
Local magnetic spin mismatch promoting photocatalytic overall water splitting with exceptional solar-to-hydrogen efficiency†
Yiyang
Li
 a,
Zihan
Wang
b,
Yiqi
Wang
b,
András
Kovács
c,
Christopher
Foo
a,
Rafal E.
Dunin-Borkowski
c,
Yunhao
Lu
d,
Robert A.
Taylor
e,
Chen
Wu
*b and
Shik Chi Edman
Tsang
*a
a,
Zihan
Wang
b,
Yiqi
Wang
b,
András
Kovács
c,
Christopher
Foo
a,
Rafal E.
Dunin-Borkowski
c,
Yunhao
Lu
d,
Robert A.
Taylor
e,
Chen
Wu
*b and
Shik Chi Edman
Tsang
*a
aWolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk; chen_wu@zju.edu.cn
bSchool of Materials Science and Engineering, State Key Laboratory of Silicon Materials, Zhejiang University, Hangzhou, 310027, China
cErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons and Peter Grünberg Institute, Forschungszentrum Jülich, 52425 Jülich, Germany
dZhejiang Province Key Laboratory of Quantum Technology and Device, Department of Physics, Zhejiang University, Hangzhou, 310027, China
eClarendon Laboratory, Department of Physics, University of Oxford, Oxford, OX1 3PU, UK
First published on 1st December 2021
Abstract
The photocatalytic overall water splitting (POWS) reaction using particulate catalysts is considered as an ideal approach for capturing solar energy and storing it in the form of hydrogen, however, current POWS systems are hindered by the slow separation but fast recombination of the photo-generated charge carriers, hence giving unsatisfactory performances. Here we report a dramatically improved POWS system for a Au-supported Fe3O4/N-TiO2 superparamagnetic photocatalyst promoted by local magnetic field effects. Strong local magnetic flux was induced by a weak external magnetic field of 180 mT, which then resulted in a quantum efficiency of 88.7% at 437 nm at 270 °C without any sacrificial reagent. The mechanism of the magnetic field effects was explored systematically and quantitatively by time-resolved spectroscopic technique and first-principles calculations, which suggested such enhancement was due to the greatly prolonged excitonic lifetime, originating from both the Lorentz force and spin-polarisation effects. By controllable manipulation of both features using local magnetic field, an unprecedented solar-to-hydrogen conversion efficiency of 11.9 ± 0.5% and an overall energy efficiency of 1.16 ± 0.05% were achieved in a particulate POWS system under AM 1.5G simulated solar illumination, which exceeds the STH goal of 10% for practical applications of POWS systems imposed by the United States Department of Energy.
Broader contextSolar energy is attracting increasing interest as a sustainable and clean energy source, but for efficient storage and transport, solar energy is required to be converted to other energy forms such as chemical fuels. Hydrogen (H2) is considered as an ideal chemical fuel because of its highest energy-capacity and its carbon-emission-free advantage. Therefore, solar-light-driven photocatalytic overall water splitting (POWS) has become a promising approach for converting solar energy into H2. However, current POWS systems are suffering from the intrinsic slow generation but fast recombination of the photo-generated charge carriers of the semiconductor photocatalysts. Herein, we report a highly efficient POWS system for a TiO2-based superparamagnetic photocatalyst promoted by local magnetic field effects. The mechanism of the magnetic field effects was explored systematically and quantitatively by time-resolved photoluminescence technique and first-principles calculations, which suggested such enhancement was due to the greatly prolonged excitonic lifetime, originating from the Lorentz force and spin-polarisation effects. An unprecedented solar-to-hydrogen (STH) conversion efficiency of 11.9 ± 0.5% was achieved in a particulate POWS system under AM 1.5G simulated solar illumination. The magnetic field effects demonstrated here could presumably contribute to other solar conversion systems. |
Introduction
Photocatalytic overall water splitting (POWS) reaction using particulate nano-catalysts has attracted increasing attention in recent decades, and is recognised as a potential strategy for harnessing solar energy to supply clean hydrogen fuel. Although extensive efforts have been made, the solar-to-hydrogen (STH) efficiencies of currently reported particulate POWS systems are still hindered by the intrinsic slow generation but fast recombination of the photo-generated charge carriers of the semiconductor materials used. The United States Department of Energy (US DoE) imposed a goal of 10% STH efficiency for practical applications of the particulate POWS systems,1 however, STH efficiencies of less than 5% have been achieved for particulate POWS systems so far.2–4 It is recently recognised that a broadened absorption would not necessarily lead to improved photocatalytic performance, and the challenge is to improve QEs by facilitating the slow intrinsic charge separation.5 The photo-generated charge carriers must have sufficient lifetimes to migrate to the surface active centres to allow chemical reactions to take place, instead of recombining to generate heat or photoluminescence irradiation.6 Conventional approaches to prolong excitonic lifetimes (e.g. fabrication of a built-in electric field in the materials and interfaces) normally need sophisticated material design but give limited enhancement.2,7–12We have recently reported that the separation of the photo-generated charge carriers and the regeneration of oxygen vacancies (VOs) can be promoted by using elevated temperatures, which also leads to enhanced water dissociation and favourable reaction kinetics.5,13–15 Such observation allowed us to further investigate the impacts of local fields: for example, it is noted that the excitonic lifetime and the POWS activity are both enhanced linearly by the local electric field introduced by polar-faceted materials at 270 °C.5,14 Building upon these previous works, we here explore systematically magnetic field effects (MFEs) on POWS performance as a novel non-contact technique. MFEs in particulate POWS system has scarcely been reported before, because it is conventionally believed that the Zeeman energy produced by a magnetic field is negligible compared to the Gibbs free energy of the POWS reaction (ca. 273 kJ mol−1), thus will not contribute to the thermodynamics.16–18 But studies on MFEs in other photocatalytic systems are emerging very recently, including nitrogen fixation,19 denitrification of contaminated water,20 CO2 conversion21 and dye degradation,16,17,22 where an external magnetic field is applied during material preparation or reaction. Such effects have been attributed to enhanced adsorption of charged species,20 regulation of internal field,19 facilitated migration of charge carriers,19,20etc. Obviously, even though the Zeeman energy is small, MFEs could still contribute to photocatalysis via other pathways. However, it should be noted that common semiconductor photocatalysts such as TiO2 and BaTiO3 actually exhibit very low magnetic flux even under strong magnetic fields,16,17,19,20 hence giving insignificant enhancement in photocatalytic performance. Moreover, the mechanism of MFEs has not been systematically studied in the reported works, thus deep understandings of such effects are still absent. Here in this work, we demonstrate that by placing nitrogen-doped TiO2 (N-TiO2) photocatalyst in proximity to superparamagnetic Fe3O4 nanoparticles (NPs) under an external magnetic field, a very strong local magnetic flux can be induced, which leads to efficient hydrogen and oxygen evolution in a stoichiometric 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio from pure water under visible light irradiation at 270 °C. An extraordinary hydrogen evolution rate (21.2 mmol g−1 h−1), QE (e.g. 88.7% at 437 nm) and an unprecedented STH efficiency of 11.9 ± 0.5% can be achieved, exceeding the practical STH target of 10%. An overall energy efficiency of 1.16 ± 0.05% has also been demonstrated. Careful investigations of the charge and spin properties of electrons suggest that this remarkable enhancement is attributed to the magnetically induced Lorentz force facilitating the charge separation, as well as the dramatic suppression of charge carrier recombination in a strong spin-polarised energy band in N-TiO2 during illumination. This facilitates alternative reaction routes to charge relaxation by water splitting at a high rate at elevated temperature. Mechanism of the MFEs was explored with the help of time-resolved photoluminescence (TRPL) technique and first-principles density-functional theory (DFT) calculations, and the contributions were studied quantitatively at different magnetic flux densities.
:1 ratio from pure water under visible light irradiation at 270 °C. An extraordinary hydrogen evolution rate (21.2 mmol g−1 h−1), QE (e.g. 88.7% at 437 nm) and an unprecedented STH efficiency of 11.9 ± 0.5% can be achieved, exceeding the practical STH target of 10%. An overall energy efficiency of 1.16 ± 0.05% has also been demonstrated. Careful investigations of the charge and spin properties of electrons suggest that this remarkable enhancement is attributed to the magnetically induced Lorentz force facilitating the charge separation, as well as the dramatic suppression of charge carrier recombination in a strong spin-polarised energy band in N-TiO2 during illumination. This facilitates alternative reaction routes to charge relaxation by water splitting at a high rate at elevated temperature. Mechanism of the MFEs was explored with the help of time-resolved photoluminescence (TRPL) technique and first-principles density-functional theory (DFT) calculations, and the contributions were studied quantitatively at different magnetic flux densities.
Experimental section
Material preparation
Fe3O4@SiO2 was prepared from reverse micelles using a previously reported procedure.23 Briefly, Fe3O4 nanoparticles (2 mg) and 100 μL of TEOS were added to a heterogeneous solution containing cyclohexane (24 mL), hexanol (4.8 mL), Triton X-100 (6 mL), and deionised water (1 mL). After 6 h of stirring, NH3·H2O (30 wt%) (100 mL) was added to initiate the hydrolysis of TEOS. The reaction was allowed to continue for another 24 h with stirring at room temperature. The product was well dispersed in ethanol and further purified by centrifugation (14000 rpm, 10 min).
Synthesis of the N-TiO2, Fe3O4/N-TiO2 and Fe3O4@SiO2/N-TiO2 photocatalysts
TiO2 NPs were synthesised via a sol–gel process: solution A was obtained by adding 5 mL of titanium tetraisopropoxide (TTIP) in 15 mL ethanol and solution B is obtained by mixing 10 mL DI water, 10 mL ethanol and 1 mL acetic acid. Then solution A was slowly added to solution B dropwise. A transparent gel forms, which was then aged overnight, following by drying in vacuum oven at 70 °C. Then obtained dry gel was then calcined in N2 atmosphere at 400 °C for 2 h, then treated in ammonia flow at 600 °C for 10 h for N-doping. After the NH3 treatment, the powder product was washed with deionised water for at least 3 time to remove any adsorbed NH3 residuals or any other –NH2, –NH– species, followed by drying in vacuum at 80 °C overnight.Fe3O4/N-TiO2 and Fe3O4@SiO2/N-TiO2 photocatalysts were synthesised following a similar procedure, but adding Fe3O4 or Fe3O4@SiO2 nanoparticles to solution A at the beginning. Photocatalysts containing different amount of Fe3O4 were also synthesised by this method by changing the amount of Fe3O4 NPs added to solution A. The Fe3O4 NPs content was calculated to be 10%, 20%, 30% and 40 wt%, and the as-obtained samples were denoted as Fe3O4/N-TiO2-1, Fe3O4/N-TiO2-2, Fe3O4/N-TiO2-3 and Fe3O4/N-TiO2-4, respectively.
Photocatalysts were all used after treatment with supporting Au nanoparticles (1.0 wt%) via a photo-deposition method: 50 mg of as-obtained photocatalysts was suspended in 60 mL methanol aqueous solution (50 vol%) under vigorous stirring, and a certain amount of solution containing Au precursor was then added into the above suspension. This suspension was irradiated under a 300 W ultraviolet lamp (Helios Italquartz S.R.L.) for 2 hours before being filtered and washed with water and ethanol for 3 times, respectively. The final products were obtained after drying in a 70 °C oven overnight.
Photocatalytic water splitting activity test
The POWS activity was determined by measuring the amount of hydrogen and oxygen evolved from the water splitting. The reactions were carried out in a close 20 mL stainless-steel high-pressure vessel equipped with two quartz windows (10 mm in diameter and 18 mm in thickness) and a glass lining (20 mm i.d. × 24 mm o.d. × 52 mm height). 1 wt% of Au was deposited on all the photocatalysts via the photo-reduction method before testing. In a typical experiment, a certain amount of photocatalyst which contained 5 mg of TiO2 was added to 8 mL of Milli-Q H2O under vigorous magnetic stirring (600 rpm); then the high-pressure vessel was purged with continuous Ar gas flow for 5 min to get rid of the dissolved air (i.e., O2 and N2) in water after being well sealed. The vessel was finally pressurised with 6 bar of Ar gas as the inert environment. The particulate suspension in the reactor was then heated up to 270 °C by electric heaters, establishing the saturated equilibrium pressure of water at the mean time. Tungsten light (70W, Glamox Professional 2000) was then applied through the quartz windows to provide visible-light irradiation after the vessel reached 270 °C. External magnetic field was provided by two paralleled identical magnets. The field strength was modified by changing the distance between the magnets and measured by a Gauss/Tesla meter (Dexing Magnet, DX-150). Generally, the reaction was performed for 2 hours, except the stability tests and recyclability tests, where the reaction time is specified in the Results and discussion. After the reaction, the high-pressure vessel was allowed to cool down naturally to room temperature, and the amounts of hydrogen and oxygen were measured by a gas chromatograph (GC) equipped with two thermal conductivity detectors (TCD) for better sensitivity: H2 was measured by the TCD with N2 as the carrier gas and O2 was measured by the TCD with He as the carrier gas. Each TCD was calibrated for the certain gas separately before the measurements. GC analyses were also performed before reaction to make sure that the O2 dissolved in water was completely removed. The hydrogen evolution rates were calculated based on the amount of N-TiO2 contained in the photocatalysts instead of the total amount, since it is the photocatalytically active component.Isotopic studies were also carried out using deuterium oxide (D2O) under the same conditions as those describe above. The gaseous products were analysed using mass spectrometry (Hiden Analytical) after the reactions.
QE and STH conversion efficiency measurements and calculation
The apparent QE was measured in the same vessel following the same procedure and the POWS performance was evaluated by adding a certain amount of photocatalyst which contained 20 mg of TiO2 to 5 mL of Milli-Q H2O under vigorous magnetic stirring (600 rpm), then the autoclave was irradiated by a 300 W Xenon lamp (Newport) equipped with band-pass filters of 385 ± 20, 437 ± 10, 575 ± 25, 650 ± 20, 750 ± 20 and near infrared (NIR, 800–1200 nm), respectively. Generally, the incident photons were corrected by subtracting the scattered and transmitted light from the incident light: there are two silica windows parallelly equipped on the both sides of the batch reactor, which are facing each other. Thus, the incident light was firstly measured using a light metre in the centre of the batch reactor; and then the scattered and transmitted light was also measured outside the opposite window when the reaction suspension is present. Subsequently, the light coming out of reactor was subtracted from the incident light, and the absorbed light was worked out. The light inside the reactor may also be scattered by the photocatalyst particles, but most of it will be reflected by the stainless-steel surface and finally absorbed by the photocatalyst. Subsequently, the relevant number of incident photons was calculated from the irradiation powers at each wavelength. The apparent QE can be calculated by using the equation: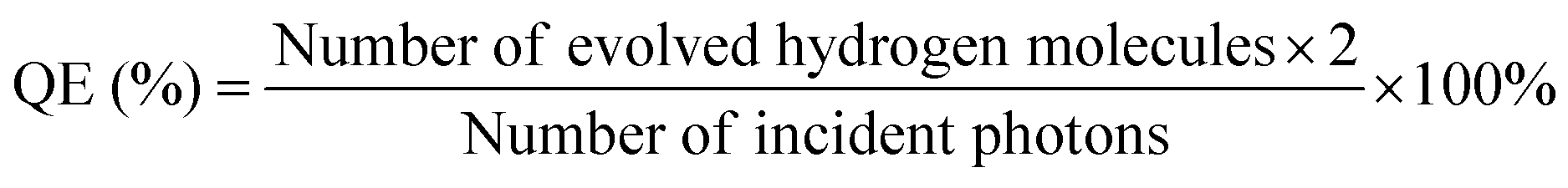 | (1) |
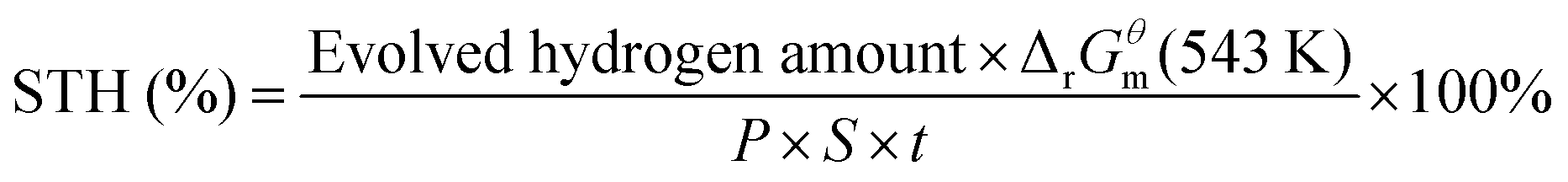 | (2) |
QE and STH measurements were repeated for at least 3 times and the average values and standard deviations were calculated. Examples of QE and STH calculation are given in the ESI† (Notes S1 and S2).
Characterisation
X-ray diffraction (XRD) measurements were performed on a Bruker D8 Advance diffractometer with LynxEye detector and Cu Kα1 radiation (λ = 1.5406 Å); X-ray photoelectron spectroscopy (XPS) measurements were carried out on the Thermo Scientific model Nexsa. Survey scans were obtained at a pass energy of 200 eV, 5 scans with step size 1 eV, whereas for those detailed spectra 50 eV pass energy, 10 scans with 0.1 eV step size were used; Continuous-wave Electron paramagnetic resonance (EPR) spectra were obtained by using an X-band (9.4 GHz) Bruker EMX EPR spectrometer; Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) was performed on a PerkinElmer Lambda 750S UV-visible spectrometer at room temperature. Time-resolved photoluminescence (TRPL) spectroscopy and corresponding lifetimes of excitons were obtained from a bespoke micro-photoluminescence setup, in which a Ti-Sapphire laser (λ = 266 nm, pulse duration = 150 fs, repetition rate = 76 MHz) is directed onto the sample. Time-resolved measurements are performed using the spectrometer as a monochromator before passing the selected signal to a photomultiplier tube (PMT) detector with an instrument response function width of ∼150 ps connected to a time-correlated single-photon counting module. Magnetisation curve measurements (M–H curve) were measured using a superconducting quantum interference device (SQUID, Quantum Design -XL-5).Density functional theory (DFT) calculations
First-principles calculations were carried out within the density functional theory formalism, as implemented in the Vienna ab initio simulation package (VASP)24,25 and generalised gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE)26 was used for the exchange–correlation functional with a projector augmented-wave (PAW) pseudopotentials, treating the N-2s 2p, O-2s 2p, Ti-3p 3d 4s as valence electrons. The kinetic-energy cut-off for the plane-wave basis was set above 400 eV in all calculations and a 3 × 3 × 3 Monkhorst–Pack k-space grid was used for the sampling of the Brillouin zone.27 The convergence criteria electronic and geometric optimisation is 10–5 eV and 0.01 eV Å−1, respectively. The effective on-site Hubbard U (U = 3.0 eV) was employed for Ti 3d orbitals.28 The simulation of the spin states under the influence of the magnetic field was achieved by setting the degeneracy of two spin channels. In the absence of a magnetic field, the degeneracy of two spin channels is preserved for all calculations and the magnetic moments of N-doped TiO2 is not aligned, with both spin channels existing in the bandgap. When a magnetic field is present, the break of the degeneracy is allowed, and the magnetic ordering is set as ferromagnetism to simulate the electronic structure under a magnetic. The partial charge density (the wave function density of spin-up and spin-down electrons) of the CBM was represented by yellow bubbles to see the electron distribution in the supercell. Besides, in the defect band introduced by the N-doping, the charge density of spin-up and spin-down electrons varies in different positions. Therefore, spin density (the wave function density of spin-up/spin-down electrons) could help to investigate the spin-polarisation effect. The yellow bubble means the spin-up states play a leading role at this position, while the blue bubble means that the spin-down states play the leading role.Results and discussion
Spectroscopic and morphological properties
To enhance the local magnetic flux applied to nano-photocatalyst under external magnetic field, magnetic Fe3O4 nanoparticles (NPs) were initially synthesised using the method adopted from our previous study23 (Fig. 1a). High-resolution transmission electron microscopy (HRTEM) images confirmed that well-dispersed but high crystallinity Fe3O4 NPs were synthesised with a lattice spacing of 0.298 nm corresponding to the (220) plane of the Fe3O4 structure (Fig. 1b and Fig. S1a, ESI†). The Fe3O4 NPs were successfully encapsulated in silica (denoted as Fe3O4@SiO2) (Fig. 1a and Fig. S1b, ESI†), which unfortunately, did not show photocatalytic activity under irradiation by visible light due to the large bandgap of silica. The synthesis of Fe3O4 NPs in different particle sizes were also attempted by controlling the amount of the surfactant, which leads to the coexistence of Fe2O3 and Fe3O4 phases. The two phases were carefully differentiated and quantified by Mössbauer spectroscopy (Fig. S1, ESI†). We noted that a larger particle size gives rise to a higher proportion of Fe2O3 due to partial oxidation. Consequently, Fe3O4 NPs with a mean size of 8 nm were used for further study. X-ray diffraction pattern of pure magnetic Fe3O4 NPs gives several characteristic peaks at 2θ values of 30.40°, 35.58°, 53.72°, 56.58°, and 62.74°, representing well the single phase crystalline fcc structure.29 As mentioned before, our previous study demonstrated that N-TiO2 showed an outstanding performance for the POWS reaction at elevated temperatures.5,15 Therefore, the Fe3O4 NPs were then combined with TiO2 using a similar method, followed by a high-temperature ammonia treatment for N-doping, where the designated content of Fe3O4 was 20 wt% (denoted as Fe3O4/N-TiO2-2). The Fe3O4 NPs does not show any structural change nor magnetic change before and after the ammonia treatment. XRD indicated the existence of the anatase phase TiO2, and no phase transition during ammonia treatment was observed (Fig. 1c). It has been reported that high-temperature NH3 treatment may lead to formation of TiN phase, which shows metallic properties. As shown in our recent work, a treatment temperature of higher than 660 °C would result in the formation of TiN; while in the current work, the NH3 treatment temperature is 600 °C, in which case the TiN phase is absent and a new oxynitride phase (TiOxNy) could be observed, as supported by our recent variable-temperature synchrotron XRD studies.30 HRTEM images and energy dispersive X-ray spectroscopy (EDS) mappings showed that the Fe3O4 NPs were mixed in the TiO2 matrix with a certain degree of aggregation (Fig. 1d and Fig. S1, ESI†). Depth-profiling X-ray photoelectron spectroscopy (XPS) was performed to investigate the distribution of the chemical species of Fe3O4/N-TiO2-2. The sample was etched by an ion-beam for different time periods so as to obtain chemical information from the surface/subsurface to the inner region. A typical XPS survey spectrum shows peaks of N 1s, O 1s, Ti 2p and Fe 2p, as shown in Fig. S2 (ESI†). We previously demonstrated that the N-doping only exists on the surface and sub-surface regions with the help of low energy ion scattering spectroscopy.5 Thus, the surface N-doping concentration can be investigated quantitatively, as summarised in Table S1 (ESI†). Moreover, the Fe 2p peaks show an increasing trend when the sample is sputtered (Fig. 1f), indicating that the Fe3O4 NPs are mostly encapsulated in the N-doped TiO2. EPR was performed at room temperature on the N-TiO2 with and without magnetic NPs (Fig. S3, ESI†). The Fe3O4 NPs give a very broad and strong resonance signal at a B-field of around 3150 Gauss, due to the unpaired electrons of the paramagnetic Fe(II) and Fe(III) species and the dipolar interaction between the nanoparticles.31 The EPR experiments were also carried out on the Fe3O4/N-TiO2-2 photocatalysts before and after the nitrogen doping, both of which showed a much smaller EPR signal compared with that of the Fe3O4 NPs due to the magnetic dilution of the TiO2. Since the strong and broad Fe3O4 NPs signal makes the signal changes of the TiO2 species undistinguishable, measurements were then performed on pure anatase TiO2 and N-TiO2 which was synthesised following the same sol–gel method and nitrogen doping treatment. As shown in Fig. S3 (ESI†), pure TiO2 was silent on the EPR, while after N-doping, a peak centred at a g-factor of 2.003 could be observed which could be attributed to the VOs created during N-doping. We have previously demonstrated that these surface VOs in N-TiO2 could be replenished by O2 or H2O at room temperature in 24 hours; while they could be regenerated at elevated temperatures.5 Therefore, further EPR measurements were carried out on the N-TiO2. The N-TiO2 were calcined in Ar flow at different temperatures for 1 h and allowed to naturally cool down to room temperature. Then the sample was exposed to air for 10 min before EPR measurements. As can be seen in Fig. S3a (ESI†), it showed an anisotropic g tensor (gzz = 2.026, gyy = 2.006, gxx = 1.998), which is originated from the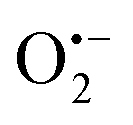 radicals (resulted from the O2 molecules stabilised on the surface VOs);32 besides, another signal of g = 2.012 was also observed which can be attributed to the doped interstitial N species. Clearly, the signal of
radicals (resulted from the O2 molecules stabilised on the surface VOs);32 besides, another signal of g = 2.012 was also observed which can be attributed to the doped interstitial N species. Clearly, the signal of 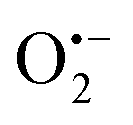 radicals indicative to the surface VOs increases with the calcination temperature; while the signal of doped N remains the same. These results suggest that the surface oxygen vacancies can be regenerated and created at elevated temperatures. Similar experiments have also been performed in other environments.5 Then in Fig. S3b (ESI†), to better distinguish the EPR signals of
radicals indicative to the surface VOs increases with the calcination temperature; while the signal of doped N remains the same. These results suggest that the surface oxygen vacancies can be regenerated and created at elevated temperatures. Similar experiments have also been performed in other environments.5 Then in Fig. S3b (ESI†), to better distinguish the EPR signals of 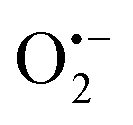 radicals and the interstitial N, more EPR experiments were carried out. N-TiO2 was calcined in Ar flow at 300 °C for 1 h and after cooling down, the sample was measured under Ar protection, which shows only the EPR signal of interstitial N (g = 2.012). Then the same sample was exposed to air for 10 min and measured again, where both signals at g = 2.012 and g = 2.006 are visible. This confirms the existence of both doped-N and O vacancy in the sample, and the signals can be clearly differentiated. It should be mentioned that recent studies on the lattice oxygen-mediated mechanism for oxygen evolution reaction have also demonstrated the importance of the VOs.33
radicals and the interstitial N, more EPR experiments were carried out. N-TiO2 was calcined in Ar flow at 300 °C for 1 h and after cooling down, the sample was measured under Ar protection, which shows only the EPR signal of interstitial N (g = 2.012). Then the same sample was exposed to air for 10 min and measured again, where both signals at g = 2.012 and g = 2.006 are visible. This confirms the existence of both doped-N and O vacancy in the sample, and the signals can be clearly differentiated. It should be mentioned that recent studies on the lattice oxygen-mediated mechanism for oxygen evolution reaction have also demonstrated the importance of the VOs.33
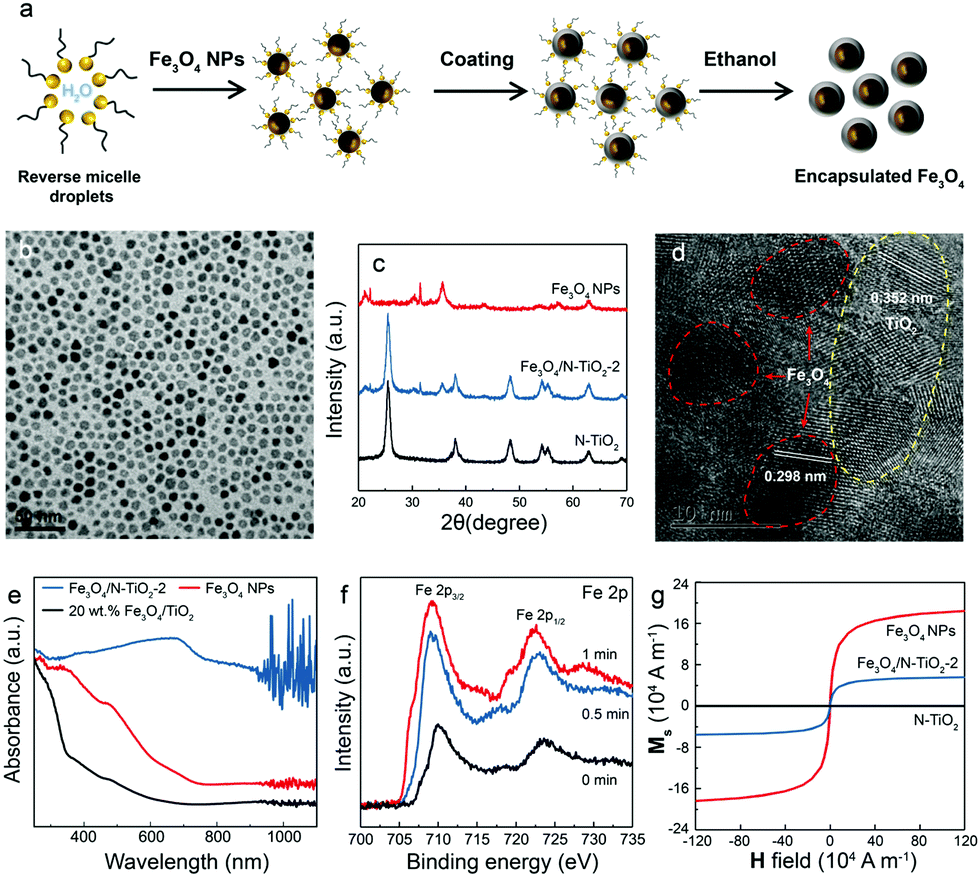 | ||
| Fig. 1 (a) Simplified schematic illustration of the synthesis procedure of Fe3O4 NPs and the encapsulated Fe3O4 photocatalysts. (b) TEM images of Fe3O4 NPs (8 nm). (c) XRD patterns of Fe3O4 NPs, Fe3O4/N-TiO2-2 and N-TiO2 obtained on a Bruker D8 Advance diffractometer. (d) HR-TEM of the Fe3O4/N-TiO2-2 photocatalyst. Red circles indicate the 8 nm Fe3O4 NPs and the yellow circle indicates the region of N-TiO2 with d-spacings shown in the image. (e) UV-vis DRS spectra of Fe3O4 NPs, Fe3O4/N-TiO2-2 and 20 wt% Fe3O4/TiO2 without N-doping; (f) Fe 2p XPS spectra of Fe3O4/N-TiO2-2 obtained at different ion-sputtering times; (g) magnetisation curves of Fe3O4 NPs, Fe3O4/N-TiO2-2 and N-TiO2. | ||
As mentioned before, the VOs were created during the ammonia treatment, and our previous study has shown that VOs can better harness the visible light and thus facilitate the oxygen evolution reaction.5,34 The visible light absorption was also substantially enhanced after nitrogen doping because of the introduction of an extra intraband energy level, which is indicated by UV-vis spectroscopy (Fig. 1e and Fig. S3e, ESI†). Apparently, the absorption edge of pristine TiO2 of around 390 nm was greatly extended after N-doping, even up to the near infrared (NIR) regime. The magnetic properties of the as-synthesised Fe3O4, Fe3O4/N-TiO2-2 and N-TiO2 were investigated with a SQUID magnetometer, and the magnetisation curves of the materials are shown in Fig. 1g. The saturation magnetisation (Ms) values of Fe3O4 and Fe3O4/N-TiO2-2 are 1.82 × 105 and 6.85 × 104 A m−1 (43.01 and 16.19 emu g−1 in CGS unit system). The saturated magnetisation value of Fe3O4/N-TiO2-2 is smaller than Fe3O4 NPs because of the inclusion of the N-TiO2. The as-prepared samples exhibit superparamagnetic feature as seen in Fig. 1g since the Fe3O4 around 8 nm is smaller than the critical size of ca. 20 nm. Due to the lack of magnetic coupling, the materials can be magnetised under an external magnetic field but will not retain residual magnetism upon removal of the external field.
MFEs on photocatalytic performance
The POWS performances of the as-prepared photocatalysts were then evaluated in a batch reactor at an elevated temperature. Although most POWS systems are investigated at room temperature, there are prior limited works (including ours) in the literature where elevated temperatures are used.5,14,15 We previously reported that the POWS activity over N-doped TiO2 at elevated temperatures was substantially higher than that of other particulate POWS systems at room temperature reported in literature.5 Interestingly, the POWS activity of N-TiO2 is highly dependent on temperature, which does not rise linearly with temperatures where more surface VOs are formed. Instead, it reaches a maximum value for H2 production when the temperature rises to 270 °C but declines rapidly on further increasing the temperature. Such trend coincides with the temperature-dependent ionic dissociation of water, which peaks at around 260–270 °C and then rapidly declines.35 Moreover, as mentioned before, the charged surface VOs play an important role in the POWS reaction and its regeneration appears to be a rate limiting step of this photocatalytic process. EPR spectra indicate the unpaired electrons from both the nitrogen doping and the associated VOs (for adsorption of O2 from air to form O2− in the photocatalysts, see Fig. S3a and b, ESI†). It is shown that after the N-doped sample was exposed to air at ambient conditions, the EPR signal, which is indicative of the presence of VO, gradually disappeared. This is attributed to the fact that oxygen sources in the air can gradually replenish the surface VO and redistribute the electrons. This could also explain the fact that N-doped TiO2 or hydrogenated TiO2 do not necessarily show good photocatalytic water splitting activity at room temperature under visible light illumination even though the remaining oxygen defects in bulk (require to diffuse to surface but the rate is very slow) can exert strong visible light absorption. Thus, it is difficult for photocatalytic water splitting to take place under ambient conditions, limited by the slow VO regeneration process. We also noticed that re-calcining the N-doped TiO2 in a N2 atmosphere at elevated temperatures would lead to regeneration of VO (see Fig. S3a and b, ESI†). Obviously, the surface VO formed in N-doped TiO2 are vulnerable to oxygen sources at room temperature, but at elevated temperatures, the faster subsequent reactions can regenerate them in both bulk and surface to sustain the surface photocatalytic processes, and therefore overcome the rate limiting regeneration of the lattice VO.5 Besides, our recent work also showed that the oxygen mobility on the surface and in sub-surface regions are facilitated at elevated temperatures. Consequently, under such conditions, other effects such as polarisation and charge separation can be studied, which would help us to understand this photocatalytic system more deeply. More details about photocatalysis at elevated temperatures can be found in our recent reports (ref. 5, 14 and 30), and the potential practical applications of such system are demonstrated in ESI† (Discussion S1). Therefore, all POWS performance tests were carried out at 270 °C which was proved to be the optimal temperature for TiO2-based photocatalysts.5,30 Different metal NPs were deposited on the Fe3O4/N-TiO2-2 photocatalyst via a photo-reduction method, since previous studies have shown that deposition of noble metal such as Au and Pt can greatly improve the H2 evolution performance.5,36–38 According to the volcano curve for the hydrogen evolution relationship, Pt has the optimal adsorption energy with hydrogen, followed by Pd and Ru.39 However, as shown in Fig. S4a (ESI†), Au-decorated Fe3O4/N-TiO2-2 showed the highest POWS activity. It is presumably due to the surface plasmon resonance effect of Au NPs, which has been extensively studied in Au/TiO2 system in literature.40,41 Then the loading amount of Au NPs was optimised, as shown in Fig. S4b (ESI†). Clearly, the POWS activity levels off when the loading amount is increased beyond 1 wt%; Moreover, the activity shows a small drop when the loading reaches 4 wt%, which is presumably due to the shielding effect of the NPs, resulting in less photons reaching the N-TiO2.42 Consequently, 1 wt% Au NPs were used for all the photocatalysts as a hydrogen evolution co-catalyst. N-TiO2 and Fe3O4/N-TiO2-2 photocatalysts both showed similar hydrogen evolution rates of around 7000 μmol g−1 h−1 from pure water without any external magnetic field (Table S2, ESI†). Control experiments were also performed, which confirmed that Fe3O4 NPs, SiO2 and Fe3O4@SiO2 all showed no detectable hydrogen evolution, indicating that N-TiO2 is the only photocatalytically active component in the catalysts under these conditions. Moreover, no activity could be observed at elevated temperatures if either the light illumination or a photocatalyst is absent. The stoichiometry of products (H2 and O2) was investigated quantitatively by gas chromatograph, which indicates a H2:O2 ratio of 2.01 ± 0.04 throughout this work. N2 was not detected in the gaseous products, which excludes the influence of mixed air in the system. The isotopic studies were then carried out by using D2O rather than H2O to confirm that H2 is generated from the POWS reaction instead of the photocatalyst itself or any other possible organic contaminants (Fig. S4c, ESI†). Also, the influence of electron transfer between N-TiO2 and Fe3O4 NPs was excluded by introducing an insulated silica layer in between (Fe3O4@SiO2/N-TiO2-2), which showed similar POWS activity as that of Fe3O4/N-TiO2-2 without the silica layer. The amounts of Fe3O4 and N-TiO2 were maintained the same in each catalyst and neither SiO2 nor Fe3O4@SiO2 showed any activity toward the POWS reaction. Thus, the following studies were carried out with the Fe3O4/N-TiO2 materials without the silica layer.
Subsequently, to study the MFE on the POWS system, an external static magnetic field of 180 mT was applied by two paralleled permanent magnets near the reactor. The applied external magnetic flux density Bexternal in the centre of the two magnets was measured by a Gauss metre. Excitingly, the POWS activity of Fe3O4/N-TiO2-2 increases to 12210 μmol g−1 h−1, showing an enhancement of 76%. The POWS activity of the N-TiO2 however, remains almost the same (Table S2, ESI†). The M–H curves of the samples in Fig. 1g exhibit that the Fe3O4 NPs are crucial for providing sufficient local magnetic field induced by the external field. Moreover, the POWS performance shows a decreasing trend as the Bexternal is reduced (Fig. 2a). Moreover, the POWS performance of the other metal-decorated Fe3O4/N-TiO2-2 were also evaluated. As shown in Fig. S4a (ESI†), upon the application of an external magnetic field of 180 mT, all the photocatalysts showed a substantial increase in the activity. But the enhancement percentage of Au-decorated photocatalyst did not show big difference compared with the others. Clearly, the plasmonic resonance effect introduced by Au NPs does not significantly change with the magnetic field, thus, it does not influence our study of the magnetic field effects in this POWS system.
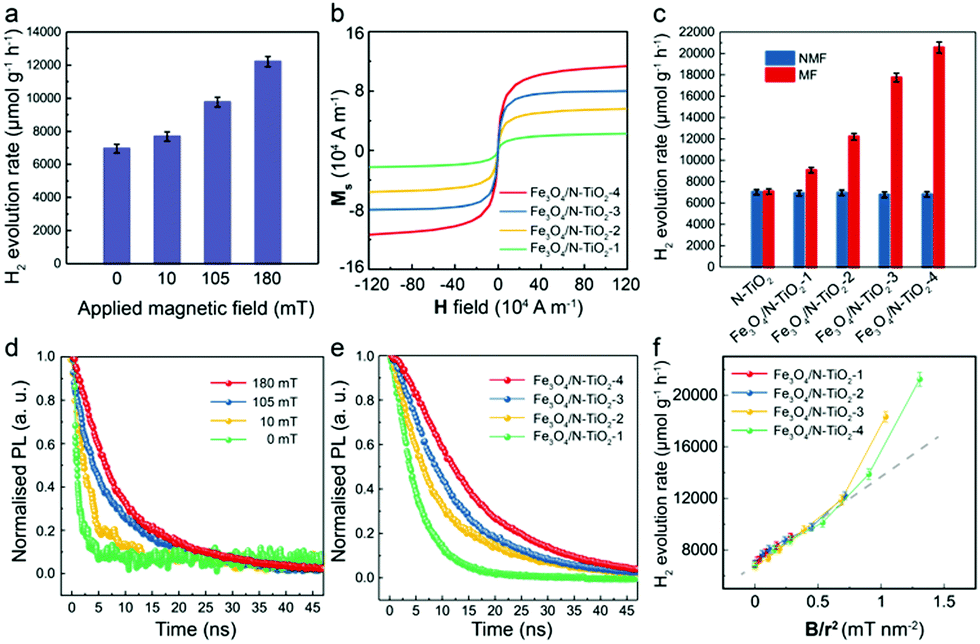 | ||
| Fig. 2 POWS activity tests at 270 °C and characterisation of the Fe3O4/N-TiO2 photocatalysts. (a) POWS activity tests of Fe3O4/N-TiO2-2 under different external magnetic flux densities (Bexternal); (b) magnetisation curves of Fe3O4/N-TiO2 photocatalysts; (c) POWS activity tests of N-TiO2 and Fe3O4/N-TiO2 photocatalysts with and without external magnetic field (180 mT) (NMF = no magnetic field, MF = magnetic field). 1 wt% Au was deposited onto the photocatalysts via the photo-reduction method before use. (d) Time-resolved PL spectra of Fe3O4/N-TiO2-2 under different Bexternal; (e) time-resolved PL spectra of Fe3O4/N-TiO2 photocatalysts with an external magnetic field of 180 mT. The excitonic lifetimes of the photocatalysts are summarised in Table S3 (ESI†). (f) the relationship between the POWS performance and Blocal/r2, where Blocal is the local magnetic flux density obtained from the M–H curve in (b) and the measured external magnetic flux density; and r is the distance from the Fe3O4 NPs to the surface of the photocatalyst particle derived from the TEM images. All error-bars indicate the standard deviation. | ||
Similarly, a series of Fe3O4/N-TiO2 photocatalysts with increasing Fe3O4 NPs contents were fabricated (the amounts of the Fe3O4 NPs in Fe3O4/N-TiO2-1, Fe3O4/N-TiO2-2, Fe3O4/N-TiO2-3 and Fe3O4/N-TiO2-4 are 10 wt%, 20 wt%, 30 wt% and 40 wt%, respectively), of which the magnetisation curves in Fig. 2b show enhanced saturation magnetisation with increased content of the Fe3O4 NPs. We have demonstrated in our previous work that the N-doping concentration could influence the visible light absorption, surface VO concentration, excitonic lifetime and POWS performance of N-TiO2 based materials.5,30 Thus, here in this work, we carefully controlled the temperature and duration of the high-temperature ammonia treatment, in order to ensure that the N-doping concentration in the N-TiO2 maintains the same among different photocatalysts. The POWS activities were then tested likewise at 270 °C under visible light irradiation with an external magnetic field of 180 mT. Fig. 2c clearly indicates that the magnetic photocatalysts become more sensitive to the external magnetic field as the Fe3O4 NPs content increases, giving a 3-fold POWS activity over Fe3O4/N-TiO2-4. Similar work has also been undertaken on the commercially available P25 TiO2 to simplify the catalyst design. P25 consists of ca. 80 wt% anatase and 20 wt% rutile, which has been widely used in various photocatalytic systems. It was doped with N firstly and then combined with the Fe3O4 NPs. The as-obtained photocatalyst contains 40 wt% of Fe3O4 NPs, the same as that in the Fe3O4/N-TiO2-4. Excitingly, the simple mixture of N-doped P25 and Fe3O4 NPs results in a significantly enhanced POWS performance in the external magnetic field (180 mT), making such magnetic field promoted system more practical for further application. It is noteworthy that in this case, the Fe3O4 NPs have not experienced any high-temperature ammonia treatment. Although this catalyst exhibited slightly lower enhancement under external magnetic field due to geometric longer distance between the magnetic phase from catalytic sites, the high-temperature ammonia treatment causing insurmountable issue on the Fe3O4 nanoparticles is not evidenced. Similar work has also been attempted on commercial ST-01, which exhibits a similar conclusion. The potential thermal effect and kinetic changes originating from the local magnetic field are discussed in the ESI† (Discussions S2 and S3).
TRPL spectroscopy was then used to understand the mechanism of the MFEs on the POWS performance. The PL spectra give very broad signals ranging from 450 nm to 850 nm (Fig. S5, ESI†). It has been reported that the broad PL emission band from 400 to 600 nm is from the intrinsic bandgap emission and intrinsic defects (such as oxygen vacancies) of the pure anatase TiO2 material,43–46 while the additional broad emission peak centred at ca. 800 nm can be attributed to the additional energy levels introduced by the defects, such as N-doping and surface oxygen vacancies, etc.47 Clearly, the major PL emission still originates from the intrinsic bandgap emission, and it is also possible that some electron transitions involving the defect states may not give detectable PL signals considering the low density of states compared to those of the conduction band and valence band; and they might also take the non-radiative recombination pathways instead. Moreover, the PL spectra did not change upon the application of an external magnetic field, which indicated that the magnetic field did not change the electron transitions in the N-TiO2, but only influenced the dynamics of the charge recombination processes (Fig. S5, ESI†). Further investigations on the probing-wavelength-dependence were carried out and the results show that the TRPL spectra does not obviously change with the probing wavelengths, giving almost identical TRPL spectra (Fig. S5, ESI†). Obviously, both Fe3O4/N-TiO2-2 and N-TiO2 showed similar exciton lifetimes without an external magnetic field, while the exciton lifetime of Fe3O4/N-TiO2-2 was prolonged substantially whilst the magnetic field existed, and that of the N-TiO2 remained unchanged (Fig. S5 and Table S3, ESI†). Such a difference in the response to an external magnetic field coincided well with what was observed in the POWS activity tests. Subsequently, further TRPL experiments showed that the exciton lifetime of Fe3O4/N-TiO2-2 increases with Bexternal, confirming the exciton lifetime changes in response to the applied magnetic field. This induces stronger local magnetic flux density Blocal and supresses the charge separation process (Fig. 2d). A series of Fe3O4/N-TiO2 with varying Fe3O4 content were then tested. Apparently, all samples showed similar TRPL spectra without an external magnetic field (Fig. S5f, ESI†); whilst, when measured in a magnetic field of 180 mT, the exciton lifetime was prolonged more substantially for a higher content of Fe3O4, which is in accordance with the magnetisation experiments (Fig. 2b and e) and the POWS activity tests (Fig. 2c). Our previous reports showed that elevated temperatures solely would not obviously influence the exciton lifetime in our system.5,14 All the above observations indicate that the charge separation process is dominated by the locally induced magnetic field, therefore the N-TiO2 alone showed no significant response to the magnetic field. In addition, the MFEs become more substantial when the external magnetic field is stronger, which can induce stronger local magnetic fields as shown in the magnetisation experiments (Fig. 2b). This could also explain why for a fixed external field, samples that contain more Fe3O4 show a larger response to the field. The energy barrier of the POWS reaction and the formation of surface VOs were investigated previously, which demonstrated that the performance of the POWS reaction on anatase TiO2 based materials is mainly limited by the diffusion of VOs.30 When the POWS reaction is operated at elevated temperatures, the VOs formation and regeneration are greatly promoted, then the performance would presumably be limited by the lifetime of the photo-generated electron–hole pairs. Turnover number (TON) and turnover frequency (TOF) are also evaluated, as shown in Table S2 (ESI†), which showed that the TOF was greatly enhanced from 131 h−1 to 434 h−1 for Fe3O4/N-TiO2-4 upon the application of an external magnetic field of 180 mT. Along with the TRPL results, it is clearly that due to the greatly prolonged lifetime of the photo-generated electron–hole pairs, more electrons could migrate from the bulk to the surface and accumulate on the Au NPs, which enables more catalytic cycles at a specific catalytic centre in a given time.
Combined correlative off-axis electron holography in TEM and energy dispersive X-ray spectroscopy spectrum imaging in STEM has been studied. It is well-established that the off-axis electron holography can recover the electron optical phase shift of the electrons that interact with the in-plane components of the electromagnetic field present within and around the sample.48 The EDX chemical measurements were to map the Fe3O4 within the agglomerates and correlate to the phase measurements of the N-TiO2. Agglomerated areas of the Fe3O4/N-TiO2-2 and Fe3O4/N-TiO2-4 samples were briefly examined using both off-axis electron holography and EDX imaging, as shown in Fig. S6 (ESI†). The electrostatic (φE) and magnetic phase shifts (φM) were carefully separated by turning the sample over inside the electron microscope and taking the difference of two experiments.48 These preliminary electron holography studies indicated the superparamagnetic nature of the photocatalysts (Fig. S6, ESI†).
It is known that charged bodies in motion can be affected by the Lorentz force produced by a magnetic field, and the photo-generated electrons and holes in the Fe3O4/N-TiO2 experience Lorentz forces to opposite directions according to the left-hand rule when they are moving together as excitons, accounting for the prolonged excitonic lifetimes. Therefore, the correlation between the enhancement of POWS performance and the local induced magnetic flux density Blocal was further investigated to study the Lorentz force effect (Fig. S7, ESI†). The Blocal of the Fe3O4/N-TiO2 samples can be derived from the M–H curves (Fig. 1g and 2b), as summarised in Table S4 (ESI†). The Lorentz force is given by F = q(v × B), where the F changes linearly with B. For the N-TiO2 sample, the external magnetic field alone shows no obvious effect on the POWS performance, when the induced local magnetic field is absent. Therefore, only the local magnetic flux generated by the Fe3O4 NPs is considered in the correlation. The photocatalytic activity was plotted against the local magnetic flux density (Blocal) of the Fe3O4 NPs. Surprisingly, the H2 evolution rate, although showing positive correlation with the local magnetic induction Blocal, did not change linearly. Also, Fe3O4/N-TiO2 with a higher Fe3O4 content always gives higher POWS performance even the local magnetic flux density is similar (Fig. S7a, ESI†). It is noticed that the average distance (r) from the core of Fe3O4 NPs to the catalyst surface (where the chemical reactions take place) decreases as the concentration of Fe3O4 NPs increases and the corresponding magnetic field strength is inversely proportional to r2. The distances were measured from the TEM images and then the POWS performance was plotted against Blocal/r2 (bearing in mind that the local magnetic flux density decays rapidly with the change of distance, only those Fe3O4 NPs near the surface were considered. See Fig. 2f and Fig. S7d, e, ESI†). Interestingly, upon calibration of the local magnetic flux density using these distances, all the samples appeared to show a similar response to Blocal/r2, exhibiting an overall positive correlation. Further scrutiny indicates that the POWS performance increases linearly until Blocal/r2 = ∼0.7, after which point it increases much more sharply, deviating from a linear relationship (Fig. 2f). The linear influence originated from the Lorentz force effect could be described as follows:
| H2 evolution rate = 7042.22 Blocal/r2 + 6988.42 (R2 = 0.9818) | (3) |
Additionally, 40 wt% Fe3O4/N-TiO2 with different nitrogen-doping concentrations were also prepared by controlling the temperature of the NH3 treatment during N-doping5. Thus, the only difference between the resulted photocatalysts is the N-doping concentration: The N-doping concentration increases with the temperature of the NH3 treatment, which is supported by XPS results (Table S1, ESI†). Then the POWS performance was evaluated with and without the external magnetic field (180 mT) with the irradiation of a 300 W Xe lamp which included UV light, considering the pure TiO2 barely absorbs the visible light. The enhancement from MFE was defined as follows:
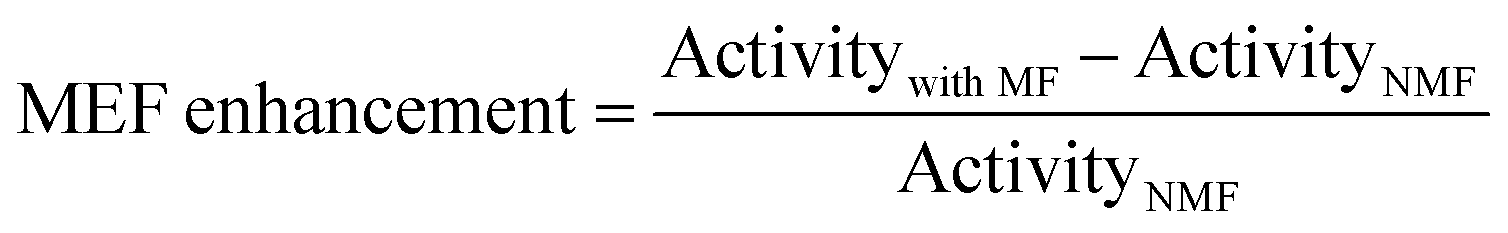 | (4) |
Evaluation of the spin-polarisation effect
Apart from the electromagnetic properties, the quantum mechanical properties of the electron, such as angular momentum (spin) has long been neglected in photocatalytic studies. It is noted that magnetic moments exist in the N-TiO2 (Fig. S8a, ESI†), implying that the cooperative alignment of such moments under a magnetic field may influence the POWS performance together with the above-discussed Lorentz force effect. It has been reported that N-substitution of O atoms in N-TiO2 could generate defective holes, and the hole-mediated double exchange through the strong p–p interaction between N and O can give spins correlation to this material.49 Considering the XPS spectra showed the doped N were dominantly in the substitutional sites (Fig. S2d, ESI†), a Ti16O28N4 supercell based on the structure of anatase TiO2 with ca. 4.6 wt% substitutional N-doping derived from our recent study (ref. 30) was constructed and studied by the first-principles DFT calculations, as shown in Fig. 3a. The calculated total density of states (DOS) of N-TiO2 clearly showed that the defect states were created within the bandgap near the Fermi level, which originated from the N-doping, while such an additional band was absent in the pure TiO2 model (Fig. 3b and Fig. S8b, ESI†). These defect states are not localised in contrast to the previous literature of the low N content, but form a wide band extended to the valence band (VB) instead, due to the strong interaction between the N atoms at our high N-doping concentration, which also accounts for the enhanced visible light absorption.50 The defect states can be distributed on two spin channels (spin-up and spin-down), but only one spin channel occurs in the bandgap region of N-TiO2 under a magnetic field (Fig. 3b), indicative that the magnetic moments can be well-aligned in the material. The calculated spatial spin distribution also indicated a strong spatial spin polarisation: only the blue surface that represented the spin-down channel was observed, whist the spin-up channel was absent (Fig. 3c). On the other hand, as shown in the Fig. S8c (ESI†), the total DOS clearly indicates that the defect band is not polarised, and the magnetic moments are distributed on the two spin channels when a magnetic field is absent. The presence of both yellow and blue surfaces in Fig. S8d (ESI†) also suggests the existence of both spin channels in the defect band without the influence of a magnetic field. Meanwhile, the magnetic field has little effect on the conduction band (CB): the calculated DOS of the CBM is almost the same no matter whether a magnetic field is present or not (Fig. 3b and Fig. S8b, ESI†). Clearly, in the presence of an induced local magnetic field from Fe3O4 NPs, the cooperative magnetic moments in N-TiO2 align in parallel in the high spatial spin polarised environment. When an electron is excited to the CB, it is likely to undergo spin relaxation during the transfer, losing its original spin characteristics and changing to the other state (spin-up), due to spin–orbital coupling, hyperfine interaction, etc.51 Upon recombination, the lack of a spin-up channel makes it more difficult for the spin-up electrons to come back to the VB. Overall, both the slow spin flips and the opposing Lorentz forces result in a prolonged exciton lifetime, as observed from TRPL measurements (mechanism illustrated in Fig. 4a). Considering the photocatalyst particles are highly dispersed in the suspension during the reaction, the interaction of local magnetic fields between different particles would be negligible. The prolonged lifetimes of the photo-generated charge carriers greatly reduce the chance of recombination, therefore enhance the diffusion to the surface to facilitate the H+ reduction and OH− oxidation reactions from water (Fig. S7c, ESI†). Such observation can also explain the relationship seen in Fig. S7b (ESI†): as the N concentration increases, the magnetic moments in the defect band also increase, forming an extended energy band, which accounts for the dramatic spin-polarisation effect. While at low doping levels, the defect band is too narrow and localised to show any spin-polarisation effect in an external magnetic field (Fig. S8e, ESI†). Therefore, the spin-polarisation effect dominates at high N-doping concentration under high magnetic flux. Since the Lorentz force effect can be described by eqn (3), the contribution of the spin-polarisation effect can be evaluated quantitatively. Analyses of the results shown in Fig. 2f shows that at Blocal/r2 = 0.71, the spin-polarisation accounted for only 8.9% of the MFEs, while the contribution increased to 22.6% at stronger local magnetic flux (Blocal/r2 = 1.31). These observations imply the opportunity of rational design of photocatalysts by modifying different aspects of the MFEs (i.e., charge and spin features) in order to control the charge separation and exciton lifetime, which presumably have profound meanings to both academic study and practical application of photocatalytic technologies. | ||
| Fig. 3 DFT calculations of the local MFE on N-doped TiO2 photocatalyst. (a) The constructed Ti16O28N4 supercell for DFT calculation (O: red; Ti: blue; N: purple); (b) calculated total DOS of Ti16O28N4 supercell (N-TiO2) and Ti16O32 supercell (pure TiO2) with the local magnetic field alignment; (c) 3D spatial distributions of spin polarisation of N-doped Ti16O28N4 model in defect band with the local magnetic field. The blue surfaces indicate the charge density of spin-down electrons; (d) calculated partial charge density of CBM of N-doped Ti16O28N4 model with the magnetic field. The yellow surfaces represent the distribution of the total electron densities in the CBM near each atom. The value of iso-surface is set to be 0.001 eV Å−1 both in (c) and (d). | ||
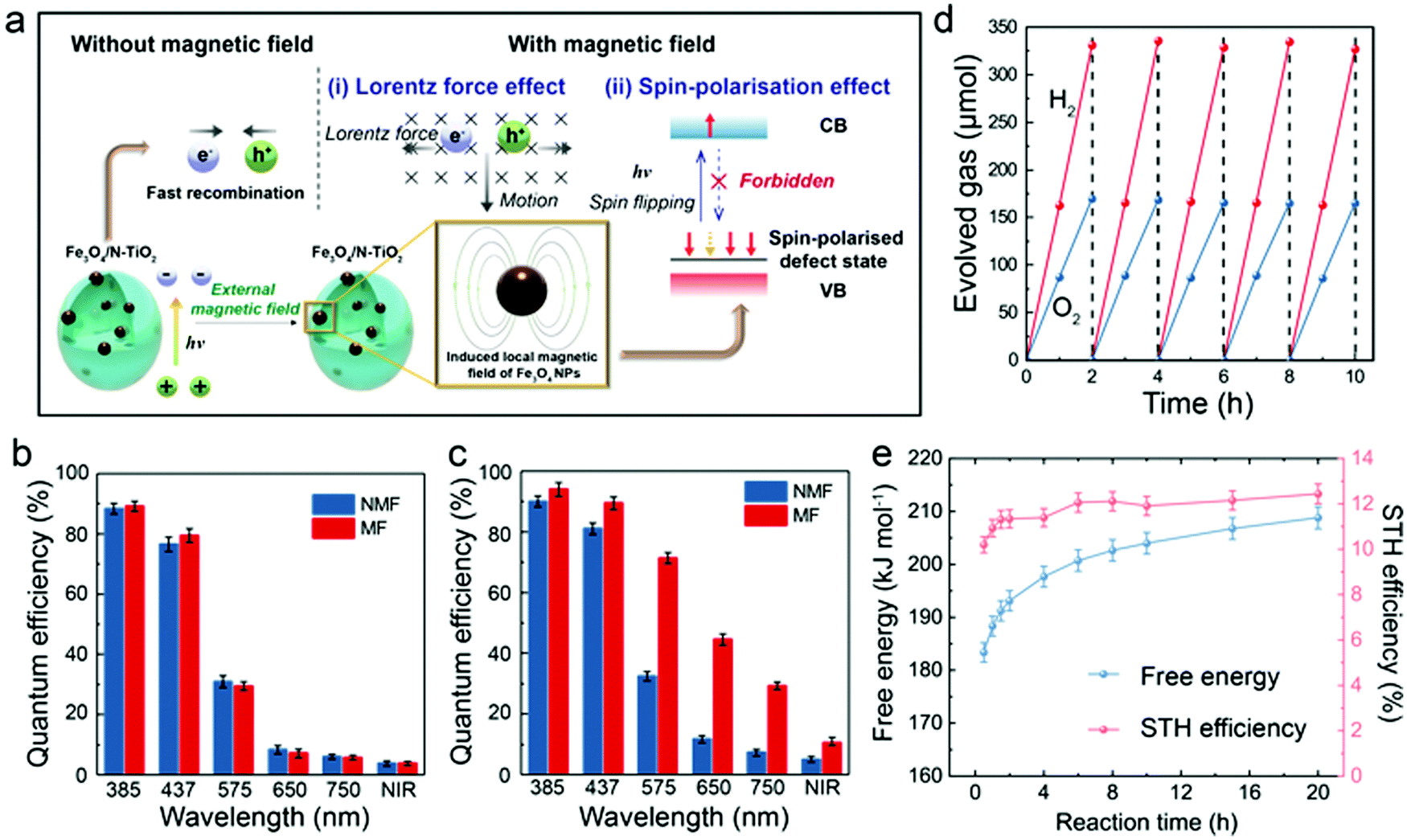 | ||
| Fig. 4 Proposed mechanism and QE tests of the POWS system. (a) Schematic illustration of the magnetic field promoted POWS system, in which the charge separation process is facilitated by the induced local magnetic field; Internal QE of N-TiO2 (b) and Fe3O4/N-TiO2-4 (c) photocatalysts with and without external magnetic field. (NIR = near infrared (800–1200 nm); NMF = no magnetic field; MF = magnetic field of 180 mT) Error bars indicate the standard deviations; (d) repeatable tests of Fe3O4/N-TiO2-4 photocatalyst at 270 °C and 180 mT under simulated solar irradiation. The head space of the batch reactor was purged with Ar gas at the end of each cycle. (e) Time-dependent free energy and STH conversion efficiency of the POWS reaction at 270 °C under our operating conditions. More details are shown in Table S7 (ESI†). | ||
The application potential of this POWS system was further evaluated by QEs and STH conversion efficiency, both of which are widely recognised as key parameters for solar conversion systems.52Fig. 4b clearly shows that the internal QEs of N-TiO2 were not influenced by an external magnetic field, while Fig. 4c shows that Fe3O4/N-TiO2-4 displays a sensitive response to the magnetic field, giving impressive QEs of 96.2% at 385 nm and 88.7% at 437 nm (Note S1, ESI†). The measurement of the internal QE in the near infrared (NIR) regime has also been attempted by using a bandpass filter which allows a wavelength range of 800–1200 nm to pass through (the UV-vis spectra of the bandpass filters can be found in Fig. S9, ESI†). Excitingly, it gives an enhanced internal QE of 10.8% even in this NIR regime on the Fe3O4/N-TiO2-4 photocatalyst under a magnetic field of 180 mT. The theoretical efficiency limit of a solar conversion system was carefully analysed by Shockley and Queisser (known as Shockley–Queisser limit or detailed-balance limit).53 Such limit was then further considered for photochemical systems by Ross and Hsiao.54 It has been demonstrated that there is always a part of the photon energy that cannot do useful work, thus, for a given reaction, the threshold energy that is capable of driving the reaction must exceed the minimum thermodynamic requirement (free energy) of the reaction by 0.2–0.3 eV.53–56 Such energy difference could be minimised by different approaches, such as engaging light trapping techniques, concentrated light irradiation, high-purity light absorbers, etc.56 Detailed evaluation and discussions of this threshold wavelength are shown in Note S1 (ESI†). Moreover, the STH conversion efficiency has been evaluated for our POWS system. As demonstrated by Domen et al. recently, it is quite common that the POWS reaction is carried out under various non-standard conditions including reduced pressure, cooling, or heating, etc., thus the changes of the free energy must be considered by default.57 Also should be noted is that in our batch process, the partial pressures of the evolved H2 and O2 change as the reaction proceeding. Also, in our system, the reaction is performed at a constant volume, thus, the Helmholtz free energy is used instead of the Gibbs free energy. Therefore, the free energy has been corrected for the operating temperature and the partial pressures in the system (Note S2, ESI†). As a result, the time-dependent STH efficiencies is plotted against the reaction duration, as show in Fig. 4e; also, a time-averaged STH efficiency of 11.9 ± 0.5% has been achieved from this local magnetic field promoted POWS system under a magnetic field of 180 mT (Note S2, ESI†). Recyclability of the Fe3O4/N-TiO2-4 photocatalyst was evaluated by recycling the catalysts for 5 times, showing no obvious change in photocatalytic activity (Fig. 4d), and it is presumably that Fe3O4 NPs are protected by the N-TiO2 coating from oxidation. The Au-decorated Fe3O4/N-TiO2-4 photocatalyst also shows a good stability for stoichiometric H2 and O2 evolution over 20 hours (Fig. S9, ESI†). The catalyst was also recycled after this long-term stability test and used for another 20 hour reaction, which showed no obvious drop of activity (Fig. S9, ESI†). It is also noteworthy that our POWS reaction could still proceed even in the presence of considerable pressures of H2 and O2 that make the reaction less favourable thermodynamically, and drive the reverse reaction more considerably (Table S7, ESI†). This has then been further confirmed by performing the POWS reaction with pre-pressurised H2, and the POWS reaction also takes place with a similar activity, as show in Fig. S10 (ESI†). Subsequently, an overall energy conversion efficiency of 1.16 ± 0.05% has been demonstrated when the energy input for heating is considered (details can be found in Note S2, ESI†). Although this overall energy efficiency of 1.16% appears to be quite low, it should be emphasised this is not the situation we anticipate. It is apparent that solar light could also provide heat by irradiation, photothermal effect, etc. Thus, in an ideal future design, the energy for heating is not provided by external source but from concentrated solar light (Discussion S1, ESI†), therefore, the energy consumption for heating the water and reactor could be excluded in that scenario, and a higher overall energy efficiency could be expected. Here in this work, we attempted to demonstrate the reasonability of solar heating: a light concentrating furnace was used to mimic the solar light concentrator without any other direct electrical heating devices (Fig. S12, ESI†). This experiment was performed to illustrate that the concentrated light could supply the required heat and visible photons for photocatalytic splitting of water in our system. The reactor temperature of 270 °C can be maintained by this light source alone, and a H2 evolution rate of about 7600 μmol g−1 h−1 is achieved on Fe3O4/N-TiO2-4 for up to 20 h when a magnetic field is absent. Also, we have previously demonstrated that such energy required for heating the system could come from the waste heat from other processes, such as the exothermic chemical reactions like ammonia synthesis or CO2 hydrogenation reactions when these processes are coupled together.5 In addition, the superheated water carries quite large thermal energy, which may subsequently be used for additional energy generation, i.e. H2 by steam turbine-electrolyser. Thus, by considering the availability of solar heating and additional H2 generation, the actual overall energy efficiency could be much higher upon further optimisation. Nevertheless, the current value is still superior to the recently reported values in literature in the related fields.58,59
Conclusions
In conclusion, a H2 evolution rate of 21230 μmol g−1 h−1 and QE of 88.7% at 437 nm are obtained at 270 °C over 40 wt% Fe3O4/N-TiO2 under an external magnetic field of 180 mT due to the greatly prolonged exciton lifetimes over this photocatalyst. An outstanding STH efficiency of 11.9 ± 0.5% can be demonstrated by our simple set-up particulate POWS system with anticipated low capex cost (Table S5, ESI†). In addition to the Lorentz force effect, spin-polarisation at elevated temperature is for the first time demonstrated systematically in this N-doped TiO2 structure under an external magnetic field: the Lorentz effect contributes dominantly when the magnetic flux is weak, but when the local magnetic flux density is strong enough (Blocal/r2 > 0.7), the spin-polarisation effect can be triggered to further improve the photocatalytic activity, accounting for 22.6% contribution at Blocal/r2 = 1.31. Both effects lead to prolonged exciton lifetime and facilitate photocatalysis over the N-TiO2 based material. As such this work provides insights on the use of the two fundamental features of electrons including charge and spin in the magnetic field promoted photocatalysis. To unlock the full potential of MFEs, not only can various photocatalytic systems be designed with tuned charge and/or spin characteristics, but also the flux density of the local magnetic field can be tuned by engineering related photocatalysts in the proximity of stronger superparamagnetic alloy shells or under a stronger external field, further contributing to novel responsive solar energy conversion technologies and the rational design of various photocatalytic systems with high efficiency.
Data availability
The source data are all shown in Fig. S1–S12, Tables S1–S7 and Notes S1–S2, ESI†. All relevant data are available from the authors.Author contributions
Y. Li prepared, characterised and tested the photocatalysts with the help of C. F.; Y. Li, C. W., Z. W., A. K. and R. E. D.-B. studied the magnetic properties of the materials; Y. W. and Y. Lu performed and investigated the DFT calculations; Y. Li collected and analysed the TRPL data with the help of R. A. T.; Y. Li and S. C. E. T. wrote the paper in discussion with C. W.; S. C. E. T. supervised the overall project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support for this project from the EPSRC in the United Kingdom (Grant EP/K040375/1), the Zhejiang University Education Foundation Global Partnership Fund (100000-11320) and European Union's Horizon 2020 Research and Innovation Programme (Grant No. 823717, project “ESTEEM3”) are kindly acknowledged.Notes and references
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- B. Tian, B. Tian, B. Smith, M. C. Scott, R. Hua, Q. Lei and Y. Tian, Nat. Commun., 2018, 9, 1397 CrossRef PubMed.
- Y. Li, Y.-K. Peng, L. Hu, J. Zheng, D. Prabhakaran, S. Wu, T. J. Puchtler, M. Li, K.-Y. Wong, R. A. Taylor and S. C. E. Tsang, Nat. Commun., 2019, 10, 4421 CrossRef PubMed.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- L. Shang, B. Tong, H. Yu, G. I. N. Waterhouse, C. Zhou, Y. Zhao, M. Tahir, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1501241 CrossRef.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- Z. Zhang, J. Huang, Y. Fang, M. Zhang, K. Liu and B. Dong, Adv. Mater., 2017, 29, 1606688 CrossRef PubMed.
- Q. Wang, S. Okunaka, H. Tokudome, T. Hisatomi, M. Nakabayashi, N. Shibata, T. Yamada and K. Domen, Joule, 2018, 2, 2667–2680 CrossRef CAS.
- Y. Li and S. C. E. Tsang, Acc. Chem. Res., 2021, 54, 366–378 CrossRef CAS PubMed.
- Y. Li, S. Wu, J. Zheng, Y.-K. Peng, D. Prabhakaran, R. A. Taylor, S. Chi and S. C. E. Tsang, Mater. Today, 2020, 41, 34–43 CrossRef CAS.
- Y. Li and S. C. E. Tsang, Mater. Today Sustain., 2020, 9, 100032 CrossRef.
- J. Li, Q. Pei, R. Wang, Y. Zhou, Z. Zhang, Q. Cao, D. Wang, W. Mi and Y. Du, ACS Nano, 2018, 12, 3351–3359 CrossRef CAS PubMed.
- W. Gao, J. Lu, S. Zhang, X. Zhang, Z. Wang, W. Qin, J. Wang, W. Zhou, H. Liu and Y. Sang, Adv. Sci., 2019, 6, 1901244 CrossRef CAS PubMed.
- C. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2020, 60, 16309–16328 CrossRef PubMed.
- Z. Zhao, D. Wang, R. Gao, G. Wen, M. Feng, G. Song, J. Zhu, D. Luo, H. Tan, X. Ge, W. Zhang, Y. Zhang, L. Zheng, H. Li and Z. Chen, Angew. Chem., 2021, 133, 12017–12025 CrossRef.
- J. Zhao, N. Li, R. Yu, Z. Zhao and J. Nan, Chem. Eng. J., 2018, 349, 530–538 CrossRef CAS.
- M. P. Jiang, K. K. Huang, J. H. Liu, D. Wang, Y. Wang, X. Wang, Z. Da Li, X. Y. Wang, Z. Bin Geng, X. Y. Hou and S. H. Feng, Chem, 2020, 6, 2335–2346 CAS.
- C. Zhao, L. Zhou, Z. Zhang, Z. Gao, H. Weng, W. Zhang, L. Li and Y. Y. Song, J. Phys. Chem. Lett., 2020, 11, 9931–9937 CrossRef CAS PubMed.
- Y. K. Peng, C. N. P. Lui, Y. W. Chen, S. W. Chou, E. Raine, P. T. Chou, K. K. L. Yung and S. C. E. Tsang, Chem. Mater., 2017, 29, 4411–4417 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 78, 1396 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed.
- P. Mishra, S. Patnaik and K. Parida, Catal. Sci. Technol., 2019, 9, 916–941 RSC.
- C. Foo, Y. Li, K. Lebedev, T. T. Chen, S. Day, C. Tang and S. C. E. Tsang, Nat. Commun., 2021, 12, 661 CrossRef CAS PubMed.
- Y. Köseoǧlu, J. Magn. Magn. Mater., 2006, 300, e327–e330 CrossRef.
- N. Feng, H. Lin, H. Song, L. Yang, D. Tang, F. Deng and J. Ye, Nat. Commun., 2021, 12, 4652 CrossRef CAS PubMed.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- C. Di Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414–11419 CrossRef CAS PubMed.
- A. V. Bandura and S. N. Lvov, J. Phys. Chem. Ref. Data, 2006, 35, 15–30 CrossRef CAS.
- S. Mubeen, G. Hernandez-Sosa, D. Moses, J. Lee and M. Moskovits, Nano Lett., 2011, 11, 5548–5552 CrossRef CAS PubMed.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- C. Zhan, Z. Y. Wang, X. G. Zhang, X. J. Chen, Y. F. Huang, S. Hu, J. F. Li, D. Y. Wu, M. Moskovits and Z. Q. Tian, J. Am. Chem. Soc., 2020, 141, 8053–8057 CrossRef PubMed.
- A. R. Zeradjanin, A. Vimalanandan, G. Polymeros, A. A. Topalov, K. J. J. Mayrhofer and M. Rohwerder, Phys. Chem. Chem. Phys., 2017, 19, 17019–17027 RSC.
- D. B. Ingram and S. Linic, J. Am. Chem. Soc., 2011, 133, 5202–5205 CrossRef CAS PubMed.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS PubMed.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1–24 Search PubMed.
- S. P. Lim, A. Pandikumar, H. N. Lim, R. Ramaraj and N. M. Huang, Sci. Rep., 2015, 5, 11922 CrossRef PubMed.
- J. Zhang, X. Chen, Y. Shen, Y. Li, Z. Hu and J. Chu, Phys. Chem. Chem. Phys., 2011, 13, 13096–13105 RSC.
- N. H. S. Nasralla, M. Yeganeh and L. Šiller, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 1–7 CrossRef.
- X. Xiang, X.-Y. Shi, X.-L. Gao, F. Ji, Y.-J. Wang, C.-M. Liu and X.-T. Zu, Chin. Phys. Lett., 2012, 29, 027801 CrossRef.
- C. Jin, B. Liu, Z. Lei and J. Sun, Nanoscale Res. Lett., 2015, 10, 95 CrossRef PubMed.
- A. Kovács and R. E. Dunin-Borkowski, Handbook of Magnetic Materials, 2018, pp. 59–153 Search PubMed.
- J. G. Tao, L. X. Guan, J. S. Pan, C. H. A. Huan, L. Wang, J. L. Kuo, Z. Zhang, J. W. Chai and S. J. Wang, Appl. Phys. Lett., 2009, 95, 93–96 Search PubMed.
- W. Wang, M. O. Tadé and Z. Shao, Prog. Mater. Sci., 2018, 92, 33–63 CrossRef CAS.
- L. Pan, M. Ai, C. Huang, L. Yin, X. Liu, R. Zhang, S. Wang, Z. Jiang, X. Zhang, J. J. Zou and W. Mi, Nat. Commun., 2020, 11, 418 CrossRef CAS PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- R. T. Ross and T. L. Hsiao, J. Appl. Phys., 1977, 48, 4783–4785 CrossRef.
- J. R. Bolton, S. J. Stricklert and J. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- A. Polman and H. A. Atwater, Nat. Mater., 2012, 11, 174–177 CrossRef CAS PubMed.
- Z. Wang, T. Hisatomi, R. Li, K. Sayama, G. Liu, K. Domen, C. Li and L. Wang, Joule, 2021, 5, 344–359 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02222a |
| This journal is © The Royal Society of Chemistry 2022 |