly, the as-prepared HPDAu showed negligible toxicities on both cell lines comparing to DOX treatment that exhibited a concentration-dependent cytotoxicity. These results indicate that HPDAu nanoparticles might minimize DOX-induced toxicity towards healthy cells and tissues.
In vivo deep tumor penetration of HPDAu nanoparticles
To further evaluate their in vivo penetrating ability, the intratumoral microdistribution and accumulation rate of HPDAu nanoparticles were evaluated at 24 h post-injection of 4T1 tumor-bearing mice, and compared to that of free DOX. Meanwhile, the tumor vasculature was labeled using FITC-labeled Lycopersicon esculentum lectin, and the corresponding microdistribution profile of DOX in tumor sections was plotted as a function of the distance from blood vessels. Obviously, stronger and more uniform red fluorescence signals (DOX) were noticed with the HPDAu nanoparticles, while free DOX resulted in barely detectable signals (Fig. 6A). Comparable results were obtained with the quantitative analysis of blue, green and red fluorescence intensities (Fig. 6B and C), wherein DOX signal in the HPDAu group remained detectable up to 200 μm from the blood vessel. In accordance with the spheroid experiment, these results provide clear evidence of the effective accumulation and deep penetrating ability of HPDAu nanoparticles into tumor interstitium.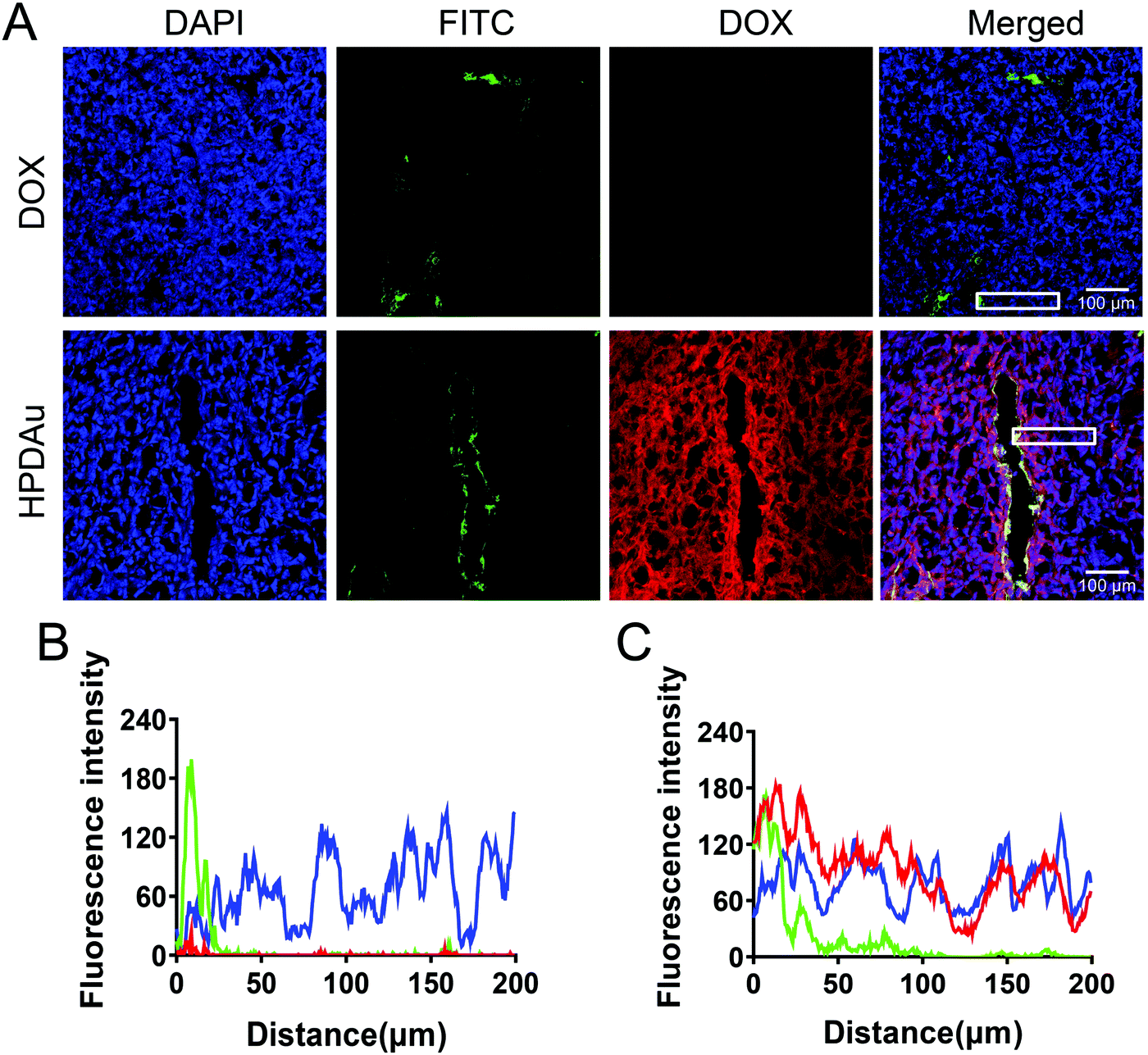 | ||
| Fig. 6 In vivo penetrating efficacy of HPDAu nanoparticles: (A) confocal images of 4T1 tumor frozen sections at 24 h post-injection of DOX and HPDAu nanoparticles. Intratumoral microdistribution profiles of blue, green and red fluorescence signals in representative zones (white rectangles in A) of tumor sections from DOX (B) and HPDAu groups (C) as a function of relative distance from blood vessels as analyzed using ImageJ. Tumor cell nuclei (4′,6-diamidino-2-phenylindole (DAPI), blue), blood vessels (fluorescein isothiocyanate (FITC), green), and nanoparticles (DOX, red). The scale bars indicate 100 μm in length. | ||
In vivo targetability evaluation of HPDAu nanoparticles
The targeting capability and biodistribution profile of free IR780 and IR780/HPDAu nanoparticles were estimated using a non-invasive optical imaging technique in the 4T1 murine breast cancer model. Quantitative evaluation of the tumor fluorescence signals from different groups as a function of post-injection time was acquired using ROI analysis of the corresponding tumor regions. As displayed in Fig. 7A and B, compared to IR-780, the IR780/HPDAu group showed significantly stronger fluorescence signals at the tumor site, which remained detectable over 48 h post-injection with the highest point at 24 h. The mice were sacrificed after 48 h; tumors and major organs were subsequently harvested and analyzed using the same imaging system. As expected, evidently increased signals were noticed in IR780/HPDAu tumor tissues as compared to major organs and the free IR780 group (Fig. 7C and D). The semi-quantitative analysis showed that tumor tissues from the IR780/HPDAu group displayed a 10-fold higher fluorescence signal as compared to the IR780 group, and a 1.9-fold and 5.9-fold increase compared to the liver and kidneys (IR780/HPDAu), respectively. These data further validate the superior tumor accumulation and targeting capability of HPDAu nanoparticles in vivo.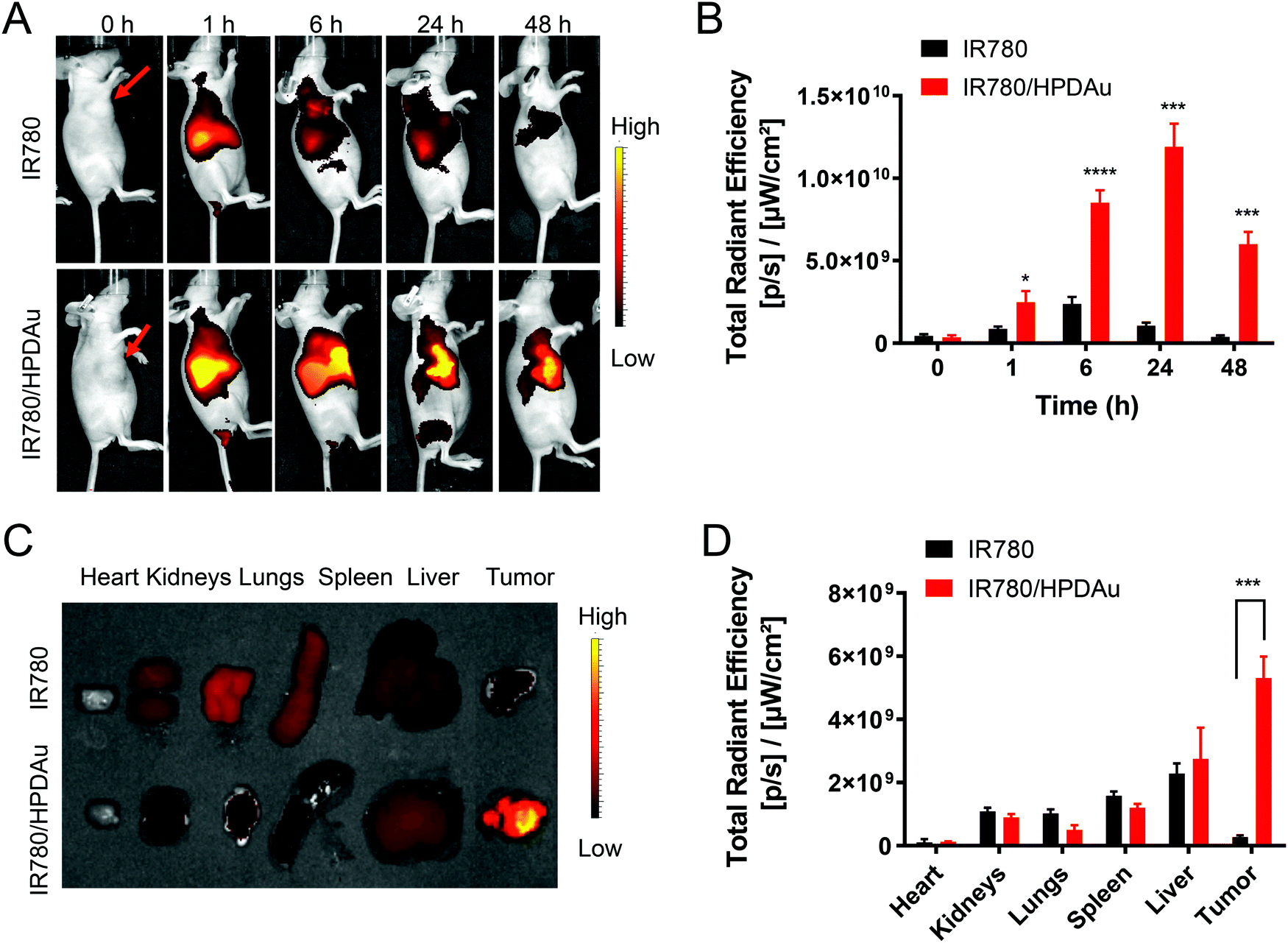 | ||
| Fig. 7 In vivo targeting evaluation: (A) near-infrared (NIR) fluorescence imaging of the 4T1 tumor-bearing mice at 0, 1, 6, 24 and 48 h post-injection of IR780 and IR780-labeled HPDAu (IR780/HPDAu). Red arrows indicate the tumor regions. (B) Quantitative estimation of fluorescence signals in tumor regions from different groups as a function of time post-injection. (C) Ex vivo fluorescence images of the dissected major organs and tumors at 48 h post-injection of IR780 and IR780/HPDAu. (D) Semi-quantitative evaluation of the fluorescence signals from the harvested tumors and major organs. Both in vivo and ex vivo data show that IR780/HPDAu achieved stronger fluorescence signals at the tumor sites as compared to IR780. All data are presented as mean ± SD (n = 4). P values were determined versus the IR780 group. *P < 0.05, ***P < 0.001, and ****P < 0.0001. | ||
In vivo antitumor efficacy
To further support their applicability, the radio-/chemotherapeutic performance of HPDAu nanoparticles (HPDAu + RT) was evaluated in vivo using the 4T1 orthotropic tumor model, and compared to those of free DOX, RT, DOX + RT and HPDAu. As shown in Fig. 8A, compared to the saline group, RT treatment slightly retarded the tumor growth, whereas a more obvious suppression was observed with DOX, DOX + RT, and HPDAu. Comparatively, HPDAu + RT significantly delayed the tumor growth (P < 0.0001 vs. saline group). After 18 days, tumor tissues from different groups were harvested, weighed and photographed (Fig. 8B–D). Likewise, comparable results were noticed, wherein HPDAu + RT achieved the highest inhibition ratio (95.60%) as compared to RT, DOX, DOX + RT and HPDAu (IR = 30.51%, 52.18%, 61.90% and 73.09%, respectively).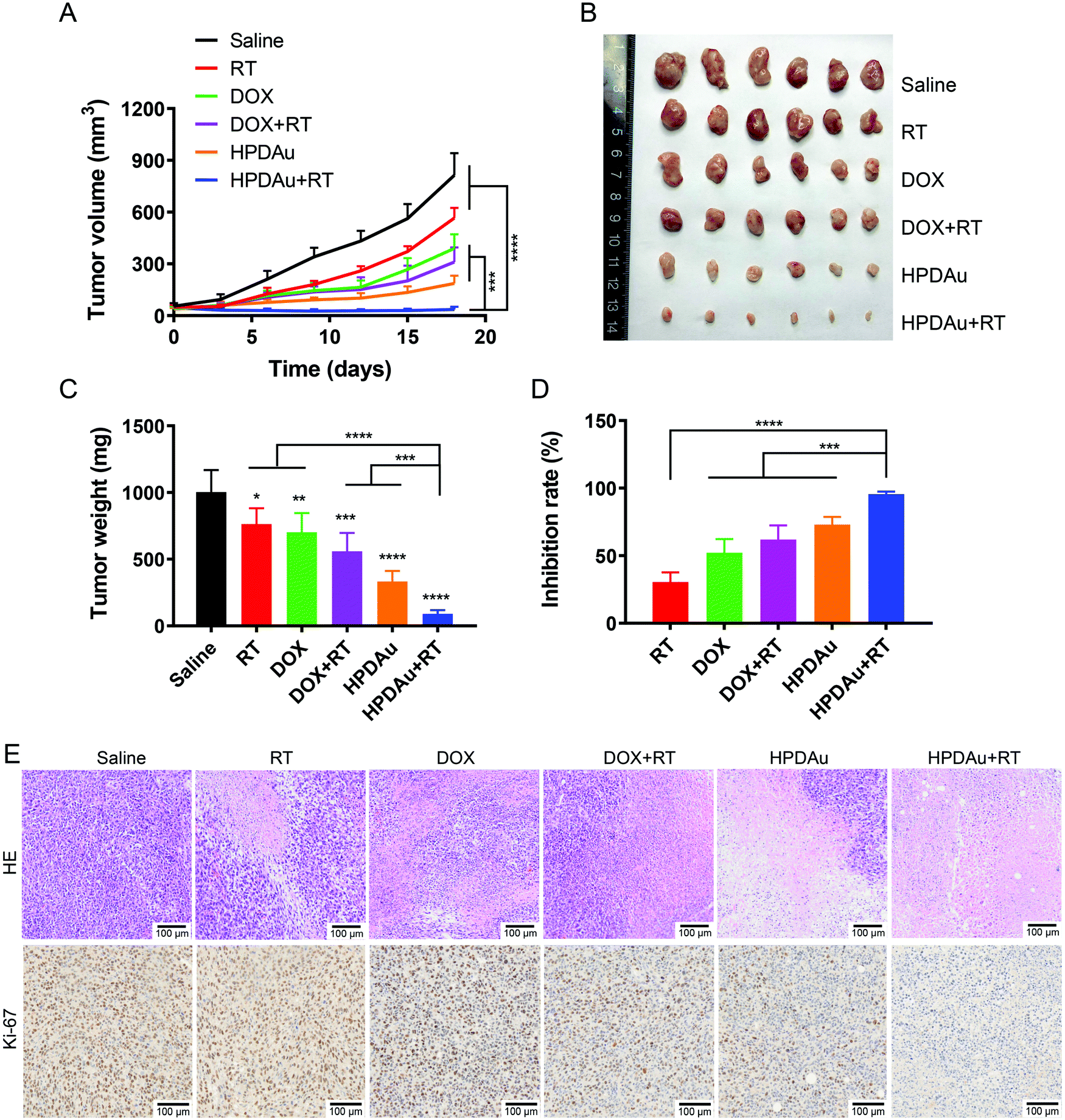 | ||
| Fig. 8 In vivo tumor inhibitory effect of HPDAu plus RT (HPDAu + RT) in the 4T1 xenograft model. (A) Tumor growth curves of saline (control), radiotherapy (RT), DOX, DOX plus RT (DOX + RT), HPDAu, and HPDAu + RT groups at a dose of 5 mg kg−1 DOX and 20 mg kg−1 Au (n = 6). (B) Representative images of dissected tumor tissues on day 14. (C) Corresponding tumor weights and (D) inhibition rates after various treatments (day 14). (E) Hematoxylin and eosin (HE) and Ki-67-stained tissue sections of harvested 4T1 tumors from different groups. The scale bars indicate 100 μm in length. All data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. | ||
The harvested tumor tissues were further analyzed using HE-staining. The corresponding histopathological images from different groups are shown in Fig. 8E. In comparison to the control group (saline), different extents of tumor cell remission, characterized by nucleus and cytosol degradation, were observed with the treated groups. Importantly, the most dramatic increase in necrotic areas was noticed in tumor sections from HPDAu + RT as compared to those of the other groups. Meanwhile, the altering effect of different treatments on tumor cell proliferation was further assessed using Ki-67 staining. As expected, an evidently reduced proliferating activity was noted with HPDAu + RT treatment.
In agreement with the above-stated results, these data further demonstrate the in vivo efficacy of HPDAu + RT, thereby providing strong evidence of the radio-/chemotherapeutic effect of HPDAu nanoparticles. Such superior efficacy could be explained by their higher tumor accumulation owing to the combination of passive and active targeting capabilities of HPDAu towards the tumor cells (via EPR effect and CD44-mediated internalization, respectively). Furthermore, due to their size-shrinking ability and surface charge reversal upon reaching the tumor microenvironment, the released deep-penetrating ultrasmall nanoparticles (PD and AuNPs) could directly bind to plasma membranes via electrostatic interactions, thereby further promoting drug internalization efficiency.42 Thence, the association of chemotherapeutic effect (DOX) and AuNP/DOX-induced radiosensitization would result in prominent antitumor efficacy.18–21
Preliminary toxicity analysis
The preliminary evaluation of drug systemic toxicity was performed through observing the body weight change, mice behavior and histopathological examination of major organs (heart, liver, kidney, lung, and spleen). Additionally, the nanoparticle toxicity was further evaluated by the determination of serum concentrations of the liver (AST and ALT), kidney (BUN and Cr), and heart (CK) enzymatic markers in different groups. As displayed in Fig. 9A, a slight weight loss with abnormal activity was noticed with DOX and DOX + RT treated mice as compared to the other groups, which is likely due to the free drug's systemic toxicity.51–53 As expected, these treatments resulted in increased serum levels of the detected enzymatic markers (CK, AST, ALT, and BUN), whereas no obvious impact was noticed after HPDAu and HPDAu + RT treatments compared to that of the control group. Besides, histopathological examination revealed some dissolved and broken cardiac muscle fibers (Fig. 9B–F),54,55 liver inflammatory injuries (moderate hepatocyte necrosis and inflammatory cell infiltration) and renal tubular epithelial cell necrosis with the DOX and DOX + RT groups (Fig. 10),56 while imperceptible effects were noted in lungs and spleen tissues (Fig. S9, ESI†). This might be attributed to the poor tumor accumulation, lack of targeting capability and non-specific distribution of the free drug.51 In comparison, no noticeable outcome was observed with the nanoparticle groups. Taken together, these results indicate that HPDAu markedly reduced DOX-induced cardiomyopathy, and hepato- and nephrotoxicity, indicating their improved safety profile, which would further support their applicability for targeted tumor therapy.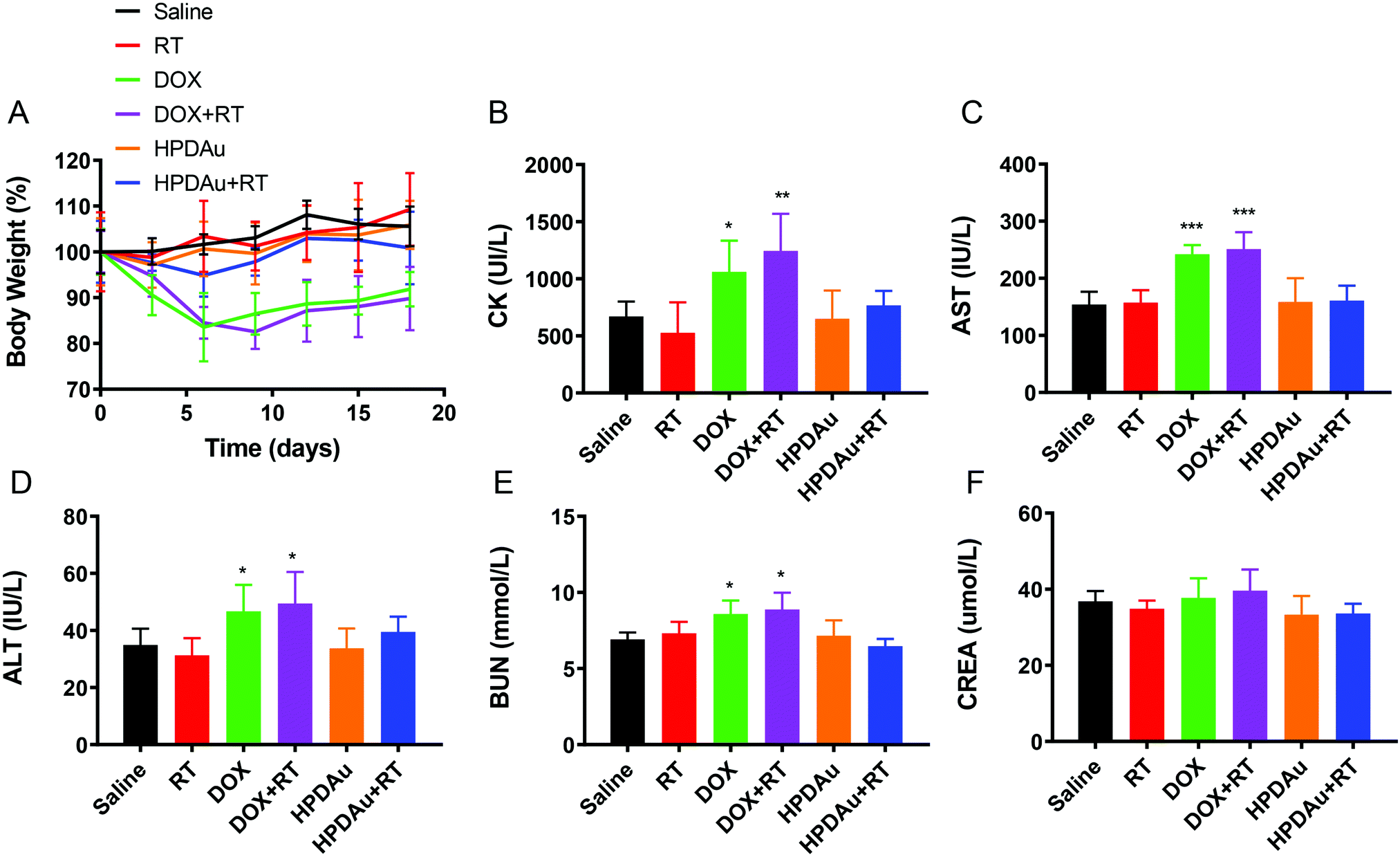 | ||
| Fig. 9 Preliminary toxicity evaluation: (A) change in body weight percentage after treatment with different formulations as a function of time. (B–F) Serum concentrations of creatine kinase (CK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine (CREA) in treated mice, respectively (day 14). P values were determined versus saline (control) group. All data are presented as mean ± SD (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
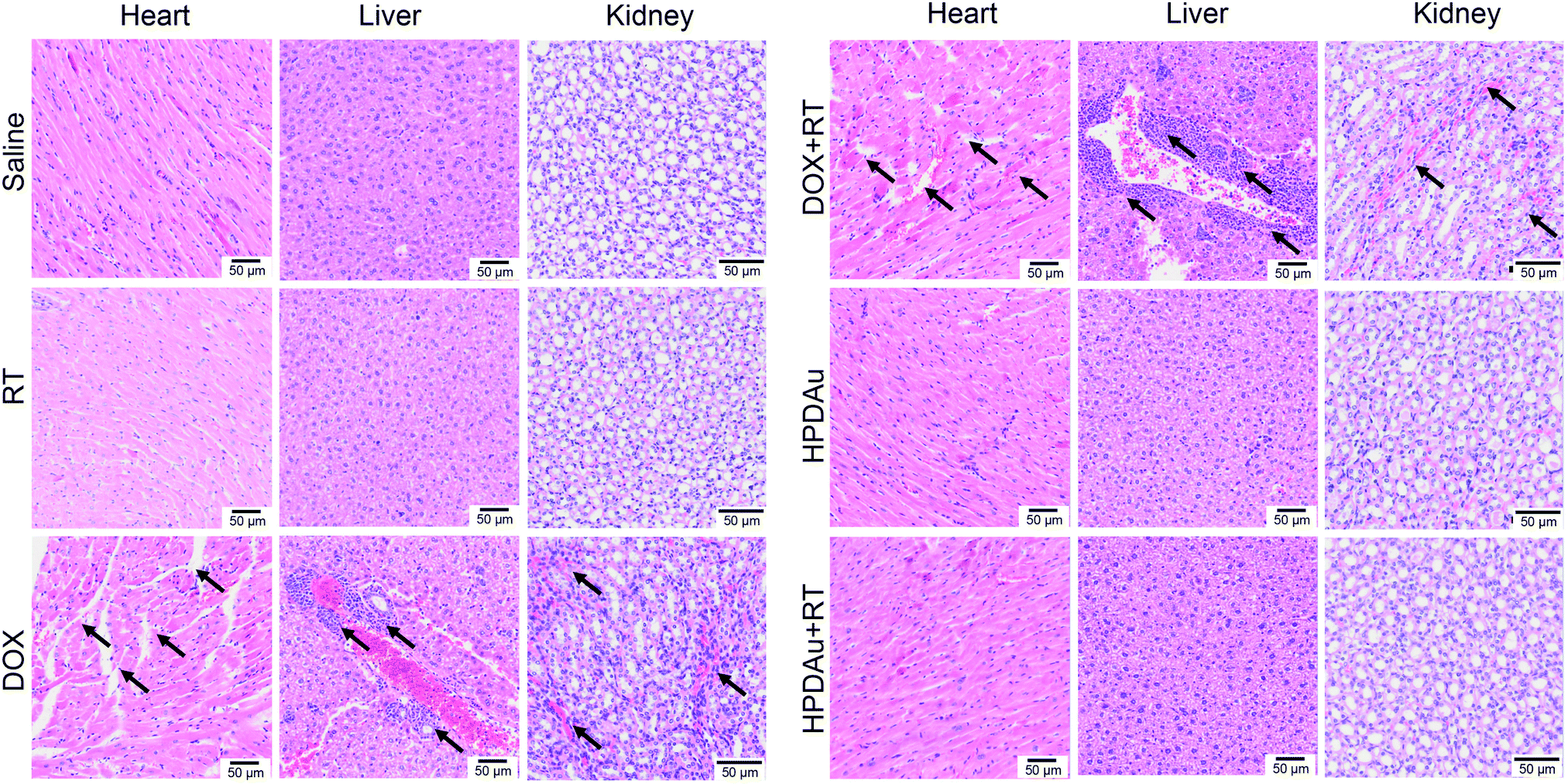 | ||
| Fig. 10 Histopathological examination of excised organs from different groups: saline (control), radiotherapy (RT), DOX, DOX plus RT (DOX + RT), HPDAu, and HPDAu plus RT (HPDAu + RT). The scale bars indicate 50 μm in length. The black arrows indicate the sites of injury. | ||
Conclusions
In summary, we successfully prepared a multistimuli-responsive and deep penetrating nanosystem co-delivering PAMAM-stabilized gold nanoparticles (AuNPs) and pH-responsive DOX prodrugs (PD conjugate) that were incorporated into a HA-based nanoshell. The developed HPDAu nanoparticles could sequentially respond to the endogenous features of tumor tissue (HAase) to form ultrasmall cationic nanostructures, and showed a burst drug-release upon incubation under tumor microenvironment-mimicking conditions (HAase degradation and acidic conditions). Taking advantage of the different features of the PD, AuNPs and HA nanoshell, the feasibility of HPDAu nanoparticles was evidenced both in vitro and in vivo by achieving a remarkable intratumoral penetration and synergistic radio–chemotherapeutic effect. This strategy will pave the way for the investigation of more refined nanomedicines by exploiting both extracellular and intracellular signals and synergistically promoting a targeted combinative anticancer therapy.Abbreviations
| HA | Hyaluronic acid |
| DOX | Doxorubicin |
| PAMAM | Poly(amidoamine) |
| AuNPs | PAMAM-stabilized gold nanoparticles |
| PD | DOX–PAMAM conjugate |
| CAD | cis-Aconityl-doxorubicin |
| HPDAu | HA-coated PD/AuNPs |
| ECM | Extracellular matrix |
| HAase | Hyaluronidase |
| HPLC | High performance liquid chromatography |
| 1H NMR | Proton nuclear magnetic resonance |
| DLS | Dynamic light scattering |
| PDI | Polydispersity index |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| EDS | Energy-dispersive X-ray spectroscopy |
| CLSM | Confocal laser scanning microscope |
| HE | Hematoxylin and eosin staining |
| RT | Radiotherapy |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFC0115701) and the National Natural Science Foundation of China (No. 81671748).References
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef.
- J. Chen, J. Ding, Y. Wang, J. Cheng, S. Ji, X. Zhuang and X. Chen, Adv. Mater., 2017, 29, 1701170 CrossRef PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- J. Ji, F. Ma, H. Zhang, F. Liu, J. He, W. Li, T. Xie, D. Zhong, T. Zhang, M. Tian, H. Zhang, H. A. Santos and M. Zhou, Adv. Funct. Mater., 2018, 28, 1801738 CrossRef.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS.
- H. J. Li, J. Z. Du, X. J. Du, C. F. Xu, C. Y. Sun, H. X. Wang, Z. T. Cao, X. Z. Yang, Y. H. Zhu, S. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS.
- Z. Zhang, H. Wang, T. Tan, J. Li, Z. Wang and Y. Li, Adv. Funct. Mater., 2018, 28, 1801840 CrossRef.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed.
- R. Tong, H. H. Chiang and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19048–19053 CrossRef CAS.
- C. Hu, X. Cun, S. Ruan, R. Liu, W. Xiao, X. Yang, Y. Yang, C. Yang and H. Gao, Biomaterials, 2018, 168, 64–75 CrossRef CAS.
- S. Ruan, X. Cao, X. Cun, G. Hu, Y. Zhou, Y. Zhang, L. Lu, Q. He and H. Gao, Biomaterials, 2015, 60, 100–110 CrossRef CAS.
- H. Yim, S. J. Park, Y. H. Bae and K. Na, Biomaterials, 2013, 34, 7674–7682 CrossRef CAS.
- H.-X. Wang, Z.-Q. Zuo, J.-Z. Du, Y.-C. Wang, R. Sun, Z.-T. Cao, X.-D. Ye, J.-L. Wang, K. W. Leong and J. Wang, Nano Today, 2016, 11, 133–144 CrossRef CAS.
- Y. R. Lawrence, B. Vikram, J. J. Dignam, A. Chakravarti, M. Machtay, B. Freidlin, N. Takebe, W. J. Curran, Jr., S. M. Bentzen, P. Okunieff, C. N. Coleman and A. P. Dicker, J. Natl. Cancer Inst., 2013, 105, 11–24 CrossRef CAS PubMed.
- R. A. Sharma, R. Plummer, J. K. Stock, T. A. Greenhalgh, O. Ataman, S. Kelly and R. Clay, Nat. Rev. Clin. Oncol., 2016, 13, 627–642 CrossRef CAS.
- G. S. Higgins, S. M. O'Cathail, R. J. Muschel and W. G. McKenna, Cancer Treat. Rev., 2015, 41, 105–113 CrossRef PubMed.
- L. Tian, Q. Chen, X. Yi, G. Wang, J. Chen, P. Ning, K. Yang and Z. Liu, Theranostics, 2017, 7, 614–623 CrossRef CAS.
- T. S. Lawrence, B. G. Haffty and J. R. Harris, J. Clin. Oncol., 2014, 32, 1173–1179 CrossRef CAS PubMed.
- H. J. Mamon and J. E. Tepper, J. Clin. Oncol., 2014, 32, 367–369 CrossRef PubMed.
- S. Her, D. A. Jaffray and C. Allen, Adv. Drug Delivery Rev., 2017, 109, 84–101 CrossRef CAS.
- Y. Zhang, F. Huang, C. Ren, J. Liu, L. Yang, S. Chen, J. Chang, C. Yang, W. Wang, C. Zhang, Q. Liu, X. J. Liang and J. Liu, Adv. Sci., 2019, 6, 1801806 CrossRef PubMed.
- G. Liang, X. Jin, S. Zhang and D. Xing, Biomaterials, 2017, 144, 95–104 CrossRef CAS PubMed.
- A. R. Fernandes, J. Jesus, P. Martins, S. Figueiredo, D. Rosa, L. M. Martins, M. L. Corvo, M. C. Carvalheiro, P. M. Costa and P. V. Baptista, J. Controlled Release, 2017, 245, 52–61 CrossRef CAS.
- K. E. Ibrahim, M. G. Al-Mutary, A. O. Bakhiet and H. A. Khan, Molecules, 2018, 23, 1848 CrossRef.
- Z. Yu, M. Wang, W. Pan, H. Wang, N. Li and B. Tang, Chem. Sci., 2017, 8, 4896–4903 RSC.
- M. Hossain and M. Su, J. Phys. Chem. C, 2012, 116, 23047–23052 CrossRef CAS PubMed.
- S. J. McMahon, W. B. Hyland, M. F. Muir, J. A. Coulter, S. Jain, K. T. Butterworth, G. Schettino, G. R. Dickson, A. R. Hounsell, J. M. O'Sullivan, K. M. Prise, D. G. Hirst and F. J. Currell, Sci. Rep., 2011, 1, 18 CrossRef PubMed.
- N. Ma, F. G. Wu, X. Zhang, Y. W. Jiang, H. R. Jia, H. Y. Wang, Y. H. Li, P. Liu, N. Gu and Z. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 13037–13048 CrossRef CAS.
- J. Li, H. Liang, J. Liu and Z. Wang, Int. J. Pharm., 2018, 546, 215–225 CrossRef CAS.
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274–5300 CrossRef CAS.
- N. J. Boylan, A. J. Kim, J. S. Suk, P. Adstamongkonkul, B. W. Simons, S. K. Lai, M. J. Cooper and J. Hanes, Biomaterials, 2012, 33, 2361–2371 CrossRef CAS PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- M. Swierczewska, H. S. Han, K. Kim, J. H. Park and S. Lee, Adv. Drug Delivery Rev., 2016, 99, 70–84 CrossRef CAS.
- S. Mizrahy and D. Peer, Chem. Soc. Rev., 2012, 41, 2623–2640 RSC.
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS.
- E. Kim, J. Yang, J. Park, S. Kim, N. H. Kim, J. I. Yook, J. S. Suh, S. Haam and Y. M. Huh, ACS Nano, 2012, 6, 8525–8535 CrossRef CAS.
- T. Jiang, R. Mo, A. Bellotti, J. Zhou and Z. Gu, Adv. Funct. Mater., 2014, 24, 2295–2304 CrossRef CAS.
- P. S. Lai, P. J. Lou, C. L. Peng, C. L. Pai, W. N. Yen, M. Y. Huang, T. H. Young and M. J. Shieh, J. Controlled Release, 2007, 122, 39–46 CrossRef CAS PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS.
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS.
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Adv. Drug Delivery Rev., 2016, 97, 204–236 CrossRef CAS PubMed.
- M. Gotte and G. W. Yip, Cancer Res., 2006, 66, 10233–10237 CrossRef PubMed.
- W.-Y. Huang, J.-N. Lin, J.-T. Hsieh, S.-C. Chou, C.-H. Lai, E.-J. Yun, U. G. Lo, R.-C. Pong, J.-H. Lin and Y.-H. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 30722–30734 CrossRef CAS PubMed.
- Y. Liu, M. K. Shipton, J. Ryan, E. D. Kaufman, S. Franzen and D. L. Feldheim, Anal. Chem., 2007, 79, 2221–2229 CrossRef CAS.
- X. Yi, L. Chen, J. Chen, D. Maiti, Z. Chai, Z. Liu and K. Yang, Adv. Funct. Mater., 2018, 28, 1705161 CrossRef.
- T. Li, M. Zhang, J. Wang, T. Wang, Y. Yao, X. Zhang, C. Zhang and N. Zhang, AAPS J., 2016, 18, 146–155 CrossRef CAS.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef CAS.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- C. Carvalho, R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira, Curr. Med. Chem., 2009, 16, 3267–3285 CrossRef CAS.
- S. Zhang, X. Liu, T. Bawa-Khalfe, L. S. Lu, Y. L. Lyu, L. F. Liu and E. T. Yeh, Nat. Med., 2012, 18, 1639–1642 CrossRef CAS PubMed.
- G. Curigliano, D. Cardinale, S. Dent, C. Criscitiello, O. Aseyev, D. Lenihan and C. M. Cipolla, Ca-Cancer J. Clin., 2016, 66, 309–325 CrossRef PubMed.
- R. Injac, M. Perse, M. Cerne, N. Potocnik, N. Radic, B. Govedarica, A. Djordjevic, A. Cerar and B. Strukelj, Biomaterials, 2009, 30, 1184–1196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including in vitro release and responsiveness of HPDAu nanoparticles, cellular uptake assessment, in vitro apoptosis detection and MTT assay. Synthesis routes of CAD and PD, physicochemical characterization, HPLC chromatograms, NMR spectra, EDS spectrum, changes in particle size and zeta potential under different conditions, in vitro cytotoxic effect towards normal cells, and histopathological evaluation. See DOI: 10.1039/c9tb00716d |
| This journal is © The Royal Society of Chemistry 2019 |