
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of molecular conformation on the photophysics of organic room temperature phosphorescent luminophores†
Rongjuan
Huang
a,
Jonathan S.
Ward
b,
Nadzeya A.
Kukhta
b,
João
Avó
c,
Jamie
Gibson
d,
Thomas
Penfold
*d,
João C.
Lima
 e,
Andrei S.
Batsanov
b,
Mário N.
Berberan-Santos
c,
Martin R.
Bryce
b and
Fernando B.
Dias
*a
e,
Andrei S.
Batsanov
b,
Mário N.
Berberan-Santos
c,
Martin R.
Bryce
b and
Fernando B.
Dias
*a
aPhysics Department, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: f.m.b.dias@durham.ac.uk
bChemistry Department, Durham University, South Road, Durham, DH1 3LE, UK
cCQFM-IN and IBB-Institute for Bioengineering and Biosciences, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal
dChemistry-School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: tom.penfold@newcastle.ac.uk
eREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
First published on 17th August 2018
Abstract
A series of novel donor–acceptor–donor (D–A–D) compounds featuring dibenzothiophene (DBT) and phenothiazine (PTZ) units are presented. A different degree of steric hindrance between the donor and acceptor fragments is achieved by the systematic changes of donor substituents (methyl, iso-propyl, tert-butyl groups). This leads to the tuning of photophysical properties by conformational control. The unsubstituted DPTZ–DBT molecule exists in both equatorial and axial forms in the ground state, due to the ability of PTZ to form H-extra and H-intra folded conformers that allow formation of parallel quasi-axial (ax) and perpendicular quasi-equatorial (eq) conformers, respectively. However, the equatorial conformer prevails in the excited state. This leads to strong room temperature phosphorescence (RTP) in the green region with high phosphorescence quantum yield (60 ± 8%). Under the influence of bulky substituents, the alkyl-DPTZ–DBT derivatives change molecular conformation, preventing formation of the excited charge transfer state. Hence, blue, but much weaker, phosphorescence is observed. The less bulky methyl substituent on the donor results in dual RTP (blue and green), apparently violating Kasha's rule imposed by the modulation of the barriers between excited states. The experimental results are supported by DFT calculations in the ground and excited state. Control of conformation with substituents is an effective strategy for tuning the excited state properties of D–A–D molecules for RTP emission.
Introduction
Room-temperature phosphorescent (RTP) emitters have attracted great attention in recent years due to their potential for optoelectronic and photonic applications, such as in organic light-emitting diodes (OLEDs),1–4 luminescence labels,5 imaging,6 sensing7,8 and optical thermometry.9,10 The emission from the triplet excited state involves a quantum mechanically forbidden spin flip, which is uncompetitive with the non-radiative relaxation via thermal and collisional processes.11 The triplet state is, therefore, highly sensitive to temperature and strongly affected by the presence of molecular oxygen. However, achieving efficient RTP is possible when the intersystem crossing (ISC) from the lowest excited singlet (S1) to the triplet state (T1) is enhanced, and the non-radiative relaxation from T1 to the ground state due to molecular vibrations is suppressed.3Efficient RTP is obtained in organic–metal complexes due to the presence of heavy metals or halogens.12 In these compounds the spin–orbit coupling (SOC) is enhanced due to the effect of heavy-atoms promoting ISC by mixing the molecular orbital configurations of singlet and triplet states. However, halogen–carbon bonds are weak and prone to photolytic cleavage, which could lead to less stable emitters. Additionally, RTP has been reported in compounds that do not contain heavy-metals, by using additional strategies to reduce vibrational quenching of the triplet population.13 This is typically achieved by using rigid hosts, molecular crystals and molecular aggregation.14,15 However, molecular design strategies to enhance ISC and RTP in metal-free molecules are still limited and relatively inefficient compared to heavy metals. This reflects the lack of detailed understanding of how alterations of molecular conformation, nuclei vibrations and substituents may affect the radiative rate and the non-radiative internal conversion (IC), and ISC rates, as well as the corresponding yields for these processes. This information is relevant not only for RTP molecules, but also for emitters showing thermally activated delayed fluorescence (TADF), which are now routinely used in OLEDs.16,17 In the latter, controlling the ISC and reverse ISC (RISC) rates has direct impact on device efficiency and stability. Fast ISC/RISC rates are preferable to promote efficient triplet harvesting and obtain shorter-lived excited states.18 This is key to suppressing the efficiency roll-off observed in OLEDs.19,20 Detailed investigations of molecular structure–property relationships are thus needed to guide synthetic strategies for the design of more efficient RTP and TADF emitters.
Herein is reported the synthesis, computation and photophysics of four donor–acceptor–donor (D–A–D) metal-free, and halogen-free, organic molecules, which are based on phenothiazine (PTZ) and dibenzothiophene (DBT) moieties as the electron donor (D) and acceptor (A) units, respectively. These are the unsubstituted parent molecule, DPTZ–DBT, and the three analogues, DPTZ–Me–DBT, DPTZ–iPr–DBT, and DPTZ–tBu–DBT, substituted with methyl, iso-propyl and tert-butyl groups at the C1-position of each PTZ unit. These molecules show profoundly different excited state properties due to the different conformations (axial vs. equatorial)21 imposed by the effect of steric hindrance around the D–A bonds. These equatorial and axial conformers have been identified recently in related compounds, showing different RTP and TADF responses.22
Results and discussion
Molecular design, synthesis and structures
Fig. 1 shows the molecular structures of the four D–A–D compounds studied in this work. These molecules were rationally designed based on the following considerations. (i) PTZ is a donor unit in many luminescent materials,23–26 and emits phosphorescence at room temperature in solid zeonex films; (ii) DBT is also a standard luminophore, although usually serving as an electron donor in combination with acceptor units, e.g. fluorene,27,28 anthracene,29 or benzoyl.30 Its use as an acceptor in the present series of compounds is, therefore, unusual, although there is precedent with the bis((3,6-di-t-butyl)N-carbazolyl) derivative reported previously.3,31 The DBT unit has been previously identified as promoting fast RISC;3 (iii) these four molecules are direct analogues of the dibenzothiophene-S,S-dioxide series which possess an unusual combination of TADF and RTP.32 The key distinction with the present series is the different oxidation state of the thiophene sulfur (S in the present work; SO2 in the previous study).32 The SO2 unit imparts strong acceptor properties with a much lower LUMO and increased charge-transfer properties compared to the present dibenzothiophene series. The 2,8-disubstitution of DBT (PTZpara to the sulfur) was chosen to maximize the interaction of the D and A units, compared to the isomeric 3,7-derivatives (PTZ units meta to sulfur);3,31,33 (iv) both PTZ and DBT units on their own have potential to show strong triplet formation properties, due to the presence of the sulfur and nitrogen atoms;3,32 (v) the increasing steric hindrance imparted by the alkyl substituents on the PTZ units serves as a probe of steric effects on the conformation of the D–A–D system and its effect on the ISC/RISC rates as discussed in more detail below. | ||
| Fig. 1 Molecular structures of the compounds studied in this work. | ||
The syntheses of DPTZ–DBT, DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT are presented in the ESI.† Their structures and high purity were unambiguously established by a combination of NMR spectroscopy, mass spectrometry, elemental analysis and single-crystal X-ray analysis (ESI†).
The X-ray molecular structures of DPTZ–DBT, DPTZ–Me–DBT and DPTZ–iPr–DBT revealed interesting conformational properties of the molecules in the solid state. The structures of DPTZ–DBT and DPTZ–iPr–DBT show that the PTZ units are exclusively in equatorial and axial conformations, respectively, see Fig. 2. This is in agreement with the calculations of the relative energies of each conformer for all 4 molecules shown in Table S1 (ESI†). However, in the polymorphic structures of DPTZ–Me–DBT there is conformational disorder of the PTZ units. This is consistent with the dual phosphorescence observed in DPTZ–Me–DBT which is ascribed to the low energy barrier to subtle molecular vibrations (see discussion below). Additional figures, discussion and tables of dihedral angles in the X-ray structures are shown in Section S6 (ESI†).
 | ||
| Fig. 2 X-ray molecular structures of DPTZ–DBT·CDCl3 (left) and DPTZ–iPr–DBT (right) showing equatorial and axial PTZ groups, respectively. Thermal ellipsoids are drawn at the 50% probability level; H atoms are omitted; primed atoms are generated by the twofold axis. | ||
Photophysics
In Fig. 3a the absorption and emission spectra of DPTZ–DBT and DPTZ–iPr–DBT are compared in toluene solution. The absorption spectrum of DPTZ–iPr–DBT shows three well-defined transitions. These consist of low energy bands around 380 nm and 320 nm, and a higher energy transition below 300 nm, which is not entirely observed in toluene, due to the solvent cut-off. Similar bands are observed in the absorption spectra of the other substituted molecules, DPTZ–Me–DBT and DPTZ–tBu–DBT (Fig. S2, ESI†). The onset of the absorption spectrum of the unsubstituted DPTZ–DBT is around 400 nm, with the absorption intensity growing smoothly, up to the point where the peak of the lower energy band at 320 nm is reached. A second absorption band is also observed below 300 nm, as in the substituted molecules. However, in clear contrast with the substituted molecules, no well-defined band peaking at 380 nm is observed in DPTZ–DBT. The two transitions at 320 nm and below 300 nm, also identified in the substituted compounds, are the signatures of the individual PTZ and DBT units, respectively, as shown in Fig. S2 (ESI†). The low energy band is observed only in the absorption spectra of the substituted molecules and not in the spectra of either PTZ or DBT. This low energy band is also not a charge transfer (CT) transition, as the CT state is clearly not formed in the substituted compounds (see Fig. S3, ESI†). Instead, as shown by the HOMO and LUMO plots in Fig. 4, this transition corresponds mostly to a transition centred on the DBT unit, but due to the axial conformation DBT mixes with PTZ. It is this mixing which explains why the band is absent for the absorption spectra of the two fragments.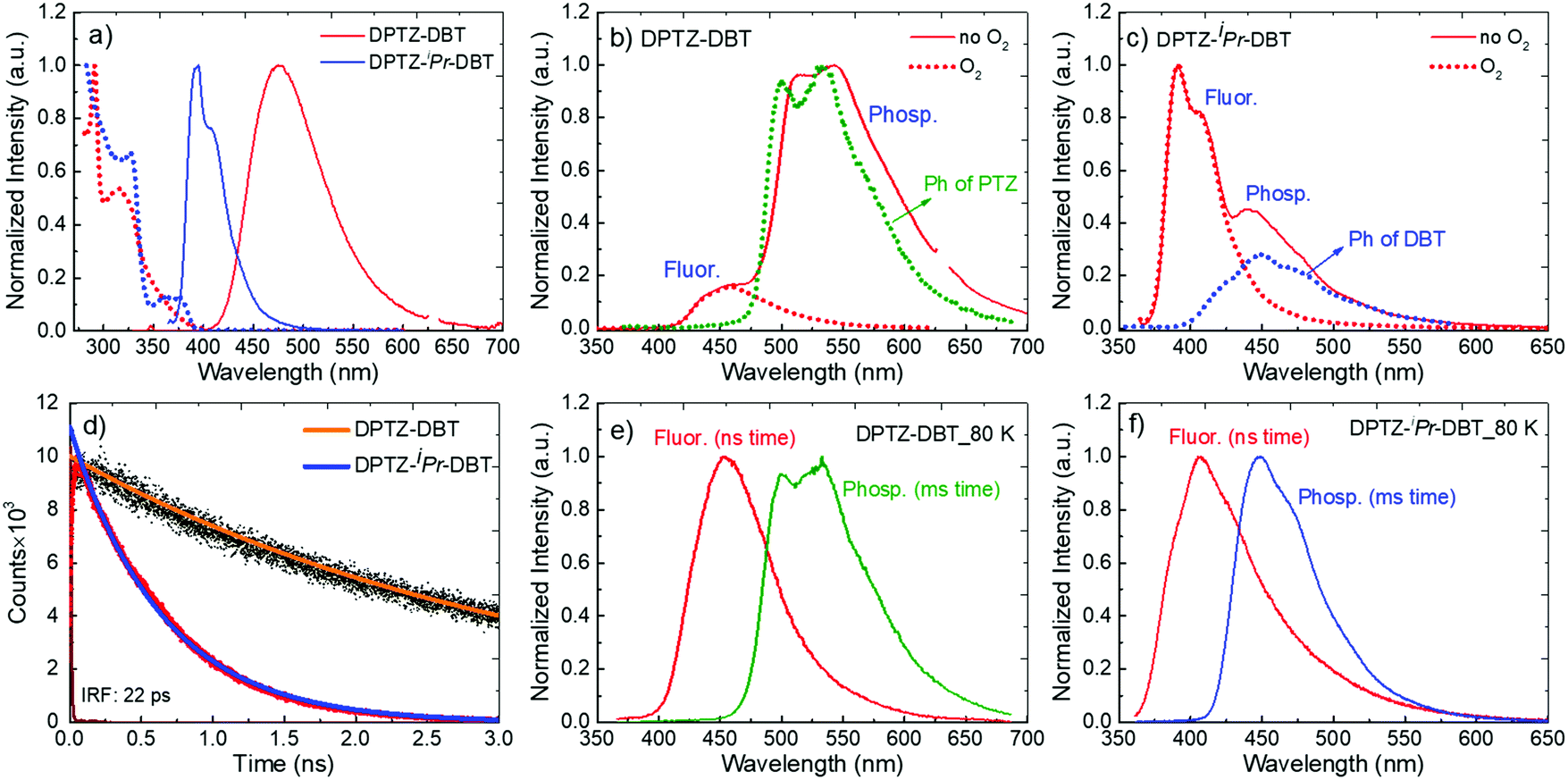 | ||
| Fig. 3 (a) Absorption and steady-state emission spectra of DPTZ–DBT and DPTZ–iPr–DBT in toluene at RT. (b) Steady-state emission spectra in zeonex at RT, in air and under vacuum for DPTZ–DBT. The phosphorescence of PTZ is also shown matching the phosphorescence of DPTZ–DBT. (c) Steady-state emission spectra in zeonex at RT, in air and under vacuum for DPTZ–iPr–DBT. The phosphorescence of DBT unit is also shown matching the phosphorescence of DPTZ–iPr–DBT; (d) fluorescence decays of DPTZ–DBT and DPTZ–iPr–DBT, in toluene solution at RT. (e and f) Time-resolved fluorescence and phosphorescence spectra at 80 K for (e) DPTZ–DBT; (f) DPTZ–iPr–DBT. The fluorescence and phosphorescence for e and f were collected with delay times of 1.1 ns and 50.1 ms, respectively. | ||
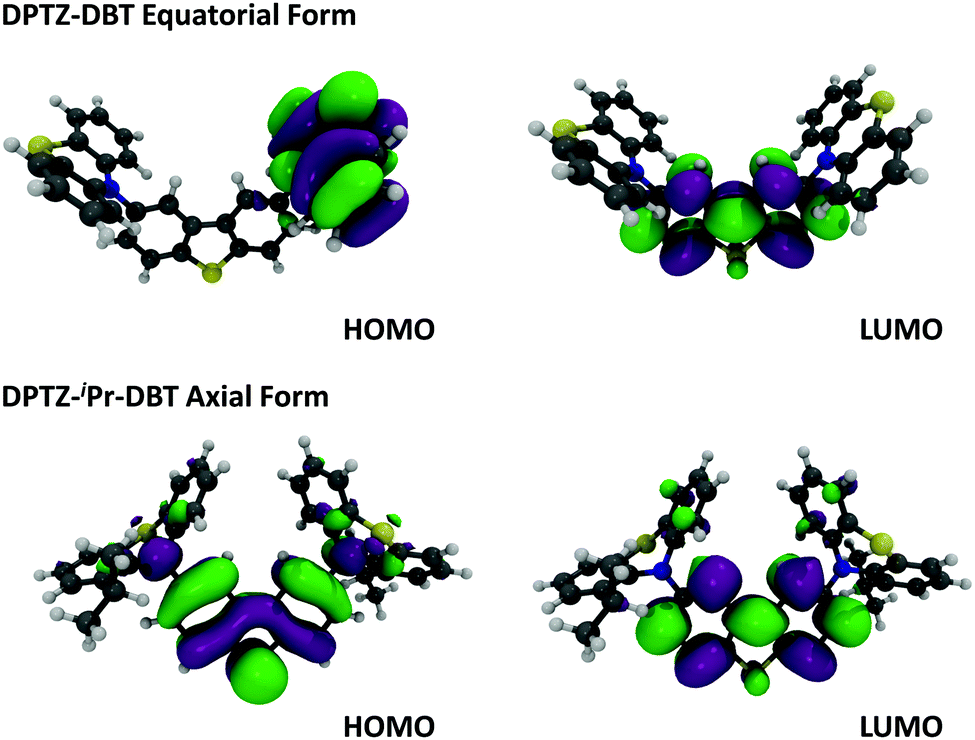 | ||
| Fig. 4 Molecular orbitals, HOMO and LUMO of DPTZ–DBT (equatorial) and DPTZ–iPr–DBT (axial) forms at DFT (M062X)/Def2SVP level of theory. | ||
Clear and defined differences are observed in the emission spectra in this molecular series. DPTZ–DBT shows broad emission in toluene, peaking at 480 nm, which further red-shifts with increasing solvent polarity (Fig. S3a, ESI†). In contrast, the substituted compounds DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT show well-resolved, blue-shifted emission, which is entirely independent of solvent polarity (Fig. S3, ESI†). These observations are consistent with the CT character of the lowest singlet excited state in DPTZ–DBT, as expected from the HOMO and LUMO orbitals in Fig. 4. In the substituted compounds the excited state has no CT character, based on the absence of solvatochromism in the emission (Fig. S3, ESI†). This observation is also confirmed by the calculations shown in Table S1 (ESI†).
The unsubstituted DPTZ–DBT exhibits axial and equatorial forms in the ground state with only a small energy difference between the two conformers (Table S1, ESI†). The axial form shows no shift with the polarity of the solvent in agreement with the calculations shown in Table S1 (ESI†). However, the equatorial form consistently shifts to lower energies with increasing solvent polarity. In DPTZ–DBT the lowest singlet excited state, S1, is equatorial and the calculated emission is 446, 490 and 566 nm in toluene, anisole, and ethanol, respectively. This strong solvatochromism reflects the stronger CT character of the equatorial form (vs. axial) in agreement with the experimental observations (Fig. S3, ESI†). In contrast, the axial conformer has significant local excitonic character on the DBT unit at 334 nm, and therefore is unaffected by solvent polarity. The substituted molecules DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT in the ground state exist only in the axial form, which is identified by the absorption at 380 nm. However, they show both axial and equatorial forms in the excited state, although the former is more stable due to the steric hindrance caused by the alkyl groups (Table S1, ESI†).
Fig. 3b shows that the emission of DPTZ–DBT is dominated by phosphorescence, peaking around 540 nm. The fluorescence band is also observed, peaking at 450 nm, at a similar energy of the fluorescence of the donor PTZ, (Fig. S2a, ESI†). The phosphorescence band in DPTZ–DBT matches with the phosphorescence of PTZ, showing that the lowest energy triplet state is localized in the PTZ unit. In the presence of oxygen, the phosphorescence is quenched and only the fluorescence band at 450 nm is observed.
As for DPTZ–DBT, the substituted molecules, DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT, show dual-emission in zeonex (Fig. 3c and Fig. S7, ESI†). Fluorescence is clearly observed peaking around 390 nm, and is accompanied by the phosphorescence band, peaking at 450 nm. This contrasts with DPTZ–DBT as the phosphorescence in DPTZ–iPr–DBT and DPTZ–tBu–DBT matches perfectly with the phosphorescence of the DBT unit, and not of PTZ. The phosphorescence behaviour in DPTZ–Me–DBT is more complex and is discussed in detail below.
The fluorescence lifetime of DPTZ–DBT emission in toluene is 10.3 ns, and fluorescence is observed up to 60 ns (Fig. 5a). This is in clear contrast with the fluorescence decay of the substituted molecules DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT (Fig. 5), which show fluorescence lifetimes of approximately 600 ps, and where the fluorescence is observed only up to 2.5 ns. The fluorescence decay in the substituted molecules is, therefore, 17 times faster than that of DPTZ–DBT.
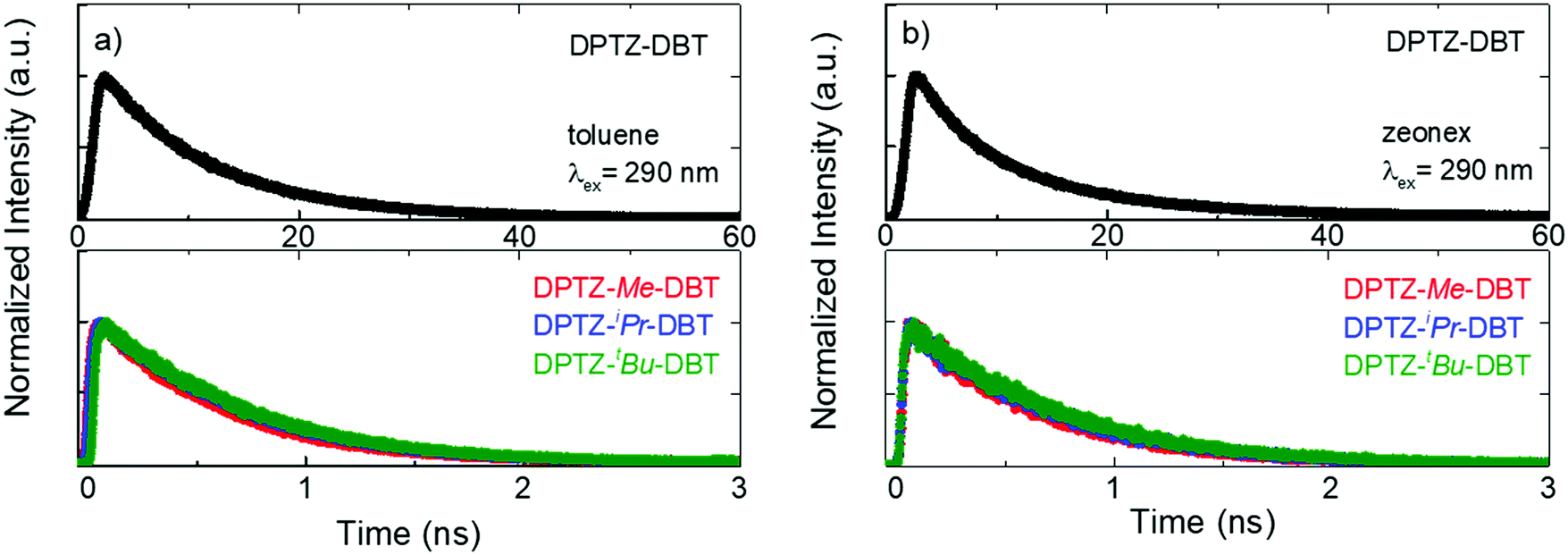 | ||
| Fig. 5 Emission decays of DPTZ–DBT, DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT collected at the fluorescence bands in air at RT in (a) toluene solution and (b) zeonex film. | ||
The fluorescence and phosphorescence bands from DPTZ–DBT and DPTZ–iPr–DBT in zeonex are observed independently at early and later times, respectively, due to their different lifetimes (Fig. 3e and f). Remarkably, both fluorescence and phosphorescence in DPTZ–iPr–DBT are blue-shifted, when compared with the corresponding spectra of DPTZ–DBT. However, while the fluorescence does not match entirely the fluorescence of either DBT or PTZ, the phosphorescence originates from the high energy triplet state that matches the triplet state localized on the DBT unit. DPTZ–Me–DBT and DPTZ–tBu–DBT show similar emission properties when compared to DPTZ–iPr–DBT, and also have a higher triplet energy level, with phosphorescence matching that of the DBT unit (Fig. 6, Fig. S4 and S7, ESI†).
 | ||
| Fig. 6 DPTZ–Me–DBT in zeonex film: (a) steady-state emission spectra in air and in vacuum at RT. The phosphorescence of the DBT and the PTZ units are also shown, matching the two phosphorescence bands in DPTZ–Me–DBT. (b) Time-resolved fluorescence and phosphorescence spectra at RT, collected at 1.1 ns and 50 ms, respectively. (c) Phosphorescence spectra at RT and 80 K, compared with the phosphorescence spectra of DBT and PTZ fragments. (d) Phosphorescence spectra at 50 ms delay-time collected as a function of temperature. | ||
The observation of blue-shifted fluorescence and phosphorescence spectra is not the simple effect of substitution on the PTZ unit. The absorption, fluorescence and phosphorescence spectra of PTZ and 1-alkyl-PTZ derivatives are compared in Fig. S5 (ESI†). Only minimal variations in the absorption and emission spectra are observed upon 1-alkyl substitution. Therefore, the substitution of the PTZ molecule alone cannot explain the observations made from the molecules in Fig. 1.
Noteworthy, the methyl substituted DPTZ–Me–DBT exhibits dual phosphorescence emission properties. The steric effect of the substituent is less pronounced, compared to DPTZ–iPr–DBT and DPTZ–tBu–DBT and dual phosphorescence is observed at RT in zeonex film. The steady-state emission spectra of DPTZ–Me–DBT (Fig. 6a) show the fluorescence band peaking at 390 nm, as in the case of the other two substituted compounds. However, in DPTZ–Me–DBT the fluorescence is accompanied by phosphorescence from the accepting DBT unit, peaking at 450 nm, as well as phosphorescence from the donating PTZ–Me at 540 nm. The observation of dual phosphorescence is an apparent violation of Kasha's rule.34
The fluorescence spectrum of DPTZ–Me–DBT, peaks at 390 nm, and was collected at early time (1.1 ns, Fig. 6b). As in the case of DPTZ–iPr–DBT and DPTZ–tBu–DBT, dual phosphorescence is observed at 50 ms delay-time, composed of low and high energy bands, matching the phosphorescence of PTZ–Me and DBT respectively (see Fig. 6c). With decreasing temperature, the contribution of PTZ–Me phosphorescence progressively disappears and only the high-energy DBT phosphorescence remains at 80 K (Fig. 6c and d).
The phosphorescence behaviour of DPTZ–Me–DBT strongly indicates the presence of two conformers in all four DPTZ–DBT derivatives, which are separated by a considerable energy barrier. The height of this barrier is very sensitive to the length of the bond between the donor and acceptor groups18 and therefore while it is high in the ground state, may decrease in the excited state. The axial and equatorial forms are identified in the ground and excited states of DPTZ–DBT (see Table S1, ESI†). However, both fluorescence and phosphorescence occur from the most stable state, which is isoenergetic with the PTZ singlet and triplet states, and no emission is observed from the high energy state, even at low temperature (Fig. S6, ESI†). Therefore, the excited state in DPTZ–DBT is able to relax to the most stable state, showing no activation barrier. In DPTZ–Me–DBT only the axial form is identified in the ground-state. However, both axial and equatorial forms exist in the excited-state (Table S1, ESI†). Interestingly, fluorescence occurs entirely from the higher energy state (when compared with DPTZ–DBT). The short lifetime of the singlet excited state does not allow the relaxation to the low energy singlet state to occur. Remarkably, as the triplet state is long-lived, the relaxation to the low energy triplet is possible. Therefore, at high temperatures, DPTZ–Me–DBT is able to cross the barrier and gives origin to dual phosphorescence. DBT-like phosphorescence is observed peaking at 450 nm, and PTZ–Me-like phosphorescence is observed around 530 nm. At low temperatures this relaxation is not possible and consequently only DBT-like phosphorescence is observed. This shows that the relaxation between the two triplet states involves an energy barrier separating the two states, which is likely to increase with the bulkiness of the substituents. The fluorescence and phosphorescence from the iso-propyl and tert-butyl substituted compounds, DPTZ–iPr–DBT and DPTZ–tBu–DBT, confirms this scenario. In these two compounds only the high energy fluorescence and the DBT-like phosphorescence are observed, even at RT, as there is insufficient energy available to surmount the barrier. The excited state dynamics in the DPTZ–DBT derivatives studied here is summarized in a simplified scheme given in Fig. 7.
 | ||
| Fig. 7 Schematic representation of the excited state relaxation in the unsubstituted DPTZ–DBT and substituted DPTZ–Me–DBT and DPTZ–tBu–DBT. | ||
Further evidence supporting the switching between the two excited states that are probably associated with the axial and equatorial forms of DPTZ–DBT derivatives is obtained from time-resolved phosphorescence decays shown in Fig. 8. In DPTZ–DBT the switching involves a vanishingly small energy barrier and the phosphorescence decays mono-exponentially with a 47 ms time constant from the triplet isoenergetic with the PTZ phosphorescence (Fig. 8a). However, for the substituted analogues the addition of the alkyl side groups makes switching between the high and low energy excited states sequentially more difficult as the steric hindrance increases. Therefore, the phosphorescence decay of DPTZ–Me–DBT shows double exponential profile with a fast component of 6 ms and a long decay component of 37 ms. The fast component progressively disappears with decreasing temperature, with the decay becoming single exponential at low temperature. The phosphorescence decays of DPTZ–iPr–DBT and DPTZ–tBu–DBT show similar behaviours, although the fast component becomes less important in the most restricted DPTZ–tBu–DBT. For the methyl and iso-propyl substituted compounds the dual component of the phosphorescence decays is clearly observed showing a 30% contribution of the fast component with a time constant around 6 ms, and 70% contribution for the long decay component of around 35 ms. The amplitude of the fast component is stronger in the compound carrying the bulkier iso-propyl substituent, relative to the methyl substituted compound. In the more sterically hindered compound substituted with tert-butyl groups the longer decay component contributes above 91% of the phosphorescence decay, and this is practically single exponential decaying with a time constant around 85 ms.
 | ||
| Fig. 8 Time resolved phosphorescence decays in zeonex obtained as a function of temperature for (a) DPTZ–DBT, (b) DPTZ–Me–DBT, (c) DPTZ–iPr–DBT and (d) DPTZ–tBu–DBT. | ||
Rates and luminescence yields
Tables 1 and 2, show the photophysical data collected for DPTZ–DBT, DPTZ–Me–DBT, DPTZ–iPr–DBT, and DPTZ–tBu–DBT in zeonex solid film and in toluene solution, respectively. The marked decrease on the fluorescence lifetimes (see Fig. 5) are observed due to the pronounced increase on the ISC and IC rates (kISC and kIC) along the series. In solution, the fluorescence lifetime decreases from 10.3 ns in DPTZ–DBT to approximately 600 ps in the substituted DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT, and in zeonex the behaviour is similar. This is consistent with the increasing oscillator strength of the S1 state obtained from the calculations, which is 0.0017 for DPZ–DBT, and 0.0390, 0.0442 and 0.0451 for DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT, respectively, and is also indicative of an increase in the locally excited state character of the emitting state. The IC rate (kIC) increases consistently in the series, 1.0 × 107 s−1 in DPTZ–DBT, 3.7 × 108 s−1 in DPTZ–Me–DBT and DPTZ–iPr–DBT, and 7.4 × 108 s−1 in DPTZ–tBu–DBT. The radiative rate kf also increases, but shows no significant variation within the different substituents.| Compound | Φ fl ±0.02 | τ fl ±0.01 (ns) | k f ±0.002 (×109 s−1) | k ISC ±0.01a (×109 s−1) | k IC ±0.01 (×109 s−1) | Φ Ph ±0.08 | Φ Lum (Fluo + Phos) |
|---|---|---|---|---|---|---|---|
| a Calculated using ΦT determined in solution. | |||||||
| DPTZ–DBT | 0.06 | 12.60 | 0.005 | 0.07 | 0.004 | 0.60 | 0.66 |
| DPTZ–Me–DBT | 0.03 | 0.60 | 0.050 | 1.26 | 0.37 | 0.01 | 0.04 |
| DPTZ–iPr–DBT | 0.04 | 0.65 | 0.061 | 1.11 | 0.37 | 0.02 | 0.06 |
| DPTZ–tBu–DBT | 0.03 | 0.67 | 0.045 | 0.69 | 0.74 | 0.02 | 0.05 |
| Compound | Φ fl ±0.02 | Φ T ±0.1 | τ fl ±0.01 (ns) | τ T ±0.1 (μs) | k f ±0.002 (×109 s−1) | k ISC ±0.01 (×109 s−1) | k IC ±0.01 (×109 s−1) |
|---|---|---|---|---|---|---|---|
| a Measured in benzene. | |||||||
| DPTZ–DBT | 0.04 | 0.85 | 10.29 | 63.3 | 0.004 | 0.08 | 0.01 |
| DPTZ–Me–DBT | 0.03 | 0.75 | 0.61 | 27.6 | 0.049 | 1.24 | 0.36 |
| DPTZ–iPr–DBT | 0.02 | 0.72 | 0.64 | 6.2 | 0.031 | 1.13 | 0.41 |
| DPTZ–tBu–DBT | 0.02 | 0.47 | 0.66 | 3.5 | 0.030 | 0.72 | 0.77 |
The triplet lifetime in solution also decreases sequentially from 63.3 μs in DPZ–DBT to 27.6 μs for DPTZ–Me–DBT, 6.2 μs for DPTZ–iPr–DBT, and 3.5 μs for DPTZ–tBu–DBT (Table 2). This trend is consistent with the enhancement of the IC rate, and the decrease of the phosphorescence efficiency in the substituted molecules, giving indication for the presence of additional vibrational modes in the substituted molecules, which promote more efficient quenching of the triplet excited state population in DPTZ–Me–DBT, DPTZ–iPr–DBT and DPTZ–tBu–DBT. As a result, the phosphorescence in these compounds is relatively short-lived than in the unsubstituted DPZ–DBT, and shows pronounced variation with temperature (Fig. 7).
The total luminescence yield determined in zeonex at RT is much higher in DPTZ–DBT (≈66%) than in the substituted compounds, for which it is <6%. This is the result of the fast ISC rate and slower IC rate in DPTZ–DBT. However, the fluorescence yield is low, even in DPTZ–DBT (6%), due to the significantly faster kISC, when compared to the radiative rate (kf). In the substituted compounds the ISC rate increases significantly and becomes approximately two orders of magnitude faster than the radiative rate, and is surprisingly fast in the methyl substituted DPTZ–Me–DBT. The ISC rate in solution, for example, is 8.0 × 107 s−1 for DPZ–DBT, 1.24 × 109 s−1 in DPTZ–Me–DBT, 1.13 × 109 s−1 in DPTZ–iPr–DBT, and 7.2 × 108 s−1 in DPTZ–tBu–DBT. And in zeonex the behaviour is similar, for DPTZ–DBTkISC = 7.0 × 107 s−1 and in DPTZ–Me–DBTkISC = 1.3 × 109 s−1. The rate decreases slightly with increasing bulkiness of the substituent, along with increasing IC rate in the substituted compounds.
To understand the influence of the alkyl substituents on the ISC rates, we have simulated the rate of intersystem crossing using the Fermi golden rule approximation combined with a Marcus formalism to estimate the density of states;
 | (1) |
Here λ corresponds to the reorganization energy, i.e. the energy variation in the initial singlet excited state when switching from the singlet equilibrium geometry to the triplet equilibrium geometry, while the driving force ΔEST is identified as the adiabatic singlet–triplet gap.35 The S1 state is closest in energy to the T3 state and therefore we consider ISC to occur between these states. The rates to lower triplet states were calculated, but in each case ΔEST > 0.15 eV yielding very small kISC.
Using eqn (1) and the calculated data shown in Table 3, we find an ISC rate of 0.014 × 109 s−1 for DPTZ–DBT. The rate increases to 0.209 × 109 s−1 for DPTZ–Me–DBT, 5.16 × 108 s−1 in DPTZ–iPr–DBT, and 2.44 × 108 s−1 in DPTZ–tBu–DBT. This reveals calculated rates broadly consistent with the experimental observations in that kISC is larger for the substituted molecules. This is due to the smaller energy gap between the S1 and T3 states. While the trends of kISC for the substituted molecules do not exactly follow experimental observations, these differences are within the uncertainty of the calculations, which due to the exponential dependence of λ and ΔEST in eqn (1) are very sensitive to small differences in λ and ΔEST.
| Compound | Conformer | SOCME (cm−1) | λ (eV) | ΔEST (eV) | k ISC (×109 s−1) |
|---|---|---|---|---|---|
| DPTZ–DBT | Equatorial | 0.10 | 0.01 | 0.06 | 0.014 |
| Axial | 0.80 | 0.08 | 0.06 | 0.015 | |
| DPTZ–Me–DBT | Axial | 0.80 | 0.02 | 0.04 | 0.209 |
| DPTZ–iPr–DBT | Axial | 1.00 | 0.01 | 0.03 | 0.516 |
| DPTZ–tBu–DBT | Axial | 0.80 | 0.06 | 0.02 | 0.244 |
The IC rate (kIC) increases consistently in the series, kIC = 4.0 × 106 s−1, in DPTZ–DBT, kIC = 3.7 × 108 s−1 in DPTZ–Me–DBT and DPTZ–iPr–DBT, and kIC = 7.4 × 108 s−1 in DPTZ–tBu–DBT. Finally, the radiative rate kf also increases, but shows no significant variation within the different substituents.
Finally, by comparing DPTZ–DBT with previously reported carbazole substituted analogue, 3,7-di(N-carbazolyl)-dibenzothiophene (DCz–DBT),3 the influence of the sulfur atom in the PTZ units is revealed. The presence of PTZ induces a pronounced enhancement of the triplet formation yield, and shows no effect on the fluorescence yield. In the case of the PTZ H-intra conformation (equatorial form), the nitrogen lone pair is delocalized over the phenyl rings of the PTZ unit, which may explain the excellent RTP properties of the DPTZ–DBT compound. Marked differences are however observed in the rate constants for radiative decay, kf, intersystem crossing, kISC, and non-radiative internal conversion, kIC, all showing a pronounced decrease on DPTZ–DBT. The fluorescence decay, for example, decreases from ≈10 ns in DPTZ–DBT to 0.88 ns in DCz–DBT. DPTZ–DBT can also be compared with other PTZ derivatives using the strong acceptor dibenzothiophene-S,S-dioxide (DBTO2) unit, where the equatorial form leads to negligible singlet–triplet energy gap and emergence of TADF. However, when the axial form is adopted due to the steric effect of the alkyl groups that block the relaxation of the PTZ units in the excited-state, RTP instead of TADF is observed containing contributions from two triplet states matching the phosphorescence of PTZ and DBTO2.32
Conclusions
In summary, this series of DPTZ–DBT luminophores provides new insights into the design of all-organic RTP emitters and demonstrates how systematic changes in the substituents can tune photophysical properties by conformational control. DPTZ–DBT and substituted analogues exist in equatorial and axial forms in the ground state. Here, it has been demonstrated that different conformations have markedly different photophysical properties, with the axial conformer showing enhanced ISC rate. This is accompanied by similar acceleration of the radiative and non-radiative IC rates. Switching between electronic excited states occurs with a vanishingly small barrier in the unsubstituted DPTZ–DBT. However, an increasing barrier is imposed by the presence of the bulkier side groups, and this mainly prevents the relaxation of the singlet and triplet excited states to lower energy in these substituted compounds.The unsubstituted molecule DPTZ–DBT shows dual emission, fluorescence and strong green phosphorescence, in solid zeonex film at RT, with a high total luminescence yield of ≈66%. The triplet formation yield is the highest in the series (85%), and the fluorescence and phosphorescence lifetimes are relatively long-lived, 12.6 ns and 47 ms, respectively, resulting in relatively slow excited state decay rates, but dominant ISC. This behavior is markedly altered by tuning of the conformation due to the presence of bulky side groups in the C(1)-position of each PTZ unit. The substituted molecules show much shorter fluorescence and phosphorescence lifetimes due to faster radiative, IC and ISC rates, and also low luminescence yields, and notably, higher singlet and triplet energies. Interestingly, the molecule substituted with the less bulky methyl group, DPTZ–Me–DBT, in addition to fluorescence, shows also dual phosphorescence at RT. This originates from T1 in the green region, at 2.60 eV, coincident with the emissive triplet of DPTZ–DBT, and from T2 in the blue region, at 2.88 eV, approximately the same energy of the emissive triplet in DPTZ–iPr–DBT and DPTZ–tBu–DBT. This is an apparent violation of Kasha's rule imposed by the existence of different conformers. However, at low temperatures, only the high energy blue phosphorescence is observed as there is insufficient energy to drive the relaxation of the excited state. In conclusion, the strategy of selective functionalization of the donor unit is shown to be a viable approach to tuning the excited state properties of D–A molecules for RTP emission, including blue emitters.
Author contributions
R. H. performed the majority of the optical measurements under the supervision of F. B. D. J. W. and N. A. K. synthesized molecules under the guidance of M. R. B. and carried out NMR studies. J. A., J. C. L. and M. N. B.-S. carried out Flash-Photolysis studies and determined triplet formation and luminescence quantum yields. A. S. B. performed the X-ray characterization. The full physical model was devised by J. W., T. J. P. and F. B. D., and it was theoretically tested within a quantum chemistry framework by J. G. and T. J. P. F. B. D. wrote the manuscript, which was revised by all the co-authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
R. H. acknowledges a PhD grant funded by Durham University. F. B. D. acknowledges Samsung-SAIT for funding this work using their Global Research Outreach (GRO) Program, and F. B. D. and M. R. B. acknowledge the EPSRC for funding under grant numbers EP/L02621X/1 and EP/N028511/1. J. A. acknowledges FCT for funding under grant SFRH/BPD/120599/2016 and project PTDC/QUI-QFI/32007/2017.References
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef PubMed.
- S. Hirata and M. Vacha, Adv. Opt. Mater., 2017, 5, 1600996 CrossRef.
- R. Huang, J. Avó, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef PubMed.
- O. S. Wolfbeis, Adv. Mater., 2008, 20, 3759–3763 CrossRef.
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885 RSC.
- L. Jethi, M. M. Krause and P. Kambhampati, J. Phys. Chem. Lett., 2015, 6, 718–721 CrossRef PubMed.
- K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705–709 CrossRef PubMed.
- S. Reineke, N. Seidler, S. R. Yost, F. Prins, W. A. Tisdale and M. A. Baldo, Appl. Phys. Lett., 2013, 103, 93302 CrossRef.
- S. R. Forrest, M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley and M. E. Thompson, Nature, 1998, 395, 151–154 CrossRef.
- Y. Liu, G. Zhan, P. Fang, Z. Liu, Z. Bian and C. Huang, J. Mater. Chem. C, 2017, 5, 12547–12552 RSC.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 Search PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 12001 CrossRef PubMed.
- T. J. Penfold, F. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- A. S. D. Sandanayaka, T. Matsushima and C. Adachi, J. Phys. Chem. C, 2015, 119, 23845–23851 CrossRef.
- A. S. D. Sandanayaka, K. Yoshida, T. Matsushima and C. Adachi, J. Phys. Chem. C, 2015, 119, 7631–7636 CrossRef.
- A. Stockmann, J. Kurzawa, N. Fritz, N. Acar, S. Schneider, J. Daub, R. Engl and T. Clark, J. Phys. Chem. A, 2002, 106, 7958–7970 CrossRef.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef.
- S. Gan, W. Luo, B. He, L. Chen, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 3705–3708 RSC.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- F. B. Dias, S. King, A. P. Monkman, I. I. Perepichka, M. A. Kryuchkov, I. F. Perepichka and M. R. Bryce, J. Phys. Chem. B, 2008, 112, 6557–6566 CrossRef PubMed.
- L. Wang, Z. Y. Wu, W. Y. Wong, K. W. Cheah, H. Huang and C. H. Chen, Org. Electron., 2011, 12, 595–601 CrossRef.
- H. W. Lee, S. Jeong, S. E. Lee, Y. K. Kim and S. S. Yoon, Jpn. J. Appl. Phys., 2016, 55, 06GK07 CrossRef.
- J. Guo, X.-L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- X. Tang, T. Shan, Q. Bai, H. Ma, X. He and P. P. Lu, Chem. – Asian J., 2017, 12, 552–560 CrossRef PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1846156–1846161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc02987c |
| This journal is © The Royal Society of Chemistry 2018 |