
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroparticle self-assembly induced by travelling surface acoustic waves†
Ghulam Destgeer a,
Ali Hashmib,
Jinsoo Parka,
Husnain Ahmeda,
Muhammad Afzala and
Hyung Jin Sung*a
a,
Ali Hashmib,
Jinsoo Parka,
Husnain Ahmeda,
Muhammad Afzala and
Hyung Jin Sung*a
aDepartment of Mechanical Engineering, KAIST, Daejeon 34141, Korea. E-mail: hjsung@kaist.ac.kr
bInstitut de Biologie du Développement de Marseille (IBDM), France
First published on 11th March 2019
Abstract
We present an acoustofluidic method based on travelling surface acoustic waves (TSAWs) to induce self-assembly of microparticles inside a microfluidic channel. The particles are trapped above an interdigitated transducer, placed directly beneath the microchannel, by the TSAW-based direct acoustic radiation force (ARF). This approach was applied to trap 10 μm polystyrene particles, which were pushed towards the ceiling of the microchannel by 72 MHz TSAWs to form single- and multiple-layer colloidal structures. The repair of cracks and defects within the crystal lattice occurs as part of the self-assembly process. The sample flow through the first inlet can be switched with a buffer flow through the second inlet to control the number of particles assembled in the crystalline structure. The constant flow-induced Stokes drag force on the particles is balanced by the opposing TSAW-based ARF. This force balance is essential for the acoustics-based self-assembly of microparticles inside the microchannel. Moreover, we studied the effects of varying input voltage and fluid flow rate on the position and shape of the colloidal structure. The active self-assembly of microparticles into crystals with multiple layers can be used in the bottom-up fabrication of colloidal structures with dimensions greater than 500 μm × 500 μm, which is expected to have important applications in various fields.
Introduction
The self-assembly of individual microparticles into ordered crystals1 is important for the bottom-up fabrication of supra-colloidal microstructures,2 which are essential to the fields of photonics,3 sensors,4 robotics,5 and microfluidics,6 among others.7 External magnetic8,9 and electrodynamic10,11 force fields have been used to facilitate the self-assembly process and found to reduce the timescale of self-assembly and to provide better control over particle motion. However, these techniques are dependent upon the electrical and magnetic properties of the particles and the suspension media, which limits their applicability.12–14 Acoustic forces have been used to manipulate (align,15–17 pattern,18–20 separate,21–23 and concentrate24–26) microparticles based on their size differences and mechanical properties. Acoustic waves are suitable for the handling of virtually any micro-object and are not dependent on the electro-magnetic properties of the material and the suspension media. Acoustic waves have recently been used in the manipulation of colloidal particles and their self-assembly.27–30Owens et al.28 have demonstrated the assembly of particles into ordered colloidal crystallites by using standing bulk acoustic waves in a batch process. They employed two lead zirconate titanate (PZT) transducers to generate standing waves within a reflective microchamber and thus to concentrate particles at the acoustic pressure nodes in the two-dimensional plane. Akella and Juárez30 employed a continuous flow acoustofluidic device with a single PZT element to generate a region of low acoustic pressure within a microfluidic channel in order to concentrate and assemble particles flowing through the acoustic field region. Yang et al.31 used acoustic and magnetic fields in combination to control the particle concentration and inter-particle interactions respectively and thus to fabricate a range of colloidal structures. Most research to date on microparticle self-assembly with bulk acoustic waves has been based on the generation of acoustic pressure nodes in a standing wave. The size of the colloidal crystals formed with this approach is restricted by the saturation limit of the pressure nodes. Wollmann et al.32 utilized travelling surface acoustic waves (TSAWs) with frequencies two orders of magnitude higher than those of techniques using bulk acoustic waves to direct the self-assembly of colloidal crystals in an evaporating sessile droplet. Similarly, in a recent study, the separation of microparticles inside an evaporating sessile droplet has been demonstrated, although colloidal crystal formation was not explicitly studied.21 The separation of particles inside sessile droplets was achieved by making use of the difference between the acoustic radiation forces (ARFs) acting on particles of different sizes,21 in contrast to the induction of crystal growth within a sessile droplet dominated by an acoustic streaming flow (ASF) and standing waves, formed due to the internal reflection.25,32
In this article, we demonstrate a TSAW-based technique for the self-assembly of microparticles flowing inside a microfluidic channel, in which the particles are trapped above an interdigitated transducer (IDT) by a direct ARF (see Fig. 1). A polydimethylsiloxane (PDMS) microfluidic channel is placed on top of an IDT in order to couple acoustic waves directly into the fluid. Single or multiple layers of colloidal crystals self-assemble because the ARF on the particles is balanced by the flow-induced drag force. The size and number of layers of the crystalline structure can be controlled by mediating the flow of particles into the trapping zone, which is achieved by switching the sample flow through the first inlet of the microchannel with a buffer flow through the second inlet. The crystalline structure was studied for periods of several minutes in order to track crack propagation and self-repair in the colloidal crystal. The effects of varying the input voltage and the flow rate on the development of the crystalline structure within the microchannel were also studied. The proposed TSAW-based technique circumvents the saturation limits of standing-wave pressure nodes28,30 as well as the drawbacks of sample recovery in sessile-droplet-based systems.21,32 The crystalline structures formed out of the self-assembly of microparticles could be useful for creating three dimensional porous scaffolds for cell cultures and tumor models.33,34
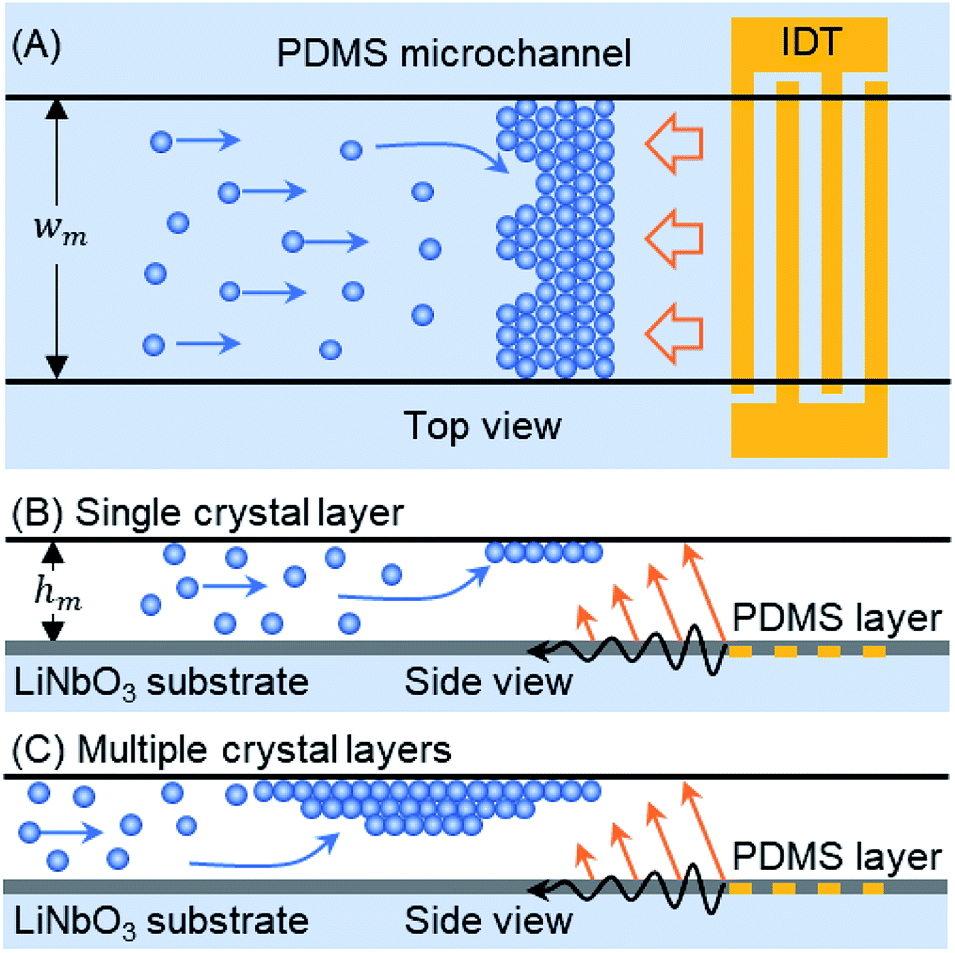 | ||
| Fig. 1 Schematic diagram of a TSAW-based trapping platform for microparticle self-assembly. (A) Top view: the TSAWs originate from an interdigitated transducer (IDT) and stop microparticles flowing inside a polydimethylsiloxane (PDMS) microchannel (width, wm). (B) Side view: the acoustic waves push the particles up towards the ceiling of the microchannel (height, hm). A single-layer crystal is formed through particle self-assembly. A thin PDMS sealing layer separates the IDT from the fluid inside the microchannel. (C) The continuous flow of particles to the trapping zone leads to the formation of a multiple-layer crystal. | ||
Methods
Device fabrication
A single-layered PDMS microchannel (wm × hm: 500 μm × 110 μm) was fabricated by using the commonly used mold replica soft lithography process.35–37 After cutting the desired PDMS microchannel, two inlet ports and one outlet port were punched through (Harris Uni-Core, I.D. 1 mm) the PDMS. A thin layer of PDMS solution (thickness, t = 38 μm) was spin-coated onto a salinized Si wafer to fabricate a sealing membrane. After curing at 65 °C for more than an hour, the PDMS microchannel was attached to the thin membrane by performing oxygen plasma bonding (Covance, Femto Science, Korea). A pair of uniformly spaced electrodes (IDT) was deposited parallel to the principal axis of a piezoelectric wafer (lithium niobate, LiNbO3, 128 Y-X cut, MTI Korea) by using E-beam evaporation and lift-off processes (IAMD, SNU, Korea). The IDT consists of 20 electrode finger pairs with a 1 mm aperture, has an actuation frequency of 72 MHz and an output with a wavelength (λTSAW) of 50 μm, and is covered with a thin layer of SiO2 (2000 Å) to protect it from mechanical damage. The thickness (t) of the sealing PDMS membrane was chosen so that t < 2λTSAW, which ensures that the ARF effect dominates the acousto-thermal effect inside the microchannel.36,38 The microchannel was placed directly on top of the IDT to reversibly bind it to the piezoelectric substrate. Therefore, a single IDT can be used with various disposable PDMS microchannels.Experimental set-up
A buffer solution was prepared by mixing deionized (DI) water and D2O (Sigma Aldrich) in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to obtain a solution density of 1.05 g cm−3, and 1% surfactant (unless otherwise mentioned) was also added. Polystyrene particles (10 μm, NIST standard, density 1.05 g cm−3, Thermo Scientific) were suspended in a density-matched buffer solution to prepare the sample (∼5 × 105 particles per ml). Two syringe pump units (neMESYS, Cetoni GmbH) were used to inject the buffer and sample solutions through the separate inlet ports. The IDT was actuated with an amplified (LZY-22+, Mini-Circuits) high frequency AC signal, produced by an RF signal generator (N5181A, Agilent Technologies). The device was mounted on a microscope (BX53, Olympus), while images were captured by using a CCD camera (DP72, Olympus) and analyzed with ImageJ software (http://imagej.nih.gov/ij/). For better visibility of the microparticles, contrast was increased with removal of background noise in the experimental images by using ImageJ (see Fig. 2, 3(A), 4(A) and (B)). In the experimental images, a single particle appears as a black disc with a bright spot at the center (see Fig. 3(B)); however, when the background noise is subtracted from the images, the particle appears as a white disc with a black spot at the center (see Fig. 3(A)).
:1 to obtain a solution density of 1.05 g cm−3, and 1% surfactant (unless otherwise mentioned) was also added. Polystyrene particles (10 μm, NIST standard, density 1.05 g cm−3, Thermo Scientific) were suspended in a density-matched buffer solution to prepare the sample (∼5 × 105 particles per ml). Two syringe pump units (neMESYS, Cetoni GmbH) were used to inject the buffer and sample solutions through the separate inlet ports. The IDT was actuated with an amplified (LZY-22+, Mini-Circuits) high frequency AC signal, produced by an RF signal generator (N5181A, Agilent Technologies). The device was mounted on a microscope (BX53, Olympus), while images were captured by using a CCD camera (DP72, Olympus) and analyzed with ImageJ software (http://imagej.nih.gov/ij/). For better visibility of the microparticles, contrast was increased with removal of background noise in the experimental images by using ImageJ (see Fig. 2, 3(A), 4(A) and (B)). In the experimental images, a single particle appears as a black disc with a bright spot at the center (see Fig. 3(B)); however, when the background noise is subtracted from the images, the particle appears as a white disc with a black spot at the center (see Fig. 3(A)).
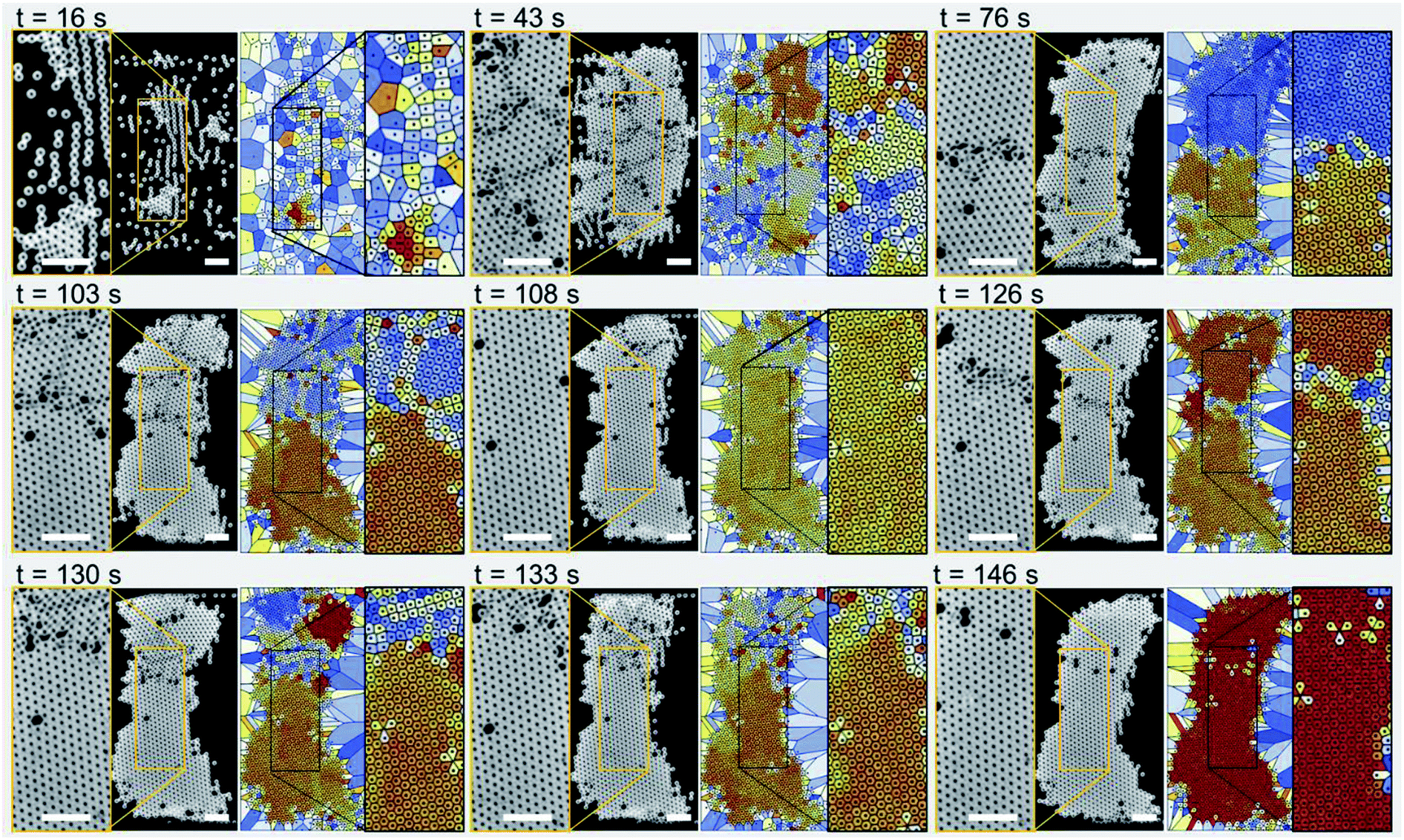 | ||
| Fig. 2 Experimental images (left panels) and Voronoi plots (right panels; the colors indicate the bond orientation angles; color homogeneity arises only for a single-crystal structure) are shown for a sequence of time steps with respect to the device actuation time. Initially, the self-assembly of the microparticles leads to the formation of small crystalline structures separated by cracks. These crystalline structures then join together as the cracks are removed, giving rise to a single-crystalline structure with a uniform crystal orientation. The sample flow rate was 250 μL h−1. The device actuation frequency and input voltage were 72 MHz and 5.1 Vpp, respectively. The scale bars are 50 μm in length. | ||
 | ||
| Fig. 3 Experimental images, with background noise removed in (A) and without background noise removal in (B), of the self-assembly of multiple-layer crystalline structures. (A) The single-layer crystal at 1.2 min has become a multiple-layer crystal at 2.7 min. The sample flow rate was 250 μL h−1 and the input voltage was 5.1 Vpp for a signal with a frequency of 72 MHz. Scale bars are 50 μm in length. (B) The predominantly bi-layer structure at 4 min has grown into a structure with more than two layers at 8 min, resulting in an overall crystal length of ~750 μm. The sample flow rate was 500 μL h−1, and the input voltage was gradually increased from 8.6 Vpp to 13.8 Vpp. Scale bars are 100 μm in length. | ||
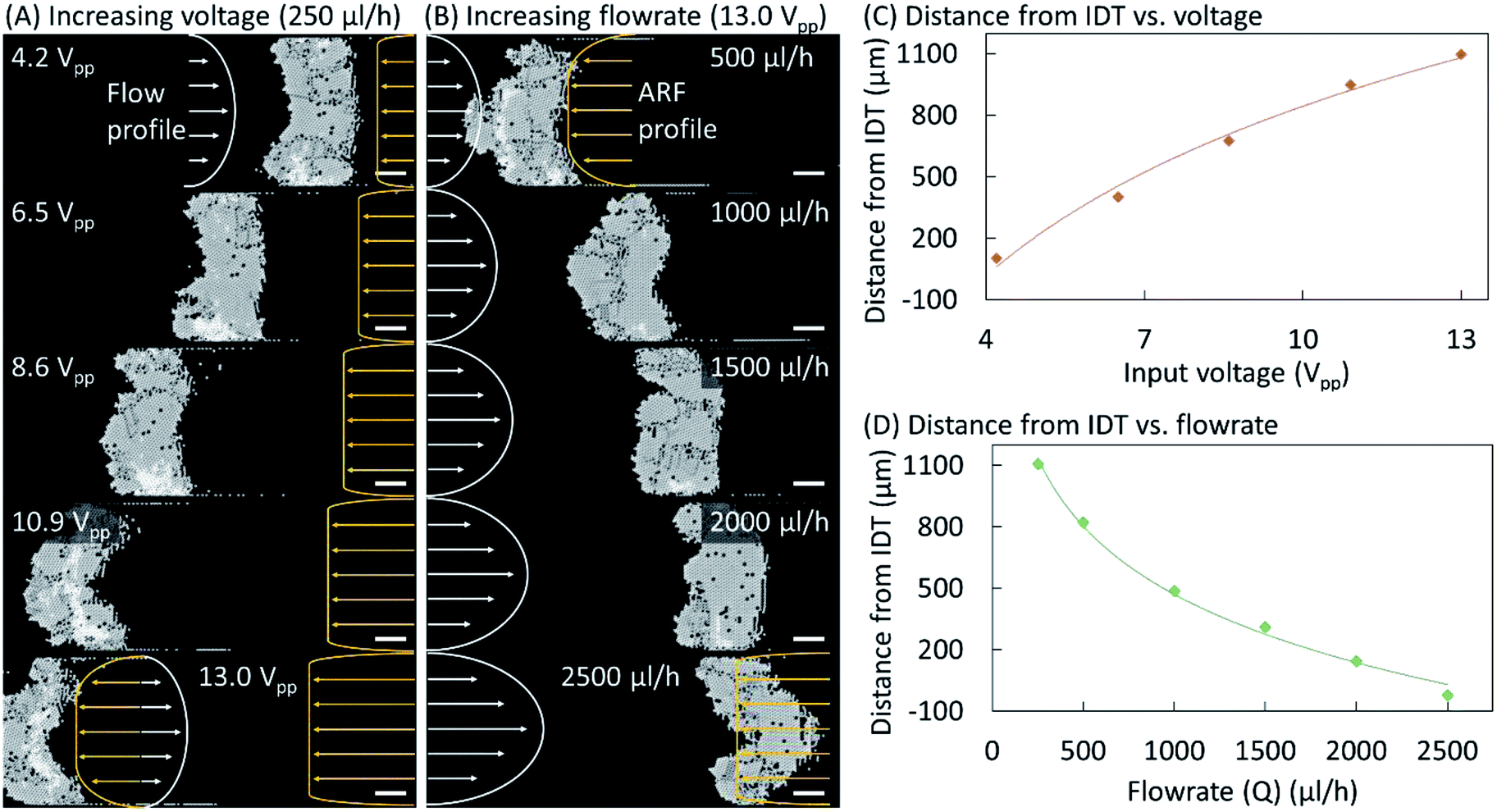 | ||
| Fig. 4 (A) For a constant sample flow rate of 250 μL h−1 the input voltage was increased from 4.2 Vpp to 13.0 Vpp to produce a signal with a frequency of 72 MHz. The crystal structure moves away from the IDT with increases in the input voltage. (B) The sample flow rate was increased from 500 μL h−1 to 2500 μL h−1 while the input voltage was kept constant at 13.0 Vpp. The crystal structure moves to the right as the sample flow rate increases. Scale bars are 100 μm in length. The distance of the crystal structure from the IDT was plotted against the variable input voltage (C) and the flow rate (D). | ||
Working mechanism
The microchannel is placed above the TSAW device so that the IDT covers the full width of the microchannel (see Fig. 1(A)). The particles pumped through the microchannel are stopped by the TSAW when the Stokes drag force (Fd) acting on the particles is balanced by the ARF (FARF).39 The two forces, FARF and Fd, determine the final position of the individual particles and of the particle clusters inside the microchannel as the crystal grows with the addition of incoming particles. The polystyrene particle diameter (dp) and TSAW frequency (f) must be chosen to ensure that the Helmholtz number (κ = πfdp/cf) is greater than one, where cf is the speed of sound in the fluid, which means that the particles will experience a strong ARF.40,41 As the TSAW attenuates away from the IDT and couples with the fluid inside the microchannel, the compressional TSAW is radiated at an angle with a strong vertical ARF component.17,24,35 Therefore, the particles are pushed up against the ceiling of the microchannel (see Fig. 1(B)). The acoustic waves attenuation lengths on the fluid–substrate interface (12.8 λTSAW) and within the fluid (3 × 106 λTSAW2) are calculated as 640 μm and 7500 μm, respectively.42,43 The microchannel height (hm) is much larger than the particle diameter (dp), so the continuous inflow of particles leads to the growth of a primary crystal layer followed by the formation of additional crystal layers underneath the primary layer (see Fig. 1(C)). Moreover, as the attenuation length within the fluid is much larger than the height of the microchannel, the effect of acoustic streaming flow was minimal. The height of the microchannel was not sufficient for the waves to be fully attenuated in the fluid, so the formation of the acoustic streaming flow was suppressed. Therefore, the microparticles were not dragged by the acoustic streaming vortices; instead, they were affected by two counter-balancing forces: drag force and acoustic radiation force.Crystalline order
To probe the arrangements of elements and defects in the crystalline lattice in a spatiotemporal manner, we employ bond-orientation angles (BOA). BOA can be determined for the individual particles by considering their adjoining neighbours using a Delaunay Mesh.44–46 For instance, the BOA for an element i to its n nearest neighbours, θi can be written as:where θi,k is the angle subtended from the line joining the centroids of the ith and the kth elements to a fixed/reference axis.
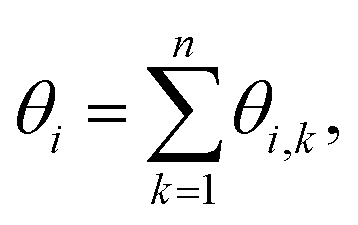
For an intuitive representation, the BOA can be delineated as a Voronoi plot (as in Fig. 2) with the Voronoi cells coloured according to the bond orientational angles. Different structures and orders within the lattice can then become apparent.
Results and discussion
Single-layer crystalline structure
Fig. 2 shows experimental images (left panels) and Voronoi plots (right panels) of a single-layer colloidal crystal (see Movie S1 in the ESI†). The particle sample solution was pumped through the microchannel at a flow rate of 250 μL h−1 while the IDT was actuated at a frequency of 72 MHz with an input voltage of 5.1 Vpp. The microparticles have a diameter of 10 μm and self-assemble mainly due to the influence of the TSAW-based direct ARF. However, note that after 16 s of device actuation, the particles started to concentrate in lines roughly parallel to the IDT because standing-wave-based pressure nodes have formed due to TSAW reflection within the microchannel.20,47 These particle lines join together to form small particle clusters that grow into larger grains as more particles flow into the particle trapping zone (at 43 s). The disordered colloidal particles (at 16 s) become relatively ordered crystalline structures (at 43 s) consisting of several segmented regions with various crystalline orientations and grain boundaries. The particle flow was stopped at this stage and a buffer solution was introduced with a similar flow rate through the second inlet into the microchannel, which ensured that the balance between the Stokes drag force and ARF was maintained so that the re-orientation and growth of the crystals into a larger single crystal could be studied. At 76 s, most of the cracks between the small crystal structures have been repaired: there are only two larger crystals present with a slightly different orientation and a dividing line in between them. The crack at the center (at 76 s) then moves upward giving rise to additional smaller cracks in the upper segment of the crystal as the crystalline structures reorient themselves (at 103 s). The self-repair of the cracks leads to a predominantly single-crystal structure with a few defects (particles missing from the crystal lattice), as confirmed by the uniform color of the Voronoi plots, which indicates the presence of a single crystal orientation (at 108 s). A new crack is then initiated just above the centerline of the microchannel, which again divides the main crystal into two parts (at 126 s). The self-repair of the crystal is initiated as the crystal structures re-orient themselves (at 130 s) and a crack moves outward from the larger crystal structure at the bottom (at 133 s). A single unified crystal with few defects and no cracks is again evident at 146 s. For additional results for the formation of single-layer crystals at different surfactant concentrations, see Fig. S1 and Movie S2 in the ESI.†Multiple-layer crystalline structure
Fig. 3 shows the self-assembly of microparticles to form multiple-layer colloidal crystals. A single-layer colloidal structure was readily formed within 1.2 min of the actuation of the TSAW-based device with an input voltage of 5.1 Vpp and a frequency of 72 MHz; the sample flow rate was 250 μL h−1 (see Fig. 3(A) and Movie S3 in the ESI†). The single-layer crystal evident in the inset (at 1.2 min) has a predominantly uniform orientation with only a few random defects (holes). The further inflow of particles leads to the formation of a secondary layer in the colloidal structure (depicted as a brighter region), and the defects present in the primary layer (at 1.2 min) are then filled by the additional particles (at 1.7 min). The inset shows the contrast between the single-layer and bi-layer colloidal structures. At 2.2 min, the secondary layer has expanded; the dark and bright shades correspond to multiple layers oriented differently with respect to the primary layer (see the inset). At 2.7 min, the crystal has grown to 450 μm in length; the extra bright regions close to the microchannel walls (see the inset) indicate the presence of multiple layers of colloidal particles that obscure the crystalline structure. Fig. 3(B) shows contrasting experimental images for multiple-layer crystal growth for which the background noise has not been removed. For a sample flow rate of 500 μL h−1, a predominantly bi-layer crystal has formed at 4 min, and the inset shows the contrast between the single-layer (bright) and bi-layer (dark) regions. As the crystal structure expands with the addition of new particles, the Stokes drag force acting on the crystal increases as well, so the input voltage was increased from 8.6 Vpp to 10.4 Vpp to impart an equivalent ARF and thus to maintain the position of the crystal (at 5 min). The input voltage was further increased to 12.2 Vpp and 13.8 Vpp at 7 min and 8 min respectively, as the crystal structure reached a length of ~750 μm. For additional results with other surfactant concentrations, see Fig. S2 in the ESI.†Input voltage and flow rate effects
Fig. 4 shows the effects of varying the input voltage and the buffer flow rate on the equilibrium position and shape of the crystal structure. Microparticles were pumped through the microchannel at 250 μL h−1 and a TSAW-based ARF with an input voltage of 4.2 Vpp was applied to induce their self-assembly into a single-layer colloidal crystal (see Fig. 4(A)). The particle flow was replaced by a buffer flow (250 μL h−1), and the input voltage was gradually increased up to 13.0 Vpp. At 6.5 and 8.6 Vpp, the crystalline structure has moved leftward while maintaining a rough rectangular shape. However, at 10.9 Vpp, the shape of the crystalline structure has become convex–concave, which was further exaggerated at 13.0 Vpp. For a constant flow rate, the Poiseuille flow profile remained constant; however, the acoustic forces increase with the voltage, resulting in a convex–concave shape at higher voltages for which the acoustic forces at the center of the microchannel dominate the Stokes drag force on the particles due to the Poiseuille flow. The ARF profile is expected to have a uniform distribution close to the IDT across the microchannel width. However, it is also expected to have a lower amplitude farther away from the IDT near the microchannel walls because of the diffraction of the waves within the microchannel and their attenuation away from the IDT.16 At 13.0 Vpp, the buffer flow rate was increased to 500 μL h−1, which pushes the crystalline structure rightward (see Fig. 4(B)). When the flow rate was further increased to 1000 μL h−1, the curvature of the convex–concave shape decreased as it regained a rectangular shape at 1500 μL h−1. A concave–convex shape also arises at 2000 μL h−1, and is particularly prominent at 2500 μL h−1. At high flow rates, the flow velocity imparts a higher drag force on the particles at the center of microchannel and thus the crystal structure adopts a concave–convex shape. The effects of varying the input voltage and the buffer flow rate on the distance between the crystalline structure and the IDT are plotted in Fig. 4(C) and (D), respectively. Additional results confirming the above behavior and the repeatability of the phenomena are presented in Fig. S3–S5 (see the ESI†).Conclusions
We have demonstrated that an external acoustic force field can be used to trap continuously flowing microparticles so that they self-assemble into a crystalline structure: the acoustic radiation force due to the travelling surface acoustic waves is balanced by the Stokes drag force on the particles. The application of a continuous buffer solution flow, which balances the ARF, induces rearrangements of the particles that repair any cracks, dislocations, or defects in a single-layer crystal lattice. The defects (or particle holes) are filled by the additional incoming particles that stack beneath the primary crystal layer to form a multi-dimensional crystal lattice. The crystalline structure re-orients itself under the influence of the external force field. However, the movement of the crystal becomes restricted when the crystal size fills the gap between the microchannel walls, which locks the crystal motion. The propagation of TSAWs through the substrate and the coupling and diffraction of the acoustic waves within the microchannel lead to the evolution of the ARF profile away from the IDT. The balance between the ARF and the drag force induced by the Poiseuille flow usually results in a rectangular crystalline structure (see Fig. S5 in the ESI†); however, at higher input voltages and lower flow rates, a convex–concave structure results, and vice versa. The TSAW-based self-assembly of the microparticles leads to the formation of larger crystalline structures than are possible with a standing-acoustic-waves-based platform because acoustic pressure nodes have a low saturation limit.Authors contributions
G. D. conceived the research. H. J. S. supervised the research. G. D., H. A., J. P. and M. A. designed and performed the experiments. G. D., A. H. and J. P. analyzed the results. All authors have contributed in preparing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) (Grant No. 2018-001483), the KUSTAR-KAIST Institute, and the Korea Polar Research Institute (KOPRI).Notes and references
- V. N. Manoharan, Sci. 80, 2015, 349, 1253751 CrossRef PubMed
.
- S. Sacanna, D. J. Pine and G.-R. Yi, Soft Matter, 2013, 9, 8096 RSC
- J. D. Joannopoulos, Nature, 2001, 414, 257–258 CrossRef CAS PubMed
- L. Basabe-Desmonts, J. Beld, R. S. Zimmerman, J. Hernando, P. Mela, M. F. García Parajó, N. F. van Hulst, A. van den Berg, D. N. Reinhoudt and M. Crego-Calama, J. Am. Chem. Soc., 2004, 126, 7293–7299 CrossRef CAS PubMed
- J. Kim, S. E. Chung, S.-E. Choi, H. Lee, J. Kim and S. Kwon, Nat. Mater., 2011, 10, 747–752 CrossRef CAS PubMed
- L. Wang and S. Sánchez, Lab Chip, 2015, 15, 4383–4386 RSC
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
- M. Wang, L. He and Y. Yin, Mater. Today, 2013, 16, 110–116 CrossRef CAS
- G. Singh, H. Chan, T. Udayabhaskararao, E. Gelman, D. Peddis, A. Baskin, G. Leitus, P. Král and R. Klajn, Faraday Discuss., 2015, 181, 403–421 RSC
- T. D. Edwards and M. A. Bevan, Langmuir, 2014, 30, 10793–10803 CrossRef CAS PubMed
- A. Azari, J. J. Crassous, A. M. Mihut, E. Bialik, P. Schurtenberger, J. Stenhammar and P. Linse, Langmuir, 2017, 33, 13834–13840 CrossRef CAS PubMed
- J. Yan, S. C. Bae and S. Granick, Soft Matter, 2015, 11, 147–153 RSC
- B. Bharti and O. D. Velev, Langmuir, 2015, 31, 7897–7908 CrossRef CAS PubMed
- N. Yanai, M. Sindoro, J. Yan and S. Granick, J. Am. Chem. Soc., 2013, 135, 34–37 CrossRef CAS PubMed
- C. D. Wood, S. D. Evans, J. E. Cunningham, R. O'Rorke, C. Wälti and A. G. Davies, Appl. Phys. Lett., 2008, 92, 044104 CrossRef
- A. Fakhfouri, C. Devendran, T. Albrecht, D. J. Collins, A. Winkler, H. Schmidt and A. Neild, Lab Chip, 2018, 18, 2214–2224 RSC
- H. Ahmed, G. Destgeer, J. Park, M. Afzal and H. J. Sung, Anal. Chem., 2018, 90, 8546–8552 CrossRef CAS PubMed
- J. Shi, D. Ahmed, X. Mao, S.-C. S. Lin, A. Lawit and T. J. Huang, Lab Chip, 2009, 9, 2890–2895 RSC
- J. Nam, J. Y. Kim and C. S. Lim, AIP Adv., 2017, 7, 015314 CrossRef
- D. J. Collins, R. O'Rorke, C. Devendran, Z. Ma, J. Han, A. Neild and Y. Ai, Phys. Rev. Lett., 2018, 120, 074502 CrossRef PubMed
- G. Destgeer, J. H. Jung, J. Park, H. Ahmed and H. J. Sung, Anal. Chem., 2017, 89, 736–744 CrossRef CAS PubMed
- G. Destgeer, J. H. Jung, J. Park, H. Ahmed, K. Park, R. Ahmad and H. J. Sung, RSC Adv., 2017, 7, 22524–22530 RSC
- A. Fakhfouri, C. Devendran, D. J. Collins, Y. Ai and A. Neild, Lab Chip, 2016, 16, 3515–3523 RSC
- H. Ahmed, G. Destgeer, J. Park, J. H. Jung, R. Ahmad, K. Park and H. J. Sung, Anal. Chem., 2017, 89, 13575–13581 CrossRef CAS PubMed
- G. Destgeer, H. Cho, B. H. Ha, J. H. Jung, J. Park and H. J. Sung, Lab Chip, 2016, 16, 660–667 RSC
- H. Li, J. R. Friend and L. Y. Yeo, Biomed. Microdevices, 2007, 9, 647–656 CrossRef PubMed
- K. Melde, E. Choi, Z. Wu, S. Palagi, T. Qiu and P. Fischer, Adv. Mater., 2018, 30, 1704507 CrossRef PubMed
- C. E. Owens, C. W. Shields, D. F. Cruz, P. Charbonneau and G. P. López, Soft Matter, 2016, 12, 717–728 RSC
- M. Caleap and B. W. Drinkwater, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6226–6230 CrossRef CAS PubMed
- M. Akella and J. J. Juárez, ACS Omega, 2018, 3, 1425–1436 CrossRef CAS
- Y. Yang, A. T. Pham, D. Cruz, C. Reyes, B. J. Wiley, G. P. Lopez and B. B. Yellen, Adv. Mater., 2015, 27, 4725–4731 CrossRef CAS PubMed
- S. Wollmann, R. B. Patel, A. Wixforth and H. J. Krenner, Appl. Phys. Lett., 2014, 105, 031113 CrossRef
- L. P. Ferreira, V. M. Gaspar and J. F. Mano, Biomaterials, 2018, 185, 155–173 CrossRef CAS PubMed
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737–744 CrossRef CAS PubMed
- H. Ahmed, G. Destgeer, J. Park, J. H. Jung and H. J. Sung, Adv. Sci., 2018, 5, 1700285 CrossRef PubMed
- J. Park, J. H. Jung, K. Park, G. Destgeer, H. Ahmed, R. Ahmad and H. J. Sung, Lab Chip, 2018, 18, 422–432 RSC
- G. Destgeer, K. H. Lee, J. H. Jung, A. Alazzam and H. J. Sung, Lab Chip, 2013, 13, 4210–4216 RSC
- J. Park, J. H. J. H. Jung, G. Destgeer, H. Ahmed, K. Park and H. J. H. J. Sung, Lab Chip, 2017, 17, 1031–1040 RSC
- G. Destgeer, B. Ha, J. Park and H. J. H. J. Sung, Anal. Chem., 2016, 88, 3976–3981 CrossRef CAS PubMed
- G. Destgeer, B. H. Ha, J. H. Jung and H. J. Sung, Lab Chip, 2014, 14, 4665–4672 RSC
- G. Destgeer, B. H. Ha, J. Park, J. H. Jung, A. Alazzam and H. J. Sung, Anal. Chem., 2015, 87, 4627–4632 CrossRef CAS PubMed
- D. J. Collins, Z. Ma, J. Han and Y. Ai, Lab Chip, 2017, 17, 91–103 RSC
- M. B. Dentry, L. Y. Yeo and J. R. Friend, Phys. Rev. E, 2014, 89, 013203 CrossRef PubMed
- D. R. Nelson and B. I. Halperin, Phys. Rev. B, 1979, 19, 2457–2484 CrossRef CAS
- J. J. Juárez and M. A. Bevan, Adv. Funct. Mater., 2012, 22, 3833–3839 CrossRef
- P. Rein ten Wolde, M. J. Ruiz-Montero and D. Frenkel, J. Chem. Phys., 1996, 104, 9932–9947 CrossRef
- M. Travagliati, R. J. Shilton, M. Pagliazzi, I. Tonazzini, F. Beltram and M. Cecchini, Anal. Chem., 2014, 86, 10633–10638 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures are provided in the form of a PDF file. Experimental results are further demonstrated in the movies. See DOI: 10.1039/c8ra09859j |
| This journal is © The Royal Society of Chemistry 2019 |