
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium ion dynamics in LiZr2(PO4)3 and Li1.4Ca0.2Zr1.8(PO4)3†
Isabel
Hanghofer
*a,
Bernhard
Gadermaier
 a,
Alexandra
Wilkening
a,
Daniel
Rettenwander
a and
H. Martin R.
Wilkening
*ab
a,
Alexandra
Wilkening
a,
Daniel
Rettenwander
a and
H. Martin R.
Wilkening
*ab
aInstitute for Chemistry and Technology of Materials, Christian Doppler Laboratory for Lithium Batteries, Graz University of Technology (NAWI Graz), Stremayrgasse 9, A-8010 Graz, Austria. E-mail: isabel.hanghofer@tugraz.at
bALISTORE – European Research Institute, CNRS FR3104, Hub de l'Energie, Rue Baudelocque, 80039 Amiens, France. E-mail: wilkening@tugraz.at
First published on 3rd June 2019
Abstract
High ionic conductivity, electrochemical stability and small interfacial resistances against Li metal anodes are the main requirements to be fulfilled in powerful, next-generation all-solid-state batteries. Understanding ion transport in materials with sufficiently high chemical and electrochemical stability, such as rhombohedral LiZr2(PO4)3, is important to further improve their properties with respect to translational Li ion dynamics. Here, we used broadband impedance spectroscopy to analyze the electrical responses of LiZr2(PO4)3 and Ca-stabilized Li1.4Ca0.2Zr1.8(PO4)3 that were prepared following a solid-state synthesis route. We investigated the influence of the starting materials, either ZrO2 and Zr(CH3COO)4, on the final properties of the products and studied Li ion dynamics in the crystalline grains and across grain boundary (g.b.) regions. The Ca2+ content has only little effect on bulk properties (4.2 × 10−5 S cm−1 at 298 K, 0.41 eV), but, fortunately, the g.b. resistance decreased by 2 orders of magnitude. Whereas, 7Li spin-alignment echo nuclear magnetic resonance (NMR) confirmed long-range ion transport as seen by conductivity spectroscopy, 7Li NMR spin–lattice relaxation revealed much smaller activation energies (0.18 eV) and points to rapid localized Li jump processes. The diffusion-induced rate peak, appearing at T = 282 K, shows Li+ exchange processes with rates of ca. 109 s−1 corresponding, formally, to ionic conductivities in the order of 10−3 S cm−1 to 10−2 S cm−1.
1. Introduction
In the years to come, all-solid-state Li or Na batteries1 are expected to replace conventional systems2 that still rely on flammable aprotic electrolytes. Ceramic cells may take advantage of highly flexible design possibilities if batteries with dimensions in the mm range are considered.3,4 Moreover, ceramic batteries are going to withstand higher temperatures than their analogous cells with liquid components.5For their realization, ceramic electrolytes with sufficiently high ionic conductivities are, however, needed.5–9 The overall performance of NaSICON-type (Na SuperIonic CONductor) conductors10,11 has attracted renewed interest to study the influence of synthesis conditions and dopants on morphology and ion dynamics. Earlier reports have shown that LiM2(PO4)3 (M: Ge, Ti, Sn, Hf, Zr) exhibits good chemical stabilities under ambient conditions. This property comes along with a wide electrochemical stability window.12,13 LiTi2(PO4)3 is known as a very good Li-ion conductor but it suffers from the fact that Ti(IV) can easily be reduced to Ti(III) when in contact with metallic Li.14
In contrast to LiTi2(PO4)3-based ceramics, including those in which Ti4+ is partly replaced by Al3+, LiZr2(PO4)3 (LZP) shows a much better stability against Li anodes. If in contact with a Li metallic anode LZP forms stable decomposing products, such as Li3P and layer-structured Li8ZrO6, that are able to conduct Li ions when present as thin layers. Importantly, this interphase also reveals sufficiently good wetting properties with respect to both Li metal and the electrolyte.15 LZP crystallizes with different structures, ionic conductivities sensitively depend on both the overall morphology and the defect chemistry involved. The ionic conductivity can be improved by incorporating different metals such as Y3+, Sc3+, Al3+, La3+ or Ca2+; this strategy is usually accompanied by a stabilization of the rhombohedral structure.13,16–22 LZP is typically prepared at calcination temperatures of ca. 1173 K, the phase appearing under these conditions is denoted as the α-phase of LZP, which is subdivided in a orthorhombic form (α) and a monoclinic one (α′). At higher sintering temperatures, that is, approximately at 1423 K, the highly conductive rhombohedral structure α-LiZr2(PO4)3 crystallizing with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c symmetry is formed. α′-LiZr2(PO4)3, obtained at lower temperatures, is usually a mixture of monoclinic LZP and triclinic LZP; its ionic conductivity is reported to be in the order of 10−8 S cm−1 whereas α-LiZr2(PO4)3 shows values around 10−5 S cm−1.15,22–25
c symmetry is formed. α′-LiZr2(PO4)3, obtained at lower temperatures, is usually a mixture of monoclinic LZP and triclinic LZP; its ionic conductivity is reported to be in the order of 10−8 S cm−1 whereas α-LiZr2(PO4)3 shows values around 10−5 S cm−1.15,22–25
In this work, the structure variation of different educts on the product formed is discussed. We analysed Li ion dynamics in LiZr2(PO4)3 and Li1+2xCaxZr2−x(PO4)3 (LCZP) by both broadband impedance spectroscopy26,27 and 7Li nuclear magnetic resonance (NMR) spectroscopy, i.e., by recording diffusion-induced spin–lattice relaxation rates.28–30 The latter are sensitive to both short-range and long-range ion transport through the crystal lattice of LZP. As we deal with powder samples with crystallite diameters in the μm range, NMR rates measured in the laboratory frame of reference are solely sensitive to bulk ion dynamics.
2. Experimental
2.1. Sample preparation
LiZr2(PO4)3 (LZP) was prepared via a classic solid-state reaction by mixing stoichiometric amounts of Li2CO3 (Sigma Aldrich ≥99%), (NH4)2HPO4 (Sigma Aldrich ≥99.9%) with either ZrO2 (Sigma Aldrich ≥99%) or Zr(CH3COO)4 (= Zr(ac)4). The starting compounds were mechanically milled for 2 hours by using a high-energy planetary ball mill (Fritsch Pulverisette 7 Premium line) at a rotation speed of 400 rpm. We used ZrO2 beakers (45 mL), which were filled with 180 balls made of ZrO2 (5 mm in diameter). The subsequent calcination process was carried out in Al2O3 crucibles at 900 °C (heating rate 10 °C min−1) for 10 hours, where the decomposition of the hydrogen phosphate starts and initiates the reaction with Li2CO3. The resulting white powder was milled again for 2 hours at 400 rpm to guarantee a good contact between the particles. Finally, the mixture was pressed with 0.4 tons into pellets with a diameter of 5 mm. The pellets were filled into Al2O3 crucibles and sintered at 1150 °C (heating rate 10 °C min−1) for 20 hours. To incorporate Ca2+ into LZP we used CaCO3 (Alfa Aesar, >99%). An excess of 10 wt% Li2CO3 should compensate the loss of lithium during the high temperature heating process leading to Li1.4Ca0.2Zr1.8(PO4)3 (LCZP). For the synthesis of LZP (and LCZP) using Zr(CH3COO)4 we needed to prepare the acetate as follows.31 20 mmol Zirconium oxochloride (Sigma-Aldrich 99.99%) was dissolved under reflux in 100 mL of a mixture of MeCOOH (Sigma-Aldrich ≥99.5%) and Me(COO)2O (Sigma Aldrich ≥99%) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). After the mixture was allowed to cool to room temperature, a white crystalline deposit appeared. Finally, it was filtered and dried at 60 °C under vacuum.
:9). After the mixture was allowed to cool to room temperature, a white crystalline deposit appeared. Finally, it was filtered and dried at 60 °C under vacuum.
2.2. X-ray powder diffraction
The sample obtained after the calcination process and the finally sintered product were characterized by X-ray powder diffraction (XRPD). We used a Bruker D8 Advance diffractometer operating with Bragg Brentano geometry and Cu Kα radiation. Diffractograms were recorded in air atmosphere and at room temperature covering a 2θ range from 20° to 100° with a step size of 0.02° (2 s per step). Rietveld analysis (X-PertHighScorePlus (PANanalytical)) was used to refine the diffraction data.2.3. Impedance spectroscopy
For the impedance measurements the sintered samples were equipped with gold electrodes by a sputtering process. Gold electrodes with a layer thickness of 100 nm were applied on both sides with a sputter coater (LEICA EM SCD 050) to ensure a good electrical contact. To avoid any influence of moisture, the samples were dried at 60 °C under vacuum prior to the impedance measurements.Impedance spectra were recorded with a Novocontrol Concept 80 broadband dielectric spectrometer equipped with a BDS 1200 cell combined with an active ZGS cell (Novocontrol). We measured complex impedances over a frequency range of ten decades (10 mHz to 10 MHz). The temperature in the sample holder was varied from 173 K to 473 K in steps of 20 K; the temperature program was automatically controlled by a QUATRO cryosystem (Novocontrol). During the measurements a dry nitrogen atmosphere was build up around the sample in the cryostat to avoid any contamination with water and/or oxygen.
2.4. Nuclear magnetic resonance measurements
For the time-domain NMR measurements, the powder samples LiZr2(PO4)3 and Li1.4Ca0.2Zr1.8(PO4)3 were sealed in Duran glass tubes (ca. 4 cm in length and 3 mm in diameter). During the sealing procedure they were kept under dynamic vacuum to safely protect them from any contact with humid air. We used 7Li NMR line shape measurements, spin lattice relaxation (SLR) experiments as well as 7Li spin alignment echo (SAE) NMR to collect information about Li activation energies and jump rates. Longitudinal NMR SLR rates (1/T1) as well as spin-lock rates (1/T1ρ) were measured with a Bruker Avance III spectrometer that is connected to a shimmed cryomagnet with a nominal magnetic field of 7 Tesla. This field corresponds to a 7Li Larmor frequency of ω0/2π = 116 MHz. For the measurements at temperatures ranging from 173 K to 583 K a ceramic temperature probe (Bruker Biospin) was used. Depending on temperature and at a power level of 180 W the π/2 pulse length ranged from 2.2 μs to 2.4 μs.7Li NMR SLR rates (1/T1 = R1) in the laboratory frame were acquired with the well-known saturation recovery pulse sequence. This sequence uses a comb of closely spaced π/2 pulses to destroy any longitudinal magnetization Mz. The subsequent recovery of Mz was detected as a function of waiting time td with a π/2 reading pulse: 10 × π/2 − td − π/2 – acquisition.32,33 To construct the magnetization transients Mz(td), we plotted the area under the free induction decays vs. td. The transients Mz(td) were parameterized with stretched exponentials, Mz(td) ∝ 1 − exp(−(t/T1)γ), to extract the rates R1. Additionally, rotating frame 7Li NMR SLRρ rates 1/T1ρ (= R1ρ) were measured by means of the spin lock technique: π/2-plock – acquisition.32 Here, we used a locking frequency ω1/2π of 20 kHz. The duration of the spin-lock pulse tlock was varied from 10 μs to 460 ms. To ensure full longitudinal relaxation between each scan the recycle delay was set to 5 × T1. The R1ρ rates were obtained by analyzing the resulting transients Mρ(tlock) with stretched exponentials with the form Mρ(tlock) ∝ exp(−(tlock/T1ρ)κ). The stretching exponent γ varied from 1 to 0.8, the exponent κ ranges from 1 to 0.6.
Finally, mixing time (tm) dependent 7Li SAE NMR decay curves were recorded with the help of the Jeener–Broekaert34,35 three-pulse sequence: (90°)X-tp–(45°)Y-tm–45°-acq. We used a constant preparation time tp of 25 μs to acquire two-time sinus–sinus single-spin correlation functions. The mixing time was varied from 30 μs up to several seconds. A suitable phase cycle34,36 was employed to suppress unwanted coherences and to eliminate, as best as possible, dipolar contributions affecting the echo that appears after the reading pulse.36 Fourier transformation of the spin alignment echoes, starting from the top of the signal, yields 7Li SAE NMR spectra useful to highlight quadrupole intensities due to the interaction of the quadrupole moment of the 7Li spin (spin-quantum number I = 3/2) and a non-vanishing electric field gradient.
3. Results and characterization
3.1. Characterization via X-ray powder diffraction
The purity of the crystalline samples synthesized was examined by XRPD. As mentioned above, diffraction patterns were collected at room temperature and under air atmosphere. The first XRPD pattern was recorded directly after the calcination process, i.e., after removal of CO2, NH3 and H2O at 900 °C, the pattern is depicted in Fig. S1.† After this calcination step (10 hours), we see that LiZr2(PO4)3 crystallizes with monoclinic structure (space group P121/c1, see Fig. S1 (ESI)†). Sintering the samples yields a crystalline material that is in agreement with the rhombohedral NaSICON structure37 (space group Rc, Fig. 1);19 the corresponding XRPD pattern is shown in Fig. 2. This phase is isostructural with the sibling compound LiTi2(PO4)3.38 The increase in conductivity before and after sintering, i.e., the difference in ionic transport properties of monoclinic and rhombohedral LZP is illustrated in Fig. S1b.†
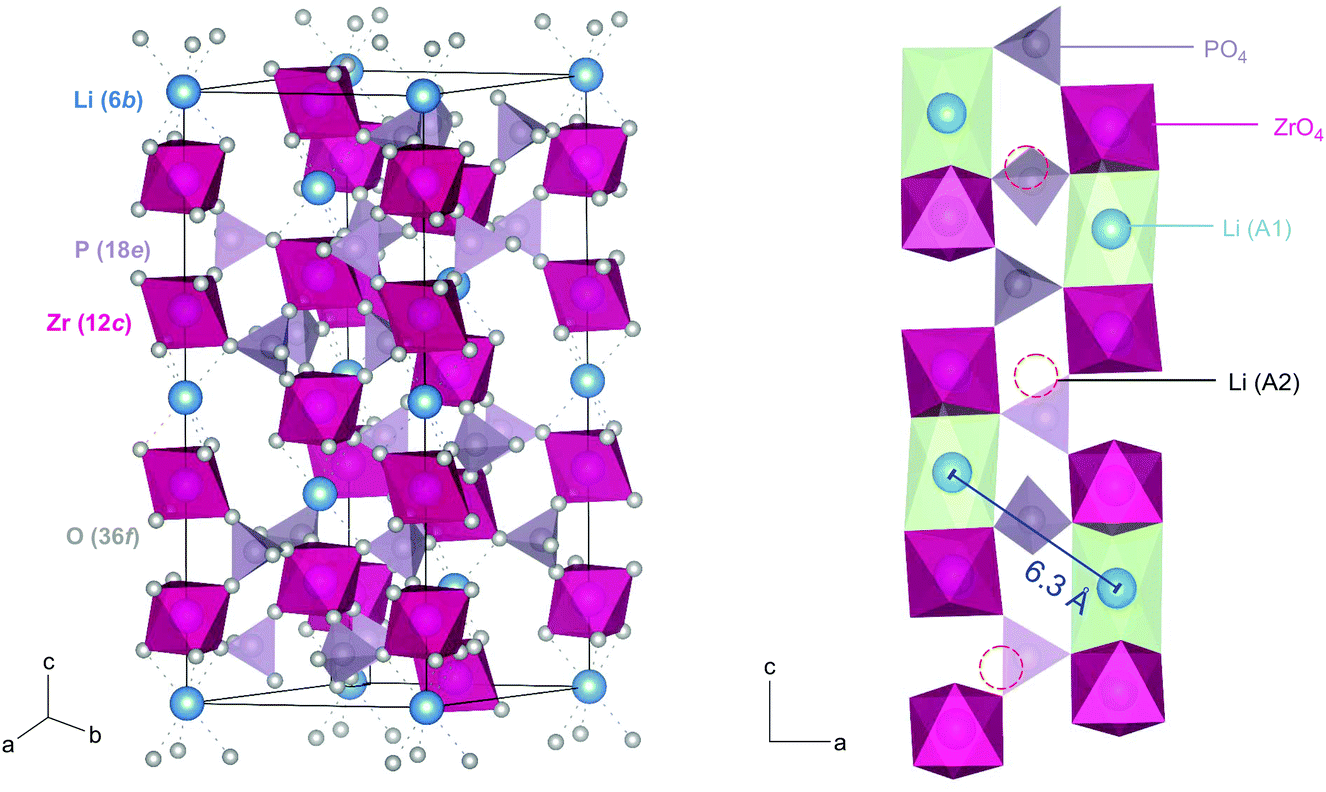 | ||
| Fig. 1 Left: Rhombohedral crystal structure of LiZr2(PO4)3. The tetrahedra in purple represent PO4-units, octahedra in pink show ZrO6, while the blue spheres denote the Li+ ions. Li+ is octahedrally coordinated by oxygen anions of the ZrO6 octahedra. Right: Section of the rhombohedral crystal structure to show interstitial sites A2; the Li ions may use to jump between the regularly occupied sites A1. The A1–A1 distance is 6.3 Å, which is, presumably, much too large for a direct jump process. | ||
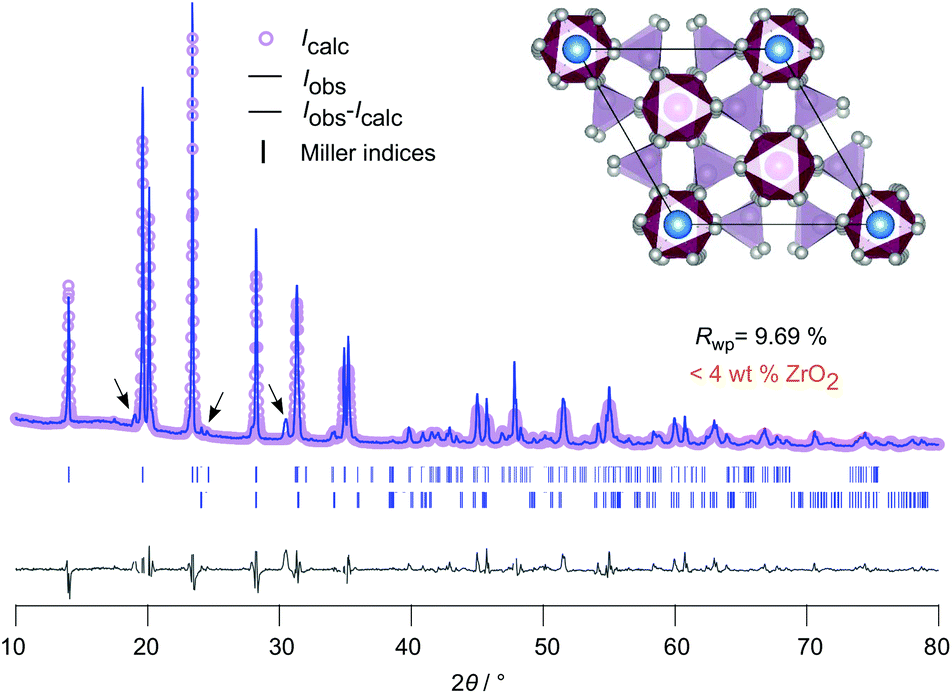 | ||
| Fig. 2 Rietveld refinement of LZP using ZrO2 as educt. The sample was sintered at 1150 °C for 20 hours in a closed Al2O3-crucible. LZP crystallizes with space group RcH, see Table S1† for further information. Selected reflections have been indexed with vertical bars. A very small amount (<4 wt%) of ZrO2 was detected. The inset shows the ab-plane of the rhombohedral NaSICON structure with the viewing direction along the c-axis. | ||
The Zr2(PO4)3 framework of rhombohedral NaSICON-type LZP consists of two ZrO6 octahedra and three PO4 tetrahedra sharing O atoms. The octahedral and tetrahedral units are alternating with the cations to form infinite chains parallel to the ternary axis of the structure. Each PO4 unit shares its oxygen with four ZrO6 octahedra of three Zr2(PO4)3 units to form the NaSICON framework. A 3D network of conduction pathways is formed that is used by the ions to diffuse through the crystal. In this case the Li ions (6b) are octahedrally coordinated by oxygen ions (36f) at the intersection of three conduction channels (A1). The A1 sites are located between pairs of ZrO6 octahedra along the c-axis, while the (vacant) interstitial sites (□) A2 can be found between O3ZrO3A1O3Zr-□-O3ZrO3A1. Since the A1–A1 distance in LZP is rather larger, we assume that interstitial sites, such as A2, are involved in Li+ diffusion. Because of the large spatial separation of Li ions in LZP, we expect rather low homonuclear dipole–dipole interactions resulting in narrow 7Li NMR lines even in the rigid lattice regime, see below.
In Fig. 2 the result of our Rietveld analysis of the diffraction pattern of LiZr2(PO4)3 synthesized by using ZrO2 as educt is shown. Our structure solution indeed points to rhombohedral symmetry characterized by the space group RcH (ICSD, no. 201935), as already found by the study of Petit et al.19Via high-temperature neutron diffraction (T ≥ 423 K), Catti et al. reported on two Li+ positions displaced from the A1 and A2 sites (see Fig. S2†).24 Here, in addition to the main rhombohedral phase, a minor impurity of ZrO2 (<4 wt%) is seen (see black bars). Rietveld analysis yields the following lattice properties a (= b) = 8.824 Å and c = 22.456 Å; V = 1514.24 Å3. By doping this sample with 5 wt% and 10 wt% Ca2+ the cell volume decreases (1512.64 Å3 (5 wt% Ca), 1506.22 Å3 (10 wt% Ca)) as expected; for further information we refer to Table S1 in the ESI.†
The preparation route with Zr(CH3COO)4 as starting material yields almost the same lattice parameters but the amount of non-reacted material is much higher and reaches values as high as 25 wt%. The incorporation of aliovalent Ca2+ ions, on the other hand, helps obtaining phase pure LZP with rhombohedral structure, as is illustrated in Fig. 3. The amount of unreacted ZrO2 continuously decreases with increasing Ca-content. This behavior is also found for LCZP prepared with the help of Zr(ac)4. Nonetheless, the amount of ZrO2 remains much higher (>15 wt%) than that in samples prepared from ZrO2 directly. Dots in Fig. 3 denote reflections belonging to ZrO2. Worth noting, here we do not find any additional Bragg reflections that belong to triclinic LZP in all diffractograms. These have been seen in earlier reports.22,39–41 We recognize that small variations in synthesis conditions, including heat treatment and cooling rates, might sensitively affect the phase purity of the final compounds.
 | ||
| Fig. 3 (a) X-ray powder diffraction pattern of the crystalline LiZr2(PO4)3 and Li1+2xZr2−xCax(PO4)3 (x = 0.1 and 0.2). All samples were prepared by a classical solid-state route with either ZrO2 as starting material (black) or Zr(CH3COO)4 (light grey). The vertical lines (in grey) at the bottom denote the reflections of rhombohedral LZP, those of monoclinic ZrO2 are indicated by green dots. (b) XRPD pattern of Li1+2xZr2−xCax(PO4)3 prepared with the help of ZrO2. The diffraction pattern reveal that the amount of the impurity phase ZrO2 decreases with increasing Ca2+-content. | ||
According to XRPD we decided to study, in detail, ion dynamics of the following samples: LiZr2(PO4)3 prepared from ZrO2 and Zr(ac)4 as well as Li1+2xZr2−xCax(PO4)3 (x = 0.1 and 0.2) prepared with the help of ZrO2. Similar to LiTi2(PO4)3 and other NaSICON-type materials α-LiZr2(PO4)3 shows low relative densities, here ranging from 78% to 84%.
3.2. Impedance spectroscopy
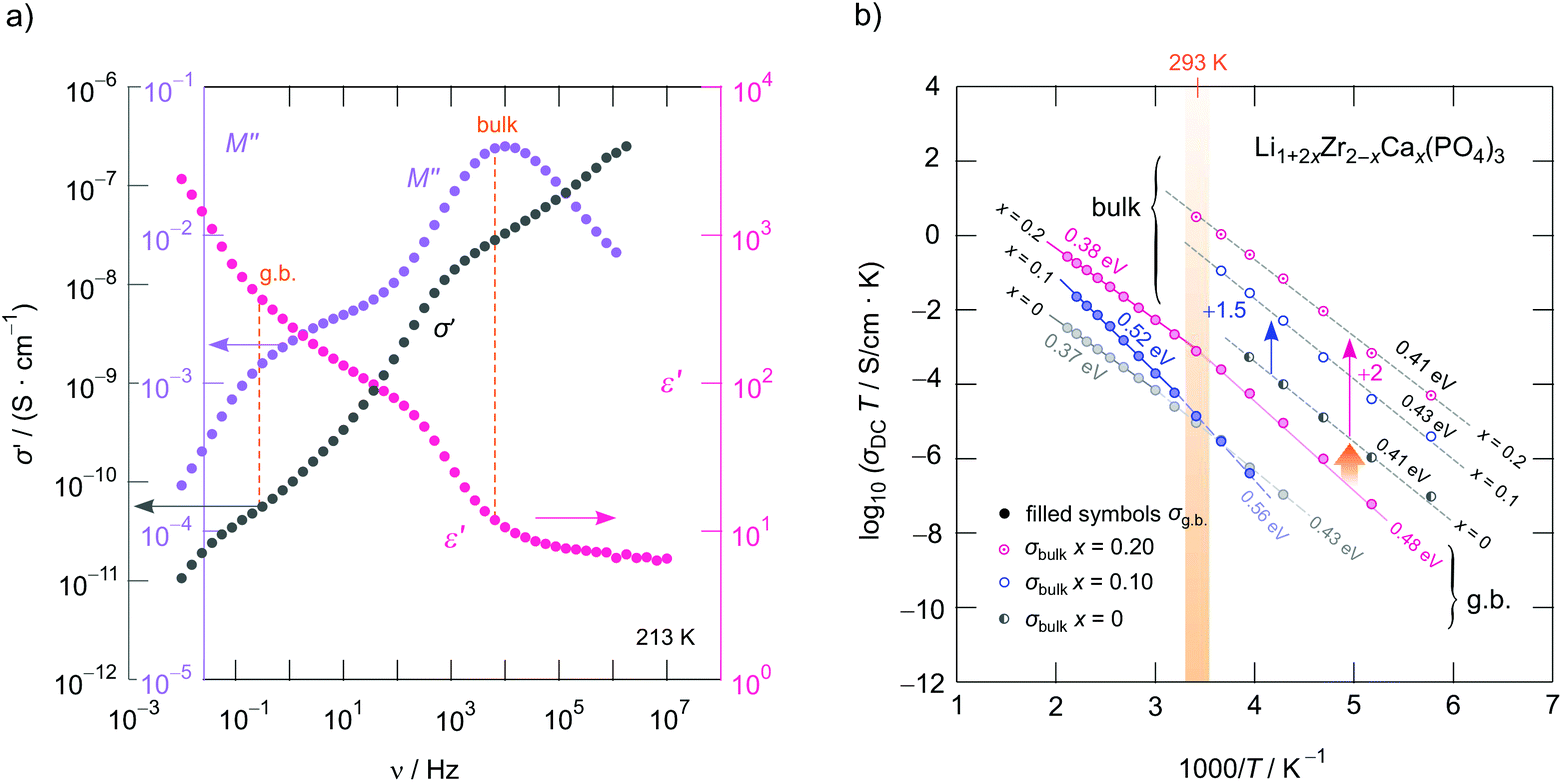
To study how Li+ ion transport is affected by x as well as to investigate whether the starting materials influence the dynamic parameters, we measured complex impedances over a large temperature and frequency range.26,42 Exemplarily, in Fig. 4a conductivity isotherms of rhombohedral LiZr2(PO4)3 are shown. Isotherms are obtained by plotting the real part, σ′, of the complex conductivity σ of LiZr2(PO4)3 as a function of frequency ν. They are composed of four regimes. (i) At low frequencies (and sufficiently high temperatures and, thus, high ionic mobility) electrode polarization (EP) appears owing to the piling-up of ions near the surface of the blocking Au electrodes applied. In many cases a stepwise decay of σ′ is seen (cf. the two arrows in Fig. 4a). (ii) The polarization regime passes into so-called conductivity plateaus (P1) governing the isotherms at intermediate temperatures and low frequencies. If this plateau is identified with a bulk response, it reflects long-range ion transport and is given by the dc-conductivity σdc. By moving to higher frequency a shallow dispersive regime with a weak frequency dependence shows up. It directly merges into another plateau (iii, P2), which finally passes over in the high-frequency dispersive regime (iv) which can roughly be approximated with Jonscher's power law.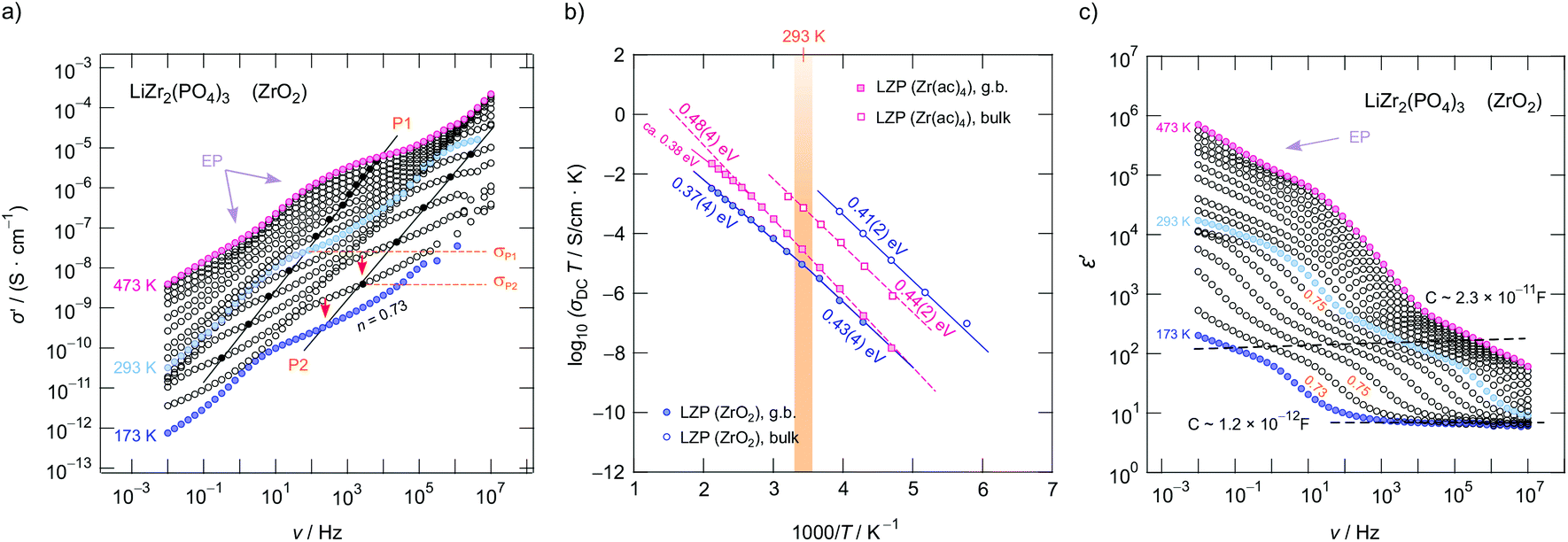 | ||
| Fig. 4 (a) Conductivity isotherms of Ca-free LZP (synthesized from ZrO2) recorded at temperatures ranging from 173 K to 473 K; isotherms have been recorded in steps of 20 K. We observed two plateaus P1 and P2 that correspond to the grain boundary (g.b., P1) and bulk response (P2). (b) Arrhenius plot of the DC conductivities associated with P1 and P2. The solid and dashed line show line fits with an Arrhenius law yielding activation energies ranging from 0.39 eV to 0.52 eV. Circles represent LZP synthesized from ZrO2 and rectangles show results of LZP that was synthesized from Zr(ac)4. LZP prepared from ZrO2 shows the highest bulk ion conductivity that is characterized by 0.41 eV. (c) Real part of the complex permittivity as a function of frequency. P1 and P2 seen in (a) produce a two-step increase of ε′ characterized by permittivities and capacities being typical for a bulk electrical response and a response including ion-blocking grain boundaries. The same characteristics are seen for LZP prepared from Zr(ac)4. Both processes can be approximated with a power law of the form ε′(ν) = ε(∞) + Asν−p with p ≠ f(T) ≈ 0.75, wherein ε(∞) represents the permittivity at very high frequencies. | ||
In Fig. 4a the inflexion points of the plateaus P1 and P2 are highlighted by filled circles; straight lines connect these circles. The dispersive region belonging to plateau P2 is best seen at 173 K, it can be approximated with Jonscher's power law:43σ′ = σdc + A0νn. Here A0 is the so-called dispersion parameter and n represents the power law exponent, which takes a value of 0.73 at 173 K. To analyse our data in terms of dimensionality effects, we also studied the frequency dependence of the real part of the complex permittivity; the corresponding isotherms (ε′ vs. ν) are plotted in Fig. 4c; the plateaus P1 and P2 produce a two-step increase of ε′ when coming from high frequencies. As for σ′, also ε′(ν) can be approximated with exponents around 0.75 for P2, see Fig. 4c. Such Jonscher exponents are expected for 3D ionic conduction.44
To determine which capacitances C govern the responses P1 and P2, we used the equation for a parallel-plate capacitor for an estimation C = ε0εrA/d. Here ε0 represents the electric field constant (8.854 × 10−12 F m−1), A the area and d the thickness of the sample. While the DC plateau (P2) at high frequencies is characterized by C = 1.2(1) × 10−12 F, for the plateau at lower ν we found C = 2.3(2) × 10−11 F. Therefore, the plateau associated with C in the pF range represents the bulk response, whereas P1 seen at lower frequencies is additionally governed by grain boundary contributions (g.b.) for which capacitances in the order of 10−11 F are typically expected.45
In the Arrhenius plot of Fig. 4b, σDCT of the two plateaus P1 and P2 is plotted vs. the inverse temperature T. σDCT does not change when several heating and cooling runs were performed. The lines in Fig. 4b refer to fits according to σDCT(P1, P2) ∝ exp(−Ea/(kBT)); kB denotes the Boltzmann's constant and Ea the corresponding activation energy. For LiZr2(PO4)3 prepared from ZrO2 we obtain Ea(P2) of 0.41(1) eV (in agreement with ref. 15 (0.40 eV) and ref. 25 (0.39 eV)), whereas for LiZr2(PO4)3, when using Zr(ac)4 as starting material, Ea increases to 0.44(1) eV (see Fig. 4b). At room temperature, the ionic conductivities of P2 are 3.6(2) × 10−5 S cm−1 and 3.4(2) × 10−6 S cm−1, respectively. For the sake of clarity, in Table 1 all activation energies Ea are listed, the table also includes values referring to Li1+2xCaxZr2−x(PO4)3 (x = 0.1, x = 0.2) that has been prepared from ZrO2.
| composition | E a/eV | log10(σ0/S cm−1 K) | Starting material |
|---|---|---|---|
| a At lower temperatures the activation energy increases to 0.43(4) eV. | |||
| LiZr2(PO4)3 | 0.44(2) (0.48(4)) | 4.97(1) (3.86(2)) | Zr(ac)4 |
| LiZr2(PO4)3 | 0.41(2) (0.37(4)a) | 4.91(2) (1.49(7)) | ZrO2 |
| Li1.2Ca0.1Zr1.9(PO4)3 | 0.43(1) (0.52(9)) | 5.44(4) (4.14(7)) | ZrO2 |
| Li1.4Ca0.2Zr1.8(PO4)3 | 0.41(2) (0.38(4)) | 5.08(2) (3.49(1)) | ZrO2 |
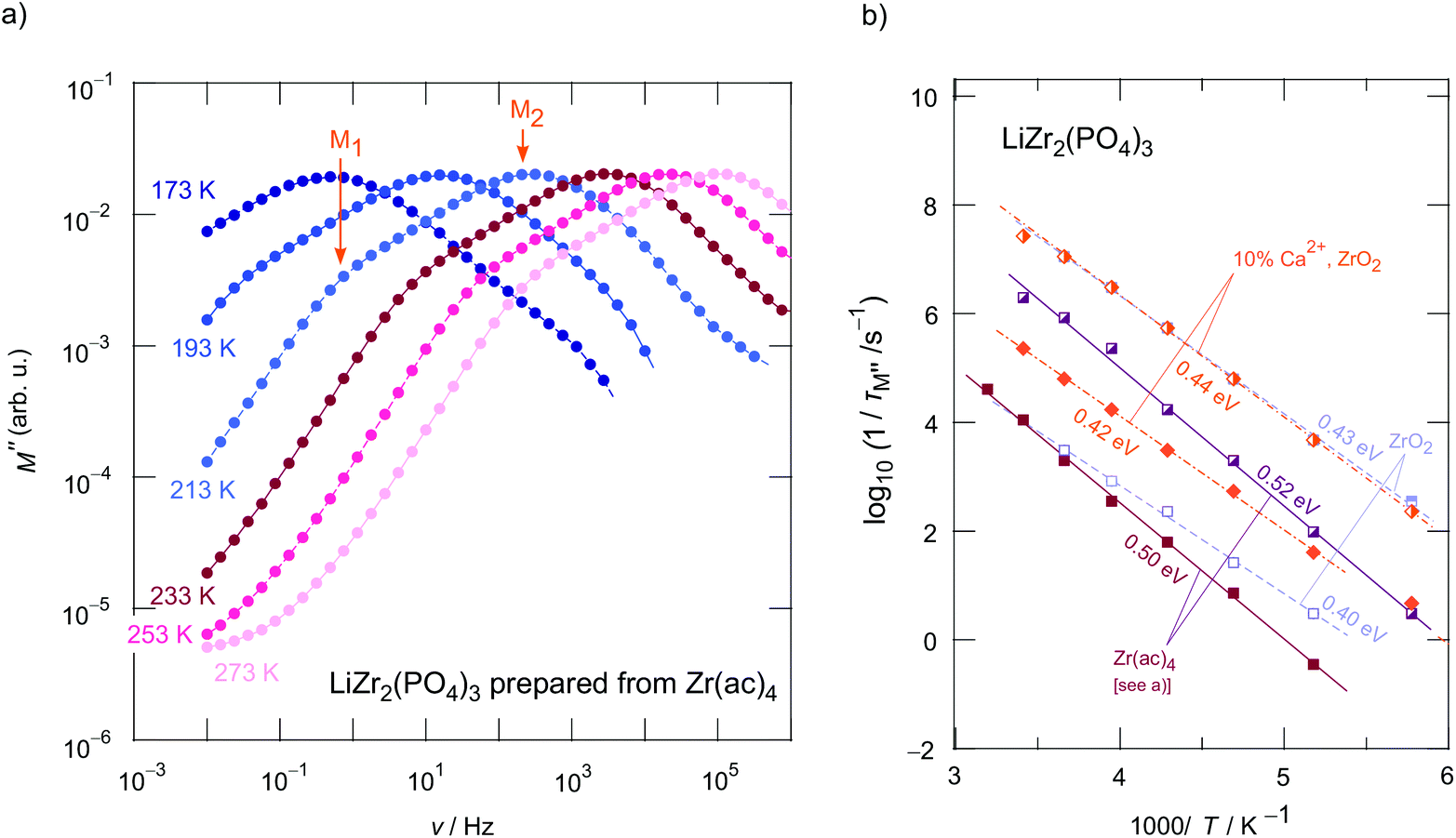
Unfortunately, by using Nyquist diagrams (see Fig. S3†), which show complex plane plots of the imaginary part Z′′ of the complex impedance, Z, vs. its real part Z′, the faster relaxation process, corresponding to P2, cannot be resolved properly. Instead, we used the complex modulus M′′(ν) representation46 to visualize the two processes, see Fig. 5a and 6 (Fig. 6b includes data for Li1+2xCaxZr2−x(PO4)3 prepared from ZrO2).
 | ||
| Fig. 5 (a) Frequency dependence of the imaginary part of the electric modulus M′′, of LiZr2(PO4)3 prepared from Zr(ac)4. Spectra were recorded at the temperatures indicated. The lines are to guide the eye. As suggested by σ′(ν), two distinct peaks are visible denoted as M1 and M2. The temperature dependence of the corresponding relaxation rate τM−1 is shown in (b). For comparison, the results of LiZr2(PO4)3 (prepared from ZrO2) and Li1.4Zr1.8Ca0.2(PO4)3 (also prepared from ZrO2) are included as well. Error margins are at least ± 0.1 eV. | ||
 | ||
| Fig. 6 (a) Frequency dependence of the imaginary part M′′ of the complex modulus, the real part, ε′, of the permittivity as well as the real part, σ′, of the complex conductivity of LiZr2(PO4)3 (prepared from ZrO2). The isotherm was recorded at 213 K. The maxima in M′′ clearly refer to the plateaus P1 and P2 seen in σ′(ν). For comparison, the change of ε′(ν) is shown, too. (b) Arrhenius plot of the ionic conductivities referring to the plateaus P1 and P2 (σDCT vs. 1000/T) of cation-mixed crystalline Li1+2xZr2−xCax(PO4)3. Solid lines and dashed lines represent fits according to an Arrhenius law yielding the activation energies Ea as indicated. While solid lines refer to conductivities influenced by the grain boundary contributions, the dashed lines represent bulk ion dynamics in Li1+2xZr2−xCax(PO4)3. For the sake of clarity, the bulk ion conductivities for the samples with x = 0.20 and x = 0.10 have been plotted using an offset of +2 and +1.5 on the logarithmic scale. These conductivities coincide with those of the Ca-free sample. Activation energies with error margins are listed in Table 1. | ||
At sufficiently low temperatures two relaxation peaks M1 and M2 appear (see the orange arrows in Fig. 5a). These peaks are separated by two orders of magnitude on the frequency scale. This distance on the frequency scale is comparable with the ratio of σDC,P2:σDC,P1; thus, they refer, as is seen in Fig. 6 to the plateaus P1 and P2 governing σ′(ν). As expected the two electrical relaxation frequencies mirror the ratio in conductivities. The peak with the larger amplitude (M2) corresponds to P2 in σ′(ν), the one with the smaller amplitude (M1) represents a relaxation process with a longer relaxation time (cf. P1), see also Fig. 6. As an estimation, M′′ is proportional to the inverse capacitance, M′′ ∝ 1/C.45,47 Thus, we expect peak M2 to be characterized by a larger amplitude (3.4 pF) than M1 (30.1 pF). C1/C2 ≈ 10 is in good agreement with the amplitude ratio seen in Fig. 5a and 6a.
In order to compare activation energies extracted from σDC(P1, P2) we determined characteristic electrical relaxation frequencies 1/τM using the modulus peaks of Fig. 5a. 1/τM refers to frequencies at which the peaks appear. A comparison of activation energies for selected compounds is shown in Fig. 5b; errors are, at least, in the order of ±0.01 eV.
As mentioned in the beginning, Ca2+ incorporation increases the ionic conductivity of LZP. Fig. 6b shows the change in σDCT(P1, P2) of Li1+2xCaxZr2−x(PO4)3 prepared from ZrO2 for x = 0, x = 0.1 and x = 0.2. Most importantly, while σDCT(P2), which refers to bulk ion dynamics, is only slightly affected by x (Ea ranges from 0.41 eV to 0.43 eV, see Fig. 6b and Table 1 that also includes the prefactors), Ca2+ incorporation mainly reduces the g.b. resistance. We clearly see that σDCT(P1) is by two orders of magnitude larger than that of the sample with x = 0, see Fig. 6b. Most likely, the sintering process benefits from a Ca-rich composition, which helps reducing the blocking nature of surface regions of the Li1+2xCaxZr2−x(PO4)3 crystallites. For x = 0.2 we obtain σDC(P2) = 4.2 × 10−5 S cm−1 at 293 K. This value is only slightly lower than that of Li et al.15 (3.8 × 10−5 S cm−1) when samples are compared that have been prepared by conventional sintering. If pellets were fired by spark plasma sintering, bulk values of 1.8 × 10−4 S cm−1 were reported.15 It would be interesting to see, in coming studies, whether Ca2+, here acting as a sintering aid, segregates in the g.b. regions; such segregation would explain the finding that ion transport in the bulk regions is only little affected by Ca2+ incorporation.
Careful inspection of σDCT(P1) reveals that, if the samples with x = 0.2 is considered as an example (Fig. 6b), a slight kink is seen for temperatures well above room temperature; the activation energy changes from 0.48 eV to 0.38 eV at higher T. Presumably, Ca2+ in the grain boundaries influences the Li+ arrangements in these regions. Li+ order/disorder phenomena might lead to such changes.
3.3. Ion dynamics as seen by NMR measurements
Fig. 7a gives an overview of the 7Li NMR SLR rate measurements performed using an Arrhenius representation plotting the R1(ρ) rates as a function of the inverse temperature. To identify the thermally activated regions, we measured R1(ρ) of Li1.4Ca0.2Zr1.8(PO4)3 over a wide temperature range to detect the maxima of diffusion-induced rate peaks.48 Here we focused on the sample with the composition Li1.4Ca0.2Zr1.8(PO4)3 as it shows the lowest bulk activation energy and the lowest amount of residual ZrO2.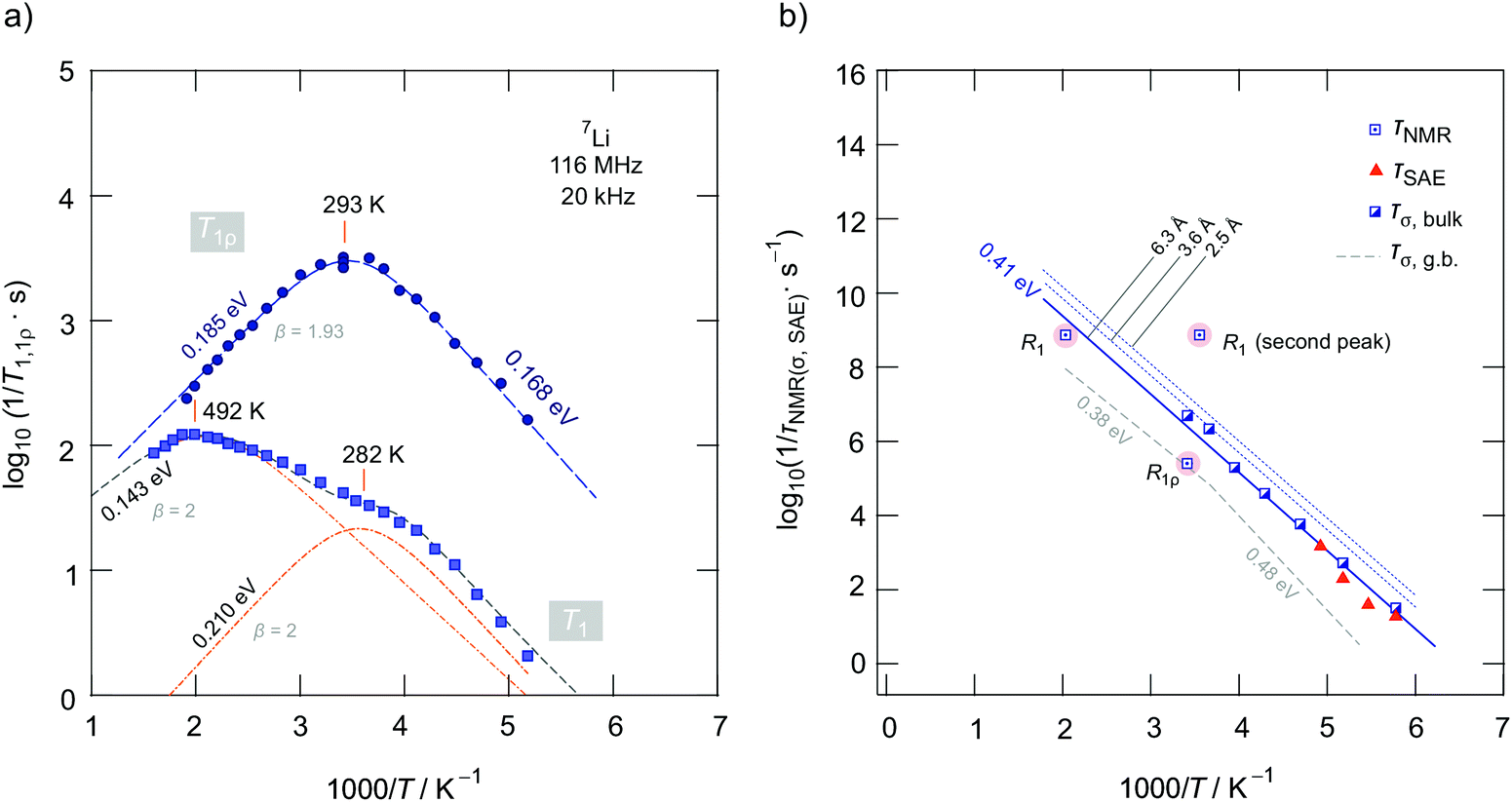 | ||
| Fig. 7 (a) Arrhenius plot of the 7Li NMR relaxation rates R1 and R1ρ of Li1.4Ca0.2Zr1.8(PO4)3 (prepared via the route using ZrO2) measured in the laboratory frame of reference (116 MHz) and in the rotating frame of reference (20 kHz, nominal locking frequency). Dashed lines represent BPP-type fits to determine activation energies, prefactors and asymmetry parameters β. Temperatures indicate Tmax of the rate peaks. The superposition of the R1 rate peaks can be approximated with two symmetric peaks (β = 2). (b) Li+ jump rates as deduced from the diffusion-induced rate peaks R1 and R1ρ. For comparison, we also included jump rates that we estimated from frequency-dependent conductivity measurements and 7Li SAE NMR. See text for further explanations and Table 2 for error margins of the activation energies shown. | ||
Below 183 K the rates R1 reveal a non-diffusive background regime. In this temperature range longitudinal relaxation is induced by lattice vibrations or coupling of the Li spins with paramagnetic impurities.48–50 At higher temperatures we expect the SLR rate to be increasingly induced by Li+ hopping processes. Such processes lead to magnetic and electric field fluctuations that cause longitudinal relaxation.48 Indeed, the rates increase with temperature and, in both cases R1 and R1ρ, characteristic diffusion-induced rate peaks appear. Importantly, we recognize that R1 passes through two maxima located at Tmax = 282 K and 492 K, respectively. In general, at Tmax the motional correlation rate 1/τc is related to ω0via the relation τcω0 ≈ 1. The so-called motional correlation rate 1/τc is identical, within a factor of two, with the Li+ jump rate 1/τ.48,50 For R1ρ, this maximum condition changes to τcω1 ≈ 0.5.51 As ω0 and ω1 differ by more than three orders of magnitude, we are able to characterize Li+ motional correlation rates in LCZP with values in both the kHz and MHz range.52
Here, we approximated the superposition of the two R1 rate peaks by a sum of two Lorentzian-shaped spectral density functions J(ω0, T) ∝ τc/(1 + (ω0τc)β) according to the concept introduced by Bloembergen, Purcell and Pound (BPP) for 3D isotropic diffusion,53,54 see dashed line in Fig. 7a that follows the R1 rates. For each peak we used a single term to deconvolute the temperature dependence of the overall R1 rates measured, cf. the dashed-dotted lines in Fig. 7a, which will be discussed below. The rate 1/τc usually obeys the Arrhenius relation 1/τc = 1/τc,0exp(−Ea/(kBT)); 1/τc,0 represents the pre-exponential factor that is typically identified as the “attempt frequency” of the jump process.55
In general, J(ω0, T) is the Fourier transform of the underlying motional correlation function G(t′).48 If G(t′) is or can be well approximated with a single exponential, β equals 2. Values smaller than 2 are expected for correlated motion, which is, e.g., seen for cations exposed to an irregularly shaped potential landscape. In such a landscape short-ranged Li+ diffusion will be different to long-range ion transport. In particular, forth-and-back jumps or, more generally speaking, localized motions will govern the rate R1 in the low-temperature regime, which is characterized by τcω0 ≫ 1. In this regime we have J(ω0) ∝ τc−1ω0−β with (1 < β ≤ 2). β < 2 produces asymmetric rate peaks which are often found for structurally complex ion conductors with a non-uniformly shaped energy landscape. Ion dynamics in this regime are anticipated to be affected by correlation effects because of both structural disorder and strong Coulomb interactions of the moving ions.56 As the peak is asymmetric, the activation energy on this side of the peak, Ea, low, is lower than that of the high-temperature flank, Ea, high; the two values are linked to each other via Ea, low = (β(ρ) − 1)Ea, high. In the regime τcω0 ≪ 1, that is, on the high-temperature side of the peak, many jump events are sensed during one Larmor precession and the probability is high that also these jumps contribute to longitudinal relaxation which are characterized by higher activation energies. Usually, on this side of the rate peak R1(1/T) long-range Li ion dynamics is sensed; in this limit we obtain J(ω0) ∝ τc.29
In the present case, approximating the rate peaks with a sum of two BPP-type spectral densities yields activation energies of Ea,1 = 0.210(5) eV and Ea,2 = 0.143(5) eV. As both rate peaks join up, information on β are difficult to obtain. Here, the fitting routine yields β = 2 for the two peaks resulting in symmetric peaks with Ea, low = Ea, high = Ea,i (i = 1, 2). Note that the peak at higher T (i = 2) is only partly visible which influences the precise determination of Ea, high. We clearly recognize that Ea,i as determined from R1 SLR NMR turned out to be significantly lower than Ea obtained from σDC measurements describing bulk ion dynamics. The fact that Ea,i < Ea, DC shows that the number of jump events seen by NMR does not include all types of jumps needed for long-range diffusion. Obviously, the spin-fluctuations sensed by NMR are already sufficient to generate a full R1 peak. Thus, we conclude that the Li+ ions in LCZP are highly mobile on a short-range length scale while long-range ion transport is, however, characterized by much larger activation energies than 0.2 eV. This situation resembles that of Li+ ion dynamics in argyrodite-type Li6PS5I, which has been studied recently by our group.42 Here, we assume that rapid forward–backward exchange processes between the sites A1 and A2 might be responsible for the peaks seen in NMR spin–lattice relaxometry. A2 sites might be occupied in samples with x > 0. Here, especially the peak appearing at Tmax = 282 K for Li1.4Ca0.2Zr1.8(PO4)3 points to rapid (localized) exchange processes with residence times in the order of several ns. According to ω0τc ≈ 1 we estimate that at Tmax the jump rate should be in the order of 1/τ = 7.3 × 108 s−1 ≈ 109 s−1. A very similar behavior has recently been seen also for NaSICON-type Na3.4Sc0.4Zr1.6(SiO4)2PO4.
To complement our R1 measurements, we carried out spin-lock NMR SLR measurements at a locking frequency of 20 kHz. As expected we detected a prominent spin-lock NMR peak R1ρ(1/T) at much lower temperature than 492 K. A single peak appears at Tmax = 293 K. It turned out to be slightly asymmetric with an activation energy of Ea, high = 0.185(5) eV and Ea, low = 0.168(5) eV. At first glance we would say that the R1ρ peak might correspond to the R1 peak seen at Tmax = 492 K. Keeping, however, both the locking frequency of only 20 kHz and the rather low activation energy of 0.185 eV in mind, the R1ρ peak belonging to R1(1/T) with Tmax = 492 K would be expected to appear at much lower temperatures than ambient. Even if we replace ω1 by an effective frequency ω1, eff (>ω1), which takes local magnetic fields into account that increase ω1, no satisfactory joint fit results that is characterized by the same Ea and the same τc,0 for the two peaks. Here, only unreliably high ω1, eff values reaching the MHz range would result in a joint fit connecting the two peaks. Table 2 shows an overview of the results obtained from analyzing the three peaks individually by BPP-type spectral density functions. It also includes the amplitudes CNMR in R1(ρ) = CNMRJ(ω0, T). We see that the amplitudes of the two R1 rate peaks differ by approximately one order of magnitude. Most likely, stronger quadrupolar relaxation governs the peak appearing at higher T. The corresponding prefactor 1/τ0 is relatively low, while that of the peak showing up at 282 K (1/τ0 = 5.9 × 1012 s−1) is consistent with frequencies typically expected for phonons.
| E a (= Ea, high) | C NMR | β (ρ) | τ 0 | |
|---|---|---|---|---|
| a The value in brackets refers to Ea, low of the R1ρ peak seen at 293 K. | ||||
| R 1 (Tmax = 492 K) | 0.143(5) eV | 1.8(1) × 1011 s−2 | 2 | 4.1(2) × 10−11 s |
| R 1 (Tmax = 282 K) | 0.210(5) eV | 2.5(1) × 1010 s−2 | 1.93 | 1.7(2) × 10−13 s |
| R 1,ρ (Tmax = 293 K) | 0.185(5) eV | 1.5(1) × 109 s−2 | 2 | 2.8(1) × 10−9 s |
| (0.168(5) eV)a | ||||
The above-mentioned jump rate derived from SLR NMR at Tmax (1/τ = 7.3 × 108 s−1) can be converted into diffusion coefficients with the help of the Einstein–Smoluchowski equation according to DNMR = a2/(6τ), which is valid for 3D diffusion.29 As an estimation, by inserting a = 6.3 Å, which is simply the shortest Li–Li distance, we obtain DNMR = a2/(6τ) = 4.8 × 10−11 m2 s−1. Assuming the interstitial sites A2 participating in exchange processes DNMR reduces to 1.8 × 10−11 m2 s−1. A distance of a = 6.3 Å is, of course, by far too long for a single hopping process. In the structural model of Catti et al. the distance reduced to 4.2 Å at 423 K.24
In the Arrhenius plot of Fig. 7b we compare the rates 1/τ from NMR (1/τNMR) with those obtained after converting σDC into jump rates by using the Nernst Einstein equation. As we do not know the exact jump distance, we calculated 1/τσ for three different values of a. 1/τσ represents an average value mainly influenced by Li+ jumps necessary for long-range ion transport. Most likely, the R1 peak appearing at 492 K is more related to jump processes enabling the ions to move over longer distances. The large discrepancy between 1/τNMR(282 K) and 1/τσ once again visualizes that the R1(1/T) peak at low T reflects localized jump processes. We do not find evidences that this peak originates from any phase transitions, as the phase-pure Ca-bearing samples show rhombohedral structure. For comparison, in Fig. 7b we also included 1/τσ rates estimated from conductivity values that characterize the influence of grain boundaries. Interestingly, 1/τNMR(293 K, R1ρ) = 2.5 × 105 s−1, which we estimated via the relation ω1τc ≈ 0.5, see above, agrees with 1/τσ, g.b. In contrast to R1, which as far as μm-sized crystallites are considered, is mainly affected by bulk processes, the corresponding spin-lock NMR rates R1ρ seems to be (also partly) sensitive to ion dynamics also influenced by interfacial processes. This behaviour is consistent with the following comparison. At frequencies in the kHz range (ω1/2π = 20 kHz) and at T = 282 K the isotherms σ′(ν) are mainly influenced by the g.b. response rather than bulk ion dynamics.
To shed more light on long-range ion dynamics, we used 7Li NMR line shape measurements and 7Li SAE NMR experiments46,57–60 to further characterize ionic transport in LCZP. Via SAE NMR we should be able to get access to diffusion parameters that characterize ion transport over longer distances as the method is sensitive to exchange processes on the time scale that is comparable to that of DC conductivity measurements.48 In Fig. 8a variable-temperature 7Li NMR spectra of Li1.4Ca0.2Zr1.8(PO4)3 are shown. Remarkably, at temperatures as low as 213 K a relatively narrow NMR line is detected whose width is only 1.4 kHz. Usually, we would expect a width in the order of several kHz due to 7Li–7Li dipolar interactions. Here, the large Li–Li distance of 6.3 Å between the A1 sites, and between A1 sites and Li ions occupying interstitial sites in samples with x > 0, leads to relatively weak dipole–dipole interactions producing a narrow line already in the rigid-lattice regime. With increasing temperature, the line undergoes a slight narrowing process because of Li diffusion that averages dipolar couplings. Finally, at even higher temperatures, i.e., in the extreme narrowing regime, its width is only governed by the inhomogeneity of the external magnetic field.
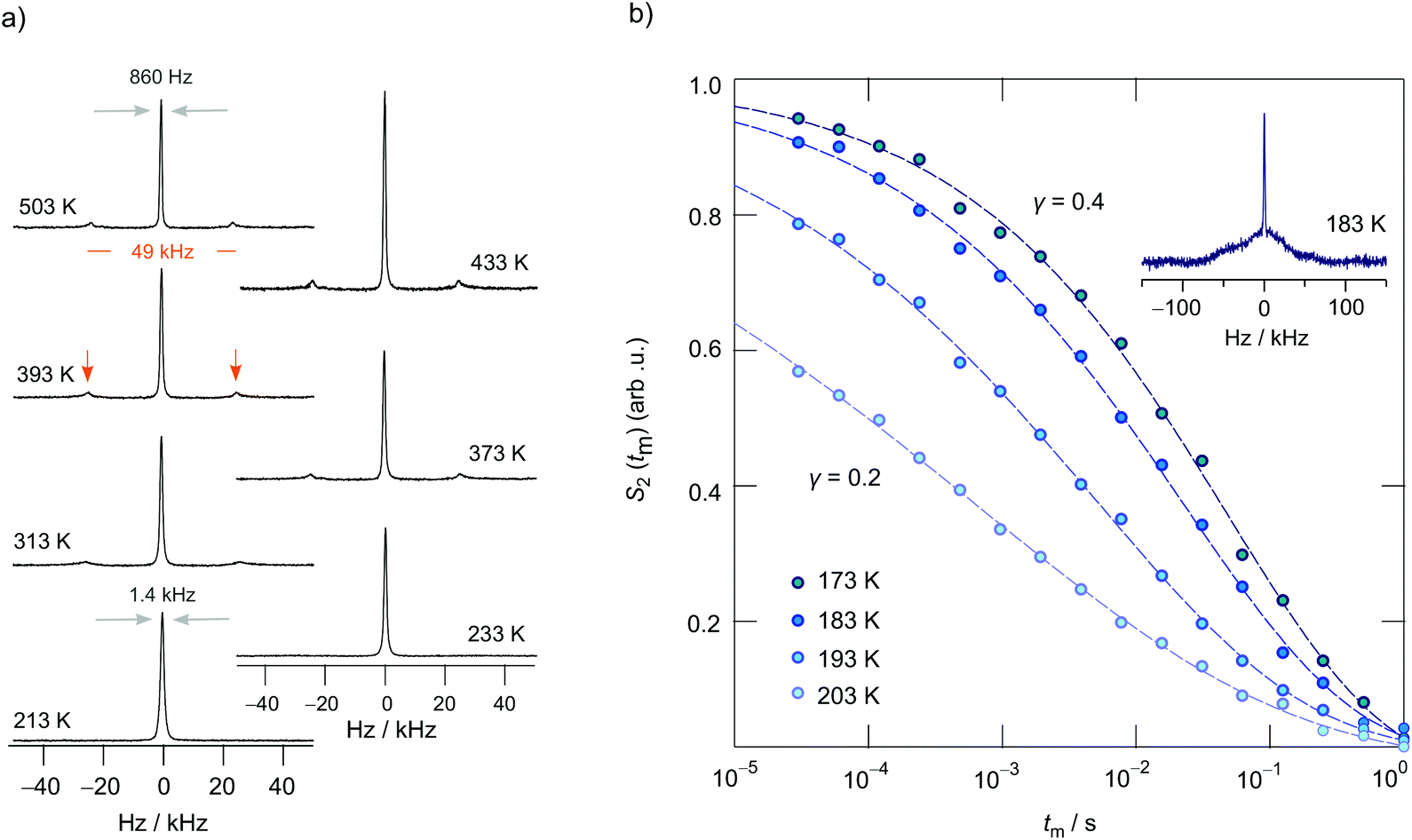 | ||
| Fig. 8 (a) 7Li NMR spectra of Li1.4Ca0.2Zr1.8(PO4)3 recorded at the temperatures indicated. The NMR line transforms from a Gaussian shape at low temperatures to a Lorentzian one at elevated T. Clearly visible and well-defined quadrupole powder patterns emerge at temperatures higher than 313 K. (b) 7Li SAE NMR decay curves of LCZP whose spectra are shown in (a). Decay curves follow stretched exponentials (0.2 < γ < 0.4) which are shown as solid lines. Data have been recorded at a Larmor frequency of 116 MHz. See text for further details. Inset: Fourier transform of a spin-alignment echo, starting from the top of the echo, which was recorded at a fixed tp of 20 μs and a short mixing time of tm = 100 μs. In addition to a “central line” a rather broad quadrupole foot is visible illustrating the distribution of EFGs seen by the Li ions in the dynamic regime of the rigid lattice. | ||
Interestingly, at 313 K two satellite lines emerge that belong to a first-order quadrupole powder pattern arising from the interaction of the quadrupole moment of the 7Li nucleus (I = 3/2) with a non-vanishing electric field gradient (EFG) at the nuclear site. The EFG is produced by the electric charge distribution in the direct neighborhood of the 7Li nucleus. This additional interaction alters the Zeeman levels and, thus, also the associated (angular) Zeeman frequency ω0 towards ω0 ± ωQ.32,54 The singularities seen in Fig. 8a correspond to the 90° satellite transitions of a powder pattern, which typically show up at sufficiently high T; this feature belongs to the universal characteristics of crystalline materials studied by NMR.32,50,61 Their distance on the frequency scale, if we simply assume an EFG with axial symmetry, corresponds to δ/2. δ is the quadrupole coupling constant. A distance of 49 kHz leads to δ = 98 kHz,58 which is in fair agreement with the value reported by Petit et al.20 Here, this value should, however, be interpreted as an average value, as we cannot exclude a small distribution of EFGs the ions are exposed to at very low temperatures. At very low T, the intensity of the singularities is too low to be detectable by single pulse experiments. Instead, echo experiments should be used that are able to avoid receiver dead time effects. In Fig. 8b (see inset) a spectrum is shown that is the Fourier transform of a stimulated echo. Indeed, a sharp central line is located on top of a broad quadrupole foot. Hence, we conclude that the Li ions are exposed to a distribution of EFGs.
Fluctuations in ωQ seen by the ions when jumping between electrically inequivalent sites can be used to record sinus–sinus two-time correlation functions. In Fig. 8a the change of the 7Li SAE amplitude S2 is shown vs. the logarithmic mixing time tm. S2 depends on both the preparation time tp and tm. Here, we measured the decay curve at fixed tp (= 20 μs) but variable mixing time. Stretched exponentials of the form S2 ∝ exp(−(tm/τSAE)λ), with a stretching factor λ ranging from 0.21 to 0.37, are best suited to describe the dependence of S2(tp = const., tm) in this temperature regime. In general, stretching factors deviating from λ = 1 indicate non-Debye-like motional process. For example, such deviations can arise from motions in disordered matrices or in confined dimensions59,62 leading to motional correlation functions whose decay slows down with increasing observation time. With increasing T the inflexion point of the echo decay curves shifts towards shorter tm. At the same time, the shape of S2 steadily becomes more stretched until a value of λ = 0.21 is reached at T = 203 K. At sufficiently long mixing times the curves S2(tp = const., tm) always reach S2,∞ = 0, which either indicates a rather large number of quadrupole frequencies involved or which points to the influence of dipolarly coupled spins, as is well-described in literature.36 The rates 1/τSAE governing the stretched decay functions are included in Fig. 7b. We recognize that they are in fair agreement with those rates, 1/τσ, bulk, which were estimated from bulk ionic conductivities of Li1.4Ca0.2Zr1.8(PO4)3.
4. Conclusion
We used a conventional solid-state reaction procedure to synthesize NaSICON-type LiZr2(PO4)3 and investigated both its ionic conductivity and Li+ diffusivity by broadband conductivity measurements and NMR spectroscopy. Ca2+ incorporation helps prepare Li1+2xZr2−xCax(PO4)3 crystallizing with rhombohedral symmetry. While bulk ion dynamics is not influenced by the Ca2+ content, the grain boundaries in LZCP turn out to be less blocking for Li+ ions as compared to the sample with x = 0. We observed an increase in the low-frequency ionic conductivity by two orders of magnitude when increasing x from x = 0 to x = 0.2. For Li1.4Ca0.2Zr1.8(PO4)37Li NMR relaxometry revealed rapid localized Li+ jump processes with activation energies of 0.21 eV and 0.14 eV. The diffusion-induced rate peak seen at 282 K points to a very high jump rate in the order of 109 s−1 at this temperature. On the other hand, 7Li spin-alignment echo spectroscopy confirmed that long-range ion transport in the bulk regions of Li1.4Zr1.8Ca0.2(PO4)3 needs to be characterized by an activation energy of 0.41 eV as determined by variable-frequency conductivity measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 769929. We thank the Deutsche Forschungsgemeinschaft for further support (FOR1277, WI3600(2-1;4-2). Further support by the K-project ‘safe battery’ (FFG) is highly appreciated.References
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- J. I. Hur, L. C. Smith and B. Dunn, Joule, 2018, 2, 1187–1201 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- Z. Z. Zhang, Y. J. Shao, B. Lotsch, Y. S. Hu, H. Li, J. Janek, L. F. Nazar, C. W. Nan, J. Maier, M. Armand and L. Q. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- Y. Inaguma, K. Funayama, A. Aimi, D. Mori, Y. Hamasaki, K. Ueda, M. Ikeda, T. Ohno and K. Mitsuishi, Solid State Ionics, 2018, 321, 29–33 CrossRef CAS.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sci. Rep., 2013, 3, 2261 CrossRef PubMed.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- M. Guin, F. Tietz and O. Guillon, Solid State Ionics, 2016, 293, 18–26 CrossRef CAS.
- H. Xie, J. B. Goodenough and Y. Li, J. Power Sources, 2011, 196, 7760–7762 CrossRef CAS.
- V. Ramar, S. Kumar, S. R. Sivakkumar and P. Balaya, Electrochim. Acta, 2018, 271, 120–126 CrossRef CAS.
- S. Hasegawa, N. Imanishi, T. Zhang, J. Xie, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2009, 189, 371–377 CrossRef CAS.
- Y. T. Li, W. D. Zhou, X. Chen, X. J. Lu, Z. M. Cui, S. Xin, L. G. Xue, Q. X. Jia and J. B. Goodenough, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13313–13317 CrossRef CAS PubMed.
- H. Xie, Y. Li and J. B. Goodenough, RSC Adv., 2011, 1, 1728–1731 RSC.
- C. R. Mariappan, P. Kumar, A. Kumar, S. Indris, H. Ehrenberg, G. Vijaya Prakash and R. Jose, Ceram. Int., 2018, 44, 15509–15516 CrossRef CAS.
- L. Xiaojuan, F. Xue, L. Wenwei, L. Haitao and Z. Yunjie, Adv. Engin. Res., International Conference on Advances in Energy, Environment and Chemical Engineering, 2015, pp. 227–230, DOI: DOI:10.2991/aeece-15.2015.45.
- D. Petit, P. Colomban, G. Collin and J. P. Boilot, Mater. Res. Bull., 1986, 21, 365–371 CrossRef CAS.
- D. Petit and B. Sapoval, Solid State Ionics, 1986, 21, 293–304 CrossRef CAS.
- M. Casciola, U. Costantino, L. Merlini, I. G. K. Andersen and E. K. Andersen, Solid State Ionics, 1988, 26, 229–235 CrossRef CAS.
- M. Catti and S. Stramare, Solid State Ionics, 2000, 136–137, 489–494 CrossRef CAS.
- M. Catti, S. Stramare and R. Ibberson, Solid State Ionics, 1999, 123, 173–180 CrossRef CAS.
- M. Catti, A. Comotti and S. Di Blas, Chem. Mater., 2003, 15, 1628–1632 CrossRef CAS.
- H. El-Shinawi, C. Greaves and J. Janek, RSC Adv., 2015, 5, 17054–17059 RSC.
- F. Preishuber-Pflügl, P. Bottke, V. Pregartner, B. Bitschnau and M. Wilkening, Phys. Chem. Chem. Phys., 2014, 16, 9580–9590 RSC.
- S. Lunghammer, Q. Ma, D. Rettenwander, I. Hanzu, F. Tietz and H. M. R. Wilkening, Chem. Phys. Lett., 2018, 701, 147–150 CrossRef CAS.
- B. Stanje, D. Rettenwander, S. Breuer, M. Uitz, S. Berendts, M. Lerch, R. Uecker, G. Redhammer, I. Hanzu and M. Wilkening, Ann. Phys., 2017, 529, 1700140 CrossRef.
- M. Uitz, V. Epp, P. Bottke and M. Wilkening, J. Electroceram., 2017, 38, 142–156 CrossRef CAS.
- V. Epp, Q. L. Ma, E. M. Hammer, F. Tietz and M. Wilkening, Phys. Chem. Chem. Phys., 2015, 17, 32115–32121 RSC.
- N. P. Kuz'mina, A. E. Altsybeev, I. P. Malkerova, A. S. Alikhanyan and I. E. Korsakov, Russ. J. Inorg. Chem., 2006, 51, 1750–1754 CrossRef.
- V. Epp, Ö. Gün, H. J. Deiseroth and M. Wilkening, Phys. Chem. Chem. Phys., 2013, 15, 7123 RSC.
- E. Fukushima and S. B. W. Roeder, Experimental Pulse NMR. A Nuts and Bolts Approach, Addison-Wesley Publ. Comp., Reading, 1981 Search PubMed.
- R. Böhmer, J. Magn. Reson., 2000, 147, 78–88 CrossRef PubMed.
- J. Jeener and P. Broekaert, Phys. Rev., 1967, 157, 232–240 CrossRef CAS.
- F. Qi, G. Diezemann, H. Böhm, J. Lambert and R. Böhmer, J. Magn. Reson., 2004, 169, 225–239 CrossRef CAS.
- C. Masquelier, C. Wurm, J. Rodríguez-Carvajal, J. Gaubicher and L. Nazar, Chem. Mater., 2000, 12, 525–532 CrossRef CAS.
- A. Aatiq, M. Ménétrier, L. Croguennec, E. Suard and C. Delmas, J. Mater. Chem., 2002, 12, 2971–2978 RSC.
- K. Arbi, M. Ayadi-Trabelsi and J. Sanz, J. Mater. Chem., 2002, 12, 2985–2990 RSC.
- I. A. Stenina, Y. A. Velikodnyi, V. A. Ketsko and A. B. Yaroslavtsev, Inorg. Mater., 2004, 40, 967–970 CrossRef CAS.
- J. Sanz, J. M. Rojo, R. Jiménez, J. E. Iglesias and J. Alamo, Solid State Ionics, 1993, 62, 287–292 CrossRef CAS.
- I. Hanghofer, M. Brinek, S. L. Eisbacher, B. Bitschnau, M. Volck, V. Hennige, I. Hanzu, D. Rettenwander and H. M. R. Wilkening, Phys. Chem. Chem. Phys., 2019, 21, 8489–8507 RSC.
- A. K. Jonscher, Nature, 1977, 267, 673–679 CrossRef CAS.
- D. L. Sidebottom, Phys. Rev. Lett., 1999, 83, 983–986 CrossRef CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- B. Ruprecht, H. Billetter, U. Ruschewitz and M. Wilkening, J. Phys.: Condens. Matter, 2010, 22, 245901 CrossRef CAS PubMed.
- S. Breuer, D. Prutsch, Q. L. Ma, V. Epp, F. Preishuber-Pflügl, F. Tietz and M. Wilkening, J. Mater. Chem. A, 2015, 3, 21343–21350 RSC.
- M. Wilkening and P. Heitjans, ChemPhysChem, 2012, 13, 53–65 CrossRef CAS PubMed.
- M. Wilkening, V. Epp, A. Feldhoff and P. Heitjans, J. Phys. Chem. C, 2008, 112, 9291–9300 CrossRef CAS.
- A. Kuhn, S. Narayanan, L. Spencer, G. Goward, V. Thangadurai and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094302 CrossRef.
- A. Kuhn, M. Kunze, P. Sreeraj, H. D. Wiemhöfer, V. Thangadurai, M. Wilkening and P. Heitjans, Solid State Nucl. Magn. Reson., 2012, 42, 2–8 CrossRef CAS PubMed.
- M. Wilkening, V. Epp, A. Feldhoff and P. Heitjans, J. Phys. Chem. C, 2008, 112, 9291–9300 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- A. Abragam, The Principles of Nuclear Magnetism, Claredon Press, Oxford, 1961 Search PubMed.
- P. M. Richards, in Topics in Current Physics, ed. M. B. Salamon, Springer, Berlin, 1979, vol. 15 Search PubMed.
- A. Bunde, P. Maass and M. Meyer, Phys. A, 1992, 191, 433–437 CrossRef CAS.
- P. Bottke, D. Rettenwander, W. Schmidt, G. Amthauer and M. Wilkening, Chem. Mater., 2015, 27, 6571–6582 CrossRef CAS.
- M. Wilkening and P. Heitjans, J. Phys.: Condens. Matter, 2006, 18, 9849–9862 CrossRef CAS.
- M. Wilkening and P. Heitjans, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 024311 CrossRef.
- M. Wilkening, A. Kuhn and P. Heitjans, Phys. Rev. B, 2008, 78, 054303 CrossRef.
- R. Bertermann, W. Müller-Warmuth, C. Jansen, F. Hiltmann and B. Krebs, Solid State Ionics, 1999, 117, 245–255 CrossRef CAS.
- P. Bottke, Y. Ren, I. Hanzu, P. G. Bruce and M. Wilkening, Phys. Chem. Chem. Phys., 2014, 16, 1894–1901 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Further X-ray powder patterns, results from structure solution and Nyquist plots. See DOI: 10.1039/c9dt01786k |
| This journal is © The Royal Society of Chemistry 2019 |