
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe researcher's guide to solid-state dye-sensitized solar cells
Iacopo
Benesperi
 ,
Hannes
Michaels
and
Marina
Freitag
*
,
Hannes
Michaels
and
Marina
Freitag
*
Department of Chemistry – Ångström Laboratory, Uppsala University, 751 20 Uppsala, Sweden. E-mail: marina.freitag@kemi.uu.se
First published on 21st September 2018
Abstract
In order to sustainably support its ever-increasing energy demand, the human society will have to harvest renewable energy wherever and whenever possible. When converting light to electricity, silicon solar cells are the technology of choice to harvest direct sunlight due to their high performance and continuously dropping price. For diffused light and indoor applications, however, silicon is not the material of choice. To power the next gizmo in your smart home, dye-sensitized solar cells (DSCs) are a viable alternative. Made from inexpensive, earth-abundant, and non-toxic materials, DSCs perform best at low light intensity. So far, issues such as leakage of the liquid electrolyte and its corrosive nature have limited the commercialization of this technology. To overcome these limitations, solid-state DSCs (ssDSCs) – in which the liquid electrolyte is replaced by a solid material – have been developed. For many years their efficiencies have been poor, preventing them from being widely employed. In the past six years, however, research efforts have led them to rival with their liquid counterparts. Here, we will review recent advancements in the field of ssDSCs. Every device component will be acknowledged, from metal oxides and new dyes to novel hole transporters, dopants, counter-electrodes and device architectures. After reviewing materials, long-term stability of devices will be addressed, finally giving an insight into the future that awaits this exciting technology.
 Iacopo Benesperi | Dr Iacopo Benesperi received his MSc in Materials Science from the Department of Chemistry, Turin University, Italy, under the supervision of Prof. Barolo. Thanks to his work on hole conducting materials for perovskite solar cells, he obtained his PhD in Chemistry from the School of Chemistry, Monash University, under the supervision of late Prof. Spiccia, Dr Simonov, Prof. Bach and Prof. Cheng. He is currently a research fellow at Uppsala University in Dr Marina Freitag's group, working on charge transport materials for thin-film solar cells. |
 Hannes Michaels | Hannes Michaels obtained his MSc degree in Nanoscience in 2017 from the University of Hamburg, Germany, after contributions to the research groups of Prof. Dr Hans-P. Oepen and Prof. Dr Wilfried Wurth in cooperation with Uppsala University, Sweden. Currently, he is a doctoral student at the Department of Chemistry in Uppsala exploring charge transport materials based on transition metal complexes for photovoltaic applications in the laboratory of Dr Marina Freitag and Prof. Dr Gerrit Boschloo. |
 Marina Freitag | Dr Marina Freitag is presently an Assistant Professor at the Department of Chemistry, Uppsala University, continuing her work in coordination chemistry and solar cells. Previously, she held a post-doc position at EPFL (Prof. Anders Hagfeldt's). She received her PhD degree with Prof. Galoppini from Rutgers University, USA, and her BSc, in Chemistry from Freie Universität Berlin, Germany. She is the inventor of the so-called Zombie-cell and she contributed to recent breakthroughs in dye-sensitized solar cells for low-power applications. |
1. Introduction
Our modern society is not the first to face an energy crisis.1 Already the ancient Greeks have faced a shortage of charcoal made from trees, after they stripped their land from the forests, resulting in wide-area erosion. Socrates described that the crisis was diverted by smart city planning, so that every household was able to take advantage of the sun.2 Certainly, the technologies for harnessing sunlight have evolved and photovoltaics have become a major focus in today's politics and research.3–7Solar cells can generate electrical power using the photovoltaic effect, which was first presented by the French physicist A. E. Becquerel in 18398 and much later implemented in a successful device by Bell Labs.9,10 The most known type of solar cells are semiconductor devices made from silicon. In these solar cells, charges are generated and separated in a p–n junction of the semiconductor. With silicon being the dominant material used in photovoltaic solar power technology, there are also a handful of competitive thin-film technologies, which are easier to realize, but rely on more expensive and toxic raw materials. Research has branched out to alternative materials for use in solar cells in laboratories around the world.
In 1966 Gerischer introduced a very different concept to the p–n junction, the approach of a photochemical cell (PEC), with a dye sensitizer bound to a metal oxide semiconductor (ZnO) and a liquid electrolyte between the anode and cathode.11,12 Only in 1991 did O'Regan and Grätzel show that the implementation of a mesoporous network of metal oxides resulted in efficient PCEs, and the concept of dye sensitized solar cells was made known.13 Unlike other solar cell technologies, here the dyes are responsible for light absorption, charge separation and injection of charge carriers into the semiconductor, which only plays the role of the electron transport layer. The redox electrolyte is responsible for regeneration of the oxidized dye and charge transport between the electrodes.14–16 Until now, efficiencies of more than 14% have been realized under full sun illumination.17–19 The DSCs promise to be made of cost efficient materials by sustainable production techniques.20–22
Currently the cost efficiency is becoming less relevant due to the tremendous drop in prices of silicon based systems.17,23 Further issues regarding the use of corrosive, toxic, flammable and volatile liquid electrolytes are still being investigated and settled.24–29 The more difficult task of sealing liquid junctions in DSCs in comparison to silicon panels, for which production has long been industrialized, is one of the major limitations for module fabrication.30,31 Solid state dye-sensitized solar cells overcome this limitation with the replacement of the liquid electrolyte by solid charge transport materials.24,32–35
It was only in 1988 that Tennakone reported for the first time a solid-state dye-sensitized heterojunction between TiO2 and CuSCN.36–39 However, dye-sensitized photocurrents were still low due to the nonporous structure of the junction. Since then, alternative approaches have been undertaken to form solid-state dye-sensitized junctions, employing either wide bandgap semiconductors or organic semiconductors. The general idea remained: in solid-state dye sensitized solar cells, the liquid redox electrolyte is replaced by a solid-state hole transporting material or a polymer electrolyte.24,40–42
Until recently, a reduction in the performance of ssDSCs in comparison to their liquid counterparts was observed and was the consequence of poor pore filling,43–48 electron recombination at the semiconductor interface49–53 and the lower conductivity54,55 of the commonly used hole transport and polymer electrolyte materials.24,33
The objective of this review is to give a broad overview of the use of the various types of charge transporting materials and the recent progress made in the field of ssDSCs. The review also highlights the performance of several ssDSCs utilizing new organic and small molecule hole transport materials.
2. Operation principles and structure
2.1. Device structure
The traditional ssDSCs (Fig. 1a) consist of two main electrodes, the photoelectrode or working electrode and the counter electrode (CE). The working electrode (WE) consists of nanoporous metal oxide deposited onto a fluorine doped tin oxide (FTO) coated glass substrate. Usually a blocking layer is deposited on top of the FTO layer to avoid direct contact with the hole transporting material (HTM). The commonly used blocking layer is a compact layer of sintered TiO2 and is usually optimized with respect to the HTL.15,56 The most commonly used semiconductor metal oxide is mesoporous anatase TiO2 with a particle size of around 20–30 nm. The high surface area of nanostructured TiO2 is a prerequisite to absorb a large amount of sensitizer on the extended surface area, as seen in the exemplary cross-section SEM picture in Fig. 1b. One of the most crucial parts of any DSC is the light absorbing sensitizer. There are a large variety of light absorbers for DSCs, which can be categorized into metalorganic complexes and organic dyes.16,57–66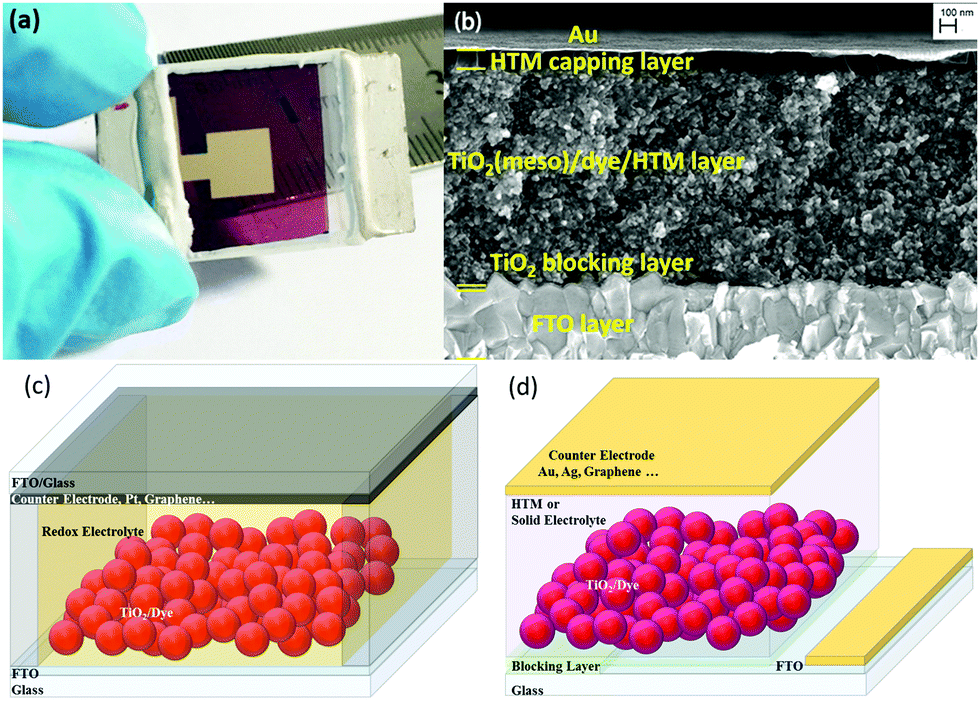 | ||
| Fig. 1 (a) A fully assembled ssDSC with a Au counter electrode. (b) Cross-sectional electron microscopy image of a ssDSC. Adapted with permission from ref. 78. Copyright 2016 American Chemical Society. (c) Schematic sketch of solar cell architectures for the assembly of liquid-junction DSCs and (d) ssDSCs. | ||
A schematic representation to distinguish between DSCs and ssDSCs is given in Fig. 1c and d. The large difference in device structure in comparison to the liquid junction is certainly the use of a solid redox electrolyte or HTM, but also redundancy of a spacer and further sealing between the electrodes. In the case of liquid-electrolyte DSCs (Fig. 1c), the charge transport between the working and counter electrodes is mediated by a redox electrolyte. In a solid-state DSC (Fig. 1d), a solid hole transporting material infiltrates the porous metal oxide (Fig. 1b). In ssDSCs, the mobility of the holes in the HTM is usually higher than the electron mobility in mesoporous TiO2. Nevertheless, it is very important to balance the thickness of the overlying HTM layer. If it is too thick, an increased series resistance arises in the solar cell. If the layer is too thin, however, pinholes could appear resulting in contact of the metal contact with the mesoporous TiO2 film and shunting of the device.67–71
The counter electrode usually consists of metal (Au, Ag) or carbon-based materials deposited by drop casting or evaporation on top of the charge transporting layer.72–77
2.2. Working principles
The conversion of light for power generation follows similar principles to those in photosynthesis. In Fig. 2, the basic working steps are shown.79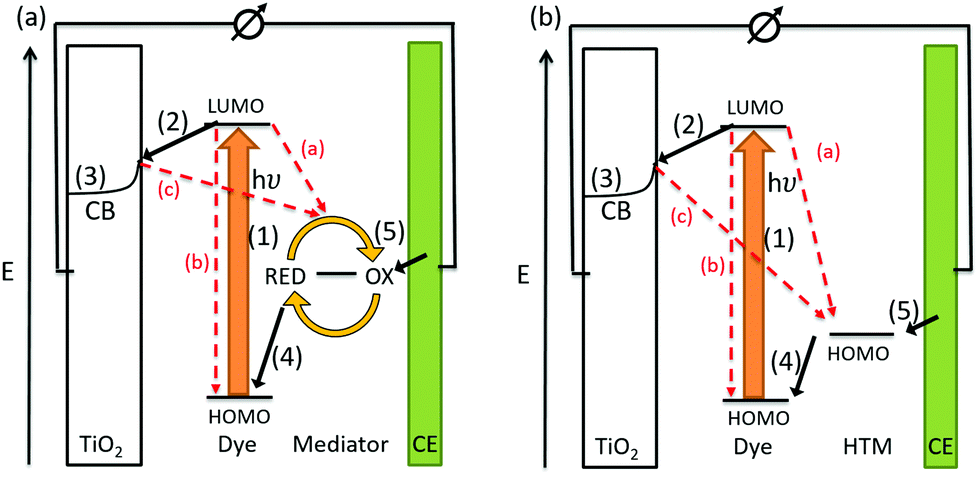 | ||
| Fig. 2 Schematic drawing of charge transport mechanisms in (a) a liquid-junction DSC and (b) a ssDSC. | ||
The dye sensitizer is bound via a functional group to a wide-band gap n-type semiconductor, most commonly TiO2 or ZnO.80,81 The sensitizer's electron in the ground state – i.e. in the highest occupied molecular orbital (HOMO) – reaches the excited state – i.e. the lowest unoccupied molecular orbital (LUMO) – upon light absorption. In the following step, the excited-state dye injects an electron into the conduction band of the metal oxide semiconductor. This is the moment of charge separation; the electrons are in the conduction band of the semiconductor and the holes are positioned in the oxidized dye. The semiconductor is responsible for fast electron transport to the FTO (fluorine doped tin oxide) electrode by diffusion.71 The redox electrolyte or in the case of ssDSCs a solid hole transport material facilitates the regeneration of the oxidized dye and manages the charge transport to the counter electrode. Redox electrolytes are ionic conductors in which charges are transported via diffusion (concentration controlled) and/or by migration (electric field controlled).26,82–84 In solid charge transport materials, the transport is electronic conduction from a solid donor species and the holes are transported to the counter electrode.85–87
The kinetics of charge transfers upon photoexcitation (1) in a liquid-junction DSC and a ssDSC are shown in Fig. 2. The ultrafast electron injection (2) from the excited state dye into the TiO2 conduction band (3) is the fastest process in a dye sensitized solar cell and occurs on a timescale of femto- to picoseconds (Fig. 3).88–92
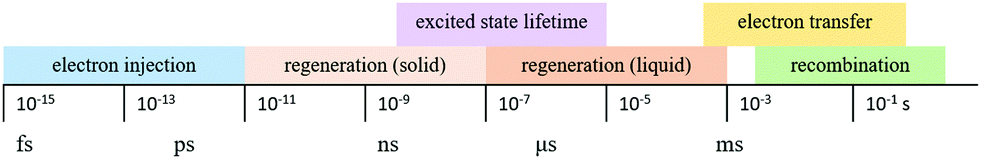 | ||
| Fig. 3 Timescales of charge transfer kinetics in ssDSCs. | ||
In ssDSCs, the dye is regenerated (4) from its oxidized state within a few hundred picoseconds, orders of magnitude faster than in the liquid junction cells, where dye regeneration occurs on the microsecond timescale. This fast regeneration process in solid-state DSCs is attributed to direct hole transfer (via an energy gradient) into the HOMO level of a solid-state hole transporter from the oxidized state of the dye molecule, whereas the redox reaction in a liquid-state system is diffusion limited. These extremely rapid regeneration dynamics lead to a rapid hole injection from the oxidized dye into the HTM essentially on the same time scale as the electron injection.46,93,94 The redox mediator or HTM is finally regenerated by charge transfer at the counter electrode (5). Recombinative charge transfers (drawn as red arrows) from the dye (a) and TiO2 (b) to the redox electrolyte/HTM or from TiO2 to the ground state of the dye impede the solar cell performance (see Section 2.4 on Limitations of ssDSCs).
Bach et al. reported 50%-hole injection within 900 ps from the oxidized N3 dye to spiro-OMeTAD.85,95–97 In fact, dye reduction (or hole injection to the HTM) may occur before electron injection into TiO2. Cappel and coworkers found that in a perylene-sensitized TiO2/ID176/spiro-OMeTAD system electron injection and dye regeneration were complete after 1 ps, based on the observation of the Stark effect, a spectral shift of the dye spectrum caused by the electric field between the electron in TiO2 and the hole in the HTM.98,99 Remarkably, much slower (millisecond) regeneration kinetics were reported for PEDOT as a polymeric HTM.40,100 Haque et al. found that a driving force of about 0.2 eV is needed for efficient (>85%) dye regeneration by the HTM.92,101 Freitag et al. and Grätzel and coworkers showed slower (μs) regeneration kinetics for copper coordination complexes as HTMs with a driving force of about 0.1 eV for sufficient dye regeneration.86,87,102–104
2.3. Characterization
A set of techniques to assess the performance of ssDSCs and their components have been developed. Thereby, the power conversion of the solar cell can be evaluated and charge transfer as well as transport processes can be monitored. Furthermore, the performance of single components of a ssDSC can be assessed. The ensuing full-device characterization techniques,• I–V characteristics
• Incident-photon-to-current conversion efficiency (IPCE)
• Electron lifetime are illustrated in the first part of this section. Consequently, component-related techniques
• Photoinduced absorption spectroscopy (PIA)
• Transient absorption spectroscopy (TAS) are introduced.
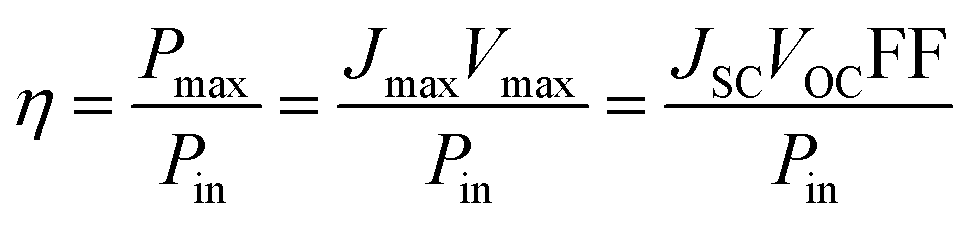 | (1) |
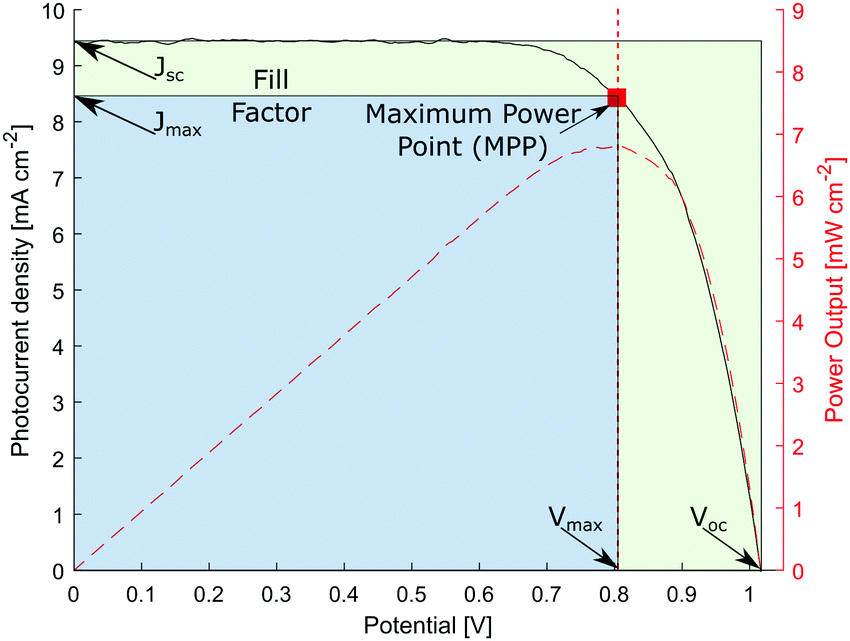 | ||
| Fig. 4 Characteristic I–V scan of a ssDSC. | ||
However, as Vmax and Jmax are not easily visible in the I–V scan, they are not commonly chosen to compare solar cell performances.
At the so-called open-circuit voltage VOC, the voltage output of the solar cell matches the applied potential and the current density vanishes (see Fig. 4). The photovoltage (VOC) is determined by the potential difference between the Fermi levels of electrons in the TiO2 film and in the hole transporting material. Similarly, the photocurrent density (JSC) is determined based on the incident light harvest efficiency (LHE), charge injection and collection efficiencies. The short-circuit current ISC is recorded at zero potential and commonly normalized by the solar cell area to give a more comparable short-circuit current density JSC. A so-called fill factor (FF) is subsequently introduced to account for ideality of the I–V curve. This fill factor represents the ratio of the actual maximum obtainable power (Pmax, blue rectangle in Fig. 4) to the product of the open circuit voltage and short circuit current (VOC·JSC, green rectangle).
| IPCE(λ) = LHE(λ)φinj(λ)φreg(λ)φcc(λ) | (2) |
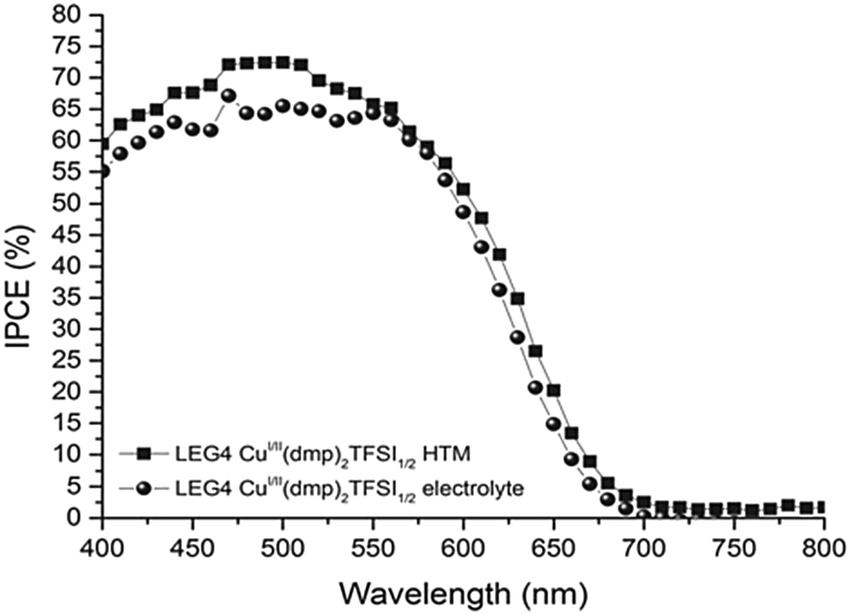 | ||
| Fig. 5 Incident-photon-to-current conversion efficiency spectra of DSCs sensitized with the organic LEG4 dye and using copper phenanthroline as a liquid-electrolyte (circles) and a solid-state HTM (squares). Reproduced from ref. 107 with permission from the Royal Society of Chemistry. | ||
Due to faster hole extraction from the dye molecules and faster conduction in the solid HTM compared to the diffusion-based liquid electrolyte, an increase in power photon-to-current conversion efficiency is observed over the entire absorption range of the LEG4 dye.
The IPCE should, in turn, be integrated with the spectral flux distribution of sunlight Φsun to match the short circuit current density
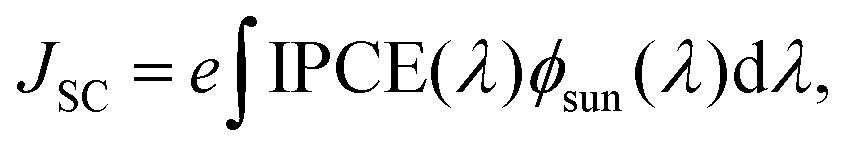 | (3) |
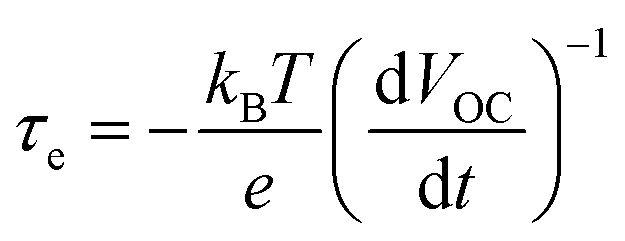 | (4) |
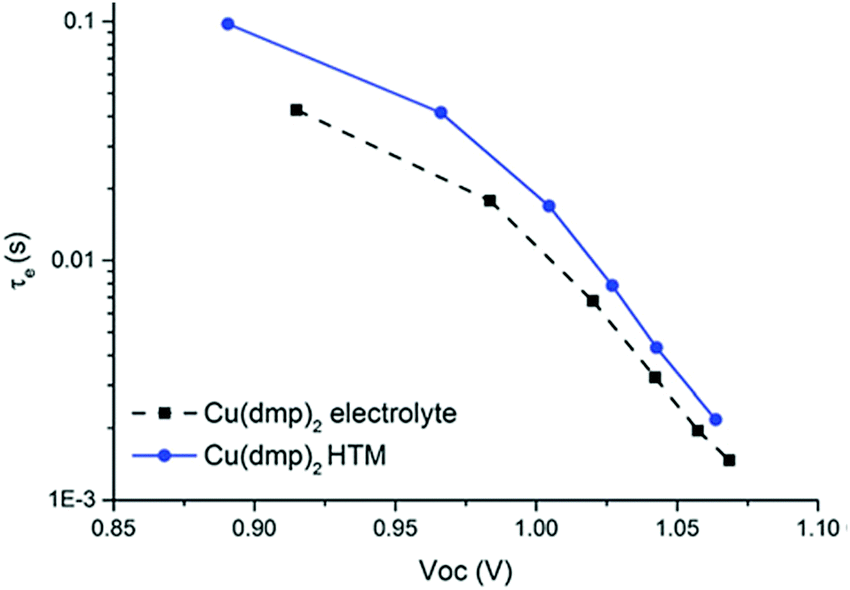 | ||
| Fig. 6 Electron lifetime in LEG4-sensitized solar cells with copper phenanthroline as a liquid electrolyte (circles) and a solid-state HTM (squares). Reproduced from ref. 107 with permission from the Royal Society of Chemistry. | ||
At high irradiation intensities (and hence high VOC), a high carrier density in the TiO2 layer increases the rate of recombination with oxidized dye molecules or the HTM. Thus, the lifetime of electrons is comparably low with respect to lower VOC.
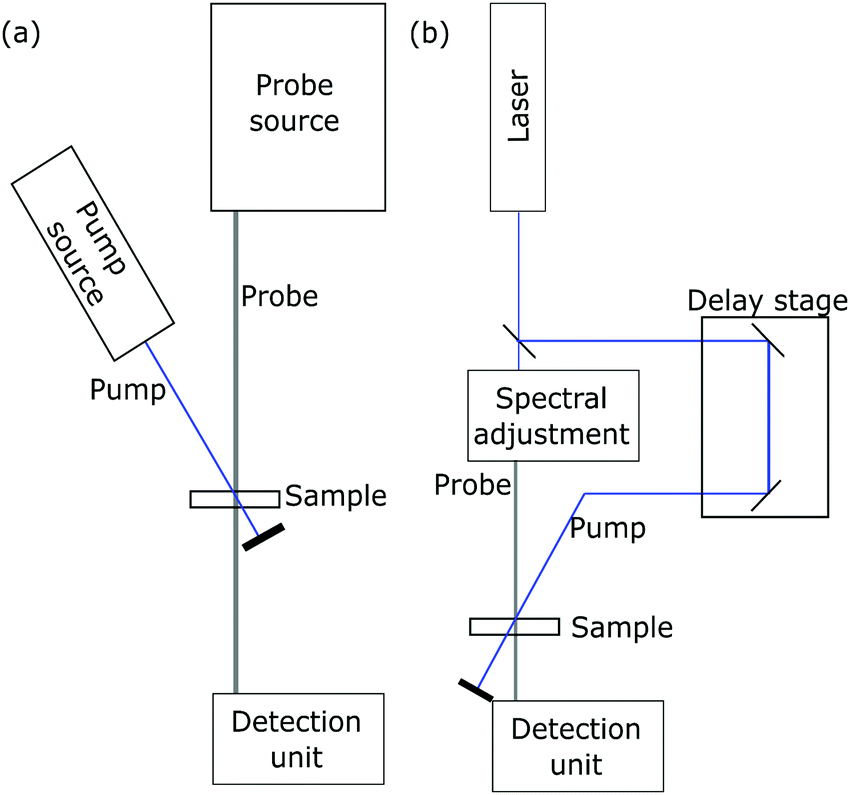 | ||
| Fig. 7 Schematic setup for (a) photoinduced absorption and (b) transient absorption measurements. | ||
An exemplary photoinduced absorption spectrum is shown in Fig. 8.
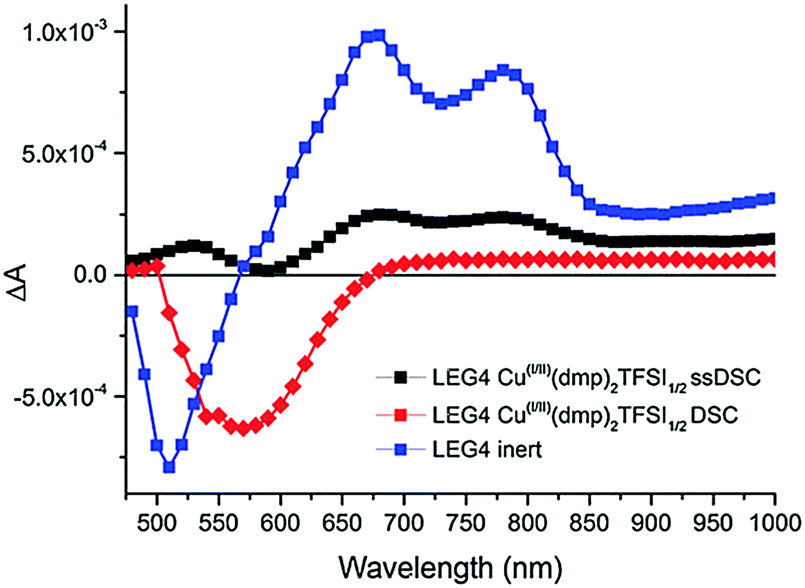 | ||
| Fig. 8 Photoinduced absorption spectra of LEG4-sensitized TiO2 photoanodes (blue) as well as in contact with copper phenanthroline complexes in liquid junction (red) and solid-state (black) DSCs. Reproduced from ref. 107 with permission from the Royal Society of Chemistry. | ||
The observed spectral change in absorption upon excitation usually closely matches the absorption spectrum of the oxidized dye species (blue in Fig. 8). In contact with an HTM, however, the dye molecules are regenerated on a nanosecond timescale. The oxidized dye molecules are ‘quenched’ and their absorption is no longer visible in the photoinduced absorption spectrum. As illustrated in Fig. 3 in Section 2.2, the dye regeneration is accelerated in ssDSCs compared to their liquid-electrolyte counterparts. Therefore, the average lifetime of the oxidized dye species is reduced and the characteristic absorption features of the dye vanish (Fig. 8). Photoinduced absorption spectroscopy can furthermore be used to investigate pore filling of the hole transport material (Section 3.2) in a ssDSC, as dye molecules which are not in contact with the HTM will remain oxidized on a microsecond timescale.
Characteristic absorption bands in the oxidized dye spectrum can then be chosen to directly determine the time constants of electron transfer with transient absorption spectroscopy (TAS).112 A schematic experimental setup is drawn in Fig. 7b. In this technique, a sensitized semiconductor oxide is excited with a nanosecond laser pulse and the decay of the oxidized dye absorption is monitored to compare regenerative charge transfer and recombinative processes.86,113 An exemplary spectrum is presented in Fig. 9.
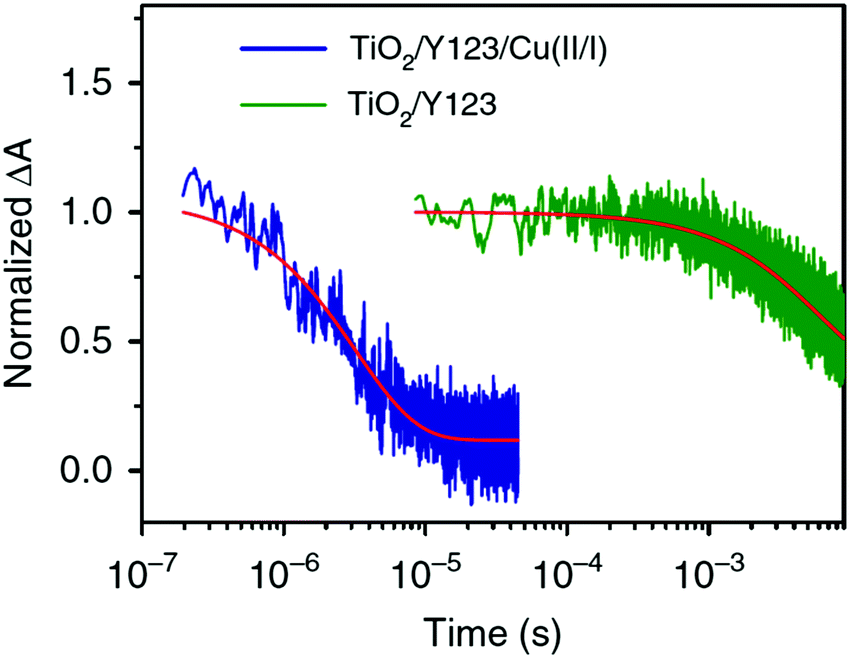 | ||
| Fig. 9 Transient absorption spectra of Y123-sensitized photoanodes inert (green) and in contact with the solid CuII/I(tmby)2 hole transport material. Reproduced from ref. 87 with permission from the Nature publishing group. | ||
Thus, it can be determined that the oxidized Y123-species is regenerated by the CuII/I(tmby)2 HTM on a nano- to microsecond timescale, while the oxidized dye species prevails up to milliseconds when electrons in the TiO2 layer recombine with the oxidized dye.
2.4. Limitations of ssDSCs
Despite continuous research and progress, the highest performing ssDSC is at 11.7%,103 lagging behind emerging hybrid organic inorganic perovskite-based solar cells or conventional silicon-based solar cells. The optimal band gap for a sensitizer dye follows the Shockley–Queisser limit, which determines the theoretical optimum band gap of a single absorbing material to be 1.1 electron volt (eV).114–116 This theoretical limit predicts a maximum conversion efficiency just above 30% with a band gap of 1.4 eV (at AM 1.5 solar spectrum) for a perfect absorber in an ideal solar cell. The limitations in performance of DSCs are largely determined by imperfect energy-level alignment between the individual components, as well as the underlying kinetics of the charge separation and charge-transfer processes.15,32,115It is well known that the main limitations in solid-state DSCs are with respect to (a) photocurrent, (b) potential and (c) fill factor.
(a) The photocurrent largely depends on the photoanode or the working electrode. Improving the light management will likely lead to higher photocurrent. This includes implementation of wide spectral range and high absorbing dyes as well as optimization of the semiconductor morphology with scattering layers, photonic crystals and plasmonics.32,117,118 Currently, the photocurrent is further limited by relatively thin semiconductor layers, where the thickness is optimized to about 2 μm to enable sufficient pore-filling and low series resistance by the HTM. This thickness is not enough to absorb and convert enough incident light to yield efficiencies above 10%. The pore-filling effect has recently been investigated, confirming that a better performing solar cell device can be obtained by increasing the amount of spiro-OMeTAD inside the pores.46,70,119
(b) The limitations in potential can be overcome by lowering the driving force for dye regeneration to increase VOC. The ideal theoretical open-circuit potential (VOC) of ssDSC is where no back flow of current exists, which in real DSCs is limited by recombination reactions, specifically between the excited dye and the HTM. The same higher rate of electron recombination also limits the device efficiency for ssDSCs based on spiro-OMeTAD. Synthetic modifications of dyes and HTMs can reduce the recombination and improve VOC.86,87,107
(c) The fill factor is limited by recombination processes between metal oxide and HTMs, resulting in dark currents as well as series resistance. Both influences are likely to be diminished by improving the morphology of the photoanode (blocking layers) as well as the choice and morphology of the counter electrode to ensure better contact with the HTM.120
3. Material development
3.1. Photoanode
 | ||
| Fig. 10 Scanning electron microscopy images of (a) bare FTO as well as with blocking layers of (b) 13 nm (c) 25 nm and (d) 50 nm titanium dioxide. Reprinted from ref. 122 with permission from Elsevier. | ||
A commonly used titanium precursor is a diluted titanium bis-(acetoacetonato)-di-(isopropanoxylate) ethanol solution. Peng and coworkers optimized the blocking layer to about 150 nm by repetition of spray cycles. Further, the thickness of the compact layer and its surface properties can have a strong impact on the performance of the ssDSC due to the lower roughness of 10 nm in comparison to the 13 nm of the FTO substrates as a result of the smaller TiO2 particles deposited.40,120,121
O’Regan and Grätzel pioneered the introduction of nanostructured titanium dioxide (TiO2) that led to DSCs in 1991, presenting a very large surface area well suited for sensitization by molecular dyes. The nanostructured electrode is crucial, as its morphology drives many physical processes that control the overall device performance: the light-harvesting properties are directly dependent on the amount of surface area available for dyes, and furthermore on the number of free charge carriers, especially electrons. The collected photocurrent is limited by the ability of photogenerated charges to flow through the nanostructured electrode.
Although several alternative metal oxide compounds have been regularly assessed, TiO2 still demonstrates the best device efficiencies, either in liquid or in solid-state device structures. In TiO2, the titanium is in oxidation state IV (d0). The oxide exists in several polymorphs of which two are more relevant: anatase (tetragonal) and rutile (tetragonal). The crystal structures of anatase and rutile are shown in Fig. 11.
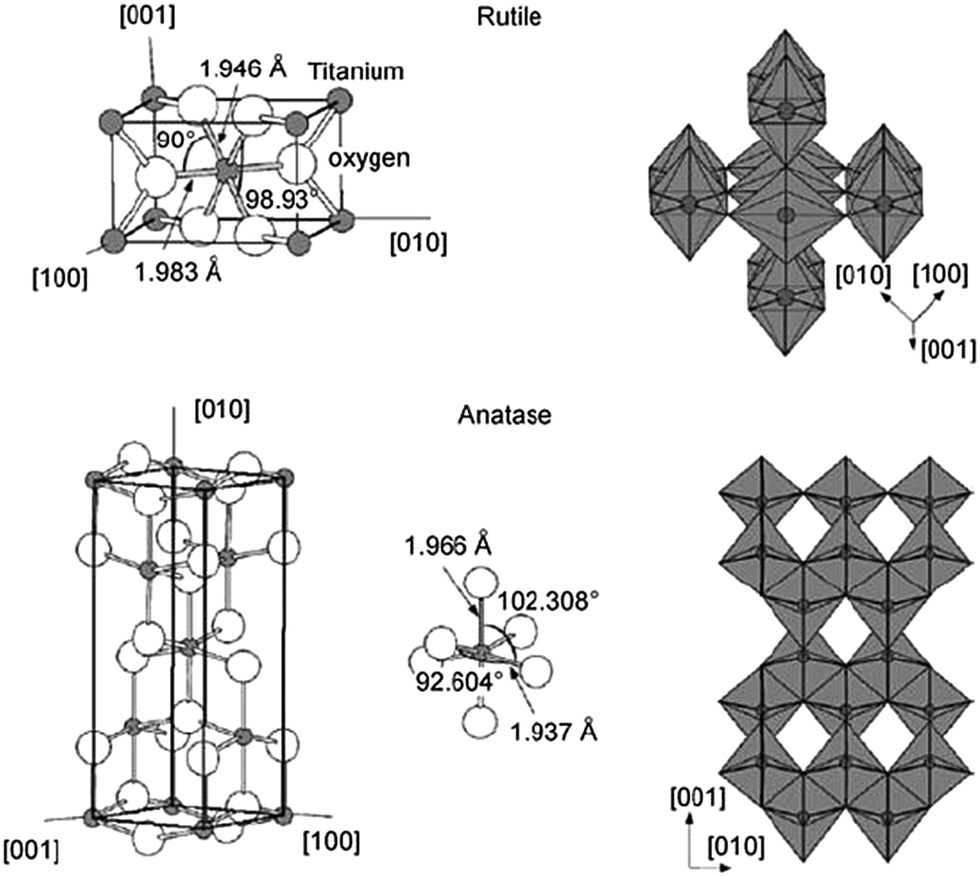 | ||
| Fig. 11 Crystal structures of rutile (top) and anatase (bottom) phases of titanium dioxide. Reprinted from ref. 123 with permission from Elsevier. | ||
For anatase, the band gap is 3.2 eV, slightly larger than that of rutile with 3.0 eV. This transition in the UV region results in an absorption at 390–400 nm. Rutile is the most stable bulk form of TiO2, but anatase is reported to have better photoactivity and is more stable in nanoparticle form. The n-type TiO2 photoelectrode nanoparticles offer some unique properties, making them an appropriate semiconductor for DSCs. They have a low intrinsic film conductivity, the small size of the nano-crystalline particles does not support a built-in electric field, and the electrolyte diffuses through the mesoporous network between the electrodes.
The high refractive index of TiO2 (n = 2.5 anatase) results in efficient diffused scattering of the light inside the porous photoelectrode, which significantly enhances light absorption. Most importantly, mesoporous TiO2 films have a high internal surface area to support the monolayer of a dye sensitizer, and their conduction band edge lies slightly below the LUMO position of many dye sensitizers.124 This enables efficient electron injection in the conduction band of semiconductors to transport the electrons through the TiO2 film by simple diffusion towards the FTO. The capability of the TiO2 anatase phase to absorb the solar spectrum in the range of ultraviolet or near-ultraviolet radiation can only capture about 4% of solar light. At the same time, the high dielectric constant of TiO2 (ε = 80 for anatase) provides good electrostatic shielding of the injected electrons from recombination with the oxidized dye.125
| Anode | Dye | HTMa | CEb | V OC (mV) | J SC (mA cm−2) | FFe | PCEf (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Hole transporting material. b Counter-electrode. c Open circuit voltage. d Short circuit current. e Fill factor. f Power conversion efficiency. g Graphite. h n.r. = not reported. | ||||||||
| Copolymer templates | D149 | Spiro-OMeTAD | Ag | 870 | 3.7 | 0.54 | 1.7 | 129 |
| Copolymer templates | D102 | Spiro-OMeTAD | Ag | 820 | 7.5 | 0.53 | 3.2 | 130 |
| Single crystal TiO2 | D102 | Spiro-OMeTAD | Ag | 760 | 6.5 | 0.63 | 3.1 | 131 |
| Columnar ZnO | Ru-phospho | CuSCN | Cg | 550 | 4.5 | 0.57 | 1.5 | 132 |
| ZnO/MgO wires | Ru-NCS-bpy | Spiro-OMeTAD | Ag | 350 | 2.3 | 0.42 | 0.3 | 133 |
| ZnO nanowires | Z907 | Spiro-OMeTAD | Au | 780 | 12.2 | 0.58 | 5.7 | 134 |
| SnO2/Al2O3 | N3 | CuI | Au | 350 | 1.7 | 0.32 | 0.3 | 135 |
| SnO2/MgO | D102 | Spiro-OMeTAD | Ag | 470 | n.r.h | n.r. | 1.26 | 136 |
After the deposition of mesoporous titanium dioxide, a scattering layer of larger particles is commonly deposited to reflect transmitted photons back into the active solar cell volume, as shown in Fig. 12.126,127
 | ||
| Fig. 12 Cross-sectional electron microscopy image of a DSC photoanode. Reprinted from ref. 128 with permission from Hindawi. | ||
Many configurations were explored to optimize the morphology and therefore electron transport of mesoporous TiO2 (Table 1). For example, the nanostructured electrodes were further modified with TiO2 nanotubes/rods to enhance electron transport through well-aligned pathways (see Fig. 13). Nonetheless, the efficiency obtained by these methods does not compete with nanoporous TiO2 in ssDSC, because of lower dye loading originating from a loss of semi-conductor/electrolyte surface area. Steiner and Snaith templated TiO2 into cylinders using block co-polymers. This allowed them to make oriented one-dimensional (1D) columnar structures as well as three-dimensional (3D) bicontinuous gyroid structures. The resulted ssDSCs with a 400 nm thick film sensitized with D149 (see Fig. 18) showed a PCE of 1.7%,129 which was later optimized to 3.2% by Docampo et al. (see Table 3) employing the indoline D102 dye (see Fig. 18).130
 | ||
| Fig. 13 Different structures of the mesoporous layer. (a) SEM image of TiO2 gyroid networks. Reprinted from ref. 129 with permission from the American Chemical Society. (b) SEM image of TiO2 mesoporous single crystals. Reprinted from ref. 131 with permission from Springer Nature. (c) TEM image of cylindrical porous TiO2. Reprinted from ref. 130 with permission from Wiley. (d) SEM image of the ZnO nanowire array. Reprinted from ref. 134 with permission from the American Chemical Society. | ||
Crossland et al. developed mesoporous single crystals of anatase TiO2, which displayed one order of magnitude higher electron mobility at the same charge density compared to conventional mesoporous TiO2 layers. Using mesoporous single crystal films processed at low temperatures, the corresponding ssDSCs attained efficiencies of over 3.1%.131
Additional modification of the mesoporous TiO2 film by an ultrathin layer of Al2O3 was carried out for ssDSCs initially with inorganic hole transport materials (CuI, CUSCN) and later also with spiro-OMeTAD. Apparently, the introduction of the ALD layer of Al2O3 improved voltages by slowing down the electron recombination from the TiO2 and HTM, sacrificing the photocurrent density.137
Optimization of the functional properties relies not only on the modulation of the shape and structure of the photoanode, but also on the application of different materials and/or composite systems, which allow a fine tuning of the electronic band structure. Nevertheless, only a few studies explored alternative materials to TiO2, but the search for increasing the power conversion efficiency (PCE) of ssDSC by incorporating n-type metal oxide semiconductors, such as ZnO, SnO2, Nb2O5, and SrTiO3, continues.138–140
Considering its electronic configuration and excellent physical properties, including the high electron mobility (∼100 cm2 V−1 s−1) and conductivity, ZnO is considered more suitable for application in ssDSCs, if the stability issue could be addressed.132,141–143
In 2000 O’Regan et al. developed a method for electrodeposition of a columnar ZnO structure, which was dye-sensitized followed by electrodeposition of p-type CuSCN to create a complete inner-surface electrical contact, resulting in ssDSC performance of 1.5%.132 Plank et al. reported on ssDSCs based on ZnO nanowires grown using hydrothermal methods, resulting in ssDSC devices with 0.3% efficiency when an ultrathin MgO or ZrO2 shell was applied.133 Gao and coworkers developed multilayer TiO2-coated ZnO nanowires, grown in a sequence fashion to reach 50 μm length, using a newly developed multistep HTM filling process. In conjunction with the Z907 dye, the resulting ssDSC attained 5.7% efficiency.134
A heterojunction of n-SnO2/Al2O3/Ru-dye/p-CuI, where the dye is coated on a thin film of Al2O3 first deposited on SnO2, was introduced by Tennakone et al. The ssDSCs delivered a short-circuit current density of 1.7 mA cm−2 and an open-circuit voltage of 350 mV.135 Docampo et al. used spiro-OMeTAD as the HTM on a SnO2-based ssDSC resulting in a performance of 1.26%. The addition of a mesoporous Al2O3 layer on top of the SnO2 mesoporous film led to suppression of recombination of the separated carriers.136
Another way to increase solar cell efficiency through modification of the mesoporous layer is to “dope” TiO2 with materials that interact with light, such as metal nanoparticles and photonic crystals. In the case of metal nanoparticles, such materials are deposited on the mesoporous TiO2 surface and they enhance dye sensitizer absorption by acting as subwavelength antennas. Through the generation of surface plasmons, these metal nanoparticles contribute to light absorption, reflection and scattering, contributing to the generation of more electrons.144 In ssDSCs, Brown et al. obtained an increase in average efficiency from 1.2% to 2.2% after depositing Au nanoparticles with a SiO2 shell on the mesoporous titania layer.145 Such devices employed Z907 as the dye and spiro-OMeTAD as the HTM. Arof and coworkers employed Ag nanoparticles in a device with the N3 dye and a polymer electrolyte to obtain an increase in efficiency from 0.78% to 1.13%.146 Photonic crystals are periodic nanostructures that can interact with light and affect its propagation. In dye-sensitized solar cells they are deposited on top of the TiO2 mesoporous layer to reflect and diffract light at specific wavelengths, increasing dye performance.147,148 In solid-state devices, photonic crystals were employed by Chung et al. with an increase in solar cell efficiency from 9.3% to 10.2%33 and by Lee et al., with an efficiency increase from 6.9% to 7.8%.149
For liquid junction dye-sensitized solar cells, high power conversion efficiencies are mostly obtained using ruthenium complexes as sensitizers, but recently a new record was set using co-sensitization of zinc porphyrin with an organic dye. Good progress is also made with pure, metal-free, organic sensitizers. While large numbers of sensitizers are being reported every year, only a few of them are routinely screened in the ssDSC configuration, leaving the overall ssDSC efficiency lagging behind that of its liquid electrolyte counterpart. The dye design plays an even greater role in the ssDSC. Not only does the dye require optimized HOMO/LUMO energy levels, but it also needs to provide high oscillator strength to be able to capture sufficient light even at thin electrode thicknesses.57 Dye molecules also need to supply sufficient surface protection on the TiO2 electrode to reduce interfacial recombination, have a significant dipole moment to permit for an upward shift in the TiO2 conduction band as well as to facilitate the wetting process of the hole transporting material within the mesoporous structure. Small molecules adsorbed on the TiO2 surface along with the dye can also affect the conduction band edge through their dipole moment.
The sensitizers can be divided into inorganic-based dyes and organic dyes according to the chemical structure. Ruthenium and osmium complex sensitizers are the representative inorganic dyes and their properties have been investigated and improved systematically.16,57,59–63 There are also many studies on purely organic dyes with good photovoltaic performance. Organic dyes are easy to synthesize, their properties can be easily tuned and they have lower cost compared with ruthenium sensitizers.118,153,155 The film thickness is a critical point in our ssDSC device, therefore a high extinction coefficient dye can enhance the photocurrent and lead to higher overall efficiency with a small amount of dye. So far, organic dyes have made it possible to reach the highest power conversion efficiencies of over 11% for ssDSCs.87,103 In general, dye molecule design makes it possible not only to selectively change the absorption range of the dye, but also to modify the energy levels.
3.1.3.1. Metal coordination complexes. The first dyes used in DSCs were ruthenium-based dyes and therefore metal coordination complexes. These ruthenium complexes have received particular interest as photosensitizers for having advantageous characteristics such as wide absorption bands, high external quantum efficiencies, favorable energy levels and relatively long excited state lifetimes. Among several Ru complexes, Ru complexes with carboxylated bi- or poly-pyridine ligands such as N3, N719, CYC-B11, Z907 and Z910 were extensively studied (Fig. 14).156
Ru-based dyes are capable of yielding conversion efficiencies greater than 10%, in liquid junction DSCs. However, the Ru-dyes have low extinction yields, which is a limitation, because the ssDSCs require thinner mesoporous layers of a semiconductor for better pore filling.
In one of the first reports of efficient ssDSCs, Bach et al. used the Ru-dye N3 in combination with the organic HTM spiro-OMeTAD and a PCE of 0.74% (Table 2) was obtained at an intensity of 9.4 mW cm−2.85 Later, the partially deprotonated form of N3, namely N719, was used and with careful optimization of the additive tert-butylpyridine (tBP) a certified efficiency of 2.56% was obtained for a solar cell device with an active area of 1.07 cm2.97 To solve the issue of rather low photocurrents, studies with addition of coadsorbents, increasing ligand length by extension of the conjugated system, cosensitization, and the use of a near-IR absorbing dye, known as a black dye, were reported. In particular, the additive tBP plays a crucial role in the inhibition of the interfacial electron recombination.
| Dye | HTMa | Additivesb | CEc | V OC (mV) | J SC (mA cm−2) | FFf | PCEg (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Hole transporting material. b List of additives: Sb = N(PhBr)3SbCl6, Li = LiTFSI, tBP = 4-tert-butylpyridine, Co = FK102, FK209 or FK269. c Counter-electrode. d Open circuit voltage. e Short circuit current. f Fill factor. g Power conversion efficiency. h n.r. = not reported. | ||||||||
| N3 | Spiro-OMeTAD | Sb, Li | Au | 340 | 0.32 | 0.62 | 0.74 | 85 |
| N719 | Spiro-OMeTAD | Sb, Li, tBP | Au | 910 | 5.1 | 0.57 | 2.6 | 97 |
| N719 | Spiro-OMeTAD | Sb, Li, tBP | Au | 930 | 4.6 | 0.71 | 3.2 | 157 |
| Z907 | Spiro-OMeTAD | Sb, Li, tBP | Au | 750 | 8.3 | 0.64 | 4.0 | 158 |
| K51 | Spiro-OMeTAD | Sb, Li, tBP | Au | 880 | 6.8 | 0.65 | 3.8 | 159 |
| CYC-B11 | Spiro-OMeTAD | Li, tBP | Au | 830 | 9.2 | 0.63 | 4.7 | 160 |
| C101 | Spiro-OMeTAD | n.r.h | Au | 800 | 8.2 | 0.69 | 4.5 | 161 |
| YD-o-C18 | Spiro-OMeTAD | n.r. | Ag | n.r. | n.r. | n.r. | 4.1 | 162 |
| KS | Spiro-OMeTAD | Li, tBP, Co | Au | 849 | 11.0 | 0.53 | 5.1 | 163 |
| FA | Spiro-OMeTAD | Li, tBP, Co | Au | 786 | 10.5 | 0.54 | 4.5 | 163 |
In 2005, the amphiphilic dye Z907 with hydrophobic tails was introduced in ssDSCs and a PCE of 4.0% was obtained. The enhanced photovoltaic performance of ssDSCs was attributed to the dense packing of dyes on the surface of TiO2 as well as the hydrophobic isolating chains which block the direct contact between spiro-OMeTAD and TiO2.158 Inspired by this, the effect of the hydrocarbon chain lengths on the Ru-dye was systematically investigated, and it was confirmed that the hydrophobic chains act as an insulating barrier between TiO2 and the HTM, and can efficiently suppress the interfacial electron recombination, as demonstrated by detailed studies via transient absorption results.
Another strategy is to improve dye packing and blocking properties towards the HTM on the mesoporous surface. This was achieved by implementation of amphiphilic, heteroleptic ruthenium sensitizers with hydrophobic spacers. Efficiencies over 7.8% were reported with an ion-coordinating sensitizer (K51) containing triethylene oxide methyl ether (TEOME) at the 4,4-position of a 2,2-bipyridine ligand; this enables the dye to prevent the ions from reaching the semiconductor interface.159
In the CYC-B11 dye, bithiophene groups were added, resulting in a good molar extinction coefficient (2.42 × 104 M−1 cm−1 at 554 nm) and a high PCE for ssDSCs of 4.7%.160 Alkylthiophene-containing groups were added to the bipyridine ligand by Peng Wang and co-coworkers in order to increase the extinction coefficient of Z907, resulting in C101 with an enhanced performance of 4.5% compared to 2.9% for Z907.161
Other issues in considering Ru-based dyes are their cost as Ru is a rare metal with high price and toxic nature. Thus, research into Ru free dyes, metal-complex porphyrin dyes, has intensified.162,163
Unlike ruthenium systems, the porphyrin exhibits strong absorption in the near-IR region due to an additional Q-band in the 500–700 nm region with good stability. The asymmetric porphyrin dye (YD2-o-C8, Fig. 15) was synthesized by introducing triarylamine as an electron donor and carboxylic acid as an acceptor. Porphyrins have been designed and synthesized based on the molecular structure of YD2-o-C8 for applications in DSCs to reach a PCE of 13% using a cobalt-based liquid-type electrolyte.19
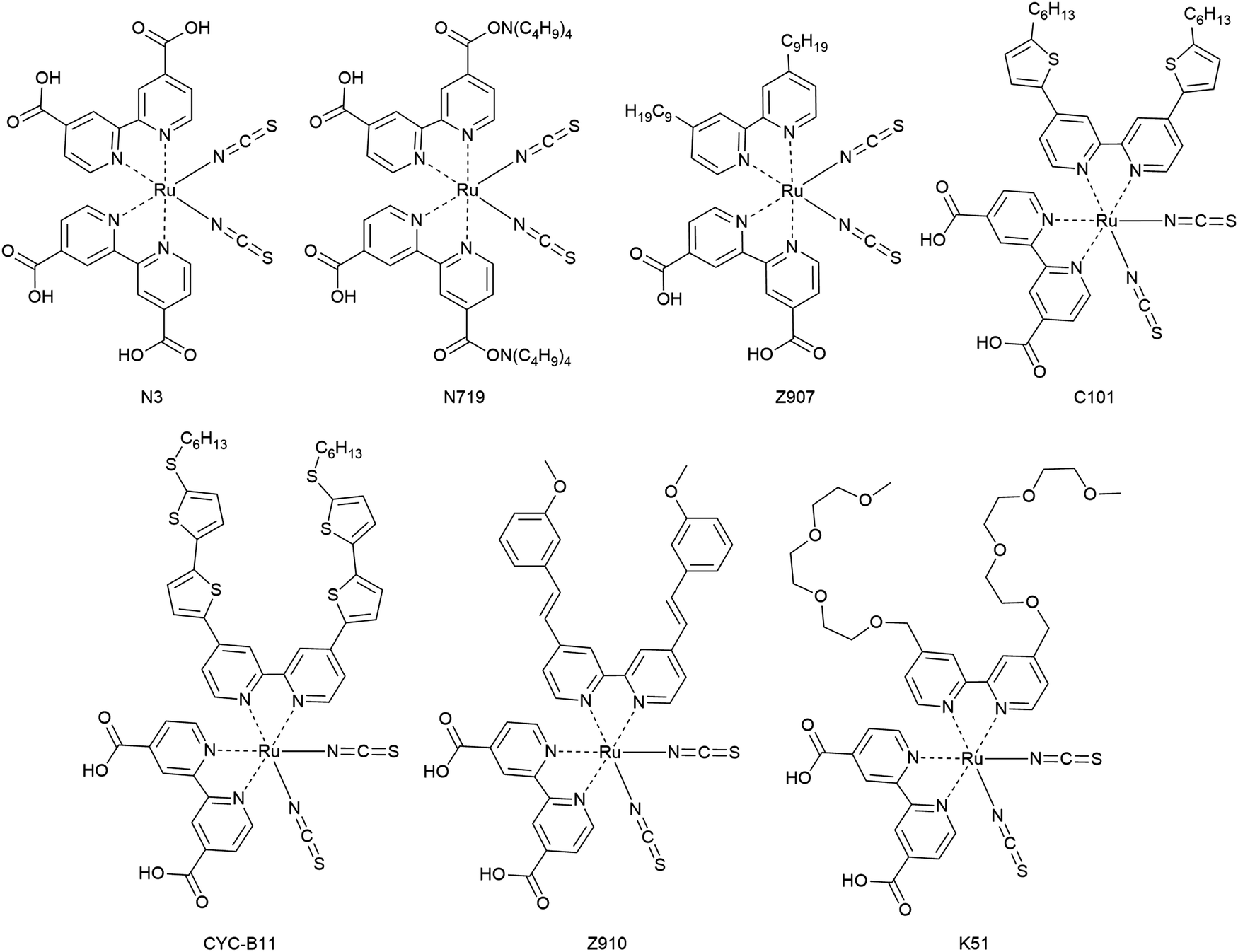 | ||
| Fig. 14 Chemical structures of ruthenium-based sensitizers for ssDSCs. | ||
The LD14 was modified for the use in ssDSC by a functional group inserted into the position between the porphyrin core and the donor group; the resulting device attained a PCE of 4.1% with spiro-OMeTAD as the HTM. The performance was still lower in comparison to LD14 in liquid junction DSCs, where a PCE of 9.2% was attained.156,164,165 Another modified push–pull porphyrin was designed having a benzothiadiazole (BTD) group between the porphyrin core and the acceptor anchoring group. The ssDSC achieved a PCE of 5.5%, when the porphyrin dye was co-sensitized with an organic dye.162 Both time-resolved transient absorption spectroscopy and frequency-domain electrochemical impedance spectroscopy techniques were applied to understand charge transport and recombination kinetics with respect to their photovoltaic performances.
Peng Qin et al. recently reported molecularly engineered weakly conjugated hybrid porphyrin dyes (KS and FA) as efficient sensitizers for solid-state dye-sensitized solar cells. By incorporating a quinolizino acridine and triazatruxene based unit as the secondary light-harvester as well as an electron-donating group at the meso-position of the porphyrin core efficiencies of 4.5% and 5.1% were achieved in ssDSCs (Table 2).163
3.1.3.2. Organic dyes. Sensitizer development for ssDSCs is largely focused on metal-free dyes, since higher efficiencies were recently reached in combination with hole transport materials based on metal coordination complexes.166 Their success, especially over the ruthenium based systems, can be attributed to the following characteristics:
(1) The organic dyes show a large variety in terms of sensitizer families (indolines (D149, ID176), triphenylamines, coumarine, arylamines (Fig. 18)).36,98,99,153,167 The ease in their design and synthesis leads to a large variety of characteristics.
(2) The organic dyes have generally higher extinction coefficients (the most efficient ssDSC incorporated 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 M−1 cm−1LEG4 and 70100 M−1 cm−1XY1 as well as 68800 M−1 cm−1XY1B).34,41,168 The high molar extinction coefficients are especially desirable for thin TiO2 films in ssDSCs. For instance, indoline dye D102169 has a very high extinction coefficient of 55800 M−1 cm−1 at 491 nm, which yields over 90% absorption over a broad spectral range when sensitizing TiO2 films are as thin as 2 μm. However, for the ruthenium sensitizers, the TiO2 film of 10 μm thickness is required to get the same absorption.38,39,158,170
800 M−1 cm−1LEG4 and 70100 M−1 cm−1XY1 as well as 68800 M−1 cm−1XY1B).34,41,168 The high molar extinction coefficients are especially desirable for thin TiO2 films in ssDSCs. For instance, indoline dye D102169 has a very high extinction coefficient of 55800 M−1 cm−1 at 491 nm, which yields over 90% absorption over a broad spectral range when sensitizing TiO2 films are as thin as 2 μm. However, for the ruthenium sensitizers, the TiO2 film of 10 μm thickness is required to get the same absorption.38,39,158,170
(3) Organic dyes lack the precious metal part, which also makes them more cost efficient and sustainable.17,171
Some general principles to design an efficient organic dye and efficient ssDSCs are as follows: a donor–π-bridge–acceptor (D–π–A, D–A–π–A) structure (Fig. 16), which can be easily modified for extending the absorption spectra, adjusting the HOMO and LUMO levels to complete the intramolecular charge separation.78,118
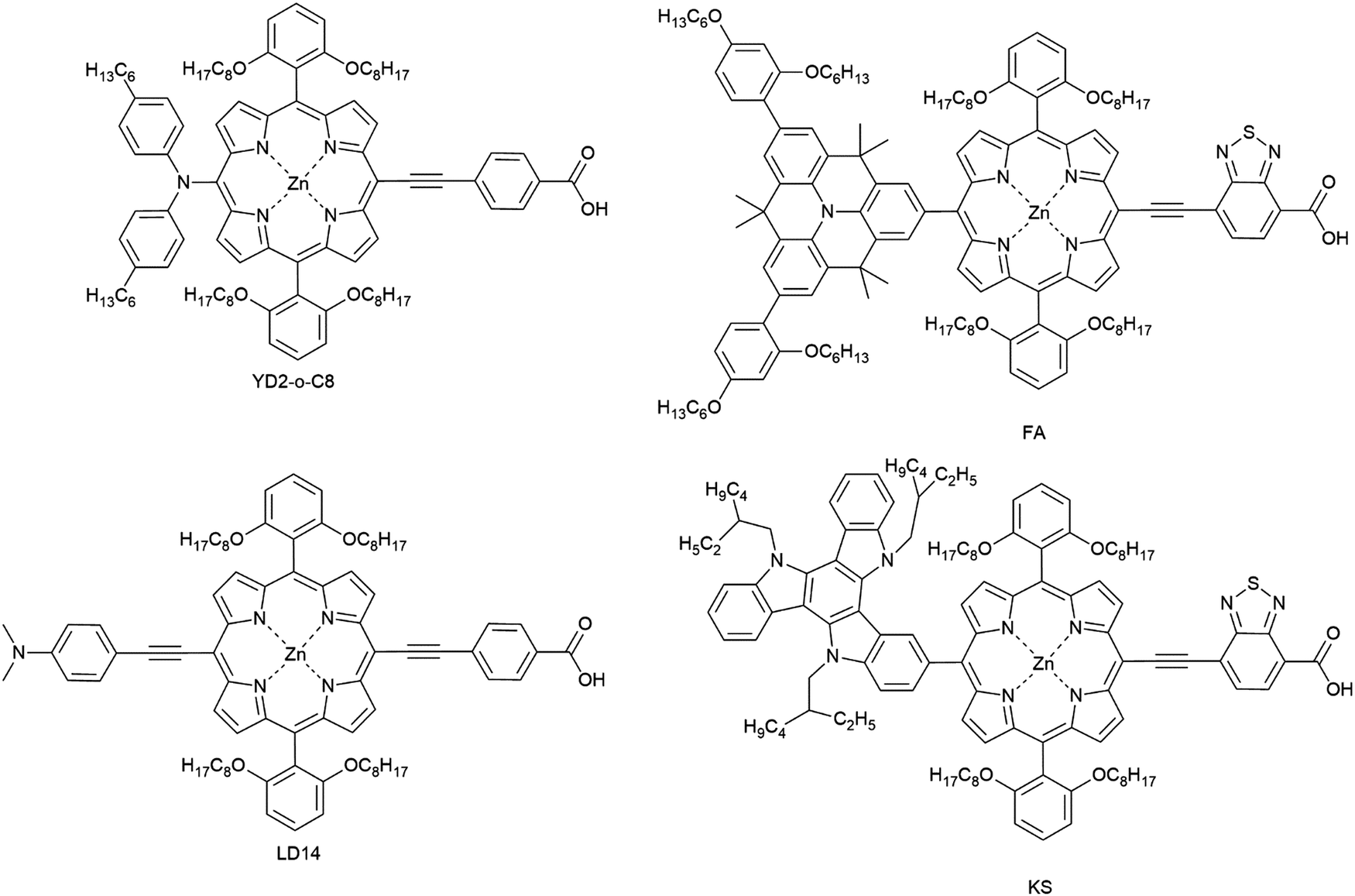 | ||
| Fig. 15 Chemical structures of porphyrin-based sensitizers for ssDSCs. | ||
Likely donors or the electron-rich groups include: coumarin, indoline, and triarylamine moieties. In particular, triarylamine dyes have been most efficient displaying good power conversion efficiencies in DSCs due to their electron donating ability and hole-transport properties.172,173 The π-bridge mostly consists of thiophene units, such as oligothiophene, thienothiophene, ethylenedioxythiophene. The favorite acceptor group is cyanoacrylic acid containing a carboxylic acid unit for binding to the metal oxide semiconductor. The electron density of the highest occupied molecular orbital (HOMO) of the dye molecule is mainly located in the donor part, whereas the density of the lowest unoccupied molecular orbital (LUMO) of the dye molecule is mostly located in the acceptor part (Fig. 17).
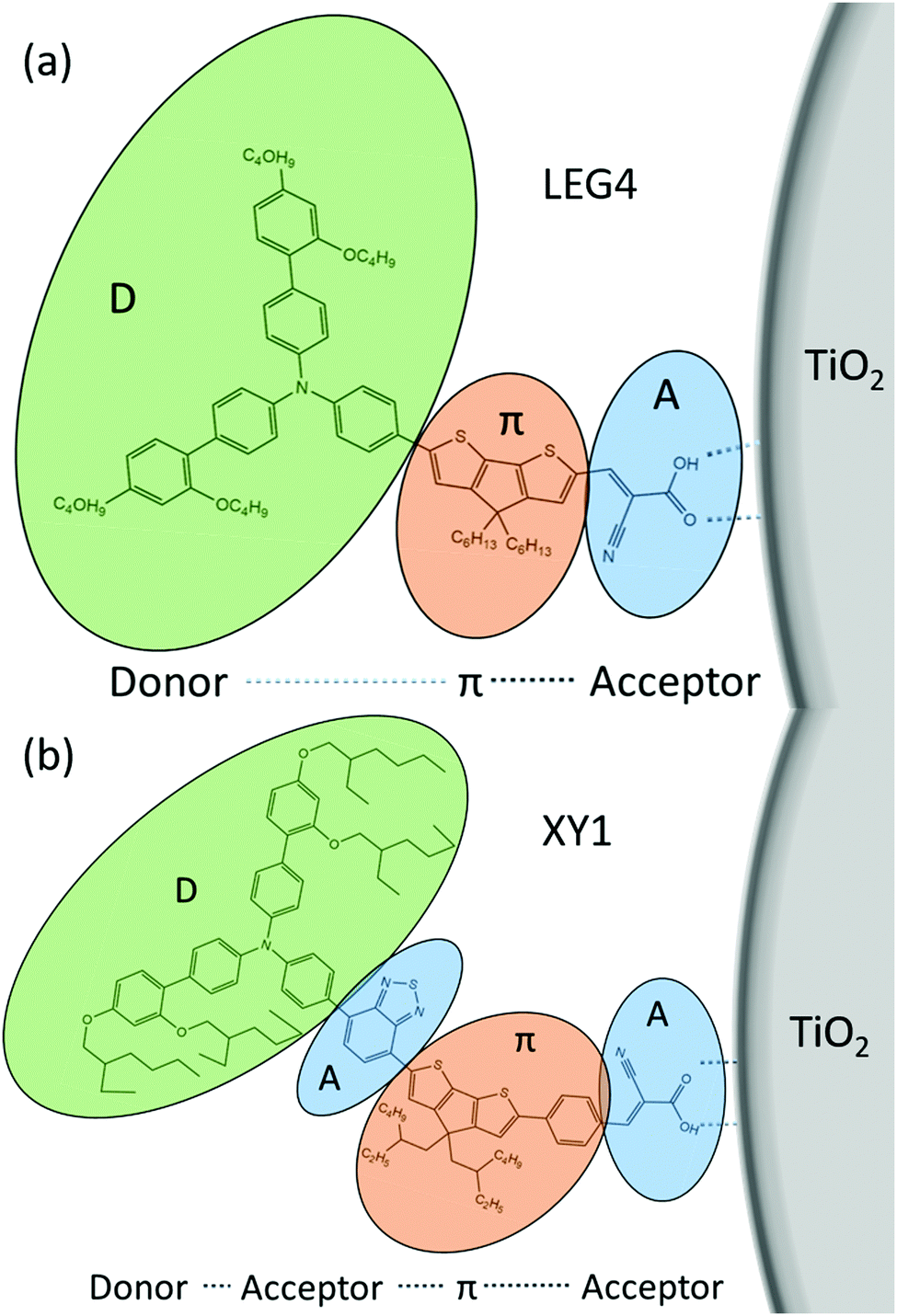 | ||
| Fig. 16 D–π–A/D–A–π–A structures of the organic dyes (a) LEG4 and (b) XY1. | ||
After the photoexcitation of the dye, the electrons subsequently transfer from the donor to the acceptor through the π-bridge. By this design, the electrons and holes are spatially separated in the dye molecule after light excitation, which could favor charge injection and dye regeneration. The properties of organic dyes can be easily tuned by suitable combination of these three parts.
Although organic dyes have many advantages as sensitizers in DSCs, the efficiency of devices prepared with them has been limited mainly by two factors: first, increasing the size of the conjugated system chain in organic dyes can cause dye aggregation or π–π stacking on the TiO2 surface due to their planar structures. Dye aggregation results in unfavorable back electron transfer and decreasing cell performance in a DSC. Second, sensitizers reported so far to give high efficiencies mostly have absorption bands below 550 nm, indicating that photons in the longer wavelength region are not absorbed efficiently. They should, however, harvest incident light in a broad spectral range to obtain a high photocurrent. Some organic dyes show a panchromatic response, but the obtained photocurrent and efficiency are not as high as expected due to their low LUMOs, which reduces injection efficiency.
The previously mentioned indoline dye D102 was also the first efficient organic sensitizer investigated for ssDSCs in 2005. It exhibits a high extinction coefficient of 55800 M−1 cm−1 at 490 nm. A PCE of over 4% was obtained by Schmidt-Mende et al. (Table 3).166 Another indoline dye, D149, was used as a sensitizer in ssDSCs with CuI as the inorganic HTM, exhibiting a PCE of 4.2%.175
| Dye | HTMa | Additivesb | CEc | V OC (mV) | J SC (mA cm−2) | FFf | PCEg (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Hole transporting material. b List of additives: Sb = N(PhBr)3SbCl6, Li = LiTFSI, tBP = 4-tert-butylpyridine, Co = FK102, FK209 or FK269. c Counter-electrode. d Open circuit voltage. e Short circuit current. f Fill factor. g Power conversion efficiency. h n.r. = not reported. | ||||||||
| D102 | Spiro-OMeTAD | Sb, Li, tBP | Au | 860 | 7.7 | 0.61 | 4.1 | 166 |
| D149 | CuI | (C2H5)3HSCN | Au | 550 | 14.1 | 0.54 | 4.2 | 175 |
| ID176 | Spiro-OMeTAD | Li, tBP | Ag | 640 | 8.7 | 0.57 | 3.2 | 98 |
| BODIPY-1 | Spiro-OMeTAD | n.r.h | Au | 800 | 2.2 | 0.37 | 0.6 | 176 |
| D35 | Spiro-OMeTAD | Li, tBP | Ag | 850 | 7.2 | 0.73 | 4.5 | 153 |
| C220 | Spiro-OMeTAD | Li, tBP | Au | 880 | 9.7 | 0.71 | 6.1 | 177 |
| Y123 | Spiro-OMeTAD | Li, tBP, Co | Ag | 930 | 9.8 | 0.75 | 6.9 | 178 |
| JD10 | Spiro-OMeTAD | Li, tBP, Co | Ag | 710 | 7.3 | 0.61 | 3.6 | 179 |
| WN3.1 | Spiro-OMeTAD | Li, tBP, Co | Ag | 870 | 9.7 | 0.75 | 6.3 | 180 |
| MK2 | Spiro-OMeTAD | Li, tBP | Ag | 700 | 6.9 | 0.58 | 2.8 | 181 |
| CPDT-3 | Spiro-OMeTAD | Li, tBP, Co | Au | 730 | 10.9 | 0.47 | 3.9 | 154 |
| XY2 | Spiro-OMeTAD | Li, tBP, Co | Au | 900 | 11.0 | 0.76 | 7.5 | 78 |
| S5 | Spiro-OMeTAD | Li, tBP, C2H2Cl4 | Ag | 830 | 12.9 | 0.73 | 7.8 | 182 |
Hagfeldt and coworkers investigated a perylene dye ID176 for ssDSCs. This dye showed a good absorption coefficient (25000 M−1 cm−1 at 590 nm) as well as a broad absorption spectrum. Devices with good performance, high photocurrent of 9 mA cm−2 and PCE of 3.2% were obtained.98 An interesting fact of this dye is that it worked well in ssDSCs but not in liquid DSCs. By detailed time-resolved absorption spectroscopy measurements it was found that (a) the dye ID176 regeneration by solid-state spiro-OMeTAD was ultrafast, and (b) lithium ions are necessary for efficient electron injection in the device.
Triphenylamine (TPA) based dyes are the most successful organic dyes due to their structural versatility and high absorption coefficients.107,182–184 The implementation of the TPA dyes started with the work by Hagfeldt and coworkers with the D35 dye and its bulky o,p-dibutoxyphenyl groups. The D35-based devices showed a promising PCE of 4.5%, due to improved electron lifetime in the device.153 Inclusion of a 4,4′-didodecyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT) conjugated linker improved the absorption spectrum of the TPA dye C220. Using such a modified dye a certified PCE of 6.1% was achieved by Cai et al. in 2011.177 With the Y123 dye a PCE of 6.9% was achieved by Dualeh et al. in a study based on the investigation of the donor influence on the VOC of the ssDSCs.178 Sellinger and coworkers obtained a PCE of 6.3% in ssDSCs based on dye WN3.1 with alkyl chains, which were instrumental in suppressing unwanted recombination processes.180
The modified, donor-free MK2 dye was introduced by Abate et al. consisting of cyanoacrylic acid-functionalized oligo (3-hexylthiophene). The modification of the dye had a long-lived oxidized state and this led to increased VOC and PCE in ssDSCs.181 Yue Hu et al. reported another series of ‘donor-free’ dyes featuring moieties of extensions of oligo(4,4-dihexyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene) (CPDT-1, CPDT-2, and CPDT-3) with molar absorption coefficients of up to 75000 M−1 cm−1. ssDSCs with CPDT-3 and spiro-OMeTAD hole transporter resulted in devices with a PCE of 3.9%.154
Very recently, He Tian and coworkers developed a series of dyes featuring benzothiadiazole (BTZ), 2,3-diphenylpyrido[3,4-b]pyrazine (PP) or 2,3-diphenylquinoxaline (QT) as the auxiliary acceptors and cyclopentadithiophene (CPDT) as the π-linker with the overall structure D–A–π–A. With the XY2 dye, having a high extinction coefficient of 6.66 × 104 M−1 cm−1, and spiro-OMeTAD as the HTM a high PCE of 7.5% was obtained.78 The XY1 dye from the same study was later used in combination with a copper coordination complex based HTM (see Section 3.2.2.3 on metal complex HTMs) to record efficiencies over 11%.87
He Tian and coworkers introduced two novel organic blue-colored dyes S4 and S5 with the donor indeno[1,2-b]thiophene functionalized triphenylamine, and the acceptors 2,3-diphenylpyrido[3,4-b]pyrazine or 2,3-diphenylquinoxaline and cyclopentadithiophene (CPDT) as the π-linker were designed and synthesized for ssDSCs. Both dyes exhibit a very high molar extinction coefficient of 6.3 × 104 M−1 cm−1 at 600 nm. The blue dye S5 resulted in devices with PCEs of 7.8% in ssDSCs.182
Organic metal-free dyes with red or near-infrared (near-IR) absorption have also been employed in ssDSCs. Grätzel and coworkers reported a near-IR absorbing squaraine dye, JD10, with strong absorption (672 nm, 2.5 × 105 M−1 cm−1). By effectively reducing the dye aggregation via adding the coadsorber chenodeoxycholic acid in the dye bath, an efficiency of 3.2% was obtained.179 Kolemen et al. developed a series of boron-dipyrromethene (BODIPY)-based molecules as red and near-IR sensitizers for ssDSCs.176 The devices with these dyes exhibited relatively low incident photon-to-current efficiency in spite of their broad absorption spectra. Further study on the kinetics of the charge transfer at the interfaces is needed to further improve their performance. It should be noted that the red or near-IR absorbing dyes provide more choices for the colors in the solar cell (such as visibly colorless) and are important to broaden the overall absorption spectrum in co-sensitized systems.
3.1.3.3. Quantum dots. Quantum dots (QDs) are inorganic nanoparticles with special electronic and optical properties due to quantum confinement of light capabilities given by their size. Orbitals and electronic behavior inside these nano-sized dots greatly vary compared to a bulk crystal of the same material. Light absorption properties are different and excited states are much longer lived, which leads to intense fluorescence upon illumination. These properties make QDs promising materials for photovoltaic applications. As it happened for the perovskite light absorber, QDs were first employed in photovoltaics as dyes in DSCs (Fig. 19), while nowadays the best-performing devices with this technology can have a planar structure and are considered p–n junctions rather than p-i-n junctions as is the case for DSCs.185–187 Nonetheless, QD-based DSCs are still actively being developed.188,189
In recent advances in solid-state quantum dot-sensitized solar cells (ssQDSCs), Jumabekov et al.190 and Park et al.191 have fabricated devices based on the PbS quantum dot. Jumabekov focused on the stability of the quantum dot by passivating its surface with L-glutathione to avoid surface oxidation. Surface passivation led to a more than doubled short circuit current, for a final device efficiency of 0.95%. Park extended absorption properties in the near-infrared of the PbS quantum dot thanks to the surface plasmon resonance effect by partly exchanging Pb2+ cations on the surface with Cu2+ cations. The inclusion of Cu produced an overall improvement of device parameters, with the PCE increasing from 2.36% to 8.07% after Cu embedding. Johansson and coworkers grew Ag2S QDs on ZnO nanowires.192 They show how the growth conditions for the QD on the nanowire greatly affect the final device. Under the best conditions, they obtained a solar cell efficiency of 0.36%. Finally, Duan et al. worked on a CdS QD in a device employing a polymer-based electrolyte.193 The best performance obtained with this setup was 0.55%.
3.2. Charge transport materials
In DSCs, the role of the redox mediator is to transport charges between the electrodes and regeneration of the oxidized dye. It is a crucial component determining the overall performance of the solar cell. The usage of the liquid solvent and especially of organic solvents, which are usually very volatile, lead to leakage and corrosion of the solar cells and degradation of the dyes.194 Even though considerable progress has been made in this field of research, fundamental stability considerations represent significant limitations for the commercialization of DSCs. Generally, the electrolytes used in DSCs can be categorized into: liquid, quasi-solid and solid states.26,195,196 The electrolyte in a liquid junction solar cell consists of a solvent, which can be organic or aqueous, with a redox mediator such as I3−/I−, copper or cobalt coordination complexes or small organic molecules.17,26 A significant part of the efforts made in this field has been devoted to the development of sustainable and efficient quasi-solid-state and solid-state hole transporting materials (HTMs) based on polymers, small organic and inorganic molecules.197 The main difference between the various charge transporting materials are the charge transport characteristics.69,82,198 In the case of polymer electrolytes it can be both ionic and electronic, whereas in others electronic transport is the dominant process.24,196The incorporation of PEs into ssDSCs made substantial steps forward in the research and development of ssDSCs. Most of the research activities are within the field of solid-state electrochemistry, in which high ion-conducting materials are developed for the energy conversion and storage applications. In this sense, PEs are a class of materials, which have been studied extensively in the last 20 years, to achieve systems with good conductivity and electrochemical stability. Polymer-based electrolytes are usually classified into solid-state polymer electrolytes, quasi-solid state gel polymer electrolytes and their composites.23,24,26,199,200
3.2.1.1. Solid polymer electrolytes. Peter V. Wright first showed in 1975 that poly(ethylene oxide) (PEO) can act as a host for sodium and potassium salts, thus producing a solid electrical conductor polymer/salt complex.201 Later, Armand et al. proposed that these systems could be used with graphite intercalation compounds for electrodes and immediately realized that lithium/PEO complexes could be employed as solid electrolytes and applied in batteries.202–204
Generally, solid polymer electrolytes (SPEs) are made from inorganic salts dispersed or dissolved in a polar polymer matrix. Most inorganic conductors consist of alkali metal salts (LiI, NaI, LiClO4, LiCF3SO3, LiSCN, NaSCN, NaClO4, LiPF6, etc.) in a host polymer (Fig. 20).
 | ||
| Fig. 17 Orbital distribution in donor–π–acceptor dyes. Reproduced from ref. 174 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 18 Chemical structures of organic dyes used in ssDSCs. | ||
The choice of polymer hosts for PEs is made based on the following characteristics: presence of groups with sufficient polarity to form strong coordinations with cations, and with low hindrance of bond rotations. Poly(ethylene oxide) (PEO) is the most commonly used host polymer, but these systems usually display low conductivity (10−8 S cm−1),205,206 which can be improved by using blends of different polymers or copolymers, as well as by synthetically modified monomers.24,207,208 This is necessary, since the required ionic conductivity values for ssDSC are around 10−3–10−4 S cm−1.196
PE-incorporating DSCs were first reported in 1999 attaining a very low IPCE of 1.3% (410 nm). The PE consisted of poly(o-methoxyaniline) (PAni) as the sensitizer and a copolymer of poly(ethylene oxide-co-epichlorohydrin) (PEO-EPI), containing NaI/I2, as the electrolyte (Table 4).209 Since then, there have been substantial research activities towards the preparation of various types of polymer electrolytes having different combinations of polymers and salts.
| Polymer | Salt | Dye | V OC (mV) | J SC (mA cm−2) | FFc | PCEd (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Open circuit voltage. b Short circuit current. c Fill factor. d Power conversion efficiency. | |||||||
| PEO-EPI | NaI/I2 | PAni | 48 | 0.012 | 0.32 | 0.00016 | 209 |
| PEO-EPI | NaI/I2 | PAni | 820 | 4.2 | 0.47 | 1.6 | 210 |
| PEO-TiO2 | Li/I2 | N3 | 664 | 7.2 | 0.58 | 4.2 | 211 |
| PNR4VPI/NR′PI/I2 | N719 | 682 | 13.4 | 0.62 | 5.6 | 212 | |
| HEII/I2 | MK2 | 733 | 14.66 | 0.69 | 7.5 | 213 | |
| CκC | NaI/I2 | N719 | 510 | 7.6 | 0.53 | 2.1 | 214 |
In 2000, Nogueira et al. prepared an elastomeric polymer electrolyte again based on copolymer poly(ethylene oxide-co-epichlorohydrin) complexes with sodium or lithium iodide salts. Unsealed prototype cells showed an efficiency of 1.6% (2.6% at 10 mW cm−2), thus demonstrating that a polymer electrolyte was an alternative as a solid-state electrolyte for ssDSCs.196,210,215 Falaras and coworkers prepared a solid polymer electrolyte consisting of PEO/TiO2, LiI, and I2. In this case, TiO2 nanoparticles were used as fillers to decrease the crystallinity leading to an increase in ionic conductivity (10−5 S cm−1) and consequently an efficiency of 4.2% at 65.6 mW cm−2 illumination.211,216,217
Wu et al. synthesized a polyelectrolyte based on polyvinyl pyridine and used it as a solid-state ionic conductor in ssDSCs. Based on a larger backbone cation poly(N-alkyl-4-vinylpyridine) (PNR4VPI) and a smaller anion I−, the polyelectrolyte showed low conductivity due to weak interactions between the cation and anion. The efficiency of the ssDSC reached 5.6% after the conductivity was immensely improved (6.41 × 10−3 S cm−1) by incorporation of I2 and N-alkylpyridine iodide (NR’PI).212,218
Li et al. introduced functionalized hydroxyethyl and ester co-functionalized imidazolium iodide (HEII) as a solid-state electrolyte and investigated the effect of substituents to the imidazolium ring on the ionic conductivity and the performance of ssDSCs. Compared to the methyl–ethyl-substituted imidazolium iodide, replacement of the methyl group with an ester group increased the ionic conductivity and the cell performance. Replacement of the ethyl group with a hydroxyethyl group further increased the ionic conductivity and cell performance significantly and the ssDSCs achieved a conversion efficiency of 7.45%.213
Bella et al. contributed to green chemistry by employing biodegradable polymers derived from seaweed as a SPE. The ssDSCs with carboxymethyl-κ-caraageenan (CκC) and NaI/I2 demonstrated a high power conversion efficiency of up to 2.06% with an ionic conductivity value of 5.53 × 10−2 S cm−1 at room temperature. The system displayed high degree of stability and even after 250 h under thermal stress (60 °C) for stability test, the cell showed only a 6% reduction of its initial photoconversion efficiency.214
Limitations associated with SPEs are still related to poor pore filling and ionic conduction resulting in a low dye regeneration rate and high electron recombination kinetics occurring in the solid polymer electrolyte and interfaces with dye and the metal oxide semiconductor. High wettability of sensitized TiO2 films is also an important factor for dye regeneration, which is attributed to a 0.2 eV decrease in the highest occupied molecular orbital energy of the dye yielding an increase in the driving force for dye regeneration. This understanding may contribute to a further increase in the energy-conversion efficiency of DSCs employing solid polymer electrolytes.
3.2.1.2. Quasi-solid-state electrolytes. Gel polymer electrolytes (GPEs) used as charge transport materials provide a viable and safe alternative to liquid electrolytes. They consist of a liquid electrolyte encapsulated in a polymer framework. These quasi-solid electrolytes combine the properties of a solid and a liquid. When combined, they suppress the leakage of volatile organic solvents, and have excellent pore filling properties. Despite the polymer being present for gelation, GPEs can hold large amounts of the electrolyte (tens to hundreds of times that of the polymer itself). Their excellent contacting and filling properties between the electrodes result in fast dye regeneration, while their high conductivity ensures fast charge transport to the counter electrode. Combined with their low vapor pressure as well as excellent thermal and long-term stability, they even provide the possibility for flexible and therefore wearable electronics.24,26,77,219–222
Common host materials are polyacrylonitrile (PAN), PEO derivatives and conducting polymers that include polypyrrole (PPy), PAni and other polymers. As organic plasticizers, dimethyl carbonate (DMC), propylene carbonate (PC) and ethylene carbonate (EC) can be used with a wide range of polar solvents, ionic liquids (ILs), and salts.223,224
Other categories of GPEs are prepared by swelling the polymeric membrane in the liquid electrolyte with a redox mediator-based electrolyte solution as illustrated. By this method, the liquid electrolyte is trapped in the polymer matrix network and a stable gel is obtained.225–227
Bella et al. conducted studies on UV-curing GPEs for quasi-solid dye-sensitized solar cells (Fig. 21).
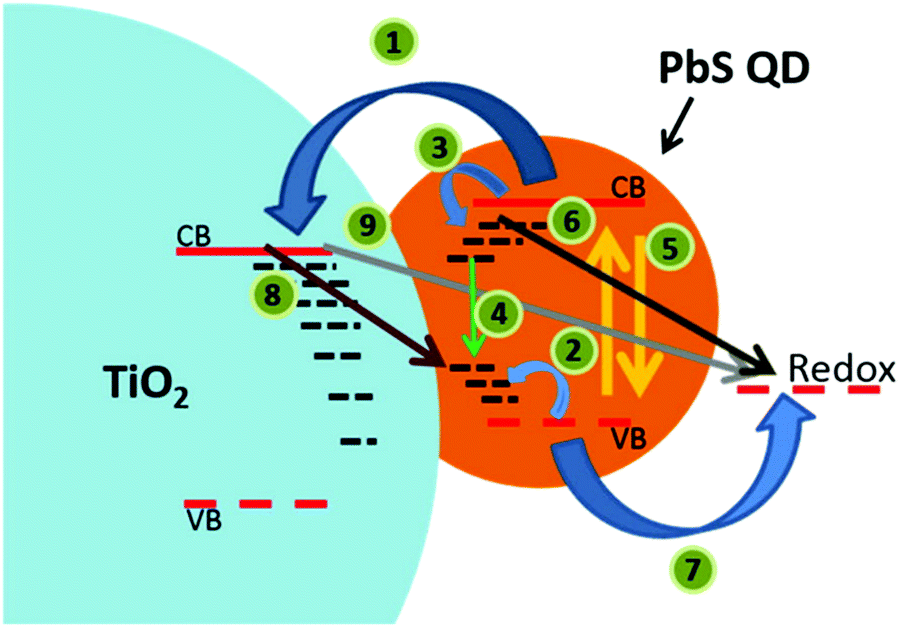 | ||
| Fig. 19 Schematic illustration of photoinduced charge-transfer processes following absorption of quanta of light by PbS QDs on TiO2. Reprinted from ref. 190 with permission from the American Chemical Society. | ||
Power conversion efficiencies of up to 4.41% (Table 5) were reported using bisphenol-A-ethoxylate dimethacrylate (BEMA) and poly(ethylene glycol)methyl ether methacrylate (PEGMA). BEMA forms a three-dimensional network and the addition of PEGMA as a copolymer influences the propagation reaction and changes the architecture of the polymeric matrix, thus affecting its properties.228–230
| Matrix | Salt | Dye | V OC (mV) | J SC (mA cm−2) | FFc | PCEd (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Open circuit voltage. b Short circuit current. c Fill factor. d Power conversion efficiency. | |||||||
| PEGMA:BEMA | NaI/I2 | N719 | 499 | 17.5 | 0.52 | 4.4 | 229 |
| Mg–MOF | NaI/I2 | N719 | 690 | 12.6 | 0.55 | 4.8 | 231 |
| BEMA:PEGMA:MFC | NaI/I2 | N719 | 760 | 15.2 | 0.61 | 7.0 | 232 |
| PMMA | BMII/I2 | N719 | 750 | 15.5 | 0.69 | 8.0 | 233 |
| Polyurethane | LiI/I2 | N719 | 740 | 15.0 | 0.55 | 6.1 | 234 |
| Polystyrene beads | BMII/I2 | N719 | 770 | 15.3 | 0.64 | 7.5 | 235 |
PEGMA was copolymerized with various bifunctional acrylate monomers which formed a three-dimensional polymer network under irradiation. The efficiency of quasi-solid DSCs ranged from 1.3% to 2.7% and was affected by the cross-link density of the membranes due to the amount of liquid electrolyte being trapped in the gel-polymer membranes. A high degree of cross-link density cannot store fair amounts of liquid electrolytes, while a low degree of cross-link density value cannot effectively trap the liquid electrolyte.
Later, a PCE of up to 4.80% was achieved by the introduction of metal–organic framework compounds (Mg–MOF) as fillers. A drastic improvement of PCE (up to 7.03%) was observed after incorporating bio-polymer organic fillers based on microfibrillated cellulose (MFC) into a 30:70 BEMA:PEGMA system.229–232,236,237
Another component for bioderived photoanodes and polymer electrodes in DSCs can be natural cellulose fibres. Simple papermaking processes using TiO2-laden paper foils can be applied to (conductive glass or plastic) substrates as alternatives for low temperature applications. The nanoscale microfibrillated cellulose is used as a reinforcing filler in acrylate/methacrylate-based thermo-set polymer electrolyte membranes prepared by UV-induced free-radical photopolymerization. PCEs between 3.55% and 5.20% were achieved for laboratory-scale DSCs.214
A PCE of 8.03% was achieved by using GPE based on poly(methyl methacrylate) (PMMA). The ionic conductivity and diffusivity of the iodine/1-butyl-3-methylimidazolium iodide (BMII) redox system were comparable to those of liquid electrolytes, and the resulting cells showed improved stability compared to traditional liquid DSCs.233 PCEs of 6.1% were achieved by using gel electrolytes based on polyurethane as the polymer matrix prepared through UV curing of a liquid electrolyte containing an aliphatic urethane acrylate.234 PCEs of 7.54% were reported for devices filled with a liquid electrolyte and controlled dissolution of polystyrene nanobeads on the CE, resulting in a gel electrolyte. The PCE of these devices is comparable to that of liquid-based DSCs (7.59%).235
Solid-state hole transporting materials introduce a new issue in the fabrication of dye-sensitized solar cells, that is pore filling of the mesoporous oxide layer.240 This issue can manifest in different forms, depending on the kind of hole transporter employed. In the case of large molecules and macromolecules, such as polymers, their size might prevent them from correctly and fully infiltrating the deepest parts of a thick mesoporous layer.241,242 Even when a compound is small enough to easily enter into all the pores, the solvent removal problem remains. HTMs are in fact deposited from solution and – to form a solid-state film – the solvent needs to be removed. The hole conductor film will have the best charge transport properties when a compact film can be formed, but the solvent needs channels and voids to escape from the mesoporous layer, eventually creating air bubbles in the HTM film. To overcome (or at least greatly mitigate) this problem, the mesoporous layer of HTM-based ssDSCs is much thinner compared to their liquid counterparts. If a hole conductor has a low melting or glass transition temperature, an alternative procedure to correctly infiltrate the compound is to melt it directly on top of the photoanode, to completely avoid the use of solvents.243
Bach et al. pioneered work on HTM for ssDSCs in 1998, when they reported for the first time a solid-state device with the hole conductor 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenyl-amine)9,9′-spirobifluorene (spiro-OMeTAD).85 That first solar cell had an efficiency of only 0.74%. However, thanks to device optimization and especially HTM layer optimization with the inclusion of additives and p-dopants to the precursor solution, in 2011 Burschka et al., also from Grätzel's research group, reported on a ssDSC featuring spiro-OMeTAD with a PCE of 7.2%.244 This work renewed research efforts towards efficient hole conductors and became a sort of benchmark for all future works. Both Bach's and Burschka's results, furthermore, contributed to elect spiro-OMeTAD as the HTM of reference, against which almost all new compounds are compared. Indeed, spiro-OMeTAD has proven to be an efficient hole conductor for solar cells. However, this material presents several drawbacks and there is a consensus on the fact that a new, efficient material will need to be found before ssDSCs become commercially viable. More in detail, spiro-OMeTAD suffers from poor conductivity and hole mobility in its pristine form, and it is not stable in the long term.54,245,246 Furthermore, its synthesis is complex and involves multiple steps, which require time- and energy-consuming purification procedures, making spiro-OMeTAD much more expensive than gold.247
In the following sections, we will review compounds belonging to several categories which aim to overcome spiro-OMeTAD as the material of choice for ssDSCs together with dopants and additives, which are of paramount importance to achieve high-efficiency devices.
3.2.2.1. Organic hole transporting materials. Most of the compounds investigated as hole transporting materials for ssDSCs belong to the family of organic molecules. Thanks to organic synthesis, in fact, their number, shape, features and properties are only limited by the researcher's creativity. These compounds can be divided into two categories – small molecules and polymers – each with their strengths and weaknesses.
Small molecules enable fine tuning of energy levels, electronic properties, film-forming properties, and solubility in different solvents by combining moieties with different characteristics. Their very well-defined composition and molecular weight allow for great synthetic reproducibility, ensuring consistent properties over different batches. Furthermore, their relatively small size allows them to infiltrate the mesoporous structure of DSCs’ photoanode more easily compared to polymers. However, their small size partially hinders inter-molecular charge transfer and they generally suffer from poor charge conductivity, especially in their pristine form.248,249
Polymers allow less control over electronic properties and the polymerization steps sometimes limit the chemical composition of the monomer itself.196 A less than perfect control over the polymerization reaction may prevent good batch-to-batch reproducibility and material properties.250 Their big size makes it difficult for them to infiltrate the thick mesoporous dye-sensitized layer, forcing researchers to find in situ polymerization techniques to overcome this issue. On the other hand, however, their long chains ensure very fast intra-molecular charge movements and their entangled nature in the solid state provides many points of contact for inter-molecular charge hopping. The combination of these two characteristics often grants conductive polymers superior charge transport properties over small molecules.196
3.2.2.1.1. Small molecules. The category of organic small molecules is that comprising the highest number of novel HTMs for ssDSCs. Most of the designed compounds share the triphenylamine moiety inside their structure. Triphenylamine is considered a good hole acceptor (electron donor) thanks to the nitrogen's lone electron pair and the presence of three phenyl rings that contribute to the delocalization of the resulting charge, stabilizing the cation. The energy level of molecules containing such a moiety is tuned by adding substituents – most often the electron donating group methoxy – on the phenyls not connected to the rest of the molecule, to destabilize the electronic cloud in the rings.251 Several of the reviewed hole conductors feature a carbazole as their core moiety, as this group is reported to have good structural and hole transporting properties.252 A list of reviewed small molecule HTMs together with their associated dye, device parameters and spiro-OMeTAD reference cell efficiency is reported in Table 6 while their chemical structures are portrayed in Fig. 22.
| HTMa | Dye | V OC (mV) | J SC (mA cm−2) | FFd | PCEe (%) | PCE with spiro-OMeTADf (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Hole transporting material. b Open circuit voltage. c Short circuit current. d Fill factor. e Power conversion efficiency. f Power conversion efficiency of a reference cell fabricated with the spiro-OMeTAD HTM. g n.r. = not reported. h Same molecule with two different names. i Same molecule with two different names. j Same molecule with two different names. | |||||||
| 3a | D102 | 860 | 0.32 | 0.44 | 0.12 | n.r.g | 253 |
| 3b | D102 | 680 | 6.32 | 0.41 | 1.75 | n.r. | 253 |
| X19 | LEG4 | 750 | 9.62 | 0.62 | 4.5 | 5.5 | 254 |
| X51 | LEG4 | 920 | 9.27 | 0.70 | 6.0 | 5.5 | 254 |
| 1 | D102 | 800 | 3.34 | 0.60 | 1.62 | n.r. | 255 |
| 2a | D102 | 760 | 4.41 | 0.48 | 1.60 | 4.44 | 256 |
| 2b | D102 | 750 | 5.25 | 0.46 | 1.81 | 4.44 | 256 |
| TCz-C3 | D102 | 690 | 6.27 | 0.51 | 2.21 | 3.03 | 167 |
| TCz-C6 | D102 | 590 | 0.86 | 0.38 | 0.20 | 3.03 | 167 |
| TCz-C12 | D102 | 660 | 0.21 | 0.34 | 0.05 | 3.03 | 167 |
| 4b | D102 | 573 | 0.75 | 0.28 | 0.12 | n.r. | 257 |
| 4d | D102 | 630 | 2.63 | 0.32 | 0.54 | n.r. | 257 |
| 5b | D102 | 531 | 1.72 | 0.35 | 0.32 | n.r. | 257 |
| 5 | D102 | 850 | 2.00 | 0.38 | 0.63 | n.r. | 258 |
| H-DATPA | D102 | 620 | 0.67 | 0.37 | 0.15 | 2.96 | 259 |
| Me-DATPA | D102 | 700 | 1.13 | 0.43 | 0.34 | 2.96 | 259 |
| MeO-DATPA | D102 | 890 | 1.93 | 0.67 | 1.16 | 2.96 | 259 |
| MeO-TPD | LEG4 | 800 | 9.5 | 0.65 | 4.9 | 4.7 | 117 |
| X1 | MKA253 | 680 | 5.8 | 0.58 | 2.3 | 6.1 | 183 |
| LEG4 | 720 | 8.8 | 0.67 | 4.3 | 5.2 | 183 | |
| LEG4 | 750 | 9.47 | 0.62 | 4.4 | 5.4 | 41 | |
| LEG4 | 880 | 9.44 | 0.69 | 5.8 | 5.9 | 260 | |
| X11 | MKA253 | 580 | 4.7 | 0.62 | 1.7 | 6.1 | 183 |
| LEG4 | 655 | 8.2 | 0.55 | 3.0 | 5.2 | 183 | |
| X2 | LEG4 | 810 | 9.79 | 0.63 | 5.0 | 5.4 | 41 |
| X3 | LEG4 | 900 | 9.70 | 0.66 | 5.8 | 5.4 | 168 |
| Z907 | 720 | 8.10 | 0.63 | 3.7 | 3.5 | 168 | |
| LEG4 | 910 | 9.52 | 0.67 | 5.8 | 5.4 | 41 | |
| X35 | LEG4 | 890 | 9.81 | 0.63 | 5.5 | 5.4 | 41 |
| X14 | LEG4 | 910 | 9.71 | 0.71 | 6.1 | 5.9 | 260 |
| HTM | Z907 | 750 | 8.5 | 0.51 | 3.3 | n.r. | 261 |
| TPAN | ZnPcOC4 | 720 | 4 | 0.42 | 1.21 | n.r. | 262 |
| HTM-1 | ID504 | 820 | 9.34 | 0.63 | 4.8 | 4.8 | 246 |
| HTM-2 | ID504 | 800 | 7.08 | 0.38 | 2.2 | 4.8 | 246 |
| HTM-3 | ID504 | 800 | 7.00 | 0.38 | 2.1 | 4.8 | 246 |
| 1(Bis) | D102 | 790 | 3.0 | 0.51 | 1.2 | n.r. | 263 |
| 2 | D102 | 870 | 1.8 | 0.29 | 0.46 | n.r. | 263 |
| 3 | D102 | 810 | 1.85 | 0.29 | 0.44 | n.r. | 263 |
| X60 | LEG4 | 890 | 11.38 | 0.72 | 7.30 | n.r. | 264 |
| HT2 | LEG4 | 920 | 9.72 | 0.71 | 6.35 | 6.36 | 247 |
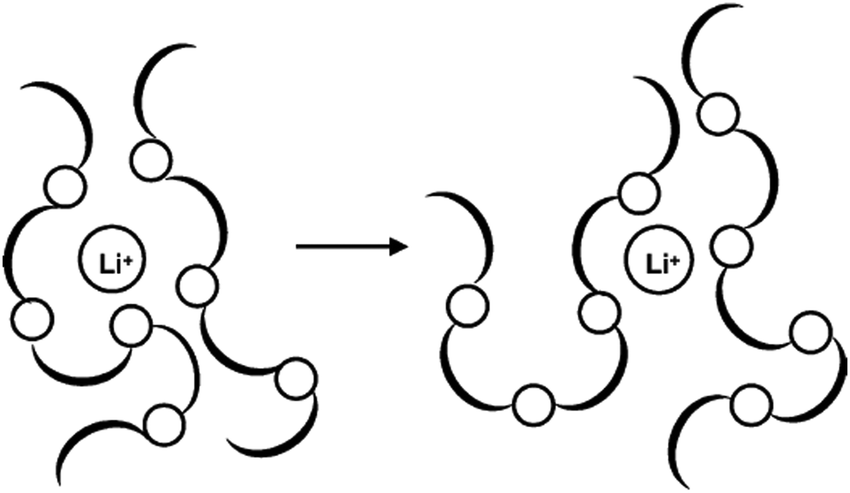 | ||
| Fig. 20 Schematic drawing of lithium ions embedded in a polymer matrix. Reprinted from ref. 196 with permission from Elsevier. | ||
Degbia et al. and Xu et al. both reported on a carbazole featuring a p-methoxyphenyl group attached to the nitrogen atom and a di(p-methoxyphenyl)amino group in para position to each benzene ring, 3b253 and X19,254 respectively. Devices with the 3b HTM were sensitized with the D102 dye, while those with X19 employed LEG4. This comparison allows understanding how important a good dye–HTM combination is in terms of charge transfer efficiency. The champion device with the 3b HTM had a VOC of 680 mV, JSC of 6.32 mA cm−2, FF of 0.41 and PCE of 1.75%. The best device with X19 featured a VOC of 750 mV, JSC of 9.62 mA cm−2, FF of 0.62 and PCE of 4.5%. While a higher current might be due to differences in light absorption profiles between the two dyes, the higher VOC and FF in the latter case is the result of a reduced series resistance (Rs) in the device based on X19. Since conductivity and hole mobility of the two HTM layers should be similar as they are based on the same molecule, it can be concluded that a great role in the Rs difference is played by the charge transfer between the dye and the HTM. In their work, Degbia et al. also explored the contribution of the p-methoxy group to charge transport properties of hole conductors by comparing 3b with compound 3a, which lacked the said groups attached to the diphenylamine moieties. The HOMO level of 3a (−4.95 eV) was slightly lower lying than that of 3b (−4.84 eV), but still sufficiently high for good dye regeneration. Despite this, the best cell with 3a had poor parameters, namely a VOC of 860 mV, JSC of 0.32 mA cm−2, FF of 0.44 and PCE of 0.12%. The authors did not perform any conductivity or hole mobility measurement of the two HTMs but, while a different pore filling cannot be excluded, due to the similar molecular structure they argued that the very low current in the case of 3a was to be attributed to poor charge transport properties caused by the lack of p-methoxy groups. In their aforementioned work, Xu et al. reported on a second carbazole-based HTM, X51.254 The constituents of X51 were similar to those of X19 but in this case two carbazole units were linked together by the two nitrogen atoms through a biphenyl linker, creating a molecule of almost double molecular weight compared to X19. X51 and X19 had similar hole mobilities of 1.51 × 10−4 and 1.19 × 10−4 cm2 V−1 s−1, respectively. However, once prepared with the additives employed in solar cell fabrication, solid-state films of the former displayed conductivities almost one order of magnitude higher compared to the latter. The higher conductivity of X51 leads to a lower Rs in the final device, as it can be inferred by cell parameters resulting in a VOC of 920 mV, JSC of 9.27 mA cm−2, FF of 0.70 and PCE of 6.0%: while current values were similar, the VOC and FF of the cell based on X51 were much higher compared to their X19 counterparts. The higher VOC was not the result of a lower-lying HOMO level of X51 compared to X19, as they were placed only 30 meV apart. The device based on X51 was also more efficient than the spiro-OMeTAD-based reference device, which had a PCE of only 5.5%.
A second comparison can be made between the work of Degbia et al. and that of Bui et al., as they both reported devices fabricated with the same dye and hole conductor, named 1255 and 2a,256 respectively. This compound is similar to 3b and X19 but it features an ethyl group attached to the nitrogen of the carbazole instead of a p-methoxyphenyl one. Device parameters for HTM 1 were a VOC of 800 mV, JSC of 3.34 mA cm−2, FF of 0.60 and PCE of 1.62% while for 2a were a VOC of 760 mV, JSC of 4.41 mA cm−2, FF of 0.48 and PCE of 1.60%. The various parameters were quite different for the two cells but they evened out, yielding a similar PCE value. It is interesting to notice that in the case of compound 1 high cell efficiencies were only reached when a final sublimation purification step was performed on the compound, while in the case of 2a a more common column chromatography final purification step was sufficient. In the same work, Bui et al. also reported on a molecule named 2b, which was similar to 2a but in which the ethyl group attached to the carbazole's nitrogen was substituted with a hexyl one. This modification was not expected to greatly change electronic properties (their HOMO levels were only 50 meV apart), but rather the film-forming properties and molecular packing in the solid state of the two molecules. Device parameters for a cell based on 2b were a VOC of 750 mV, JSC of 5.25 mA cm−2, FF of 0.46 and PCE of 1.81%. The difference between the two devices was not large, but a higher JSC was obtained with HTM 2b, leading to a slightly improved efficiency.
Similar to the work done for X51, Benhattab et al. have connected two carbazole units with alkyl chains, namely propyl (TCz-C3), hexyl (TCz-C6), and dodecyl (TCz-T12).167 Unlike X51, alkyl chains electronically separated the two carbazole units, which acted as single molecules as far as charge transfer was concerned. The presence of the alkyl linker was instead an attempt to tune the morphology of the solid-state film in the device. The champion device with TCz-C3 had a VOC of 690 mV, JSC of 6.27 mA cm−2, FF of 0.51 and PCE of 2.21%. The device based on TCz-C6 had a VOC of 590 mV, JSC of 0.86 mA cm−2, FF of 0.38 and PCE of 0.20%; while that based on TCz-C12 had a VOC of 660 mV, JSC of 0.21 mA cm−2, FF of 0.34 and PCE of 0.05%. It is clear that the longer the alkyl chain, the worse the performance of the final device, due especially to a plummet in current, a clear sign of charge transport issues. The reason behind this degradation in performance was not clear to the authors, as hole mobilities for the three compounds were similar. They suggested that lower glass transition temperatures for TCz-6 and TCz-12 might cause instability in the HTM layer during device operation. Another reason might be that – since hole mobilities were measured with a planar transistor device – the different environment provided by the DSC mesoporous layer might have affected molecular packing and ultimately the charge hopping capabilities of the studied compounds.
Tomkeviciene et al. prepared devices with three different hole transporting materials, 4b, 4d and 5b (see Fig. 22 for their structures).257 Two of them were asymmetric molecules, with substituted diphenylamino groups attached to only one of the carbazole rings (4b and 4d), while one was symmetric, in which the methoxy groups on the diphenylamino moieties were attached in the ortho position, rather than the usual para one. 4b also featured the two methoxy groups in the ortho position, which created steric hindrance in the molecule, changing the usual geometry of the relevant side groups. All devices fabricated with these compounds achieved modest results. For 4b the best cell featured a VOC of 573 mV, JSC of 0.75 mA cm−2, FF of 0.28 and PCE of 0.12%. For 4d the cell parameters were a VOC of 630 mV, JSC of 2.63 mA cm−2, FF of 0.32 and PCE of 0.54%, making it the most efficient of the three. The device based on 5b laid in between with a VOC of 531 mV, JSC of 1.72 mA cm−2, FF of 0.35 and PCE of 0.32%.
The last carbazole-based HTM – 5 – developed by Lygaitis et al. had inverted triphenylamino and carbazole groups: the former was used as the core moiety, and the latter as the side one.258 The best solar cell built with this compound featured a VOC of 850 mV, JSC of 2.00 mA cm−2, FF of 0.38 and PCE of 0.63%.
Snaith, Robertson and coworkers developed a series of rod-shaped HTMs based on two triphenylamine groups linked by a highly linear diacetylene core, namely H-DATPA, Me-DATPA, MeS-DATPA and MeO-DATPA (Fig. 23).259 The difference between them was given by the different side-groups attached in the para position to the two external rings of each triphenylamine moiety. MeS-DATPA did not form a uniform film in the device and was therefore discarded, while the other three were analyzed more in detail. As expected, the presence of side groups destabilizes the electronic cloud in the benzene rings, raising the HOMO level, whose values for H-, Me- and MeO-DATPA were −5.33, −5.23 and −5.02 eV, respectively. While hole mobility and conductivity values were similar for all compounds, the presence and nature of side groups greatly influenced their performance in ssDSCs, as seen previously for other compounds. All device parameters were improved when going from H to Me and to MeO side groups. The device fabricated with the first one achieved a VOC of 620 mV, JSC of 0.67 mA cm−2, FF of 0.37 and PCE of 0.15%, while for the other two the cell parameters were a VOC of 700 mV, JSC of 1.13 mA cm−2, FF of 0.43 and PCE of 0.34%, and a VOC of 890 mV, JSC of 1.93 mA cm−2, FF of 0.67 and PCE of 1.16%, respectively.
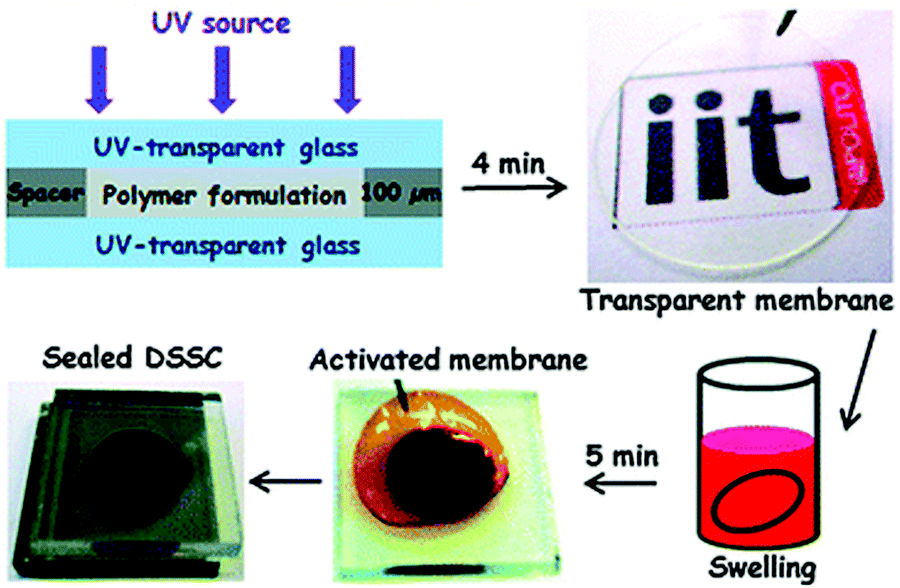 | ||
| Fig. 21 Preparation of quasi-solid polymer electrolytes by swelling techniques. Reproduced from ref. 227 with permission from the Royal Society of Chemistry. | ||
With a molecule named MeO-TPD, Johansson and coworkers showed how a light soaking treatment of complete DSC devices can greatly influence the final performance of a solar cell, suggesting that ion migration may occur in the solid-state HTM film.117 An as-prepared device with MeO-TPD attained a VOC of 750 mV, JSC of 2.7 mA cm−2, FF of 0.55 and PCE of 1.1%. However, when performing an initial light soaking treatment of 30 min under open circuit conditions, device performance was boosted to a VOC of 800 mV, JSC of 9.5 mA cm−2, FF of 0.65 and PCE of 4.9%, slightly higher than that of the spiro-OMeTAD reference device (4.7%). It is important to notice that the light soaking treatment was only required once, not each time the device was tested. Sun's and Kloo's research groups employed the same molecule – under the name of X1 – in several other publications41,183,260 (see Table 6 for all device parameters) with an efficiency of up to 5.8%, the closest to the related spiro-OMeTAD reference device (5.9%). However, in none of these publications the initial light soaking treatment was mentioned.
Yuan et al. and Liu et al. have worked on similar molecules, namely HTM261 and X11,183 composed of a fluorene core with p-methoxydiphenylamino side groups attached to each benzene ring. The hole conductor HTM, in addition, also featured long alkyl chains attached to the central fluorene ring with the aim of reducing the glass transition temperature of the compound. Unfortunately, it is not possible to make a direct comparison between the two because their devices employed different dyes and data for a spiro-OMeTAD reference cell was not always provided. A ssDSC with HTM achieved a VOC of 750 mV, JSC of 8.5 mA cm−2, FF of 0.51 and PCE of 3.3% when both the HTM solution and the substrate were pre-heated before spin-coating. The pre-heating of both solution and substrate was very important for hole conductor deposition as devices only attained an efficiency of 2.0% when just the HTM solution was heated and of 0.44% when neither the solution nor the substrate was heated. A solar cell fabricated with X11 and the MKA253 sensitizer featured a VOC of 580 mV, JSC of 4.7 mA cm−2, FF of 0.62 and PCE of 1.7%, and a VOC of 655 mV, JSC of 8.2 mA cm−2, FF of 0.55 and PCE of 3.0% when the LEG4 sensitizer was used instead.
Building on X1, Sun and coworkers synthesized a series of p-methoxy substituted triphenylamine oligomers.41 If X1 was composed of two triphenylamine groups linked linearly, with X2 and X3 the number of repeating units was extended to three and four, respectively. X35 also featured four units, but rather than being linked linearly they were connected in a star-shaped configuration, with three groups attached to a central one. Optimized devices showed that there was an increase in performance with increasing number of repeating units. Device parameters for an average of 8 cells for each compound were provided (see Table 6 for champion device details) and were as follows: for X1-based cells VOC was 725 mV, JSC was 9.26 mA cm−2, FF was 0.56 and PCE was 4.0%; for X2-based cells VOC was 800 mV, JSC was 9.51 mA cm−2, FF was 0.60 and PCE was 4.7%; for X3-based devices VOC was 880 mV, JSC was 9.23 mA cm−2, FF was 0.62 and PCE was 5.4%; for X35-based devices VOC was 875 mV, JSC was 9.62 mA cm−2, FF was 0.61 and PCE was 5.2%; lastly, spiro-OMeTAD-based reference cells had a VOC of 905 mV, JSC of 9.13 mA cm−2, FF of 0.58 and PCE of 5.2%. Both X3 and X35 had performances similar or slightly superior to spiro-OMeTAD, making them affordable candidates for a broader use. X3 was already reported by Sun's research group in a previous work with very similar performances, showing the good reproducibility of devices made with this compound (see Table 6).168
From Sun, Kloo and coworkers came another efficient hole conductor, X14.260 This molecule was also designed to improve on the X1 HTM and it featured an extended aromatic conjugation, since each of X1's methoxy groups was replaced with an o,p-dimethoxy substituted phenyl group. This substitution shifted the HOMO level by about 200 meV away from vacuum and gave X14 a hole mobility that was double that of X1 when both compounds were doped with LiTFSI. Solar cell performances were similar for the two hole transporting materials, with a small advantage in the case of X14. The best device fabricated with X1 had a VOC of 880 mV, JSC of 9.44 mA cm−2, FF of 0.69 and PCE of 5.8%; while that fabricated with X14 had a VOC of 910 mV, JSC of 9.71 mA cm−2, FF of 0.71 and PCE of 6.1%. For comparison, the best spiro-OMeTAD-based device had a PCE of 5.9%.
Aulakh et al. have developed a relatively flat compound based on the linkage of a benzimidazole and an anthracene group.262 A solar cell fabricated with a Zn porphyrin dye achieved a VOC of 720 mV, JSC of 4 mA cm−2, FF of 0.42 and PCE of 1.21%.
Malinauskas et al. have focused their work on the long-term stability of ssDSCs based on spiro-OMeTAD. They discovered that when keeping devices at 60 °C for an extended amount of time, crystalline domains were formed in the initially amorphous spiro-OMeTAD film and they proved that this was a major reason for the poor performance of such devices over time (see Section 4.1 on long-term stability).246 To overcome this issue, they modified spiro-OMeTAD's molecular structure by introducing elements of asymmetry in order to prevent crystallization. In HTM-1 a methyl group was added in the meta position to only one of the two rings of each of the four diphenylamine side groups. HTM-2 and HTM-3 had more radical substitutions, as one of the diphenylamine group was exchanged with a triphenylamine one. HTM-3 also featured the same methyl groups of HTM-1. A batch of five solar cells featuring HTM-1 managed to retain the same average efficiency of the spiro-OMeTAD reference one, with a VOC of 820 mV, JSC of 9.34 mA cm−2, FF of 0.63 and PCE of 4.8%. The more heavily substituted HTM-2 and HTM-3 proved less efficient with a VOC of 800 mV, JSC of 7.08 mA cm−2, FF of 0.38 and PCE of 2.2%; and a VOC of 800 mV, JSC of 7.00 mA cm−2, FF of 0.38 and PCE of 2.1%; respectively. More importantly, however, all three HTMs greatly outperformed spiro-OMeTAD-based devices in a long-term stress-test at 60 °C.
Hirsch, Goubard and coworkers synthesized three star-shaped hole transporters based on a triphenylamine core and diphenylamine side groups linked to the core via a thiophene bridge.263 The difference between 1 (called 1(bis) throughout this review to avoid confusion), 2 and 3 was that 2 and 3 had a methyl substitution on each thiophene bridge, on the inner or outer available carbon, respectively. Methyl substitutions in 2 and 3 appeared to be detrimental as the device with 1(bis) achieved a VOC of 790 mV, JSC of 3.0 mA cm−2, FF of 0.51 and PCE of 1.2%; while those based on 2 and 3 only achieved a VOC of 870 mV, JSC of 1.8 mA cm−2, FF of 0.29 and PCE of 0.46%; and a VOC of 810 mV, JSC of 1.85 mA cm−2, FF of 0.29 and PCE of 0.44%; respectively, indicating charge transport issues.
Xu et al. have synthesized the highest-performing organic hole conductor among those reviewed,264 and the only one capable of producing a ssDSC able to rival with Burschka's benchmark device.244X60 is based on a spiro[fluorene-9,9′-xanthene] core with p-methoxy substituted diphenylamine side groups and its core moiety was estimated to be 30 times less expensive than that of spiro-OMeTAD. A spiro-OMeTAD-based reference cell was not provided in their work but that based on X60 featured a VOC of 890 mV, JSC of 11.38 mA cm−2, FF of 0.72 and PCE of 7.30%.
To conclude this review on small molecule HTMs, Boschloo, Sun and coworkers prepared HT2, a compound based on a fluorene core featuring p-methoxy substituted diphenylamine side groups attached to its two aromatic rings and two p-methoxy substituted triphenylamine groups attached to its central ring.247 Their best device attained a VOC of 920 mV, JSC of 9.72 mA cm−2, FF of 0.71 and PCE of 6.35%, only slightly lower than the spiro-OMeTAD-based reference one (6.36%).
3.2.2.1.2. Polymers. The use of polymers in ssDSCs adds a level of complexity compared to small molecules. In fact, it is not enough to develop a highly performing compound, it is also necessary to engineer device fabrication to ensure that the polymer will penetrate the TiO2 mesoporous structure and thus regenerate the dye throughout the whole device thickness (pore filling will be discussed in greater detail in a following section). Since infiltration of large molecules is challenging, most of the studied polymers are capable of in situ polymerization. Thanks to this processing, monomers can wet the device as easily as other small molecules and – after polymerization – the generally higher conductivity of macromolecules can be exploited. For this reason, in each work of this kind there is as much attention on the polymerization process as there is on the nature and properties of the monomer itself. A summary of the champion devices reviewed in this section is presented in Table 7, while Fig. 24 contains the structure of the related units.
| HTMa | Dye | V OC (mV) | J SC (mA cm−2) | FFd | PCEe (%) | Ref. |
|---|---|---|---|---|---|---|
| a Hole transporting material. b Open circuit voltage. c Short circuit current. d Fill factor. e Power conversion efficiency. | ||||||
| PProDOT | N719 | 630 | 10.0 | 0.56 | 3.5 | 265 |
| PEDOP | D35 | 825 | 7.99 | 0.66 | 4.34 | 200 |
| D21 L6 | 645 | 7.92 | 0.59 | 3.05 | 200 | |
| Z907 | 440 | 1.97 | 0.53 | 0.46 | 200 | |
| PEDOT | LEG4 | 910 | 10.80 | 0.57 | 5.6 | 266 |
| D35 | 830 | 8.19 | 0.68 | 4.6 | 266 | |
| Z907 | 510 | 4.49 | 0.66 | 1.5 | 266 | |
| DPP07 | 770 | 11.13 | 0.65 | 5.54 | 267 | |
| PPP-b-P3HT | CYC-B11 | 810 | 8.81 | 0.65 | 4.65 | 268 |
| TPDSi2 | Z907 | 683 | 4.97 | 0.60 | 2.05 | 269 |
| P3HT | CYC-B11 | 750 | 7.71 | 0.61 | 3.53 | 268 |
| N3 | 628 | 6.29 | 0.43 | 1.70 | 270 | |
| BzTCA | 880 | 8.22 | 0.44 | 3.21 | 270 | |
| D102 | 720 | 11.37 | 0.58 | 4.78 | 271 | |
| S:DIB | LEG4 | 750 | 4.11 | 0.48 | 1.5 | 84 |
| LEG4 | 700 | 4.34 | 0.36 | 1.09 | 272 | |
| SeS2:S.DIB | LEG4 | 750 | 5.04 | 0.45 | 1.70 | 272 |
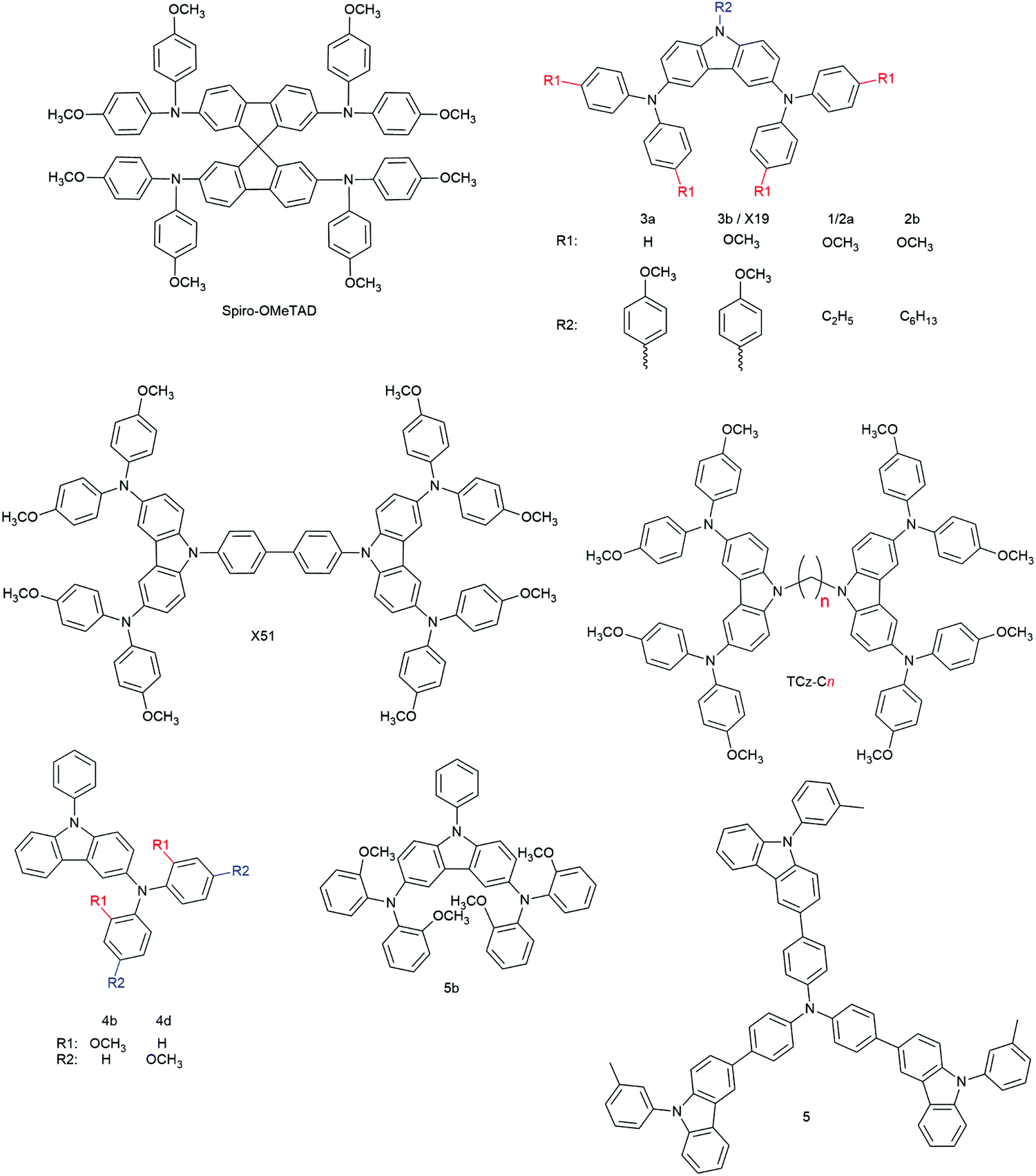 | ||
| Fig. 22 Spiro-OMeTAD and novel carbazole-based hole transport materials. | ||
Kim and coworkers proposed a polymer based on a propylenedioxythiophene monomer, ProDOT.265 The corresponding polymer, PProDOT, is similar to the more widely known poly(ethylenedioxythiophene) (PEDOT), but its monomer features a propyl alkyl chain rather than an ethyl one. They used a solid-state polymerization technique, starting with a dibrominated ProDOT monomer, to fabricate devices. This technique is very slow but also very simple. A monomer solution was drop-cast onto the photoanode. After solvent evaporation, the solid monomer was left in an oven at 25 °C for five days to allow for polymerization to spontaneously occur with evaporation of Br2 gas as a side-product. After covering this assembly with a Pt-coated FTO counter-electrode, a VOC of 630 mV, JSC of 10.0 mA cm−2, FF of 0.56 and PCE of 3.5% were achieved in the complete device.
Zhang et al. showed the performance of PEDOP (poly(ethylenedioxypyrrole)) in combination with three different dyes, demonstrating that the dye plays a crucial role in the suppression of electron recombination.200 Photoelectrochemical polymerization (PEP) was used for the in situ polymerization of EDOP. In this technique, the DSC photoanode is immersed in a solution of the HTM monomer to act as the working electrode in a three-electrode setup. When a current at a suitable potential is run through the device and light is shined on the photoanode, the hole in the HOMO orbital of the excited dye triggers the polymerization of the HTM. With this technique it is possible to achieve electropolymerization of organic compounds at much lower potentials compared to the system where a dye is not present. For this technique to work efficiently, a large enough potential difference between the HOMO of the dye and that of the monomer is required. Device performances were quite different depending on the employed dye, demonstrating once more the importance of a good dye–HTM pairing. The device with the D35 dye was the best-performing one, with a VOC of 825 mV, JSC of 7.99 mA cm−2, FF of 0.66 and PCE of 4.34%. The other organic dye, D21 L6, was slightly less performing with a VOC of 645 mV, JSC of 7.92 mA cm−2, FF of 0.59 and PCE of 3.05%; while the Ru-based dye proved to be poorly compatible with PEDOP yielding a device with a VOC of 440 mV, JSC of 1.97 mA cm−2, FF of 0.53 and PCE of 0.46%.
Zhang et al. applied the same PEP technique used for PEDOP to PEDOT as well, again testing the studied polymer in combination with three different dyes.266 Contrary to PEDOP, which was polymerized from EDOP, PEDOT was polymerized using a bisEDOT dimer, which had a lower redox potential compared to the EDOT monomer. PEP was performed with all three dyes both in organic and aqueous HTM solutions and in all cases the organic solvent proved to be a better medium for polymerization due to changes in redox potentials of both dyes and HTM precursor. The device based on the D35 dye with the PEDOT hole transporting material was slightly more efficient than that employing PEDOP, with a VOC of 830 mV, JSC of 8.19 mA cm−2, FF of 0.68 and PCE of 4.6%. LEG4 proved to be the best-performing dye, with a VOC of 910 mV, JSC of 10.80 mA cm−2, FF of 0.57 and PCE of 5.6%. Z907 proved to be more compatible with PEDOT than with PEDOP, as a solar cell with the former reached a VOC of 510 mV, JSC of 4.49 mA cm−2, FF of 0.66 and PCE of 1.5%. Two years later Zhang et al. also showed that the dye DPP07 is as efficient in combination with PEDOT as LEG4, when they fabricated a device with a VOC of 770 mV, JSC of 11.13 mA cm−2, FF of 0.65 and PCE of 5.54%.267 This device was much more efficient than a reference one based on spiro-OMeTAD (2.91%).
Wang and coworkers explored the properties of a pre-polymerized block copolymer of poly(2,5-dihexyloxy-p-phenylene), PPP and poly(3-hexylthiophene), P3HT.268 The PPP block helped the PPP-b-P3HT copolymer to self-organize in crystalline domains, which increased the hole mobility of the copolymer significantly (8.5 × 10−4 cm2 V−1 s−1) compared to the P3HT homopolymer (1.9 × 10−4 cm2 V−1 s−1) and, as the authors claimed, provided a more intimate contact with the CYC-B11 dye. The pore filling proved to be similar for both copolymer and P3HT but, thanks to the higher hole mobility and better contact with the dye, the PPP-b-P3TH-based solar cell achieved a VOC of 810 mV, JSC of 8.81 mA cm−2, FF of 0.65 and PCE of 4.65%; while those employing P3HT could only display a VOC of 750 mV, JSC of 7.71 mA cm−2, FF of 0.61 and PCE of 3.53%.
Zhou et al. relied on silanes for in situ polymerization of their hole conductor.269 The TPDSi2 monomer was composed of two triphenylamine groups linked together. In the para position to one of the phenyl rings of each group was attached a propyl chain terminated with a silane. The monomer, deposited on the substrate in an inert, glovebox atmosphere, easily infiltrated the titania mesoporous layer. Once exposed to air, the monomer's silane terminations readily underwent cross-linking, creating a rigid and thermally stable structure. It should be noted that, unlike the other reviewed polymers, the backbone of TPDSi2 is not conductive due to the alkyl chains. ssDSCs fabricated with this new HTM featured a VOC of 683 mV, JSC of 4.97 mA cm−2, FF of 0.60 and PCE of 2.05%.
Liu et al. probed the performance of P3HT with two different dyes.270 Solar cells attained a VOC of 628 mV, JSC of 6.29 mA cm−2, FF of 0.43 and PCE of 1.70% when sensitized with N3 and a VOC of 880 mV, JSC of 8.22 mA cm−2, FF of 0.44 and PCE of 3.21% when sensitized with BzTCA, once more proving that organic dyes are better-suited to work with polymeric HTMs. The best P3HT-based ssDSCs were fabricated by Clément and coworkers.271 They overcame the typical pore filling issues of P3HT by synthetizing a highly regioregular polymer with medium-range molecular weight and narrow dispersity. P3HT with such characteristics achieved high efficiency in a device with a 2 μm thick titania layer. After HTM deposition and an annealing step at 150 °C to improve film morphology, devices displayed a VOC of 720 mV, JSC of 11.37 mA cm−2, FF of 0.58 and PCE of 4.78%. For comparison, a device fabricated with spiro-OMeTAD displayed a PCE of only 3.99%.
The last two reviewed polymeric HTMs, developed by Liu, Gardner and Kloo, sit at the border between organic polymer and inorganic hole transporting materials. These two compounds are, in fact, composed of inorganic polymers (either sulfur in the case of S:DIB84 or a mix of sulfur and selenium sulfide in the case of SeS2:S:DIB272) cross-linked with the organic molecule 1,3-diisopropenylbenzene in a process called inverse vulcanization. Both polymers were soluble in organic solvents and, hence, solution-processable. The best solar cell fabricated with S:DIB displayed a VOC of 750 mV, JSC of 4.11 mA cm−2, FF of 0.48 and PCE of 1.5%. The addition of selenium sulfide increased the performance to a VOC of 750 mV, JSC of 5.04 mA cm−2, FF of 0.45 and PCE of 1.70% (compared to an efficiency of 1.09% of a device with S:DIB fabricated at the same time).
3.2.2.2. Inorganic hole transporting materials. Inorganic semiconducting materials are a class of compounds with great potential for energy conversion and transport. To date, many electronic, solar, and energy storage devices have relied on them for efficient operation.9 They possess good electronic properties, good conductivity and high temperature stability.63,80,124 In the field of dye-sensitized solar cells their use is limited, as only a few of them are soluble in solvents and at temperatures compatible with DSC fabrication. Even fewer have good energy alignment with dyes and n-type semiconductors used in this class of solar cells and it is not as easy to tune their energetics as in the case of organic compounds. The few materials that can be used in ssDSCs have better performances than most organic HTMs, although charge recombination at grain boundaries can prove to be an issue. In Table 8 all the inorganic materials that have been recently studied as hole conductors in ssDSCs can be found.
| HTMa | Dye | V OC (mV) | J SC (mA cm−2) | FFd | PCEe (%) | Ref. |
|---|---|---|---|---|---|---|
| a Hole transporting material. b Open circuit voltage. c Short circuit current. d Fill factor. e Power conversion efficiency. f n.r. = not reported. g Mix = N719 + YD2-o-C8 + RLC5, PC = photonic crystal. | ||||||
| CsSnI3 | N719 | 732 | 19.2 | 0.72 | 10.2 | 33 |
| N3 | 620 | 9 | n.r.f | 3 | 273 | |
| Cs2SnI6 | Z907 | 571 | 13.2 | 0.61 | 4.63 | 149 |
| N719 | 631 | 14.7 | 0.68 | 6.32 | 149 | |
| Mixg | 623 | 16.9 | 0.66 | 6.94 | 149 | |
| Mix + PCg | 618 | 18.6 | 0.68 | 7.80 | 149 | |
| CuI | N3 | 739 | 14.5 | 0.69 | 7.4 | 274 |
| N3 | 650 | 3.51 | 0.46 | 1.05 | 275 | |
| CuSCN | N719 | 578 | 10.52 | 0.55 | 3.39 | 276 |
Chung et al. employed the tin-based perovskite CsSnI3 as a hole transporting material in an N719-sensitized ssDSC.33 After doping with tin fluoride and engineering the device with ZnO photonic crystals their best solar cell exhibited a VOC of 732 mV, JSC of 19.2 mA cm−2, FF of 0.72 and PCE of 10.2%. From the IPCE the authors showed that the hole conductor contributed to the cell's photocurrent in the region between 550 and 700 nm. Despite the very high efficiency achieved with this material (the highest among the reviewed inorganic HTMs), CsSnI3 suffers from high instability as Sn(II) is not the most stable oxidation state of tin at room temperature in air. Peedikakkandy and Bhargava worked on the same material and their best cell attained 3% efficiency. Furthermore, they reported on the degradation mechanisms of CsSnI3.273 Their results showed that the material was stable in an inert atmosphere, but when exposed to air and moisture it first underwent a phase transition to a non-conductive, one-dimensional phase and then oxidation to Cs2SnI6.
Due to the instability of the Sn(II)-based perovskite, Chang and coworkers decided to use the air stable Cs2SnI6 compound as a hole conductor for their cells.149 This material was used to harvest holes from three differently sensitized photoanodes. The ssDSC sensitized with Z907 yielded a VOC of 571 mV, JSC of 13.2 mA cm−2, FF of 0.61 and PCE of 4.63%; while that with N719 a VOC of 631 mV, JSC of 14.7 mA cm−2, FF of 0.68 and PCE of 6.32%. The best performance was achieved with a mix of dyes, namely N719, YD2-o-C8 and RLC5. Such a device had a VOC of 623 mV, JSC of 16.9 mA cm−2, FF of 0.66 and PCE of 6.94%. The performance with the dye mix was further increased after engineering the solar cell with ZnO photonic crystals, reaching a VOC of 618 mV, JSC of 18.6 mA cm−2, FF of 0.68 and PCE of 7.80%. A sample cell with the Z907 dye proved to be quite stable for 800 h.
Sakamoto et al. worked on copper iodide, which is a well-known HTM for solar applications.274 In their study they focused on the influence that the contact materials have on the performance of the drop-cast CuI layer. Results showed that the presence of thiocyanate groups in both the dye and counter electrode was of paramount importance to achieve high efficiency. The variation of the density of SCN groups in the PEDOT:PSS based counter electrode in particular led to devices with an efficiency that was more than double that of the devices without SCN groups. The best ssDSC displayed a VOC of 739 mV, JSC of 14.5 mA cm−2, FF of 0.69 and PCE of 7.4%. Muhamad and Mahmood also fabricated devices with CuI using a mist-atomization technique, with more modest results.275 Their best device achieved a VOC of 650 mV, JSC of 3.51 mA cm−2, FF of 0.46 and PCE of 1.05%.
The last reviewed inorganic hole conductor is CuSCN, based on the work of Premalal et al.276 They doped CuSCN with triethylamine hydrothiocyanate to improve the p-type conductivity of the inorganic material. The precursor solution required an equilibration time of 20 days before deposition in a ssDSC, after which the device achieved a VOC of 578 mV, JSC of 10.52 mA cm−2, FF of 0.55 and PCE of 3.39%.
3.2.2.3. Coordination metal complex hole transporting materials. Coordination transition metal complexes are a class of materials that inherits benefits and drawbacks from both organic small molecules and inorganic compounds. They retain the ease of processing of organic compounds but with charge conductivity closer to the inorganic ones, which eliminates the need for p-dopants. More exactly, the p-dopant is embedded in the compound itself and it consists of the same complex with a higher oxidation state of its metal center. Their energy levels are easily fine-tuned by applying changes to the organic ligand or greatly varied by changing the metal center.277,278 However, they have the tendency to form crystalline domains in the solid state, either immediately upon deposition or over time.107,279 This crystallization may modify film morphology, worsening the contact between the dye or counter electrode and HTM, thus hindering charge transport; or it may create charge recombination sites at grain boundaries. Metal complexes have already contributed greatly to the advancement of liquid DSCs by replacing iodine/iodide as the electrolyte as they have simple, one-step oxidation and reduction processes (thus reducing the driving force needed for regeneration) and are much less corrosive than the latter. To date, all most-efficient liquid DSCs are based on transition metal complex electrolytes.19,104,280 In the field of solid-state dye-sensitized solar cells, however, they have only recently started to be investigated and only a few examples of their application exist. It should be noted, however, that the two best performing ssDSCs employ a metal complex as the hole conductor. Device details of metal complex-based ssDSCs are found in Table 9, while in Fig. 25 there is a graphical representation of the compounds’ molecular structures.
| HTMa | Dye | V OC (mV) | J SC (mA cm−2) | FFd | PCEe (%) | Ref. |
|---|---|---|---|---|---|---|
| a Hole transporting material. b Open circuit voltage. c Short circuit current. d Fill factor. e Power conversion efficiency. | ||||||
| Cu(dmp)2 | LEG4 | 1010 | 13.8 | 0.59 | 8.2 | 107 |
| [Co(bpyPY4)](OTf)2.33 | Y123 | 768 | 12.12 | 0.62 | 5.68 | 281 |
| [Co(bpy)3](OTf)2.33 | Y123 | 877 | 0.66 | 0.73 | 0.21 | 281 |
| Cu(tmby)2 | Y123 | 1080 | 13.87 | 0.73 | 11.0 | 87 |
| WS-72 | 1070 | 13.8 | 0.79 | 11.7 | 103 | |
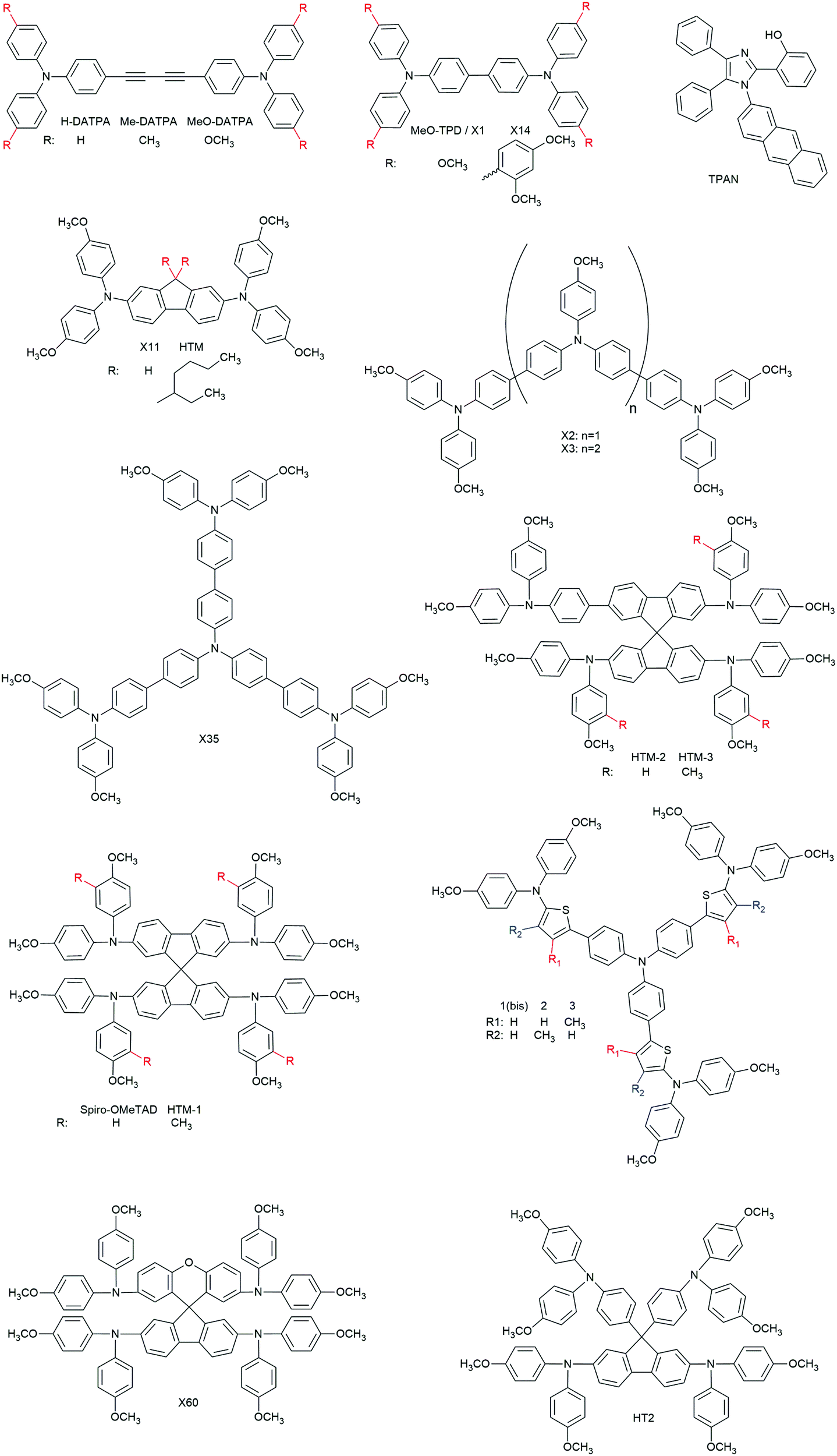 | ||
| Fig. 23 Triphenylamine-based novel organic hole transport materials. | ||
Freitag et al. were the first researchers to publish a work on solid-state dye-sensitized solar cells based on a metal complex hole conductor. They used a mix of Cu(I) and Cu(II) coordinated with a phenanthroline-based ligand (Cu(dmp)2).107 The device fabrication method was similar to that of liquid devices, but instead of sealing the cell after electrolyte injection the solvent was allowed to evaporate in air and then a new injection was performed until the space between the photoanode and the counter electrode was filled with solid HTM. Their best solar cell achieved a VOC of 1010 mV, JSC of 13.8 mA cm−2, FF of 0.59 and PCE of 8.2%, making it more efficient than the spiro-OMeTAD-based reference device (5.6%) and even more efficient than a liquid DSC based on the same electrolyte (6.0%).
Kashif et al. published a similar device based on a Co(II/III) metal center and a polypyridyl hexadentate ligand ([Co(bpyPY4)](OTf)2.33).281 As inferred by the counter-ion stoichiometry, the Co(II) to Co(III) ratio was 2:1. The device fabrication method was similar to that of Freitag et al. but in this case the HTM solvent was removed under vacuum rather than by natural evaporation in air. Kashif's best device exhibited a VOC of 768 mV, JSC of 12.12 mA cm−2, FF of 0.62 and PCE of 5.68%. In the same work, Kashif tried to fabricate ssDSCs with the Co(bpy)3 metal complex, which is known to yield high efficiency in liquid DSCs.282 However, in the solid state charge transport issues due to poor conductivity severely limited the output current, yielding a device with a VOC of 877 mV, JSC of 0.66 mA cm−2, FF of 0.73 and PCE of 0.21%. This demonstrated that not all metal complexes, not even those with a common metal center, can be employed as hole conductors in ssDSCs.
Building on the work on Cu complexes by Freitag, Grätzel and coworkers have fabricated the highest-performing ssDSCs reported so far, both based on a Cu(I/II) complex with the bipyridine-derived ligand tmby.87,103 In their first work the authors coupled Cu(tmby)2 with the Y123 dye to achieve a best-performing VOC of 1080 mV, JSC of 13.87 mA cm−2, FF of 0.73 and PCE of 11.0%. In their latest work they developed a new dye, WS-72, designed to reduce potential loss in the solar cell through better energy alignment of the various device components and reduced electron recombination. Indeed, in liquid DSCs a VOC improvement of 70 mV and a FF improvement of 0.04 were observed when moving from Y123 to the WS-72 dye. A solid-state device with Cu(tmby)2 and WS-72 was fabricated with a VOC of 1070 mV, JSC of 13.8 mA cm−2, FF of 0.79 and PCE of 11.7%. Although the VOC of this device was slightly lower compared to that of the previous work despite the better dye energy alignment, the improved FF led to a significant increase in efficiency.
3.2.2.4. Dopants for hole transporting materials. When it comes to HTM dopants, some confusion exist in the literature regarding their nomenclature, and it is important to make a distinction between additives and proper dopants (p-dopants). On the one hand, additives are compounds dissolved in the HTM precursor solution because they must be applied to the device after dye sensitization, but they do not directly affect the HTM itself. For example, additives like LiTFSI and 4-tert-butylpyridine (or other similar substituted pyridines) are known to migrate to the TiO2 surface to shift energy levels and passivate the exposed surface, in order to enable better charge injection and to reduce charge recombination at the TiO2–HTM interface.97,283–288 They are not only used in solid-state devices, but also in liquid DSCs. In the solid state, they may have the additional effect of modifying the HTM film morphology.289 On the other hand, proper dopants are characteristic of solid-state hole conductors and affect them directly by oxidizing a fraction of the material. The partial oxidation of the hole conducting material leads to the presence of vacancies in the solid-state film, which greatly increase hole mobility across the layer and, ultimately, conductivity.290 For organic compounds and small molecules in particular, which display very low hole mobility in their pristine state, the use of dopants is of paramount importance for the fabrication of efficient devices (vide infra). Compounds with a more positive redox potential with respect to that of the HTM (or strong Lewis acids) act as p-type dopants in HTM layers. Sufficient enough driving force promotes removal of electrons from the hole conductor. All the reviewed dopants have been applied to the spiro-OMeTAD molecule due to its role of reference material in ssDSCs but they can also be applied to other hole conductors to increase their conductivity.264,291,292 A summary of reviewed dopants and their effect on spiro-OMeTAD's hole mobility, and device efficiency is given in Table 10. In Fig. 26 the chemical structures of these compounds are depicted.
| Dopant | Redox potential (V vs. NHE) | Pristine conductivity (S cm−1) | Doped conductivity (S cm−1) | Pristine efficiency (%) | Doped efficiency (%) | Ref. |
|---|---|---|---|---|---|---|
| a After 5 days. b Assuming a redox potential of Fc0/+vs. NHE of 0.7 V or of NHE vs. vacuum of 4.4 V.299 c n.r. = not reported. d From ref. 244. e n.a. = not applicable. f Without LiTFSI. | ||||||
| FK102 | 1.06 | 4.4 × 10−5 | 5.3 × 10−4 | 2.3 | 5.6 | 244 |
| 2.6a | 6.1a | 244 | ||||
| FK209 | 1.02b | n.r.c | n.r. | 2.3d | 6.2 | 293 |
| FK269 | 1.28b | n.r. | n.r. | 2.3d | 6.0 | 293 |
| LiTFSI + O2 | n.a.e | 3 × 10−8f |
3 × 10−5 | ∼0f | 3 | 54 and 96 |
| F4TCNQ | 4.0b | n.r. | n.r. | 0.01f | 0.33f | 294 |
| 4.55 | 5.44 | 294 | ||||
| SnCl4 | 1.10 | ∼4-Fold increase | 2.52 | 3.40 | 295 | |
| Spiro(TFSI)2 | n.a. | 2.0 × 10−8f |
1.4 × 10−3f |
5.71 × 10−4f |
4.67f | 245 |
| 2.34 | 4.89 | 245 | ||||
| TeCA | n.r. | 1.2 × 10−6 | 7.5 × 10−5 | 5.8 | 7.7 | 296 |
| TEMPO-Br | 0.97 | 7.42 × 10−5 | 3.67 × 10−4 | 3.99 | 6.83 | 297 |
| DDQ | n.r. | 5.31 × 10−5 | 2.22 × 10−4 | 3.50 | 6.37 | 298 |
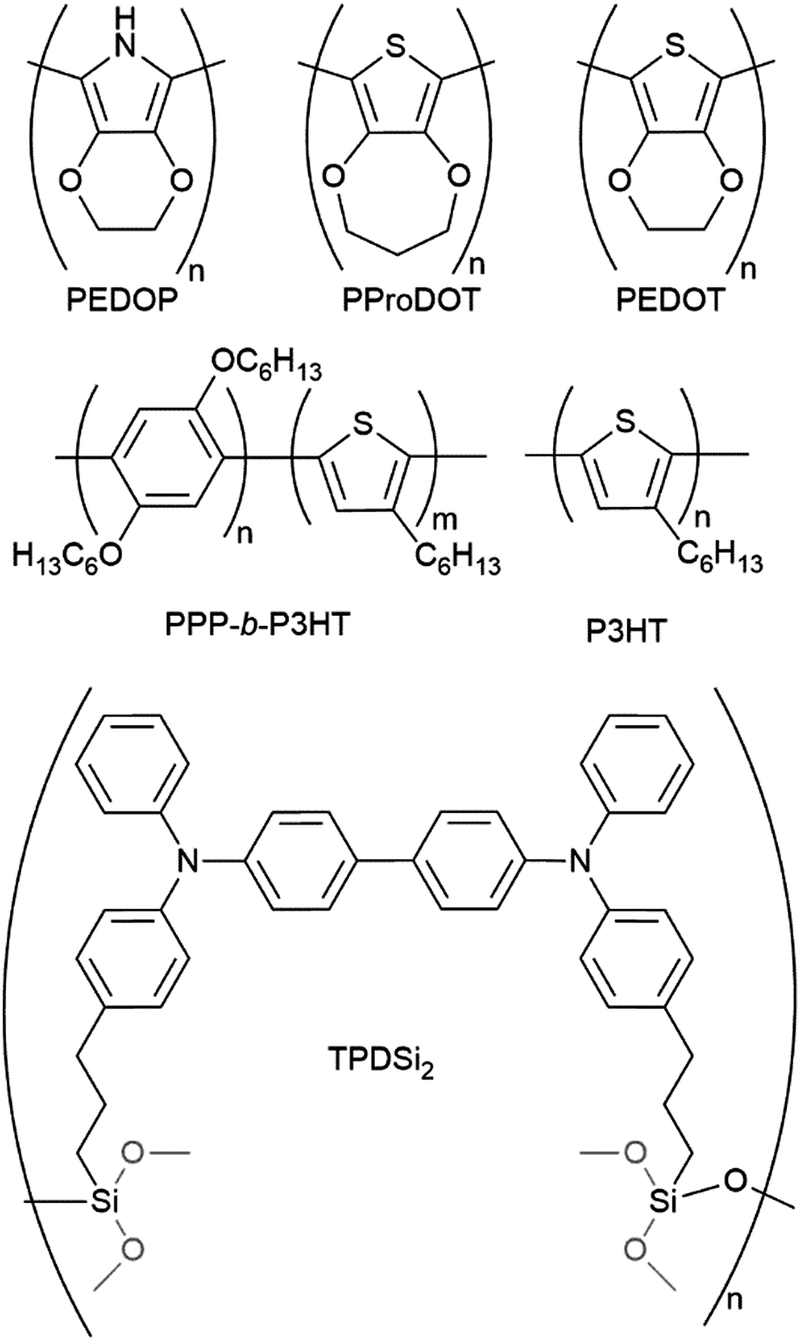 | ||
| Fig. 24 Polymer hole transport materials. | ||
Burschka et al. prepared for the first time a p-dopant – FK102 – which allowed solid-state dye sensitized solar cells to achieve relatively high efficiencies.244 Furthermore the compound – which was a Co(III)-based metal complex with three pyrazole-pyridine ligands – was soluble in the HTM precursor solution, simplifying the deposition process. The complex was able to oxidize spiro-OMeTAD and the resulting Co(II) species had a low molar extinction coefficient, which did not interfere with the effective light absorption of the dye. After doping the HTM film conductivity increased from 4.4 × 10−5 to 5.3 × 10−4 S cm−1, which in turn improved the average device efficiency from 2.3 to 5.6%. A second measurement after five days resulted in an even greater improvement as the reference cells increased their efficiency to 2.6% and those with the doped spiro-OMeTAD to 6.1%. Two years later Burschka et al. presented two more Co complex p-dopants – FK209 and FK269 – which were synthesized to improve the poor solubility of FK102 in relatively nonpolar solvents like chlorobenzene.293 In both of them PF6− counter-ions were replaced with much more soluble TFSI− ones. Furthermore, FK209 featured a ligand similar to that of FK102 but in which the pyridine was replaced with 4-tert-butylpyridine to increase solubility. FK269 was designed with a tridentate ligand instead, in which a second pyrazole moiety was added to FK102's ligand. This ligand significantly increased the redox potential of the compound (see Table 10), making it suitable to oxidize HTMs with a lower lying HOMO level than spiro-OMeTAD. The increased solubility allowed the authors to study greater concentrations – up to 5 times higher – of the dopant in the hole conductor solution. Keeping the 2.3% efficient devices of the previous study as a reference, average efficiencies of solar cells with FK209 reached 6.2% with 10% dopant concentration (6.0% with 2% concentration), while devices with 2% concentration of FK269 also attained efficiencies of 6.0%.
Cappel et al. began the study of LiTFSI's p-doping properties when devices were exposed to an air or to a N2 atmosphere in the presence of light96 and Snaith and coworkers continued their work giving a full explanation of LiTFSI's doping properties.54 The combined results of these studies showed that LiTFSI enabled molecular oxygen to oxidize spiro-OMeTAD regardless of light exposure, while LiTFSI alone was not able to oxidize the hole conductor. Conductivities after Li-catalyzed doping were increased by three orders of magnitude and the device efficiency increased from nearly 0% to 3%. Unfortunately, this redox reaction in air consumed Li+ ions due to the formation of lithium oxides, and Li+ also plays an important role as an additive on the titania surface in DSCs. For this reason, the authors recommended the use of a proper p-dopant for the oxidation of the hole transporter.
Chen et al. combined spiro-OMeTAD with the Lewis acid 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ), a strong electron acceptor.294 When they added 1.1 wt% dopant to the HTM solution a UV-Vis measurement confirmed the formation of the spiro-OMeTAD+ species. They applied pristine and doped hole transporter solutions (without LiTFSI) to ssDSCs obtaining a 3300% increase in efficiency from 0.01 to 0.33%. However, the presence of LiTFSI was required to achieve high efficiency and the two solutions with the added Li salt led to device efficiencies of 4.55 and 5.44%, respectively, confirming the beneficial effect of F4TCNQ doping.
Han and coworkers also worked on a Lewis acid, SnCl4.295 In their report they showed a figure with the I–V curves of the conductivity measurements without providing any actual number. From the picture itself, however, a ∼4-fold increase in conductivity can be inferred upon SnCl4 doping. When a 0.8% doped solution of spiro-OMeTAD was applied to solar cells, an efficiency of 3.40% was obtained as opposed to an efficiency of 2.52% for non-doped devices.
McGehee and coworkers decided to get rid of any p-dopant – which may alter film-formation or charge transfer properties of spiro-OMeTAD – from the HTM solution by pre-oxidizing the hole conductor itself.245 They reacted spiro-OMeTAD with two equivalents of AgTFSI to obtain spiro(TFSI)2 and metallic silver. After purifying the compound, they mixed it with the non-oxidized species in a ratio of 12 mol% to obtain an efficient HTM precursor solution. A solid-state film of this mixture displayed five orders of magnitude increase in conductivity, from 2.00 × 10−8 to 1.43 × 10−3 S cm−1 – two orders of magnitude higher than what was achieved with Li-induced oxygen doping by Snaith and coworkers. Devices fabricated without LiTFSI (but with tBP) showed an enormous increase in efficiency from nearly 0 to 4.67%, a much greater increase compared to the case of F4TCNQ or SnCl4 (0.35% efficiency for a SnCl4-doped device without LiTFSI). The doping beneficial effect was pronounced also in the presence of the Li salt, when solar cell efficiencies increased from 2.34 to 4.89% upon doping.
Xu et al. reported on 1,1,2,2-tetrachloroethane (TeCA), which they defined as a co-solvent, rather than a proper p-dopant.296 The reason for this is that UV light needed to be shined on the TeCA-containing HTM solution for one minute to allow spiro-OMeTAD oxidation to take place. Precipitation of AgCl upon addition of AgNO3 to the oxidized spiro-OMeTAD/TeCA solution proved that TeCA oxidized the hole conductor by forming a spiro-OMeTAD+Cl− salt. 4% TeCA-doped HTM film conductivity increased from 1.2 × 10−6 to 7.5 × 10−5 S cm−1. Similarly, device efficiencies improved from 5.8 to 7.7%. As a comparison, devices fabricated with FK209 instead of TeCA exhibited an efficiency of 6.8%.
Yang et al. oxidized spiro-OMeTAD with a compound widely used in organic synthesis – TEMPO (or, rather, its bromide salt TEMPO-Br).297 HTM films doped with 2.5% TEMPO-Br showed a smaller increase in conductivity compared to other dopants, with values increasing from 7.42 × 10−5 to 3.67 × 10−4 S cm−1. However, the dopant's effect on device efficiency was remarkable with PCEs increased from 3.99 to 6.83%.
The most recent work on p-dopants in ssDSCs was published by Sun and coworkers, who studied the effects on spiro-OMeTAD of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), another oxidant widely used in organic synthesis.298 The most notable result of their work is that only a very tiny 0.04% DDQ addition to spiro-OMeTAD was required to maximize solar cell efficiency. Conductivity of the oxidized HTM film increased from 5.31 × 10−5 to 2.22 × 10−4 S cm−1 and the device efficiency was boosted from 3.50 to 6.37%.
3.3. Counter electrode
The choice of counter electrode materials in recent ssDSC literature strongly correlates with the chosen device architecture. Liquid electrolyte-based DSCs were traditionally assembled in a sandwich structure between two glass electrodes. The counter electrode was often platinized to provide good catalytic interfaces for the reduction of the redox electrolyte species.300 Given that this sandwich design protects the HTM and other active components of the solar cell well, it is still often chosen as a layout for ssDSCs.301,302 In 2013, electropolymerized poly(3,4-ehtylenedioxythiophene) counter electrodes (Fig. 27) as a low-cost alternative to the scarce platinum were first presented by Ellis et al. with lower sheet resistance compared to platinized electrodes.74 Such organic counter electrodes have since been employed in most, especially Cu-based, ssDSCs.87,107 | ||
| Fig. 25 Transition metal complexes as hole transport materials in ssDSCs. | ||
By contrast, in a ssDSC layout where the HTM is spin-coated onto the sensitized TiO2, metal contacts are commonly evaporated on top of the HTM layer. Such thin layers of noble metals such as Au/Ag not only collect charges from the HTM but reflect unabsorbed photons back into the active part of the cell.267,303 Silver contacts generally exhibit stronger light reflection at a lower material cost. Gold contacts, however, excel with chemical stability and higher tolerance of minor defects in the HTM layer, while shunt resistances rapidly arise upon Ag penetration into the HTM layer.304 A picture as well as a cross-sectional electron microscopy image of a ssDSC employing a vacuum-evaporated gold counter contact was shown above in Fig. 1.
Occasionally, alternative approaches for counter electrode deposition are described in the literature. Chiang et al. demonstrated in 2012 that a transparent layer of ITO can be sputtered on top of spiro-OMeTAD to serve as a counter contact.305 Such bifacial transparent ssDSCs furthermore open up possible tandem designs (see Section 4.2.2) as later demonstrated by the same group.306 Aitola et al. described the deposition of carbon nanotubes as counter electrode materials in solid-state DSCs. They combined in situ photoelectropolymerized poly(3,4-ethylenedioxythiophene) onto mesoporous TiO2 sensitized with the organic LEG4 dye and achieved 4.8% power conversion efficiency with subsequently transferred single-walled carbon nanotubes doped with lithium bis(trifluoromethylsulfonyl)imide and 4-tert-butylpyridine whilst 5.2% power conversion with an evaporated Ag contact.307 Zhang et al. deposited carbon nanotubes onto a flexible conductive ITO-coated poly(ethylenetherephtalate) substrate and fabricated quasi-solid-state DSCs with 4.24% power conversion efficiency at 0.95 sun illumination, while recording 4.87% efficiency with Pt-based reference devices.308 Margulis et al. sprayed Ag nanowires on top of a poly(3,4-ethylenedioxythiophene):polystyrenesulfonate layer to obtain 3.6% power conversion efficiency and 3.7% for the reference device with evaporated silver contacts.309
Early reports indicated that power conversion efficiencies achieved with evaporated gold contacts were matched with pressed graphite.143 Even gold-coated glass substrates were tested as counter electrodes initially, but no longer pursued due to interfacial issues.310
4. Device characteristics
4.1. Stability
Ultimately, any solar cell technology is required to perform over years or even decades of operation to become a feasible investment for customers. To yield any non-negligible return-on-investment (ROI) interest for the investor, the lifetime of any solar cell needs to exceed the payback time for the production cost of the solar cell.311 However, despite the comparably low production cost of dye-sensitized solar cells, the cells need to, depending on the intended application, sustain a significant period of time to gain market appeal. Therefore, stability testing has become a crucial concern in the development of all emerging photovoltaic technologies.312,313Contrarily to silicon solar cells, no standardized stability testing protocol has been established for DSCs as well as for ssDSCs. Reports can be found in the literature from stability under dark storage conditions to operational stability under full sun illumination, strongly impeding any effective comparison of reported stabilities. Since 2008, the IEC-61646 norm has set the standard testing conditions for common solar cells with temperature cycling between −40 °C and +85 °C combined with performance tracking for 1000 hours.314 In 2016, new testing protocols were released specifically for testing of, e.g., Si, GaAs, CdTe and other thin film technology solar cells including damp-heat-tests and UV irradiation (IEC-61215-2, IEC-61215-1-2, IEC-61215-1-4).315 The most common procedure to trace the photovoltaic performance is the so-called maximum power point tracking (MPP).316
Although some studies comply with one of the aforementioned standards, milder testing conditions are commonly chosen for stability testing on a laboratory scale. Specifically, the increasing interest in employing DSCs and ssDSCs to power internet of things (IoT) devices under ambient light conditions does not call for equally robust testing procedures.102
Solid-state alternatives to liquid electrolyte-based DSCs were developed to circumvent the inherent thermal instability of volatile electrolyte solvents.317 Electrolytes based on less-volatile solvents such as 3-methoxypropionitrile were demonstrated; however, they suffered from mass transport deficiencies of the redox species in the more viscous electrolyte or aggregation effects at the TiO2/dye/electrolyte interface.318,319 Solid-state DSCs are not subjected to such degradation mechanisms in the electrolyte and do thus possess a much higher intrinsic stability compared to their liquid counterparts.
Just as in many biological ensembles, failure of a single component in ssDSCs can lead to malfunction. Generally, with the semiconductor metal oxides exhibiting excellent photo- and thermal stability, degradation pathways largely involve the dye molecules and the hole transporting material (HTM). With the bandgap of 3.2 eV in the widely used antase-TiO2 comes the absorption of UV-photons in the mesoporous layer of the cell. Consequently, high-energy carriers can lead to destructive, irreversible reactions with both absorbed sensitizers and electrolyte/HTM components.320 The degradation pathway at the TiO2/dye/HTM interface, however, has yet to be entirely revealed. While early reports by Kroon et al. pointed towards hydrophobic (co-)sensitizers to keep possible water/oxygen away from the TiO2,321 later studies indicated that molecules such as 4-tert-butylpyridine (or even acetonitrile in the case of liquid-electrolyte DSCs) can passivate reactive states at the TiO2 surface.322,323 The variety of ionic dopants (such as Li+) employed in the most-frequently used spiro-OMeTAD has been shown to draw additional humidity into the molecular ensemble.324
Different approaches for the geometrical architecture of ssDSCs have been proposed (see Section 2.1). In ssDSCs inspired by the traditional liquid electrolyte ‘sandwich’ architecture,107 the active components can be sealed, and will thus not be subjected to (e.g. humidity-induced) degradation as easily as ssDSCs with an evaporated counter contact.200 As already emphasized in the review by Docampo et al. in 2014,325 the reader of any stability report should carefully pay attention to the exclusion of UV-light from the testing protocol as well as the device architecture and sealing.
Several approaches have been reported to address the stability of ssDSCs. Already in 2003, Wang et al. observed that gelation of I−/I3− redox electrolytes with poly(vinylideneflouride-co-hexaflouropropylene) led to preservation of 94% of the initial performance after 1000 hours of storage at 80 °C, compared to only 88% for the liquid counterpart based on 3-methoxypropionitrile (MPN).221 In 2007, Zhang et al. employed a ruthenium sensitizer and CuI as the HTM to demonstrate that, upon exposure to UV-light, complete device malfunction was observed after 60 hours, while the device produced 90% of its initial power output after 500 hours when employing a 435 nm cutoff UV filter. They furthermore demonstrated that, upon passivation of the TiO2 surface with a thin layer of MgO, even under UV irradiation 70% of the initial performance was maintained after 72 hours.326 A first full long-term operational stability study on ssDSCs was carried out by the group of Grätzel and coworkers in 2010. Their devices based on ruthenium sensitizers and spiro-OMeTAD as the HTM retained 70% of the initial power output after 1000 hours of continuous full-sun illumination in an inert argon atmosphere employing a thin polyester film to cut off UV-photons below 460 nm.303 Several approaches ensued to enhance the long-term stability of quasi-solid-state DSCs by gelation of a solvent-based electrolyte and/or ionic liquid with pyrazolium derivates,327 polyvinylpyrrolidone,220 poly(1,6-hexanedioldiacrylate),302 poly(methylmethacrylate) matrices,328 polyaniline-loaded carbon black,329N,N′-1,5-pentadiylbis-dodecanamide,330 carboxymethyl-κ-caraageenan214 and poly(methylmethacrylate)-polyaniline nanotubes.301
In 2011, Grätzel and coworkers reported ssDSCs with a new cobalt-based dopant for spiro-OMeTAD in C220-sensitized ssDSCs that, employing a 420 nm UV cutoff filter, retained 80% of their initial performance after 40 days under full continuous one-sun illumination at 60 °C.244 Compelling studies by Malinauskas et al. showed that impurities in the spiro-OMeTAD layer led to the formation of large crystals, which caused complete device failure after 4 hours at 100 °C. The group instead proposed spiro-derivatives with asymmetric methylation or insertion of phenyl units that impede the crystallization (see Section 3.2.2.1 on organic hole transport materials). Devices based on their methylated spiro-OMeTAD analogue matched the photovoltaic performance of pristine spiro-OMeTAD; however, they kept over 90% of the initial power conversion after 1000 hours at 60 °C, while reference devices based on pristine spiro-OMeTAD degraded to 10% of their performance. Insertion of phenyl units into spiro-OMeTAD also led to an increased long-term stability of at least 50% performance after the same thermal stress treatment.246 Most recently, the group of Kloo and coworkers designed novel D–A–π–A dyes that allowed for preservation of 86% of the initial performance of devices with doped spiro-OMeTAD as the HTM after 170 hours in a dark ambient atmosphere.118 In 2017, Cao et al. for the first time carried out a long-term stability study on ssDSCs with the performance-leading copper complex hole transport materials. Their devices based on the Y123 dye and CuII/I(tmby)2 showed preservation of 85% power output after 200 hours of continuous illumination with a 50 mW cm−2 white LED array.87
4.2. Types of ssDSC
The power conversion efficiency of any photovoltaic device is ultimately limited by the intrinsic Shockley–Queisser limit, which was estimated to be around 33.7% in 1961.114 Briefly, in a single-junction power conversion device, only photons above the bandgap energy of the light absorber can be absorbed. However, for photons of even higher energy, the excess energy with respect to the bandgap of the photoabsorber (sometimes called overpotential) cannot be utilized and is dissipated. In order to obtain a high photovoltage, a large-bandgap material should be chosen. However, high-bandgap solar cells suffer from transmittance losses as many photons below the bandgap energy cannot be converted to photocurrent. Detailed derivations and calculations on the conversion limits of photovoltaic cells were reported by Jacobsson et al. as well as Alharbi et al. in 2015.331,332 In order to ultimately overcome the Shockley–Queisser limit, multijunction devices (or their subparts) have recently been investigated for ssDSCs and will be reviewed in the ensuing sections. Additionally, photon-upconversion materials are drawing increasing interest of the scientific community and, while implementation in all-solid-state devices is still awaited, first results on liquid-electrolyte DSCs are reported.333–336 | ||
| Fig. 26 Dopants tested to enhance the performance of spiro-OMeTAD in ssDSCs. | ||
Sensitized NiO was first studied in I−/I3−-electrolyte p-type DSCs by He et al. in 1999341 and more recently by Hammarström and coworkers.342,343 The group of Tian presented the first all-solid-state p-type cell in 2016. They sensitized mesoporous NiO with the organic P1 dye and used phenyl-C61-butyric acid methylester (PCBM) as the electron transport material to obtain 620 mV photovoltage, but with only 50 μA cm−2 photocurrent density.344 In 2017, Pham et al. sensitized NiO with a diketopyrrolopyrrole dye and measured 0.45 mA cm−2 photocurrent density.152 Tian's group recently demonstrated that the charge separation kinetics can be enhanced by deposition of a shell of titanium dioxide on top of the PB6-sensitized NiO.345 They furthermore reported that a shielding layer of Al2O3 between NiO and TiO2 suppresses carrier recombination.346
 | ||
| Fig. 27 Electropolymerized poly(3,4-ehtylenedioxythiophene) on an FTO substrate. | ||
In 2000, He et al. presented the first tandem-dye-sensitized solar cell.348 Four years later, both Dürr et al. and Kubo et al. reported solar cells consisting of two serially positioned, parallel-connected DSCs based on sensitized TiO2 and I−/I3− electrolyte.349,350 In 2009, Gibson et al. presented heterojunction devices based on a N719-sensitized TiO2 photoanode and a perylene-sensitized NiO photocathode mediated by a single cobalt redox mediator.351
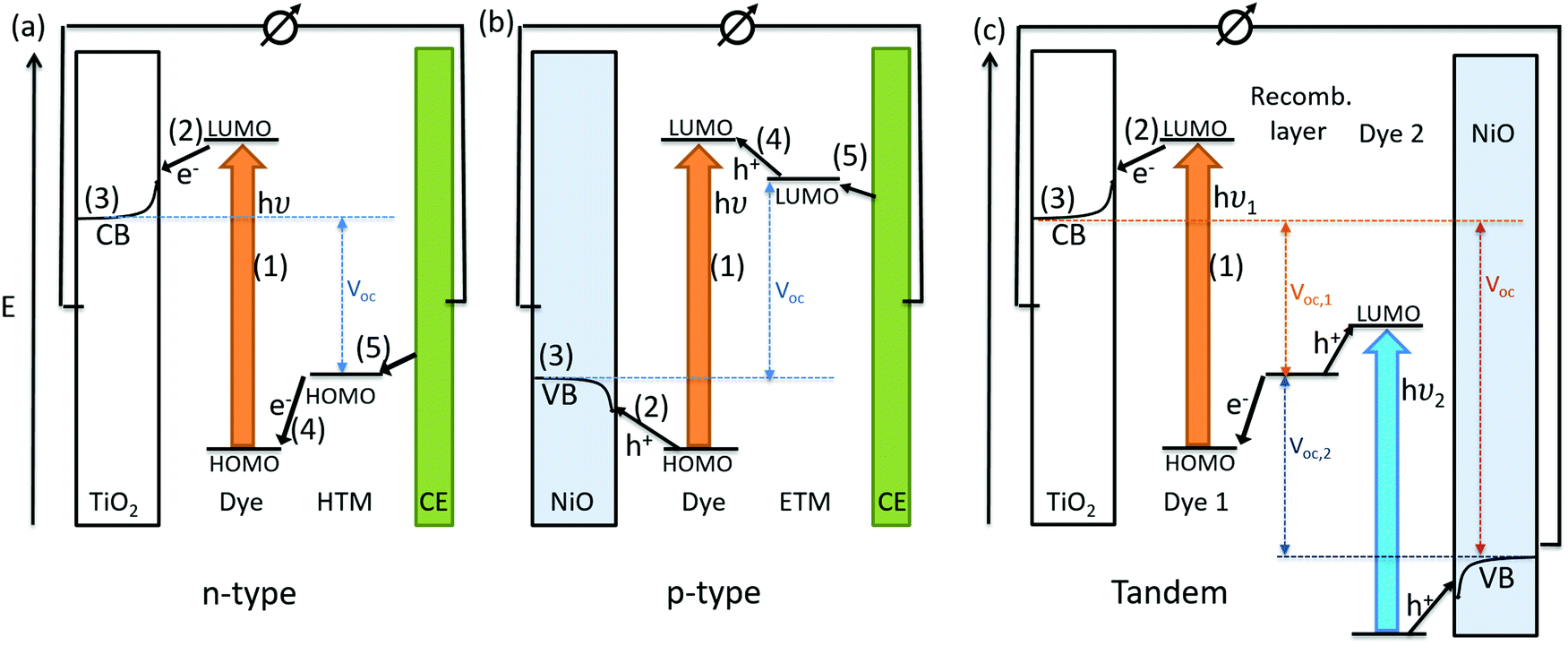 | ||
| Fig. 28 Charge transfer processes in (a) n-type, (b) p-type and (c) tandem ssDSCs. | ||
Bruder et al. reported the first all-solid-state tandem DSC in 2009. A charge recombination layer of Ag was placed between a top cell with indoline-sensitized TiO2/spiro-OMeTAD and a bottom junction of zinc phtalocyanine/C60. With each individual cell component converting sunlight at about 4.0% efficiency, they obtained (serial) tandem cells with 1.36 V open-circuit potential and 6.0% power conversion efficiency.169 In 2013, Chiang et al. used semi-transparent indium-doped tin oxide contacts to stack two n-type ssDSCs. They sensitized the bottom cell with squarine-B1, the top cell with Z907 and deposited spiro-OMeTAD as the HTM on both cells. Upon parallel connection of the cells, they recorded a 3.1% power conversion efficiency, which matched combined conversion of the two subcells.306
For solid-state DSCs, however, studies indicate that the addition of photoactive compounds can lead to an increase in, specially, photocurrent density. Electrons excited in the HTM matrix can be transferred to the TiO2-adsorbed dyes through Förster resonance energy transfer.356 In 2010, Mor et al. demonstrated a light harvesting enhancement for a squaraine-sensitized TiO2 device upon addition of a pyran dye component to the spiro-OMeTAD hole transport material.357 Unger et al. resolved the Förster resonance energy transfer contributions to the incident-photon-to-current conversion efficiency of the photoactive hole transport material tris(thienyl-vinyl-thienyl)triphenylamine in squaraine-sensitized ssDSCs.358 Grätzel and coworkers employed the absorbing poly(3-hexylthiophen-2,5-diyl) hole transport material to increase the photocurrent density of a YD2-porphyrine-sensitized ssDSC to 12.1 mA cm−2, however, with potential losses with respect to the spiro-OMeTAD device.359
5. Outlook
The ignored challenge with all emerging solar cell technologies is that they lack the stability that established technologies have; this is the largest hindrance towards their entrance into society at large. Further, in most of these systems, the development and knowledge curve of the photo-absorbing material has reached a mature state where only incremental improvements are expected for the future. The development of new charge transfer (redox mediators, HTMs, electron transport materials) systems are still far behind the efforts and understanding, which were made to develop new sensitizers or any other component of the DSCs. Large knowledge gaps still exist in the adjacent charge selective and transporting materials. Hence, it is important to address several challenges, including fundamental understanding of how various charge transport materials in solid state facilitate charge transfer and how they can be tuned to obtain ideal energy alignments in the photo-electrochemical device. Simultaneously, the new materials must be stable, easily processable, and prepared from sustainable sources of materials. Careful matching in energy alignments of panchromatic sensitizers, in combination with the new charge transporting materials with higher conductivity and charge mobility, will lead to increased charge collection efficiency and more stable hybrid solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Energy Agency (42037-1 and 43294-1), the STandUP for Energy program and the Carl Trygger Stiftelse 17:158.References
- A. A. Bartlett, Am. J. Phys., 1978, 46, 876–888 CrossRef.
- K. Butti and J. Perlin, A Golden Thread: 2500 Years of Solar Architecture and Technology, M. Boyars, 1981 Search PubMed.
- A. Goetzberger, J. Luther and G. Willeke, Sol. Energy Mater. Sol. Cells, 2002, 74, 1–11 CrossRef CAS.
- A. Cristobal, A. M. Vega and A. L. López, Next Generation of Photovoltaics: New Concepts, Springer Berlin Heidelberg, 2012 Search PubMed.
- G. F. Brown and J. Wu, Laser Photonics Rev., 2009, 3, 394–405 CrossRef CAS.
- M. A. Green, Phys. E, 2002, 14, 65–70 CrossRef CAS.
- M. A. Green, Y. Hishikawa, W. Warta, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger and A. W. H. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2017, 25, 668–676 CrossRef.
- A. Becquerel, C. R. Acad. Sci., 1839 Search PubMed.
- S. R. Wenham and M. A. Green, Prog. Photovoltaics Res. Appl., 1996, 4, 3–33 CrossRef CAS.
- J. Perlin, Silicon Solar Cell Turns 50, USA, 2004 Search PubMed.
- H. Gerischer, J. Electrochem. Soc., 1966, 113, 1174–1182 CrossRef CAS.
- H. Gerischer, Electrochim. Acta, 1990, 35, 1677–1699 CrossRef CAS.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. Grätzel, Chem. Lett., 2005, 34, 8–13 CrossRef.
- M. Freitag and G. Boschloo, Curr. Opin. Electrochem., 2017, 2, 111–119 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, Md. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- J. Gong, J. Liang and K. Sumathy, Renewable Sustainable Energy Rev., 2012, 16, 5848–5860 CrossRef CAS.
- H. Pettersson and T. Gruszecki, Sol. Energy Mater. Sol. Cells, 2001, 70, 203–212 CrossRef CAS.
- J. Goldstein, I. Yakupov and B. Breen, Sol. Energy Mater. Sol. Cells, 2010, 94, 638–641 CrossRef CAS.
- A. Hagfeldt and N. Vlachopoulos, in The Future of Semiconductor Oxides in Next-Generation Solar Cells, ed. M. B. T.-T. F. of S. O. in N.-G. S. C. Lira-Cantu, Elsevier, 2018, pp. 183–239 Search PubMed.
- M. S. Su’ait, M. Y. A. A. Rahman, A. Ahmad, M. S. Su’ait, M. Y. A. A. Rahman, A. Ahmad, M. S. Su’ait, M. Y. A. A. Rahman and A. Ahmad, Sol. Energy, 2015, 115, 452–470 CrossRef.
- J.-L. Lan, T.-C. Wei, S.-P. Feng, C.-C. Wan and G. Cao, J. Phys. Chem. C, 2012, 116, 25727–25733 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- A. Hauch and A. Georg, Electrochim. Acta, 2001, 46, 3457–3466 CrossRef CAS.
-
M. Freitag, B. Łosiewicz, T. Goryczka and J. Lel
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) tko, Application of EIS to study the corrosion resistance of passivated NiTi shape memory alloy in simulated body fluid, 2012, vol. 183 Search PubMed.
tko, Application of EIS to study the corrosion resistance of passivated NiTi shape memory alloy in simulated body fluid, 2012, vol. 183 Search PubMed. - C. Law, O. Moudam, S. Villarroya-Lidon and B. O’Regan, J. Mater. Chem., 2012, 22, 23387–23394 RSC.
- S. K. Yadav, S. Ravishankar, S. Pescetelli, A. Agresti, F. Fabregat-Santiago and A. Di Carlo, Phys. Chem. Chem. Phys., 2017, 19, 22546–22554 RSC.
- L. Kavan, P. Liska, S. M. Zakeeruddin and M. Grätzel, Electrochim. Acta, 2016, 195, 34–42 CrossRef CAS.
- M. Grätzel, MRS Bull., 2005, 30, 23–27 CrossRef.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- J.-H. Yum, P. Chen, M. Grätzel and M. K. Nazeeruddin, ChemSusChem, 2008, 1, 699–707 CrossRef CAS PubMed.
- U. Bach, PhD thesis, ÉCOLE POLYTECHNIQUE FÉDÉRALE DE LAUSANNE, 2000.
- K. Tennakone, G. R. R. A. Kumara, A. R. Kumarasinghe, K. G. U. Wijayantha and P. M. Sirimanne, Semicond. Sci. Technol., 1995, 10, 1689 CrossRef.
- K. Tennakone, A. H. Jayatissa, C. A. N. Fernando, S. Wickramanayake, S. Punchihewa, L. K. Weerasena and W. D. R. Premasiri, Phys. Status Solidi, 1987, 103, 491–497 CrossRef CAS.
- K. Tennakone, G. R. R. A. Kumara, I. R. M. Kottegoda, K. G. U. Wijayantha and V. P. S. Perera, J. Phys. D: Appl. Phys., 1999, 31, 1492–1496 CrossRef.
- G. R. R. Kumara, A. Konno, G. K. R. Senadeera, P. V. V. Jayaweera, D. B. R. De Silva and K. Tennakone, Sol. Energy Mater. Sol. Cells, 2001, 69, 195–199 CrossRef CAS.
- J. Zhang, N. Vlachopoulos, M. Jouini, M. B. Johansson, X. Zhang, M. K. Nazeeruddin, G. Boschloo, E. M. J. Johansson and A. Hagfeldt, Nano Energy, 2016, 19, 455–470 CrossRef CAS.
- X. Bo, T. Haining, L. Lili, Q. Deping, C. Hong, Z. Jinbao, V. Nick, B. Gerrit, L. Yi, Z. Fengling, H. Anders, S. Licheng, B. Xu, H. Tian, L. Lin, D. Qian, H. Chen, J. Zhang, N. Vlachopoulos, G. Boschloo, Y. Luo, F. Zhang, A. Hagfeldt, L. Sun, X. Bo, T. Haining, L. Lili, Q. Deping, C. Hong, Z. Jinbao, V. Nick, B. Gerrit, L. Yi, Z. Fengling, H. Anders and S. Licheng, Adv. Energy Mater., 2014, 5, 1401185 Search PubMed.
- D. Moia, U. B. Cappel, T. Leijtens, X. Li, A. M. Telford, H. J. Snaith, B. C. O’Regan, J. Nelson and P. R. F. Barnes, J. Phys. Chem. C, 2015, 119, 18975–18985 CrossRef CAS.
- I.-K. K. Ding, N. Tétreault, J. Brillet, B. E. Hardin, E. H. Smith, S. J. Rosenthal, F. Sauvage, M. Grätzel and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 2431–2436 CrossRef CAS.
- P. Docampo, A. Hey, S. Guldin, R. Gunning, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2012, 22, 5010–5019 CrossRef CAS.
- C. T. Weisspfennig, D. J. Hollman, C. Menelaou, S. D. Stranks, H. J. Joyce, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Funct. Mater., 2014, 24, 668–677 CrossRef CAS.
- H. J. Snaith, R. Humphry-Baker, P. Chen, I. Cesar, S. M. Zakeeruddin and M. Grätzel, Nanotechnology, 2008, 19, 424003 CrossRef PubMed.
- I. K. Ding, J. Melas-Kyriazi, N. Le Cevey-Ha, K. G. Chittibabu, S. M. Zakeeruddin, M. Grätzel and M. D. McGehee, Org. Electron., 2010, 11, 1217–1222 CrossRef CAS.
- C. D. Bailie, E. L. Unger, S. M. Zakeeruddin, M. Grätzel and M. D. McGehee, Phys. Chem. Chem. Phys., 2014, 16, 4864 RSC.
- A. S. Hoon, K. D. Jun, C. W. Seok and K. J. Hak, Adv. Funct. Mater., 2014, 24, 5037–5044 CrossRef.
- W. Cho, J. Lim, T.-Y. Kim, Y. R. Kim, D. Song, T. Park, F. Fabregat-Santiago, J. Bisquert and Y. S. Kang, J. Phys. Chem. C, 2016, 120, 2494–2500 CrossRef CAS.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162 CrossRef CAS.
- C. T. Weisspfennig, M. M. Lee, J. Teuscher, P. Docampo, S. D. Stranks, H. J. Joyce, H. Bergmann, I. Bruder, D. V. Kondratuk, M. B. Johnston, H. J. Snaith and L. M. Herz, J. Phys. Chem. C, 2013, 117, 19850–19858 CrossRef CAS.
- J. Lu, Y. C. Chang, H. Y. Cheng, H. P. Wu, Y. Cheng, M. Wang and E. W. G. Diau, ChemSusChem, 2015, 8, 2529–2536 CrossRef CAS PubMed.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo, I. McPherson and H. J. Snaith, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- T. Leijtens, I.-K. Ding, T. Giovenzana, J. T. Bloking, M. D. McGehee and A. Sellinger, ACS Nano, 2012, 6, 1455–1462 CrossRef CAS PubMed.
- M. K. Nazeeruddin, E. Baranoff and M. Grätzel, Sol. Energy, 2011, 85, 1172–1178 CrossRef CAS.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Péchy and M. Grätzel, Chem. Commun., 2008, 5194 RSC.
- S. Gauthier, B. Caro, F. Robin-Le Guen, N. Bhuvanesh, J. A. Gladysz, L. Wojcik, N. Le Poul, A. Planchat, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, Dalton Trans., 2014, 43, 11233–11242 RSC.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fré, P. Rubino, C. Choné, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS PubMed.
- H. L. Wong, C. S. K. Mak, W. K. Chan and A. B. Djurišić, Appl. Phys. Lett., 2007, 90, 20–22 Search PubMed.
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed.
- N. Alonso-Vante, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1994, 11, 1649 RSC.
- Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17011–17017 CrossRef CAS.
- C. Qin, Y. Numata, S. Zhang, X. Yang, A. Islam, K. Zhang, H. Chen and L. Han, Adv. Funct. Mater., 2014, 24, 3059–3066 CrossRef CAS.
- E. Stathatos, P. Lianos, A. Laschewsky, O. Ouari and P. Van Cleuvenbergen, Chem. Mater., 2001, 13, 3888–3892 CrossRef CAS.
- M. Kraus, S. Richler, A. Opitz, W. Brütting, S. Haas, T. Hasegawa, A. Hinderhofer and F. Schreiber, J. Appl. Phys., 2010, 107, 3–8 CrossRef.
- H. J. Snaith and M. Grätzel, Appl. Phys. Lett., 2006, 89, 262114 CrossRef.
- J. U. Wallace, Carrier mobility in organic charge transport materials: methods of measurement, analysis, and modulation, University of Rochester, 2009 Search PubMed.
- H. J. Snaith and M. Grätzel, Adv. Mater., 2007, 19, 3643–3647 CrossRef CAS.
- J. E. Kroeze, N. Hirata, L. Schmidt-Mende, C. Orizu, S. D. Ogier, K. Carr, M. Grätzel and J. R. Darrant, Adv. Funct. Mater., 2006, 16, 1832–1838 CrossRef CAS.
- K. Aitola, K. Sveinbjornsson, J.-P. Correa-Baena, A. Kaskela, A. Abate, Y. Tian, E. M. J. Johansson, M. Gratzel, E. I. Kauppinen, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2016, 9, 461–466 RSC.
- C.-E. Cheng, C.-Y. Lin, C.-H. Shan, S.-Y. Tsai, K.-W. Lin, C.-S. Chang and F. Shih-Sen Chien, J. Appl. Phys., 2013, 114, 14503 CrossRef.
- H. Ellis, N. Vlachopoulos, L. Häggman, C. Perruchot, M. Jouini, G. Boschloo and A. Hagfeldt, Electrochim. Acta, 2013, 107, 45–51 CrossRef CAS.
- A. A. Arbab, M. H. Peerzada, I. A. Sahito and S. H. Jeong, J. Power Sources, 2017, 343, 412–423 CrossRef CAS.
- Y. Hou, D. Wang, X. H. Yang, W. Q. Fang, B. Zhang, H. F. Wang, G. Z. Lu, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 1583 CrossRef PubMed.
- Y. Rong, Z. Ku, M. Xu, L. Liu, M. Hu, Y. Yang, J. Chen, A. Mei, T. Liu and H. Han, RSC Adv., 2014, 4, 9271 RSC.
- X. Zhang, Y. Xu, F. Giordano, M. Schreier, N. Pellet, Y. Hu, C. Yi, N. Robertson, J. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 10742–10745 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- K. Rajeshwar, Fundamentals of semiconductor electrochemistry and photoelectrochemistry, in Encyclopedia of Electrochemistry, ed. A. J. Bard, M. Stratmann and S. Licht, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, vol. 6, pp. 1–53 Search PubMed.
- R. Jose, V. Thavasi and S. Ramakrishna, J. Am. Ceram. Soc., 2009, 92, 289–301 CrossRef CAS.
- M. Wang, C. Grätzel, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2012, 5, 9394–9405 RSC.
- J. Cong, X. Yang, L. Kloo and L. Sun, Energy Environ. Sci., 2012, 5, 9180–9194 RSC.
- P. Liu, J. M. Gardner and L. Kloo, Chem. Commun., 2015, 51, 14660–14662 RSC.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer, M. Grätzel, F. Weissortel, J. Salbeck, H. Spreitzer, M. Gratzel, F. Weissörtel, J. Salbeck, H. Spreitzer, M. Grätzel, F. Weissortel, J. Salbeck, H. Spreitzer, M. Gratzel, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- M. Freitag, F. Giordano, W. Yang, M. Pazoki, Y. Hao, B. Zietz, M. Grätzel, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2016, 120, 9595–9603 CrossRef CAS.
- Y. Cao, Y. Saygili, A. Ummadisingu, J. J. Teuscher, J. Luo, N. Pellet, F. Giordano, S. M. Zakeeruddin, J.-E.-E. E. J.-E. Moser, M. Freitag, A. Hagfeldt and M. Grätzel, Nat. Commun., 2017, 8, 15390 CrossRef PubMed.
- F. Fabregat-Santiago, J. Bisquert, L. Cevey, P. Chen, M. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 131, 558–562 CrossRef PubMed.
- J. Bisquert and F. Fabregat-Santiago, Dye Sol. Cells, 2010, 604 Search PubMed.
- E. Palomares, J. N. Clifford, S. A. Haque, T. Lutz and J. R. Durrant, J. Am. Chem. Soc., 2003, 125, 475–482 CrossRef CAS PubMed.
- B. C. O’Regan and J. R. Durrant, Acc. Chem. Res., 2009, 42, 1799–1808 CrossRef PubMed.
- S. A. Haque, E. Palomares, B. M. Cho, A. N. M. Green, N. Hirata, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 3456–3462 CrossRef CAS PubMed.
- S. Tatay, S. A. Haque, B. O’Regan, J. R. Durrant, W. J. H. Verhees, J. M. Kroon, A. Vidal-Ferran, P. Gaviña and E. Palomares, J. Mater. Chem., 2007, 17, 3037–3044 RSC.
- J. E. Kroeze, N. Hirata, S. Koops, M. K. Nazeeruddin, L. Schmidt-Mende, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2006, 128, 16376–16383 CrossRef CAS PubMed.
- U. Bach and T. Daeneke, Angew. Chem., Int. Ed., 2012, 51, 10451–10452 CrossRef CAS PubMed.
- U. B. Cappel, T. Daeneke and U. Bach, Nano Lett., 2012, 12, 4925–4931 CrossRef CAS PubMed.
- J. Krüger, R. Plass, L. Cevey, M. Piccirelli, M. Grätzel and U. Bach, Appl. Phys. Lett., 2001, 79, 2085–2087 CrossRef.
- U. B. Cappel, M. H. Karlsson, N. G. Pschirer, F. Eickemeyer, J. Schöneboom, P. Erk, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2009, 113, 14595–14597 CrossRef CAS.
- U. B. Cappel, A. L. Smeigh, S. Plogmaker, E. M. J. Johansson, H. Rensmo, L. Hammarström, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2011, 115, 4345–4358 CrossRef CAS.
- A. J. Mozer, D. K. Panda, S. Gambhir, B. Winther-Jensen and G. G. Wallace, J. Am. Chem. Soc., 2010, 132, 9543–9545 CrossRef CAS PubMed.
- U. Bach, Y. Tachibana, J.-E. Moser, S. A. Haque, J. R. Durrant, M. Grätzel and D. R. Klug, J. Am. Chem. Soc., 1999, 121, 7445–7446 CrossRef CAS.
- M. Freitag, J. Teuscher, Y. Saygili, X. Zhang, F. Giordano, P. Liska, J. Hua, S. M. S. M. S. M. S. M. S. M. Zakeeruddin, J.-E. J.-E. M. J.-E. J.-E. E. Moser, M. Grätzel and A. Hagfeldt, Nat. Photonics, 2017, 11, 372–378 CrossRef CAS.
- W. Zhang, Y. Wu, H. W. Bahng, Y. Cao, C. Yi, Y. Saygili, J. Luo, Y. Liu, L. Kavan, J. E. Moser, A. Hagfeldt, H. Tian, S. M. Zakeeruddin, W. Zhu and M. Grätzel, Energy Environ. Sci., 2018, 11, 1779–1787 RSC.
- Y. Cao, Y. Liu, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Joule, 2018, 2, 1108–1117 CrossRef CAS.
- M. Wolf and H. Rauschenbach, Adv. Energy Convers., 1963, 3, 455–479 CrossRef.
- D. Gentilini, D. D’Ercole, A. Gagliardi, A. Brunetti, A. Reale, T. Brown and A. Di Carlo, Superlattices Microstruct., 2010, 47, 192–196 CrossRef CAS.
- M. Freitag, Q. Daniel, M. Pazoki, K. Sveinbjörnsson, J. Zhang, L. Sun, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2015, 8, 2634–2637 RSC.
- J. J. Nelson, T. J. Amick and C. M. Elliott, J. Phys. Chem. C, 2008, 112, 18255–18263 CrossRef CAS.
- T. M. W. J. Bandara, W. J. M. J. S. R. Jayasundara, H. D. N. S. Fernado, M. A. K. L. Dissanayake, L. A. A. De Silva, I. Albinsson, M. Furlani and B. E. Mellander, J. Appl. Electrochem., 2015, 45, 289–298 CrossRef CAS.
- J. Bisquert, F. Fabregat-santiago, I. Mora-Seró, G. Garcia-Belmonte and S. Gimenez, J. Phys. Chem. C, 2009, 113, 17278–17290 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Inorg. Chim. Acta, 2008, 361, 729–734 CrossRef CAS.
- T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925–16928 CrossRef CAS PubMed.
- W. Yang, N. Vlachopoulos, Y. Hao, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2015, 17, 15868–15875 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- H. J. Queisser, Mater. Sci. Eng., B, 2009, 159–160, 322–328 CrossRef CAS.
- P. K. Nayak and D. Cahen, Adv. Mater., 2014, 26, 1622–1628 CrossRef CAS PubMed.
- L. Yang, B. Xu, D. Bi, H. Tian, G. Boschloo, L. Sun, A. Hagfeldt and E. M. J. Johansson, J. Am. Chem. Soc., 2013, 135, 7378–7385 CrossRef CAS PubMed.
- P. Liu, W. Sharmoukh, B. Xu, Y. Y. Li, G. Boschloo, L. Sun and L. Kloo, ACS Omega, 2017, 2, 1812–1819 CrossRef CAS.
- L. Yang, U. B. Cappel, E. L. Unger, M. Karlsson, K. M. Karlsson, E. Gabrielsson, L. Sun, G. Boschloo, A. Hagfeldt and E. M. J. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 779–789 RSC.
- B. Peng, G. Jungmann, C. Jäger, D. Haarer, H.-W. Schmidt and M. Thelakkat, Coord. Chem. Rev., 2004, 248, 1479–1489 CrossRef CAS.
- J. Park and M. Lee, Electron. Mater. Lett., 2015, 11, 271–275 CrossRef CAS.
- H. Choi, C. Nahm, J. Kim, J. Moon, S. Nam, D.-R. Jung and B. Park, Curr. Appl. Phys., 2012, 12, 737–741 CrossRef.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- H. Yan, X. Wang, M. Yao and X. Yao, Prog. Nat. Sci.: Mater. Int., 2013, 23, 402–407 CrossRef.
- A. Wypych, I. Bobowska, M. Tracz, A. Opasinska, S. Kadlubowski, A. Krzywania-Kaliszewska, J. Grobelny and P. Wojciechowski, J. Nanomater., 2014, 2014, 124814 Search PubMed.
- S. Hore, C. Vetter, R. Kern, H. Smit and A. Hinsch, Sol. Energy Mater. Sol. Cells, 2006, 90, 1176–1188 CrossRef CAS.
- J. Wang and Y. Shang, Appl. Phys. Lett., 2013, 102, 143113 CrossRef.
- W. Maiaugree, S. Pimanpang, W. Jarernboon and V. Amornkitbamrung, Int. J. Photoenergy, 2016, 2016, 1–10 CrossRef.
- E. J. W. Crossland, M. Kamperman, M. Nedelcu, C. Ducati, U. Wiesner, D.-M. Smilgies, G. E. S. Toombes, M. A. Hillmyer, S. Ludwigs, U. Steiner and H. J. Snaith, Nano Lett., 2009, 9, 2807–2812 CrossRef CAS PubMed.
- P. Docampo, S. Guldin, M. Stefik, P. Tiwana, M. C. Orilall, S. Hüttner, H. Sai, U. Wiesner, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2010, 20, 1787–1796 CrossRef CAS.
- E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215–219 CrossRef CAS PubMed.
- B. O’Regan, D. T. Schwartz, S. M. Zakeeruddin and M. Grätzel, Adv. Mater., 2000, 12, 1263–1267 CrossRef.
- N. O. V. Plank, H. J. Snaith, C. Ducati, J. S. Bendall, L. Schmidt-Mende and M. E. Welland, Nanotechnology, 2008, 19, 465603 CrossRef CAS PubMed.
- C. Xu, J. Wu, U. V. Desai and D. Gao, Nano Lett., 2012, 12, 2420–2424 CrossRef CAS PubMed.
- K. Tennakone, V. P. S. Perera, I. R. M. Kottegoda, L. A. A. De Silva, G. R. R. A. Kumara and A. Konno, J. Electron. Mater., 2001, 30, 992–996 CrossRef CAS.
- P. Docampo and H. J. Snaith, Nanotechnology, 2011, 22, 225403 CrossRef PubMed.
- B. C. O’Regan, S. Scully, A. C. Mayer, E. Palomares and J. Durrant, J. Phys. Chem. B, 2005, 109, 4616–4623 CrossRef PubMed.
- S. Suresh, G. E. Unni, M. Satyanarayana, A. Sreekumaran Nair and V. P. Mahadevan Pillai, J. Colloid Interface Sci., 2018, 524, 236–244 CrossRef CAS PubMed.
- L. Li, Q. Lu, W. Li, X. Li, A. Hagfeldt, W. Zhang and M. Wu, J. Power Sources, 2016, 308, 37–43 CrossRef CAS.
- P. Jayabal, V. Sasirekha, J. Mayandi, K. Jeganathan and V. Ramakrishnan, J. Alloys Compd., 2014, 586, 456–461 CrossRef CAS.
- M. Saito and S. Fujihara, Energy Environ. Sci., 2008, 1, 280–283 RSC.
- J. Bouclé and J. Ackermann, Polym. Int., 2012, 61, 355–373 CrossRef.
- B. O’Regan, F. Lenzmann, R. Muis and J. Wienke, Chem. Mater., 2002, 14, 5023–5029 CrossRef.
- W.-Y. Rho, D. H. Song, H.-Y. Yang, H.-S. Kim, B. S. Son, J. S. Suh and B.-H. Jun, J. Solid State Chem., 2018, 258, 271–282 CrossRef CAS.
- M. D. Brown, T. Suteewong, R. S. S. Kumar, V. D’Innocenzo, A. Petrozza, M. M. Lee, U. Wiesner and H. J. Snaith, Nano Lett., 2011, 11, 438–445 CrossRef CAS PubMed.
- M. H. Buraidah, L. P. Teo, C. M. Au Yong, S. Shah and A. K. Arof, Opt. Mater., 2016, 57, 202–211 CrossRef CAS.
- C. T. Yip, H. Huang, L. Zhou, K. Xie, Y. Wang, T. Feng, J. Li and W. Y. Tam, Adv. Mater., 2011, 23, 5624–5628 CrossRef CAS PubMed.
- S. Guldin, S. Hüttner, M. Kolle, M. E. Welland, P. Müller-Buschbaum, R. H. Friend, U. Steiner and N. Tétreault, Nano Lett., 2010, 10, 2303–2309 CrossRef CAS PubMed.
- B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C.-Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. H. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379–6381 RSC.
- K. Kakiage, T. Tokutome, S. Iwamoto, T. Kyomen and M. Hanaya, Chem. Commun., 2013, 49, 179–180 RSC.
- T. T. T. Pham, S. K. Saha, D. Provost, Y. Farré, M. Raissi, Y. Pellegrin, E. Blart, S. Vedraine, B. Ratier, D. Aldakov, F. Odobel and J. Bouclé, J. Phys. Chem. C, 2017, 121, 129–139 CrossRef CAS.
- X. Jiang, K. M. Karlsson, E. Gabrielsson, E. M. J. Johansson, M. Quintana, M. Karlsson, L. Sun, G. Boschloo and A. Hagfeldt, Adv. Funct. Mater., 2011, 21, 2944–2952 CrossRef CAS.
- Y. Hu, A. Abate, Y. Cao, A. Ivaturi, S. M. Zakeeruddin, M. Grätzel and N. Robertson, J. Phys. Chem. C, 2016, 120, 15027–15034 CrossRef CAS.
- H. Ellis, S. K. Eriksson, S. M. Feldt, E. Gabrielsson, P. W. Lohse, R. Lindblad, L. Sun, H. Rensmo, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2013, 117, 21029–21036 CrossRef CAS.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- J. Krüger, R. Plass, M. Grätzel and H.-J. Matthieu, Appl. Phys. Lett., 2002, 81, 367 CrossRef.
- L. Schmidt-Mende, S. M. Zakeeruddin and M. Grätzel, Appl. Phys. Lett., 2005, 86, 2001–2004 CrossRef.
- H. J. Snaith, S. M. Zakeeruddin, L. Schmidt-Mende, C. C. Klein, M. Grätzel, M. Grätzel and M. Grätzel, Angew. Chem., Int. Ed., 2005, 44, 6413–6417 CrossRef CAS PubMed.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- M. Wang, S.-J. J. Moon, D. Zhou, F. Le Formal, N.-L. L. Cevey-Ha, R. Humphry-Baker, C. Grätzel, P. Wang, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2010, 20, 1821–1826 CrossRef CAS.
- E. W.-G. Diau, Meet. Abstr., 2015, MA2015-01, 1012 Search PubMed.
- P. Qin, P. Sanghyun, M. I. Dar, K. Rakstys, H. ElBatal, S. A. Al-Muhtaseb, C. Ludwig and M. K. Nazeeruddin, Adv. Funct. Mater., 2016, 26, 5550–5559 CrossRef CAS.
- J.-W. Shiu, Y.-C. Chang, C.-Y. Chan, H.-P. Wu, H.-Y. Hsu, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, J. Mater. Chem. A, 2015, 3, 1417–1420 RSC.
- J. Liu, W. Zhou, J. Liu, Y. Fujimori, T. Higashino, H. Imahori, X. Jiang, J. Zhao, T. Sakurai, Y. Hattori, W. Matsuda, S. Seki, S. K. Garlapati, S. Dasgupta, E. Redel, L. Sun and C. Wöll, J. Mater. Chem. A, 2016, 4, 12739–12747 RSC.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–815 CrossRef CAS.
- S. Benhattab, R. Nakar, J. W. Rodriguez Acosta, N. Berton, J. Faure-Vincent, J. Bouclé, F. Tran Van and B. Schmaltz, Dyes Pigm., 2018, 151, 238–244 CrossRef CAS.
- B. Xu, H. Tian, D. Bi, E. Gabrielsson, E. M. J. Johansson, G. Boschloo, A. Hagfeldt and L. Sun, J. Mater. Chem. A, 2013, 1, 14467–14470 RSC.
- I. Bruder, M. Karlsson, F. Eickemeyer, J. Hwang, P. Erk, A. Hagfeldt, J. Weis and N. Pschirer, Sol. Energy Mater. Sol. Cells, 2009, 93, 1896–1899 CrossRef CAS.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799–3802 CrossRef CAS PubMed.
- R. Grisorio, L. De Marco, C. Baldisserri, F. Martina, M. Serantoni, G. Gigli and G. P. Suranna, ACS Sustainable Chem. Eng., 2015, 3, 770–777 CrossRef CAS.
- H. Ellis, I. Schmidt, A. Hagfeldt, G. Wittstock and G. Boschloo, J. Phys. Chem. C, 2015, 119, 21775–21783 CrossRef CAS.
- A. Mahmood, Sol. Energy, 2016, 123, 127–144 CrossRef CAS.
- J. Jia, Y. Chen, L. Duan, Z. Sun, M. Liang and S. Xue, RSC Adv., 2017, 7, 45807–45817 RSC.
- A. Konno, G. R. A. Kumara, S. Kaneko, B. Onwona-Agyeman and K. Tennakone, Chem. Lett., 2007, 36, 716–717 CrossRef CAS.
- S. Kolemen, Y. Cakmak, S. Erten-Ela, Y. Altay, J. Brendel, M. Thelakkat and E. U. Akkaya, Org. Lett., 2010, 12, 3812–3815 CrossRef CAS PubMed.
- N. Cai, S.-J. Moon, L. L. Cevey-Ha, T. Moehl, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin, M. Grätzel and M. Grätzel, Nano Lett., 2011, 11, 1452–1456 CrossRef CAS PubMed.
- A. Dualeh, F. De Angelis, S. Fantacci, T. Moehl, C. Yi, F. Kessler, E. Baranoff, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2012, 116, 1572–1578 CrossRef CAS.
- A. Dualeh, J. H. Delcamp, M. K. Nazeeruddin and M. Grätzel, Appl. Phys. Lett., 2012, 100, 173512 CrossRef.
- W. H. Nguyen, C. D. Bailie, J. Burschka, T. Moehl, M. Grätzel, M. D. McGehee and A. Sellinger, Chem. Mater., 2013, 25, 1519–1525 CrossRef CAS.
- A. Abate, M. Planells, D. J. Hollman, S. D. Stranks, A. Petrozza, A. R. S. Kandada, Y. Vaynzof, S. K. Pathak, N. Robertson and H. J. Snaith, Adv. Energy Mater., 2014, 4, 1400166 CrossRef.
- Z. Shen, B. Xu, P. Liu, Y. Hu, Y. Yu, H. Ding, L. Kloo, J. Hua, L. Sun and H. Tian, J. Mater. Chem. A, 2017, 5, 1242–1247 RSC.
- P. Liu, B. Xu, K. M. Karlsson, J. B. Zhang, N. Vlachopoulos, G. Boschloo, L. C. Sun and L. Kloo, J. Mater. Chem. A, 2015, 3, 4420–4427 RSC.
- M. Suzuka, N. Hayashi, T. Sekiguchi, K. Sumioka, M. Takata, N. Hayo, H. Ikeda, K. Oyaizu and H. Nishide, Sci. Rep., 2016, 6, 28022 CrossRef CAS PubMed.
- X. Lan, O. Voznyy, F. P. García de Arquer, M. Liu, J. Xu, A. H. Proppe, G. Walters, F. Fan, H. Tan, M. Liu, Z. Yang, S. Hoogland and E. H. Sargent, Nano Lett., 2016, 16, 4630–4634 CrossRef CAS PubMed.
- M. Liu, O. Voznyy, R. Sabatini, F. P. García de Arquer, R. Munir, A. H. Balawi, X. Lan, F. Fan, G. Walters, A. R. Kirmani, S. Hoogland, F. Laquai, A. Amassian and E. H. Sargent, Nat. Mater., 2017, 16, 258–263 CrossRef CAS PubMed.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef PubMed.
- M. Ye, X. Gao, X. Hong, Q. Liu, C. He, X. Liu and C. Lin, Sustain, Energy Fuels, 2017, 1, 1217–1231 CAS.
- W. Wang, W. Feng, J. Du, W. Xue, L. Zhang, L. Zhao, Y. Li and X. Zhong, Adv. Mater., 2018, 30, 1705746 CrossRef PubMed.
- A. N. Jumabekov, N. Cordes, T. D. Siegler, P. Docampo, A. Ivanova, K. Fominykh, D. D. Medina, L. M. Peter and T. Bein, ACS Appl. Mater. Interfaces, 2016, 8, 4600–4607 CrossRef CAS PubMed.
- J. P. Park, J. H. Heo, S. H. Im and S.-W. Kim, J. Mater. Chem. A, 2016, 4, 785–790 RSC.
- X. Zhang, J. Liu, J. Zhang, N. Vlachopoulos and E. M. J. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 12786–12795 RSC.
- J. Duan, Q. Tang, Y. Sun, B. He and H. Chen, RSC Adv., 2014, 4, 60478–60483 RSC.
- A. G. Kontos, T. Stergiopoulos, V. Likodimos, D. Milliken, H. Desilvesto, G. Tulloch and P. Falaras, J. Phys. Chem. C, 2013, 117, 8636–8646 CrossRef CAS.
- C. Law, S. C. Pathirana, X. Li, A. Y. Anderson, P. R. F. Barnes, A. Listorti, T. H. Ghaddar and B. C. O′Regan, Adv. Mater., 2010, 22, 4505–4509 CrossRef CAS PubMed.
- A. F. Nogueira, C. Longo and M.-A. A. De Paoli, Coord. Chem. Rev., 2004, 248, 1455–1468 CrossRef CAS.
- H. Klauk, Organic Electronics II: More Materials and Applications, Wiley, 2012 Search PubMed.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- J. Zhang, L. Häggman, M. Jouini, A. Jarboui, G. Boschloo, N. Vlachopoulos and A. Hagfeldt, ChemPhysChem, 2014, 15, 1043–1047 CrossRef CAS PubMed.
- P. V. Wright, Br. Polym. J., 1975, 7, 319–327 CrossRef CAS.
- C. Berthier, W. Gorecki, M. Minier, M. B. Armand, J. M. Chabagno and P. Rigaud, Solid State Ionics, 1983, 11, 91–95 CrossRef CAS.
- M. Armand, Solid State Ionics, 1983, 9–10, 745–754 CrossRef CAS.
- G. C. Farrington and J. L. Briant, Science, 1979, 204, 1371–1379 CrossRef CAS PubMed.
- M. Armand, Solid State Ionics, 1994, 69, 309–319 CrossRef CAS.
- M. B. Armand, M. J. Duclot and P. Rigaud, Solid State Ionics, 1981, 3–4, 429–430 CrossRef CAS.
- M. S. Michael, M. M. E. Jacob, S. R. S. Prabaharan and S. Radhakrishna, Solid State Ionics, 1997, 98, 167–174 CrossRef CAS.
- K. M. Abraham, Z. Jiang and B. Carroll, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS.
- A. F. Nogueira, N. Alonso-Vante and M.-A. De Paoli, Synth. Met., 1999, 105, 23–27 CrossRef CAS.
- J. Nei de Freitas, A. F. Nogueira and M.-A. De Paoli, J. Mater. Chem., 2009, 19, 5279–5294 RSC.
- T. Stergiopoulos, I. M. Arabatzis, G. Katsaros and P. Falaras, Nano Lett., 2002, 2, 1259–1261 CrossRef CAS.
- J. Wu, S. Hao, Z. Lan, J. Lin, M. Huang, Y. Huang, P. Li, S. Yin and T. Sato, J. Am. Chem. Soc., 2008, 130, 11568–11569 CrossRef CAS PubMed.
- J. Li, H. Wang, G. Zhou and Z.-S. Wang, Chem. Commun., 2013, 49, 9446–9448 RSC.
- F. Bella, N. N. Mobarak, F. N. Jumaah and A. Ahmad, Electrochim. Acta, 2015, 151, 306–311 CrossRef CAS.
- A. F. Nogueira, M. A. S. Spinacé, W. A. Gazotti, E. M. Girotto and M.-A. de Paoli, Solid State Ionics, 2001, 140, 327–335 CrossRef CAS.
- G. Katsaros, T. Stergiopoulos, I. M. Arabatzis, K. G. Papadokostaki and P. Falaras, J. Photochem. Photobiol., A, 2002, 149, 191–198 CrossRef CAS.
- E. Chatzivasiloglou, T. Stergiopoulos, N. Spyrellis and P. Falaras, J. Mater. Process. Technol., 2005, 161, 234–240 CrossRef CAS.
- J. H. Seo, A. Gutacker, Y. Sun, H. Wu, F. Huang, Y. Cao, U. Scherf, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 8416–8419 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, P. Comte, I. Exnar and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 1166–1167 CrossRef CAS PubMed.
- L. Fan, S. Kang, J. Wu, S. Hao, Z. Lan and J. Lin, Energy Sources, Part A, 2010, 32, 1559–1568 CrossRef CAS.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS PubMed.
- F. Bella, N. Vlachopoulos, K. Nonomura, S. M. Zakeeruddin, M. Gratzel, C. Gerbaldi, A. Hagfeldt, M. Grätzel, C. Gerbaldi, A. Hagfeldt, M. Gratzel, C. Gerbaldi, A. Hagfeldt, M. Grätzel, C. Gerbaldi, A. Hagfeldt, M. Gratzel, C. Gerbaldi and A. Hagfeldt, Chem. Commun., 2015, 51, 16308–16311 RSC.
- J. N. de Freitas, A. de, S. Gonçalves, M.-A. De Paoli, J. R. Durrant and A. F. Nogueira, Electrochim. Acta, 2008, 53, 7166–7172 CrossRef.
- W. Kubo, K. Murakoshi, T. Kitamura, S. Yoshida, M. Haruki, K. Hanabusa, H. Shirai, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2001, 105, 12809–12815 CrossRef CAS.
- F. Bella, J. R. Nair and C. Gerbaldi, RSC Adv., 2013, 3, 15993–16001 RSC.
- F. Bella and R. Bongiovanni, J. Photochem. Photobiol., C, 2013, 16, 1–21 CrossRef CAS.
- F. Bella, D. Pugliese, J. R. Nair, A. Sacco, S. Bianco, C. Gerbaldi, C. Barolo and R. Bongiovanni, Phys. Chem. Chem. Phys., 2013, 15, 3706–3711 RSC.
- J. R. Nair, C. Gerbaldi, G. Meligrana, R. Bongiovanni, S. Bodoardo, N. Penazzi, P. Reale and V. Gentili, J. Power Sources, 2008, 178, 751–757 CrossRef CAS.
- F. Bella, E. D. Ozzello, A. Sacco, S. Bianco and R. Bongiovanni, Int. J. Hydrogen Energy, 2014, 39, 3036–3045 CrossRef CAS.
- F. Bella, A. Sacco, D. Pugliese, M. Laurenti and S. Bianco, J. Power Sources, 2014, 264, 333–343 CrossRef CAS.
- F. Bella, R. Bongiovanni, R. S. Kumar, M. A. Kulandainathan and A. M. Stephan, J. Mater. Chem. A, 2013, 1, 9033–9036 RSC.
- A. Chiappone, F. Bella, J. R. Nair, G. Meligrana, R. Bongiovanni and C. Gerbaldi, ChemElectroChem, 2014, 1, 1350–1358 CrossRef CAS.
- C.-H. Tsai, C.-Y. Lu, M.-C. Chen, T.-W. Huang, C.-C. Wu and Y.-W. Chung, Org. Electron., 2013, 14, 3131–3137 CrossRef CAS.
- H.-S. Lee, C.-H. Han, Y.-M. Sung, S. S. Sekhon and K.-J. Kim, Curr. Appl. Phys., 2011, 11, S158–S162 CrossRef.
- K. S. Lee, Y. Jun and J. H. Park, Nano Lett., 2012, 12, 2233–2237 CrossRef CAS PubMed.
- F. Bella, A. Lamberti, A. Sacco, S. Bianco, A. Chiodoni and R. Bongiovanni, J. Membr. Sci., 2014, 470, 125–131 CrossRef CAS.
- G. P. Salvador, D. Pugliese, F. Bella, A. Chiappone, A. Sacco, S. Bianco and M. Quaglio, Electrochim. Acta, 2014, 146, 44–51 CrossRef CAS.
- Y. Li, H. Li, C. Zhong, G. Sini and J.-L. Brédas, npj Flex. Electron., 2017, 1, 2 CrossRef.
- I. Yavuz and K. N. Houk, J. Phys. Chem. C, 2017, 121, 993–999 CrossRef CAS.
- J. Melas-Kyriazi, I.-K. Ding, A. Marchioro, A. Punzi, B. E. Hardin, G. F. Burkhard, N. Tétreault, M. Grätzel, J.-E. Moser and M. D. McGehee, Adv. Energy Mater., 2011, 1, 407–414 CrossRef CAS.
- Y. Zhang, K. Cao, X. Zhu, X. Li, X. Qiao, G. Tu, B. Zhang, D. Huang, Y. Shen and M. Wang, RSC Adv., 2013, 3, 14037–14043 RSC.
- M. Rawolle, K. Sarkar, M. A. Niedermeier, M. Schindler, P. Lellig, J. S. Gutmann, J.-F. Moulin, M. Haese-Seiller, A. S. Wochnik, C. Scheu and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2013, 5, 719–729 CrossRef CAS PubMed.
- M. Juozapavicius, B. C. O’Regan, A. Y. Anderson, J. V. Grazulevicius and V. Mimaite, Org. Electron., 2012, 13, 23–30 CrossRef CAS.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N.-L. L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133, 18042–18045 CrossRef CAS PubMed.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. Mcgehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
- T. Malinauskas, D. Tomkute-Luksiene, R. Sens, M. Daskeviciene, R. Send, H. Wonneberger, V. Jankauskas, I. Bruder and V. Getautis, ACS Appl. Mater. Interfaces, 2015, 7, 11107–11116 CrossRef CAS PubMed.
- Y. Hua, J. Zhang, B. Xu, P. Liu, M. Cheng, L. Kloo, E. M. J. Johansson, K. Sveinbjörnsson, K. Aitola, G. Boschloo and L. Sun, Nano Energy, 2016, 26, 108–113 CrossRef CAS.
- D.-G. Ha, J.-J. Kim and M. A. Baldo, AIP Adv., 2016, 6, 045221 CrossRef.
- H. Uratani, S. Kubo, K. Shizu, F. Suzuki, T. Fukushima and H. Kaji, Sci. Rep., 2016, 6, 39128 CrossRef CAS PubMed.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- G. Sathiyan, E. K. T. Sivakumar, R. Ganesamoorthy, R. Thangamuthu and P. Sakthivel, Tetrahedron Lett., 2016, 57, 243–252 CrossRef CAS.
- M. Degbia, B. Schmaltz, J. Bouclé, J. V. Grazulevicius, F. Tran-Van and F. Tran-Van, Polym. Int., 2014, 63, 1387–1393 CrossRef CAS.
- B. Xu, E. Sheibani, P. Liu, J. Zhang, H. Tian, N. Vlachopoulos, G. Boschloo, L. Kloo, A. Hagfeldt and L. Sun, Adv. Mater., 2014, 26, 6629–6634 CrossRef CAS PubMed.
- M. Degbia, M. Ben Manaa, B. Schmaltz, N. Berton, J. Bouclé, R. Antony and F. Tran Van, Mater. Sci. Semicond. Process., 2016, 43, 90–95 CrossRef CAS.
- T.-T. Bui, S. K. Shah, M. Abbas, X. Sallenave, G. Sini, L. Hirsch and F. Goubard, ChemNanoMat, 2015, 1, 203–210 CrossRef CAS.
- A. Tomkeviciene, G. Puckyte, J. V. Grazulevicius, M. Degbia, F. Tran-Van, B. Schmaltz, V. Jankauskas and J. Bouclé, Synth. Met., 2012, 162, 1997–2004 CrossRef CAS.
- R. Lygaitis, B. Schmaltz, R. Degutyte, J. V. Gražulevičius, M. Degbia, F. T. Van, P. Strohriegl, V. Jankauskas and J. Bouclé, Synth. Met., 2014, 195, 328–334 CrossRef CAS.
- M. Planells, A. Abate, D. J. Hollman, S. D. Stranks, V. Bharti, J. Gaur, D. Mohanty, S. Chand, H. J. Snaith and N. Robertson, J. Mater. Chem. A, 2013, 1, 6949–6960 RSC.
- P. Liu, B. Xu, Y. Hua, M. Cheng, K. Aitola, K. Sveinbjörnsson, J. Zhang, G. Boschloo, L. Sun and L. Kloo, J. Power Sources, 2017, 344, 11–14 CrossRef CAS.
- W. Yuan, H. Zhao and G. L. Baker, Org. Electron., 2014, 15, 3362–3369 CrossRef CAS.
- R. K. Aulakh, S. Sandhu, Tanvi, S. S. Kumar, A. Mahajan, R. K. Bedi and S. S. Kumar, Synth. Met., 2015, 205, 92–97 CrossRef CAS.
- T.-T. Bui, S. K. Shah, X. Sallenave, M. Abbas, G. Sini, L. Hirsch and F. Goubard, RSC Adv., 2015, 5, 49590–49597 RSC.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt, L. Sun, M. Gratzel, L. Kloo, A. Hagfeldt, L. Sun, M. Grätzel, L. Kloo, A. Hagfeldt, L. Sun, M. Gratzel, L. Kloo, A. Hagfeldt, L. Sun, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- B. Kim, J. K. Koh, J. H. J. J. H. J. Kim, W. S. Chi, J. H. J. J. H. J. Kim and E. Kim, ChemSusChem, 2012, 5, 2173–2180 CrossRef CAS PubMed.
- J. Zhang, L. Yang, Y. Shen, B.-W. W. Park, Y. Hao, E. M. J. Johansson, G. Boschloo, L. Kloo, E. Gabrielsson, L. Sun, A. Jarboui, C. Perruchot, M. Jouini, N. Vlachopoulos and A. Hagfeldt, J. Phys. Chem. C, 2014, 118, 16591–16601 CrossRef CAS.
- J. Zhang, N. Vlachopoulos, Y. Hao, T. W. Holcombe, G. Boschloo, E. M. J. J. Johansson, M. Grätzel and A. Hagfeldt, ChemPhysChem, 2016, 17, 1441–1445 CrossRef CAS PubMed.
- W.-C. Chen, Y.-H. Lee, C.-Y. Chen, K.-C. Kau, L.-Y. Lin, C.-A. Dai, C.-G. Wu, K.-C. Ho, J.-K. Wang and L. Wang, ACS Nano, 2014, 8, 1254–1262 CrossRef CAS PubMed.
- N. Zhou, B. Lee, A. Timalsina, P. Guo, X. Yu, T. J. Marks, A. Facchetti and R. P. H. Chang, J. Phys. Chem. C, 2014, 118, 16967–16975 CrossRef CAS.
- Q. Liu, C. Li, K. Jiang, Y. Song and J. Pei, Particuology, 2014, 15, 71–76 CrossRef CAS.
- M. Chevrier, H. Hawashin, S. Richeter, A. Mehdi, M. Surin, R. Lazzaroni, P. Dubois, B. Ratier, J. Bouclé and S. Clément, Synth. Met., 2017, 226, 157–163 CrossRef CAS.
- P. Liu, L. Kloo and J. M. Gardner, ChemPhotoChem, 2017, 1, 363–368 CrossRef CAS.
- L. Peedikakkandy and P. Bhargava, Mater. Sci. Semicond. Process., 2015, 33, 103–109 CrossRef CAS.
- H. Sakamoto, S. Igarashi, M. Uchida, K. Niume and M. Nagai, Org. Electron., 2012, 13, 514–518 CrossRef CAS.
- M. Nur Amalina and M. Mohammad Rusop, Adv. Mater. Res., 2013, 667, 317–323 Search PubMed.
- E. V. A. Premalal, N. Dematage, G. R. R. A. Kumara, R. M. G. Rajapakse, M. Shimomura, K. Murakami and A. Konno, J. Power Sources, 2012, 203, 288–296 CrossRef CAS.
- K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed.
- M. Wałęsa-Chorab, R. Banasz, D. Marcinkowski, M. Kubicki and V. Patroniak, RSC Adv., 2017, 7, 50858–50867 RSC.
- Z. Wei, J. Fan, C. Dai, Z. Pang and S. Han, ACS Omega, 2018, 3, 6874–6879 CrossRef CAS.
- A. Yella, H.-W. H.-W. H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. K. Nazeeruddin, E. W.-G. W.-G. W.-G. Diau, C.-Y. C.-Y. C.-Y. C.-Y. Yeh, S. M. Zakeeruddin, M. Grätzel, M. Gratzel and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- M. K. Kashif, R. A. Milhuisen, M. Nippe, J. Hellerstedt, D. Z. Zee, N. W. Duffy, B. Halstead, F. De Angelis, S. Fantacci, M. S. Fuhrer, C. J. Chang, Y.-B. Cheng, J. R. Long, L. Spiccia and U. Bach, Adv. Energy Mater., 2016, 6, 1600874 CrossRef.
- M. K. Kashif, M. Nippe, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 5527–5531 CrossRef CAS PubMed.
- J.-Y. Kim, J. Y. Kim, D.-K. Lee, B. Kim, H. Kim and M. J. Ko, J. Phys. Chem. C, 2012, 116, 22759–22766 CrossRef CAS.
- L. Yang, R. Lindblad, E. Gabrielsson, G. Boschloo, H. Rensmo, L. Sun, A. Hagfeldt, T. Edvinsson and E. M. J. Johansson, ACS Appl. Mater. Interfaces, 2018, 10, 11572–11579 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- J. Krüger, R. Plass, M. Grätzel, P. J. Cameron and L. M. Peter, J. Phys. Chem. B, 2003, 107, 7536–7539 CrossRef.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS PubMed.
- E. J. Juarez-Perez, M. R. Leyden, S. Wang, L. K. Ono, Z. Hawash and Y. Qi, Chem. Mater., 2016, 28, 5702–5709 CrossRef CAS.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- J. Luo, J. Xia, H. Yang, L. Chen, Z. Wan, F. Han, H. A. Malik, X. Zhu and C. Jia, Energy Environ. Sci., 2018, 11, 2035–2045 RSC.
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H.-P. Steinrück, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358, 1192–1197 CrossRef CAS PubMed.
- J. Burschka, F. Kessler, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2013, 25, 2986–2990 CrossRef CAS.
- D.-Y. Chen, W.-H. Tseng, S.-P. Liang, C.-I. Wu, C.-W. Hsu, Y. Chi, W.-Y. Hung and P.-T. Chou, Phys. Chem. Chem. Phys., 2012, 14, 11689–11694 RSC.
- M. Xu, Y. Rong, Z. Ku, A. Mei, X. Li and H. Han, J. Phys. Chem. C, 2013, 117, 22492–22496 CrossRef CAS.
- B. Xu, E. Gabrielsson, M. Safdari, M. Cheng, Y. Hua, H. Tian, J. M. Gardner, L. Kloo and L. Sun, Adv. Energy Mater., 2015, 5, 1402340 CrossRef.
- X. Yang, W. Wang, Y. Zhang and L. Sun, Sol. Energy, 2018, 170, 1001–1008 CrossRef CAS.
- W. Wang, X. Yang, J. Li, H. Wang, J. An, L. Zhang, X. Jiang, Z. Yu and L. Sun, Energy Technol., 2018, 6, 752–758 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- Z. Tang, J. Wu, M. Zheng, J. Huo and Z. Lan, Nano Energy, 2013, 2, 622–627 CrossRef CAS.
- K. Mohan, A. Bora, B. C. Nath, P. Gogoi, B. J. Saikia and S. K. Dolui, Electrochim. Acta, 2016, 222, 1072–1078 CrossRef CAS.
- D. Qin, Y. Zhang, S. Huang, Y. Luo, D. Li and Q. Meng, Electrochim. Acta, 2011, 56, 8680–8687 CrossRef CAS.
- M. Wang, S.-J. Moon, M. Xu, K. Chittibabu, P. Wang, N.-L. Cevey-Ha, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Small, 2010, 6, 319–324 CrossRef CAS PubMed.
- J. Zhang, M. Freitag, A. Hagfeldt and G. Boschloo, Molecular Devices for Solar Energy Conversion and Storage, 2018, pp. 151–185 Search PubMed.
- Y. F. Chiang, C. H. Tsai, P. Chen and T. F. Guo, Sol. Energy, 2012, 86, 1967–1972 CrossRef CAS.
- Y.-F. F. Chiang, R.-T. T. Chen, A. Burke, U. Bach, P. Chen and T.-F. F. Guo, Renewable Energy, 2013, 59, 136–140 CrossRef CAS.
- K. Aitola, J. Zhang, N. Vlachopoulos, J. Halme, A. Kaskela, A. G. Nasibulin, E. I. Kauppinen, G. Boschloo and A. Hagfeldt, J. Solid State Electrochem., 2015, 19, 3139–3144 CrossRef CAS.
- J. Zhang, J. Feng, Y. Hong, Z. Hu, K. Xia, P. Wang and Y. Zhu, J. Renewable Sustainable Energy, 2014, 6, 043116 CrossRef.
- G. Y. Margulis, M. G. Christoforo, D. Lam, Z. M. Beiley, A. R. Bowring, C. D. Bailie, A. Salleo and M. D. McGehee, Adv. Energy Mater., 2013, 3, 1657–1663 CrossRef CAS.
- G. R. A. Kumara, A. Konno, K. Shiratsuchi, J. Tsukahara and K. Tennakone, Chem. Mater., 2002, 14, 954–955 CrossRef CAS.
- I. Celik, A. B. Philips, Z. Song, Y. Yan, R. J. Ellingson, M. J. Heben and D. Apul, IEEE J. Photovoltaics, 2018, 8, 305–309 Search PubMed.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 RSC.
- Cincinnati Sub-Zero Industrial, Photovoltaic Module Solar Panel Environmental Testing Guide, 2016 Search PubMed.
- Fraunhofer Institute for Solar Energy Systems, PV Module certification for new standards and new technologies, 2016 Search PubMed.
- S. Jain and V. Agarwal, IEEE Power Electron. Lett., 2004, 2, 16–19 CrossRef.
- W. Yang, Y. Hao, P. Ghamgosar and G. Boschloo, Electrochim. Acta, 2016, 213, 879–886 CrossRef CAS.
- R. Jiang and G. Boschloo, Inorganics, 2018, 6, 60 CrossRef.
- M. Flasque, A. N. Van Nhien, J. Swiatowska, A. Seyeux, C. Davoisne and F. Sauvage, ChemPhysChem, 2014, 15, 1126–1137 CrossRef CAS PubMed.
- D. Bari, N. Wrachien, G. Meneghesso and C. Andrea, Reliab. Phys. Symp. (IRPS), 2013 IEEE Int., 2013, pp. 4B.3.1–4B.3.7.
- J. M. Kroon, N. J. Bakker, H. J. P. Smit, P. Liska, K. R. Thampi, P. Wang, S. M. Zakeeruddin, M. Grätzel, A. Hinsch, S. Hore, U. Würfel, R. Sastrawan, J. R. Durrant, E. Palomares, H. Pettersson, T. Gruszecki, J. Walter, K. Skupien and G. E. Tulloch, Prog. Photovoltaics Res. Appl., 2007, 15, 1–18 CrossRef CAS.
- S. Yu, S. Ahmadi, C. Sun, P. T. Z. Adibi, W. Chow, A. Pietzsch and M. Göthelid, J. Chem. Phys., 2012, 136, 154703 CrossRef PubMed.
- K. Schwanitz, U. Weiler, R. Hunger, T. Mayer and W. Jaegermann, J. Phys. Chem. C, 2007, 111, 849–854 CrossRef CAS.
- A. K. Jena, Y. Numata, M. Ikegami and T. Miyasaka, J. Mater. Chem. A, 2018, 6, 2219–2230 RSC.
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013–4030 CrossRef CAS PubMed.
- X.-T. Zhang, T. Taguchi, H.-B. Wang, Q.-B. Meng, O. Sato and A. Fujishima, Res. Chem. Intermed., 2007, 33, 5–11 CrossRef CAS.
- T. He, Y. F. Wang and J. H. Zeng, J. Mater. Chem. C, 2016, 4, 8235–8244 RSC.
- S.-H. Park, J. Lim, I. Y. Song, J.-R. Lee and T. Park, Adv. Energy Mater., 2014, 4, 1300489 CrossRef.
- C. P. Lee, P. Y. Chen, R. Vittal and K. C. Ho, J. Mater. Chem., 2010, 20, 2356–2361 RSC.
- L. Tao, Z. Huo, Y. Ding, Y. Li, S. Dai, L. Wang, J. Zhu, X. Pan, B. Zhang, J. Yao, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem. A, 2015, 3, 2344–2352 RSC.
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Sol. Energy Mater. Sol. Cells, 2015, 138, 86–95 CrossRef CAS.
- F. H. Alharbi and S. Kais, Renewable Sustainable Energy Rev., 2015, 43, 1073–1089 CrossRef.
- G. Xie, J. Lin, J. Wu, Z. Lan, Q. Li, Y. Xiao, G. Yue, H. Yue and M. Huang, Chin. Sci. Bull., 2011, 56, 96–101 CrossRef CAS.
- L. Liang, Y. Yulin, Z. Mi, F. Ruiqing, Q. LeLe, W. Xin, Z. Lingyun, Z. Xuesong and H. Jianglong, J. Solid State Chem., 2013, 198, 459–465 CrossRef.
- J. S. Lissau, J. M. Gardner and A. Morandeira, J. Phys. Chem. C, 2011, 115, 23226–23232 CrossRef CAS.
- C. P. Ponce, R. P. Steer and M. F. Paige, Photochem. Photobiol. Sci., 2013, 12, 1079–1085 RSC.
- P. Tiwana, P. Docampo, M. B. Johnston, H. J. Snaith and L. M. Herz, ACS Nano, 2011, 5, 5158–5166 CrossRef CAS PubMed.
- A. K. Chandiran, M. Abdi-Jalebi, M. K. Nazeeruddin and M. Grätzel, ACS Nano, 2014, 8, 2261–2268 CrossRef CAS PubMed.
- A. Kay and M. Grätzel, Chem. Mater., 2002, 14, 2930–2935 CrossRef CAS.
- B. Onwona-Agyeman, S. Kaneko, A. Kumara, M. Okuya, K. Murakami, A. Konno and K. Tennakone, Jpn. J. Appl. Phys., Part 2, 2005, 44, L731–L733 CrossRef CAS.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- L. D’Amario, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. C, 2014, 118, 19556–19564 CrossRef.
- L. D’Amario, L. J. Antila, B. Pettersson Rimgard, G. Boschloo and L. Hammarström, J. Phys. Chem. Lett., 2015, 6, 779–783 CrossRef PubMed.
- L. Zhang, G. Boschloo, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2016, 18, 5080–5085 RSC.
- L. Tian, J. Föhlinger, P. B. Pati, Z. Zhang, J. Lin, W. Yang, M. Johansson, T. Kubart, J. Sun, G. Boschloo, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2018, 20, 36–40 RSC.
- L. Tian, J. Föhlinger, Z. Zhang, P. B. Pati, J. Lin, T. Kubart, Y. Hua, J. Sun, L. Kloo, G. Boschloo, L. Hammarström and H. Tian, Chem. Commun., 2018, 54, 3739–3742 RSC.
- T. J. Jacobsson, J. P. Correa-Baena, M. Pazoki, M. Saliba, K. Schenk, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1706–1724 RSC.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- M. Dürr, A. Bamedi, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2004, 84, 3397–3399 CrossRef.
- W. Kubo, A. Sakamoto, T. Kitamura, Y. Wada and S. Yanagida, J. Photochem. Photobiol., A, 2004, 164, 33–39 CrossRef CAS.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, J. Fortage, G. Boschloo, E. Blart, Y. Pellegrin, F. Odobel, A. Hagfeldt and L. Hammarström, Angew. Chem., Int. Ed., 2009, 48, 4402–4405 CrossRef CAS PubMed.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- N. Papageorgiou, M. Grätzel and P. P. Infelta, Sol. Energy Mater. Sol. Cells, 1996, 44, 405–438 CrossRef CAS.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. A. B. A. B. A. B. Munoz-García, S. M. S. M. S. M. S. M. S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. J.-E. E. Moser, M. Grätzel, A. Hagfeldt, M. Freitag, A. B. Munoz-Garcia, S. M. S. M. S. M. S. M. S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. J.-E. E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CrossRef CAS PubMed.
- T. Förster, Naturwissenschaften, 1946, 33, 166–175 CrossRef.
- G. K. Mor, J. Basham, M. Paulose, S. Kim, O. K. Varghese, A. Vaish, S. Yoriya and C. A. Grimes, Nano Lett., 2010, 10, 2387–2394 CrossRef CAS PubMed.
- E. L. Unger, A. Morandeira, M. Persson, B. Zietz, E. Ripaud, P. Leriche, J. Roncali, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2011, 13, 20172 RSC.
- S. J. Moon, E. Baranoff, S. M. Zakeeruddin, C. Y. Yeh, E. W. G. Diau, M. Grätzel and K. Sivula, Chem. Commun., 2011, 47, 8244–8246 CAS.
| This journal is © The Royal Society of Chemistry 2018 |