Oligothiophene quinoids containing a benzo[c]thiophene unit for the stabilization of the quinoidal electronic structure†
Received
17th April 2018
, Accepted 17th May 2018
First published on 18th May 2018
Abstract
Bis(dicyanomethylene)-substituted quinoidal π-conjugated systems possess a high electron-accepting nature and thus have been extensively investigated for application as n-type semiconductors. We focus on the utilization of benzene-annelation for the stabilization of the quinoidal electronic structure against the biradicaloid structure by designing quinoidal thiophenes 3-mer (BTQ) and 6-mer (BTQ-6) that have benzo[c]thiophene units. We also develop quinoidal oligothiophenes (BTQ-F) consisting of both benzo[c]thiophene and fluorinated thiophene units. The influence of benzo[c]thiophene on the quinoidal electronic structure is investigated by theoretical studies and property measurements. The molecular structure of BTQ-F is unambiguously confirmed through single-crystal X-ray diffraction. Analyses of cyclic voltammetry reveal that the lowest unoccupied molecular orbital energy levels of these compounds lie below −4.0 eV, leading to good electron-transporting characteristics even under ambient conditions in organic field-effect transistors (OFETs). Due to an increased highest occupied molecular orbital energy level, ambipolar transport is observed in BTQ-6, indicating the versatility of quinoidal π-conjugated systems incorporating benzo[c]thiophene.
Introduction
Significant research interest continues to be focussed on structurally well-defined π-conjugated oligomers, not only for the elucidation of fundamental structure–property relationship, but also for their applications in organic thin-film electronics such as organic field-effect transistors (OFETs) and organic photovoltaics.1,2 π-Conjugated compounds bearing quinoidal structures have been extensively investigated not only as a model structure of doped conducting arylene polymers, but also as a Wurster-type multi-redox system in Hünig's classification.3 Among them, bis(dicyanomethylene)-substituted quinoidal compounds, which generate an anionic aromatic state by accepting one or two electrons, exhibit a high electron-accepting ability for instance the well-known compound tetracyano-p-quinodimethane.4 Therefore, the extension of the π-conjugation of such quinoidal systems, typically tetracyano quinoidal oligothiophenes, has attracted great attention for promising electron-transporting (n-type) semiconductors.5–13 Another unique feature of the quinoidal systems is the equilibrium between quinoidal and biradicaloid electronic structures,14 which causes a configurational instability of quinoidal oligothiophenes. A representative approach to increase the contribution of the quinoid character in oligothiophenes involves the utilization of aromatic stabilization in the quinoidal form, which was accomplished by the introduction of a fused aromatic ring on the thiophene [c]bond, as observed first in the synthesis of poly(isothianaphthene) with a small band gap by Wudl et al.15 In this context, quinoidal oligothiophenes containing thieno[3,4-b]thiophene have been extensively developed by Zhu et al.16 We envisioned that if the benzo[c]thiophene (isothianaphthene) quinoid unit can be incorporated into π-extended quinoidal structures, the high aromatic stabilization energy of this unit compared to that of benzo[c]thiophene would allow us to create quinoidal oligothiophenes that have potential as air-stable semiconducting materials. To pursue this possibility, we designed new bis(dicyanomethylene)-substituted quinoidal oligothiophenes (BTQ and BTQ-6) containing 5,6-dialkylbenzo[c]thiophene units (Fig. 1). Furthermore, in order to increase the electron-accepting ability of BTQ, fluorine substituents were introduced at the β-position of the quinoidal terthiophene (BTQ-F). In this contribution, we report on the synthesis, properties, structures, and OFET characteristics of these compounds. Although the parent bis(dicyanomethylene)benzo[c]thiophene was synthesized and characterized by Cava et al. in 1993,17 its π-extended derivative has not been reported so far.
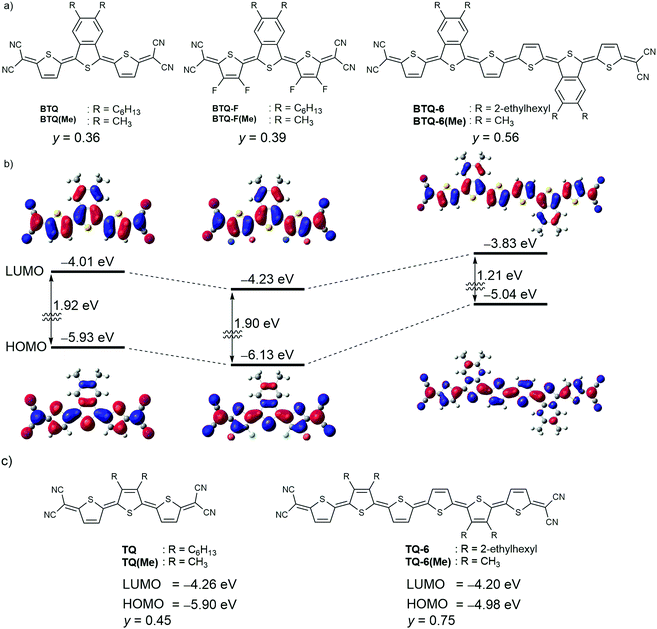 |
| | Fig. 1 (a) Chemical structures of compounds investigated in this study. (b) Calculated energy levels and orbitals for model compounds BTQ(Me) (left), BTQ-F(Me) (center), and BTQ-6(Me) (right). (c) Chemical structures of reference compounds. | |
Results and discussion
Theoretical studies
To estimate the molecular structure and frontier orbital energies, we performed density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory. All alkyl groups were replaced with methyl groups for the ease of the calculation. Optimized geometries of BTQ(Me), BTQ-F(Me), and BTQ-6(Me) showed a planar conformation which indicates that the introduction of the benzene annelation in the central thiophene ring of terthiophene does not increase the distortion with respect to the dihedral angle (Fig. 1(b)). The lowest unoccupied molecular orbital (LUMO) energy level of BTQ(Me) is higher compared to that of TQ(Me) (Fig. 1(b) and (c)) but is still located at a low level of −4.01 eV. The substitution of hydrogen atoms in the thiophene rings with fluorine atoms for BTQ-F(Me) led to a further decrease of the LUMO energy level to −4.23 eV, while maintaining a similar highest occupied molecular orbital (HOMO)–LUMO energy gap. It should be mentioned that the LUMO of both compounds is delocalized over the entire π-conjugated backbone including the annelated benzene ring, which is favorable for the intermolecular overlap of LUMOs between neighboring molecules in the solid state.16b On the other hand, the extension of π-conjugation from BTQ(Me) to BTQ-6(Me) mainly contributes to an increase of the HOMO energy level from −5.93 eV to −5.04 eV. The singlet biradical character (y) of these quinoidal compounds was calculated by the natural orbital occupation number of the lowest unoccupied natural orbital with spin-unrestricted calculations at the UHF/6-31G(d) level.18 Compounds BTQ(Me) and BTQ-F(Me) gave y values of 0.36 and 0.39, respectively. As we expected, these values were smaller than that of the reference TQ(Me) (y = 0.45). Similarly, BTQ-6(Me) showed a lower y value of 0.56 compared to TQ-6(Me) (0.75). These results indicate that the introduction of the benzo[c]thiophene unit is effective in increasing the contribution of the quinoidal electronic structure in the ground state.
Synthesis and characterization
Since 5,6-dialkyl derivatives of benzo[c]thiophene are not easily accessed from commercially available compounds, we applied a new synthetic route, which relies on the construction of the benzo[c]thiophene core from tetrasubstituted benzene (Scheme 1). Methyl diester compound 1 was converted to 2-pyridinyl ester 2, which then reacted with Grignard reagents, generated from 2-bromothiophene or 2-bromo-3,4-difluoro-5-trimethylsilylthiophene, to give key intermediates 3a and 3b. The treatment of 3a and 3b with Davy's reagent led to the formation of the benzo[c]thiophene core, and the obtained terthiophene derivatives were then treated with N-bromosuccinimide (NBS) to afford 4a and 4b. Finally, palladium-catalyzed coupling reactions of 4a and 4b with sodium dicyanomethanide followed by oxidation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) afforded BTQ and BTQ-F in 40% and 30% yield, respectively. The hexyl groups in the benzo[c]thiophene moiety ensured the good solubility of the molecules. The synthetic route to BTQ-6 from 5 prepared similarly to 4 is also shown in Scheme 1. Note that the 2-ethylhexyl groups on the benzo[c]thiophene core are essential for ensuring appropriate solubility of the quinoidal thiophene 6-mer. On the other hand, we cannot obtain pure TQ-6 due to low chemical stability. The chemical structures of BTQ, BTQ-F, and BTQ-6 were fully characterized by NMR spectroscopy and MS. Detailed synthesis and characterization are summarized in the ESI.†
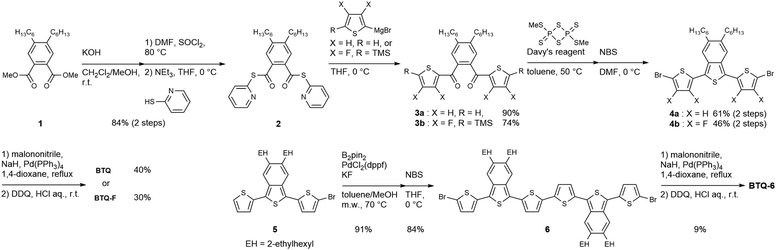 |
| | Scheme 1 Synthetic route to BTQ, BTQ-F, and BTQ-6. | |
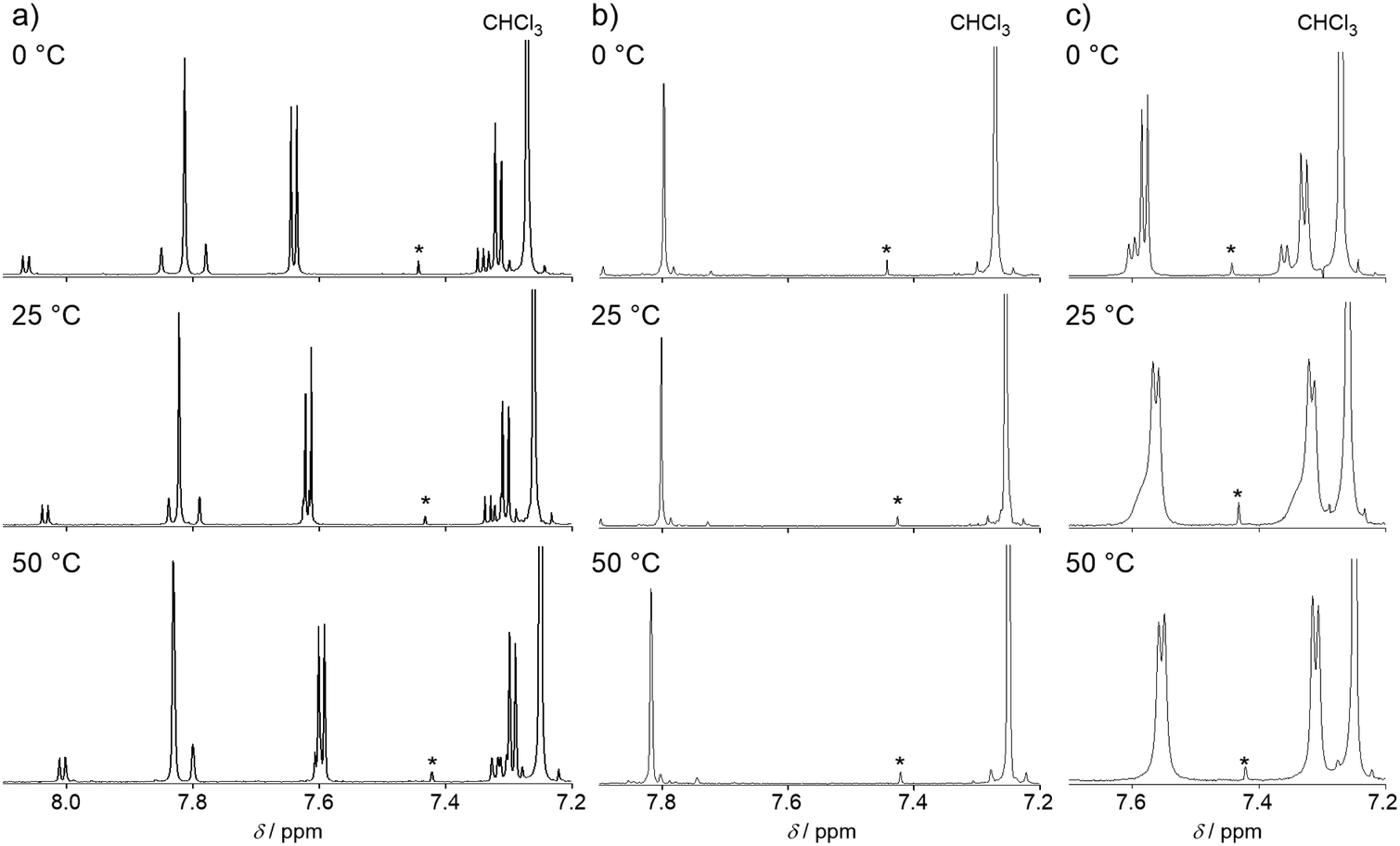
In the 1H NMR spectrum of BTQ at 25 °C, two sets of signals with an integration ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were observed in the aromatic region (Fig. 2),5 and all the two-set peaks were assigned to anti–anti and anti–syn isomers of the quinoidal terthiophene backbone, respectively, by nuclear Overhauser effect measurements (Fig. S1 in the ESI†). On the other hand, almost one singlet signal was observed for BTQ-F. In order to investigate the isomerization behavior of BTQ, BTQ-F, and TQ, variable temperature (VT)-NMR measurements were performed at 0, 25, and 50 °C. As shown in Fig. 2, the spectra of BTQ and BTQ-F showed little temperature dependence. Furthermore, the isomer ratio of BTQ is constant, irrespective of the temperature. On the other hand, although the spectrum of TQ showed two sets of signals at 0 °C similarly to that of BTQ, small peaks originating from the anti–syn isomer broadened at 25 °C and merged with the main peaks at 50 °C, implying the equilibrium between the two isomers probably via an aromatic biradical form. Similar anti–syn equilibrium was also reported on tetracyano bithiophene quinoid at 25 °C.19 These results indicate that the quinoidal electronic structures of BTQ and BTQ-F are stabilized owing to the reduced contribution of the biradicaloid structure by benzene annulation. In addition, the presence of fluorine atoms on the thiophene rings in BTQ-F induces not only steric congestion in the syn form but also intramolecular S–F attractive interactions, forming fixed anti configurations between adjacent thiophene rings.20 In contrast, BTQ-6 showed broad signals even under a low temperature of −45 °C (Fig. S2, ESI†), indicating the presence of equilibrium between the isomers.
:1 were observed in the aromatic region (Fig. 2),5 and all the two-set peaks were assigned to anti–anti and anti–syn isomers of the quinoidal terthiophene backbone, respectively, by nuclear Overhauser effect measurements (Fig. S1 in the ESI†). On the other hand, almost one singlet signal was observed for BTQ-F. In order to investigate the isomerization behavior of BTQ, BTQ-F, and TQ, variable temperature (VT)-NMR measurements were performed at 0, 25, and 50 °C. As shown in Fig. 2, the spectra of BTQ and BTQ-F showed little temperature dependence. Furthermore, the isomer ratio of BTQ is constant, irrespective of the temperature. On the other hand, although the spectrum of TQ showed two sets of signals at 0 °C similarly to that of BTQ, small peaks originating from the anti–syn isomer broadened at 25 °C and merged with the main peaks at 50 °C, implying the equilibrium between the two isomers probably via an aromatic biradical form. Similar anti–syn equilibrium was also reported on tetracyano bithiophene quinoid at 25 °C.19 These results indicate that the quinoidal electronic structures of BTQ and BTQ-F are stabilized owing to the reduced contribution of the biradicaloid structure by benzene annulation. In addition, the presence of fluorine atoms on the thiophene rings in BTQ-F induces not only steric congestion in the syn form but also intramolecular S–F attractive interactions, forming fixed anti configurations between adjacent thiophene rings.20 In contrast, BTQ-6 showed broad signals even under a low temperature of −45 °C (Fig. S2, ESI†), indicating the presence of equilibrium between the isomers.
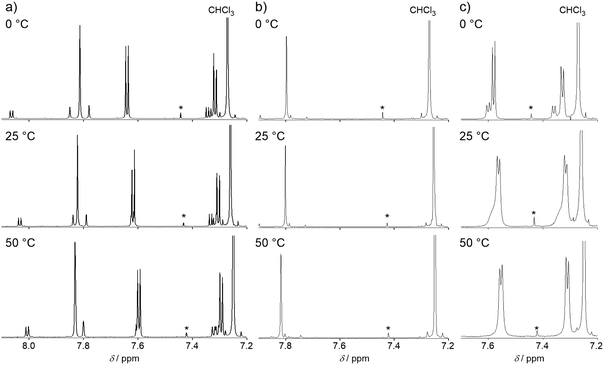 |
| | Fig. 2 VT-NMR spectra of (a) BTQ, (b) BTQ-F, and (c) TQ in the aromatic regions in CDCl3. Signals marked by an asterisk are satellite peaks in CHCl3. | |
Photophysical and electrochemical properties
The UV-vis-NIR absorption spectra of BTQ, BTQ-F, BTQ-6, and TQ in CH2Cl2 solutions are shown in Fig. 3, and the photophysical data are summarized in Table 1. The absorption spectra of BTQ and BTQ-F showed similar shapes with an absorption maximum (λmax) at around 640 nm, which are slightly blue-shifted compared to that of TQ (λmax = 670 nm) (Fig. 3(a)). These intense bands are mainly assigned to the HOMO–LUMO transition by time-dependent (TD)-DFT calculations at the B3LYP/6-31G(d,p) level of theory (see the ESI†). The qualitative trend of molar extinction coefficients (ε) for these compounds is in good accordance with the estimated oscillator strength. As summarized in Table 1, the optical HOMO–LUMO energy gaps (ΔEoptg) of these compounds estimated from the onsets in the absorption spectra are found to be ca. 1.7 eV. The absorption spectrum of BTQ-6 in CH2Cl2 solution exhibited an absorption band at the near-infrared region (λmax = 978 nm) with a large ε of 2.6 × 105 M−1 cm−1. This behavior is in contrast to a previous report where quinoidal sexithiophene showed a reduced molar extinction coefficient with concomitant appearance of marked absorptions in the visible region, compared to the corresponding quinoidal quinquethiophene. The reduced extinction coefficient was attributed to the high contribution of the biradical character,14b which indicates that the high ε of BTQ-6 is rationalized by the higher contribution of the quinoidal electronic structure owing to the presence of benzo[c]thiophene units in the framework. Compared to the solution spectra, well-structured shoulders with red-shifted λmax were observed for BTQ and BTQ-F in thin films (Fig. 3(b)). This phenomenon is attributed to the intermolecular electronic interactions of π−π stacked backbones, which are favorable for carrier transport in thin-film devices. The absorption spectrum of BTQ-6 in thin films showed a broad absorption with the onset reaching 1800 nm. Such a large bathochromic shift was observed for quinoidal quaterthiophene.9 In addition to the intermolecular electronic interactions, the existence of the biradical character may have influence on this bathochromic shift.
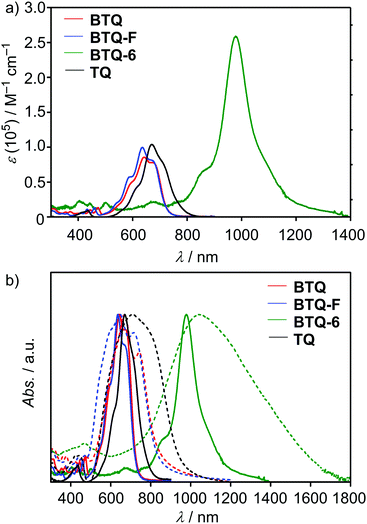 |
| | Fig. 3 (a) UV-vis-NIR absorption spectra of BTQ (red) and BTQ-F (blue), TQ (black), and BTQ-6 (green) in CH2Cl2 and (b) in CH2Cl2 (solid line) and thin films (dashed line). | |
Table 1 Photophysical and electrochemical properties
| Compound |
λ
max
(ε × 105)/nm (M−1 cm−1) |
ΔEoptga/eV |
E
ox1/2
/V |
E
red1/2
/V |
E
HOMO
/eV |
E
LUMO
/eV |
|
In CH2Cl2.
In CH2Cl2 containing 0.1 M TBAPF6. V vs. Fc/Fc+.
E
HOMO = −Eox1/2 − 4.8.
E
LUMO = −Ered1/2 − 4.8.
|
|
BTQ
|
642 (0.9) |
1.70 |
0.86 |
−0.74 |
−5.66 |
−4.06 |
|
BTQ-F
|
636 (1.0) |
1.71 |
1.05 |
−0.50 |
−5.85 |
−4.30 |
|
BTQ-6
|
978 (2.6) |
1.14 |
0.08 |
−0.54 |
−4.88 |
−4.26 |
| 0.35 |
−0.61 |
|
TQ
|
670 (1.1) |
1.60 |
0.81 |
−0.58 |
−5.61 |
−4.23 |
The electrochemical properties of these compounds were investigated by cyclic voltammetry (CV) measurements in CH2Cl2 containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. All potentials are calibrated against a ferrocene/ferrocenium (Fc/Fc+) couple as the standard, and these values are listed in Table 1. As presented in Fig. 4, the cyclic voltammograms of BTQ, BTQ-F, and TQ showed one reversible oxidation wave and a reduction wave. The observed good reversibility indicates the stable formation of both cationic and anionic species. Integration of each peak area for these compounds implied the participation of one electron and two electrons in oxidation and reduction processes, respectively. As shown in Table 1, due to the electron-withdrawing nature of fluorine atoms, both the half-wave oxidation potential (Eox1/2) and reduction potential (Ered1/2) of BTQ-F are positively shifted compared to those of BTQ. Based on the assumption that the energy level of Fc/Fc+ is −4.8 eV below the vacuum level,21–23 the LUMO energy levels (ELUMO) of BTQ and BTQ-F were estimated to be −4.06 and −4.30 eV, respectively. These values are in good agreement with those estimated from the theoretical calculation. For BTQ-6, two reversible oxidation and reduction processes were observed. The ELUMO and EHOMO of BTQ-6 estimated from the first Ered1/2 and Eox1/2 were −4.26 and −4.88 eV, respectively, indicating that the extension of the quinoidal π-system leads to a significant increase of the HOMO energy level. This qualitative trend of HOMO and LUMO energy levels is also in good agreement with the theoretical estimation.
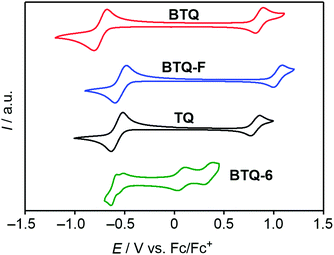 |
| | Fig. 4 Cyclic voltammograms of BTQ (red) and BTQ-F (blue), TQ (black), and BTQ-6 (green) in CH2Cl2 containing 0.1 M TBAPF6. | |
X-Ray single-crystal structures
The structure of BTQ-F was unambiguously determined using X-ray crystallographic analysis of the crystals that were grown by slow evaporation from a solution in ethyl acetate (EtOAc) and a MeOH mixed solvent. As shown in Fig. 5(a), the molecular structure of BTQ-F adopts all anti and planar conformations with dihedral angles of less than 2.7°. On the other hand, the distance of the intramolecular S–F contact (2.75 Å) is smaller than the sum of van der Waals radii of S and F atoms (3.27 Å), indicating the presence of nonbonding attractive interactions between these atoms. In the packing diagram, BTQ-F molecules are oriented in a face-to-face fashion with minimum intermolecular π–π distances of 3.55 Å and 3.42 Å (a and b in Fig. 5(b), respectively). On the basis of the calculation by the Amsterdam Density Functional program at the PW91/TZP level, the transfer integrals between facial stacked structures of BTQ-F in the crystal were estimated. All the transfer integrals for electron transport (tLUMO) and hole transport (tHOMO) are shown in Fig. 5(c). BTQ-F showed a large tHOMO and tLUMO of 75.5 and 99.9 meV, respectively, which is expected to construct a charge-carrier transporting pathway along the stacking direction.
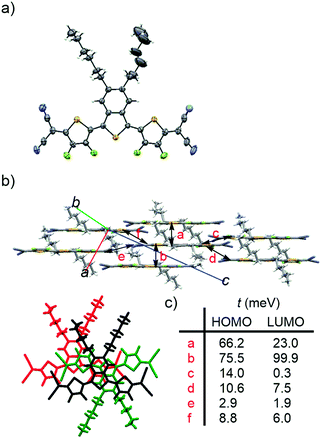 |
| | Fig. 5 (a) ORTEP drawing of the molecular structure, (b) packing diagram from side view (top) and top view (bottom), and (c) estimated transfer integrals of HOMOs and LUMOs for BTQ-F. | |
Charge carrier transport and film morphologies
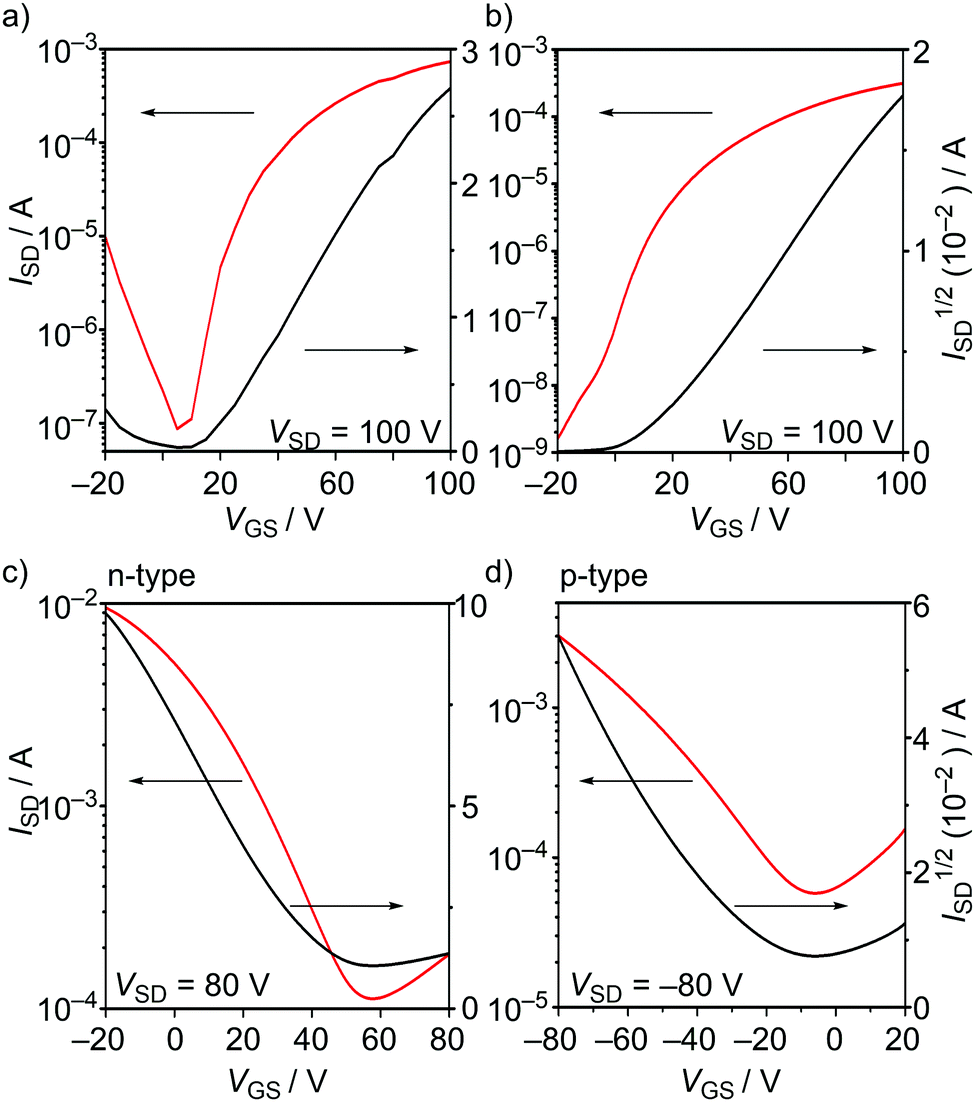
To evaluate the semiconducting charge-transport characteristics of BTQ, BTQ-F, BTQ-6, and TQ, OFET devices based on thin films were fabricated. Although there have been several reports on high or finely tuned ambipolar OFETs of ter- and quater-thiophene quinoids,5 the compounds used in this work were characterized using a conventional bottom-gate bottom-contact transistor configuration with similar fabrication conditions for all devices. The active layer was prepared by spin-coating from a 0.3 wt% CHCl3 solution onto hexamethyldisilazane (HMDS)-modified Si/SiO2 substrates. Thermal annealing was effective for improving the performance for BTQ and BTQ-F, and the current–voltage characteristics of best-performance devices are shown in Fig. 6 and Fig. S3, S4 (ESI†). The hole (μh) and electron mobilities (μe) were extracted from the transfer characteristics at constant source–drain voltages. The corresponding device parameters such as mobility, threshold voltage (Vth), and current on/off ratio (Ion/Ioff) are summarized in Table 2. Both BTQ and BTQ-F revealed solely an electron transport with μe in the order of 10−3 cm2 V−1 s−1 due to their low-lying LUMO energy levels. Furthermore, OFETs based on these compounds retained the same order of μe under air-exposed conditions. While the TQ-based device exhibited a higher μe compared with BTQ and BTQ-F, the Ion/Ioff ratio was quite low due to a high off source–drain current (Fig. S4(d), ESI†). This might indicate that the ambipolar character of the TQ framework affects the increase of carrier density even in the off state.6b
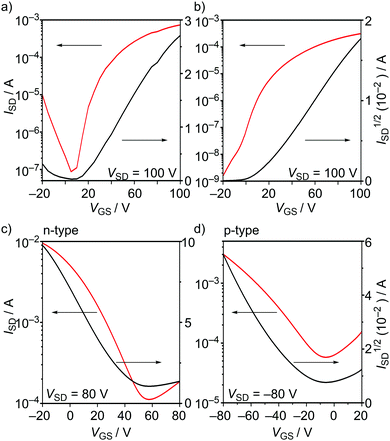 |
| | Fig. 6 (a) Transfer characteristics of OFETs using (a) BTQ, (b) BTQ-F, (c) BTQ-6, and (d) BTQ-6. VGS, ISD, and VSD denotes gate voltage, source–drain current, and source–drain voltage, respectively. | |
Table 2 Field-effect characteristics under vacuum and in air
| Compounds |
Anneal temp./°C |
V
th/V |
I
on/Ioff |
μ
e/cm2 V−1 s−1 (μh/cm2 V−1 s−1) |
|
Under vacuum.
In air.
Hole-transporting characteristics are shown in parenthesis.
|
|
BTQ
|
170 |
15 |
104 |
3.4 × 10−3 |
|
BTQ
|
170 |
18 |
105 |
2.5 × 10−3 |
|
BTQ-F
|
170 |
13 |
105 |
1.2 × 10−3 |
|
BTQ-F
|
170 |
−9.4 |
103 |
3.0 × 10−4 |
|
TQ
|
As cast |
35 |
101 |
3.3 × 10−2 |
|
TQ
|
150 |
22 |
102 |
3.4 × 10−2 |
|
BTQ-6
|
As cast |
78 |
101 |
8.9 × 10−4 |
| (As cast)c |
(−29)c |
(102)c |
(3.1 × 10−2)c |
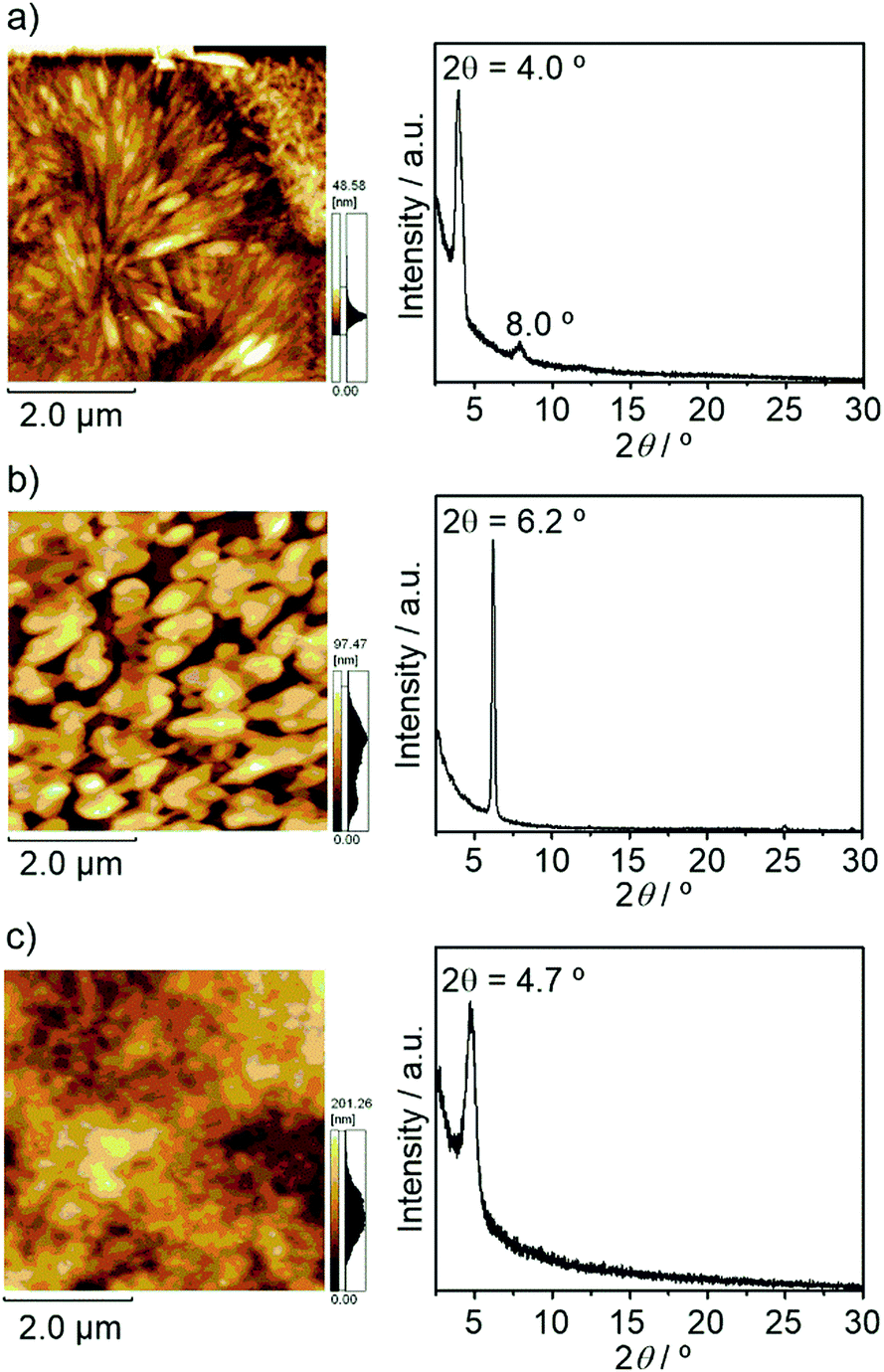
In order to investigate the film properties of these compounds, X-ray diffraction (XRD) and atomic force microscopy (AFM) measurements of these thin films on HMDS-modified Si/SiO2 substrates were performed. As shown in Fig. 7 and Fig. S5 (ESI†), these compounds exhibited clear diffraction peaks in X-ray diffractogram (XRD), indicating the formation of crystalline structures in thin films. The peak of BTQ-F at 2θ = 6.2° can be indexed as the (011) diffraction peak with a d spacing of 14.2 Å according to the above-mentioned X-ray crystal structure analysis. Thus, the BTQ-F molecules are edge-on arranged with the stacking direction parallel to the SiO2 surface. While the AFM image of BTQ revealed crystal-shaped micrometer-sized grains, BTQ-F showed a rough film surface with large crystal grains (Fig. 7). Both BTQ and BTQ-F maintained OFET responses under air-exposed conditions, irrespective of the difference of the film morphologies. This result indicates that the observed air-stability mainly originates from the thermodynamic stability, i.e., a low LUMO energy level of less than −4.0 eV. As discussed above, the introduction of benzene annelation led to the isomerization behavior different from that of TQ against temperature change. Therefore, to investigate the influence of the benzene annelation on the device characteristics, temperature-dependent OFET measurements of BTQ- and TQ-based devices were performed. For these measurements, we performed the surface modification of the source and drain gold electrodes using hexadecanethiol to minimize the contact resistance between Au electrodes and active layer.24 The decreased contact resistance was confirmed by the output characteristics (Fig. S6, ESI†).
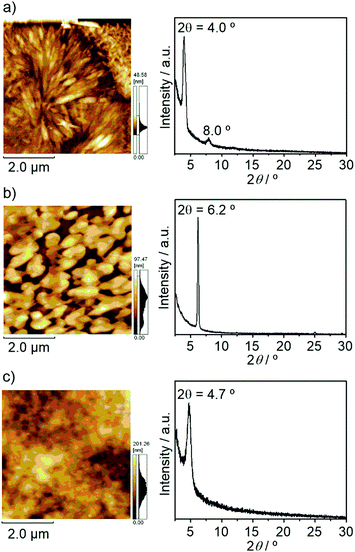 |
| | Fig. 7 AFM height images and XRD data of (a) BTQ, (b) BTQ-F, and (c) BTQ-6. | |
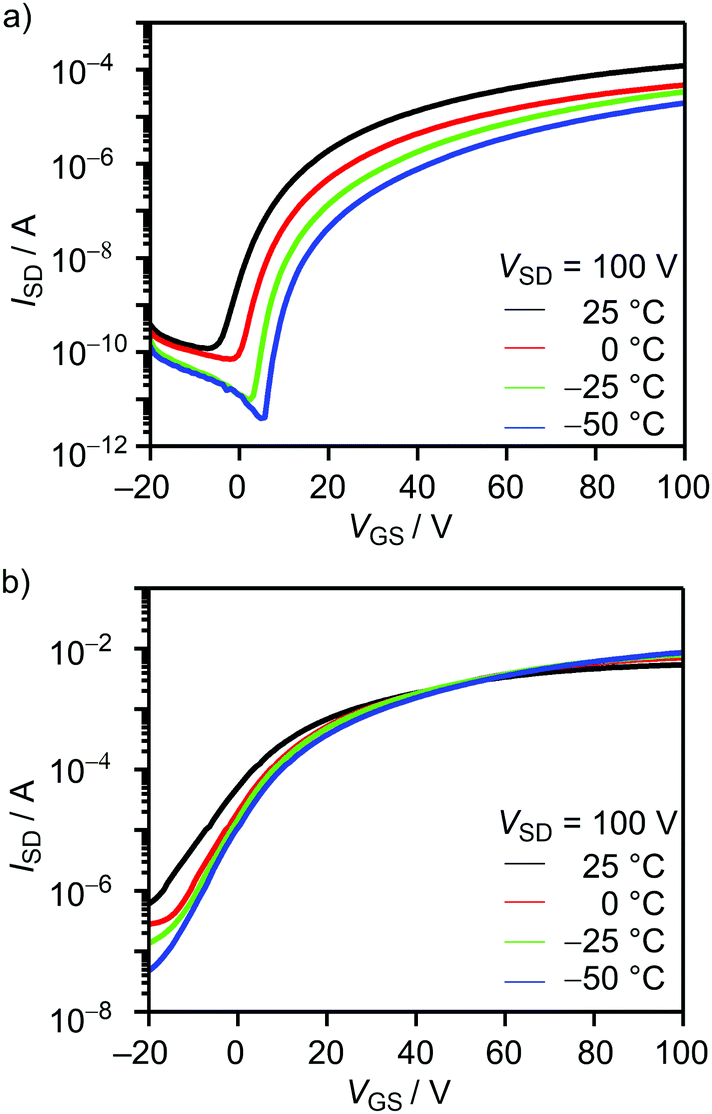
As shown in Fig. 8(a), OFETs using BTQ showed a decrease of electron mobility with decreasing temperature. This phenomenon is in agreement with the temperature dependence of hopping dominated carrier transport.25 The activation energy of BTQ was estimated to be 0.13 eV (Fig. S7, ESI†). On the other hand, the TQ-based device showed a reverse trend with a decrease in the off current (Fig. 8(b)). We speculate that the diminution of the biradical character for BTQ might facilitate the inherent electron transport, which means that biradicals act as carriers. Although the presence of the sterically bulky 2-ethylhexyl groups may prevent dense π–π stacking, XRD results indicate the crystalline structures in thin films (Fig. 7(c)). In fact, OFET devices based on BTQ-6 showed not only electron-transporting characteristics (8.9 × 10−4 cm2 V−1 s−1) but also hole-transporting characteristics (3.1 × 10−2 cm2 V−1 s−1). The appearance of p-type behavior can be explained by the increased HOMO energy levels. Note that the field-effect response for the quinoidal thiophene 6-mer is not reported so far. The OFET devices based on BTQ, TQ, and BTQ-6 were stored and measured periodically under the air-exposed conditions. As shown in Fig. S8 (ESI†), compared to TQ, a decrease of carrier mobility is suppressed for benzo[c]thiophene-containing semiconductors. We have considered that the introduction of the benzene annelation to form a stabilized quinoidal electronic structure becomes an effective strategy in developing new semiconducting materials.
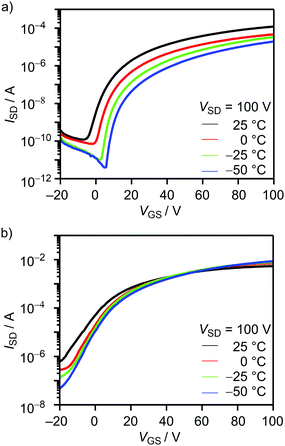 |
| | Fig. 8 Temperature-dependent transfer characteristics of the 1-hexadecanethiol-treated OFETs using (a) BTQ and (b) TQ. | |
Conclusions
In summary, to investigate the influence of benzene-annelation on the stabilization of the quinoidal electronic structure, we successfully synthesized new bis(dicyanomethylene)-substituted quinoidal oligothiophenes containing benzo[c]thiophene units. We also developed quinoidal oligothiophenes containing both a benzo[c]thiophene unit and fluorinated thiophenes. Theoretical calculations as well as experimental results indicate that the introduction of benzo[c]thiophene into the oligothiophene π-conjugated quinoid system is effective in stabilizing quinoidal electronic structures. These molecules showed good electron-transporting characteristics, and the extension of π-conjugation from thiophene 3-mer to 6-mer led to the appearance of both hole- and electron-transporting characteristics. These results show that quinoidal π-conjugated systems containing benzo[c]thiophene units show potential as candidates for semiconducting materials using stabilized quinoidal structures. Further investigations toward expansion of the π-conjugation are currently underway in our group.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (16H04191) and Innovative Areas (JP25110004) and “Dynamic Alliance for Open Innovation Bridging Human, Environmental and Materials” from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and ACT-C programs from the Japan Science and Technology Agency Japan. Thanks are extended to the Comprehensive Analysis Center (CAC), ISIR, for assistance in elemental analysis and HRMS. Open Access funding provided by the Max Planck Society.
Notes and references
-
(a) A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef PubMed;
(b) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef PubMed;
(c) Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef PubMed;
(d) J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef PubMed;
(e) S. M. McAfee, J. M. Topple, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2015, 3, 16393–16408 RSC.
-
(a) J. Yang, D. Yan and S. T. Jones, Chem. Rev., 2015, 115, 5570–5603 CrossRef PubMed;
(b) O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef PubMed.
- K. Deuchert and S. Hünig, Angew. Chem., Int. Ed., 1978, 17, 875–886 CrossRef.
-
(a) J. B. Torrance, Acc. Chem. Res., 1979, 12, 79–86 CrossRef;
(b) N. Martín, J. L. Segura and C. Seoane, J. Mater. Chem., 1997, 7, 1661–1676 RSC.
- J. Casado, R. P. Ortiz and J. T. L. Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 RSC.
-
(a) T. M. Pappenfus, R. J. Chesterfield, C. D. Frisbie, K. R. Mann, J. Casado, J. D. Raff and L. L. Miller, J. Am. Chem. Soc., 2002, 124, 4184–4185 CrossRef PubMed;
(b) R. J. Chesterfield, C. R. Newman, T. M. Pappenfus, P. C. Ewbank, M. H. Haukaas, K. R. Mann, L. L. Miller and C. D. Frisbie, Adv. Mater., 2003, 15, 1278–1282 CrossRef;
(c) D. E. Janzen, M. W. Burand, P. C. Ewbank, T. M. Pappenfus, H. Higuchi, D. A. da Silva Filho, V. G. Young, J.-L. Brédas and K. R. Mann, J. Am. Chem. Soc., 2004, 126, 15295–15308 CrossRef PubMed.
- S. Handa, E. Miyazaki, K. Takimiya and Y. Kunugi, J. Am. Chem. Soc., 2007, 129, 11684–11685 CrossRef PubMed.
-
(a) J. C. Ribierre, T. Fujihara, S. Watanabe, M. Matsumoto, T. Muto, A. Nakao and T. Aoyama, Adv. Mater., 2010, 22, 1722–1726 CrossRef PubMed;
(b) J.-C. Ribierre, S. Watanabe, M. Matsumoto, T. Muto, A. Nakao and T. Aoyama, Adv. Mater., 2010, 22, 4044–4048 CrossRef PubMed;
(c) J. C. Ribierre, S. Watanabe, M. Matsumoto, T. Muto and T. Aoyama, Appl. Phys. Lett., 2010, 96, 083303 CrossRef.
- J. Li, X. Qiao, Y. Xiong, W. Hong, X. Gao and H. Li, J. Mater. Chem. C, 2013, 1, 5128–5132 RSC.
-
(a) Y. Qiao, Y. Guo, C. Yu, F. Zhang, W. Xu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2012, 134, 4084–4087 CrossRef PubMed;
(b) Y. Qiao, J. Zhang, W. Xu and D. Zhu, J. Mater. Chem., 2012, 22, 5706–5714 RSC;
(c) Q. Wu, S. Ren, M. Wang, X. Qiao, H. Li, X. Gao, X. Yang and D. Zhu, Adv. Mater., 2013, 23, 2277–2284 Search PubMed.
- Y. Xiong, J. Tao, R. Wang, X. Qiao, X. Yang, D. Wang, H. Wu and H. Li, Adv. Mater., 2016, 28, 5949–5953 CrossRef PubMed.
-
(a) Q. Wu, R. Li, W. Hong, H. Li, X. Gao and D. Zhu, Chem. Mater., 2011, 23, 3138–3140 CrossRef;
(b) Q. Wu, S. Ren, M. Wang, X. Qiao, H. Li, X. Gao, X. Yang and D. Zhu, Adv. Funct. Mater., 2013, 23, 2277–2284 CrossRef;
(c) J. Li, X. Qiao, Y. Xiong, H. Li and D. Zhu, Chem. Mater., 2014, 26, 5782–5788 CrossRef.
- T. Mori, N. Yanai, I. Osaka and K. Takimiya, Org. Lett., 2014, 16, 1334–1337 CrossRef PubMed.
-
(a) V. Hernández, S. C. Losada, J. Casado, H. Higuchi and J. T. L. Navarrete, J. Phys. Chem. A, 2000, 104, 661–672 CrossRef;
(b) T. Takahashi, K.-i. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928–8929 CrossRef PubMed;
(c) R. P. Ortiz, J. Casado, V. Hernández, J. T. L. Navarrete, P. M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Angew. Chem., Int. Ed., 2007, 46, 9057–9061 CrossRef PubMed;
(d) E. V. Canesi, D. Fazzi, L. Colella, C. Bertarelli and C. Castiglioni, J. Am. Chem. Soc., 2012, 134, 19070–19083 CrossRef PubMed;
(e) R. Kishi, M. Dennis, K. Fukuda, Y. Murata, K. Morita, H. Uenaka and M. Nakano, J. Phys. Chem. C, 2013, 117, 21498–21508 CrossRef;
(f) L. Colella, L. Brambilla, V. Nardone, E. Parisini, C. Castiglioni and C. Bertarelli, Phys. Chem. Chem. Phys., 2015, 17, 10426–10437 RSC;
(g) S. Ray, S. Sharma, U. Salzner and S. Patil, J. Phys. Chem. C, 2017, 121, 16088–16097 CrossRef.
- F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382–3384 CrossRef.
-
(a) C. Zhang, Y. Zang, E. Gann, C. R. McNeill, X. Zhu, C.-a. Di and D. Zhu, J. Am. Chem. Soc., 2014, 136, 16176–16184 CrossRef PubMed;
(b) C. Zhang, Y. Zang, F. Zhang, Y. Diao, C. R. McNeill, C.-a. Di, X. Zhu and D. Zhu, Adv. Mater., 2016, 28, 8456–8462 CrossRef PubMed;
(c) L. Ren, H. Fan, D. Huang, D. Yuan, C.-a. Di and X. Zhu, Chem. – Eur. J., 2016, 22, 17136–17140 CrossRef PubMed;
(d) L. Ren, D. Yuan, E. Gann, Y. Guo, L. Thomsen, C. R. McNeill, C.-a. Di, Y. Yi, X. Zhu and D. Zhu, Chem. Mater., 2017, 29, 4999–5008 CrossRef;
(e) C. Zhang, D. Yuan, H. Wu, E. Gann, L. Thomsen, C. R. McNeill, C.-a. Di, X. Zhu and D. Zhu, J. Mater. Chem. C, 2017, 5, 1935–1943 RSC.
- D. Lorcy, K. D. Robinson, Y. Okuda, J. L. Atwood and M. P. Cava, J. Chem. Soc., Chem. Commun., 1993, 345–347 RSC.
- R. Kishi, S. Ochi, S. Izumi, A. Makino, T. Nagami, J.-y. Fujiyoshi, N. Matsushita, M. Saito and M. Nakano, Chem. – Eur. J., 2016, 22, 1493–1500 CrossRef PubMed.
- H. Higuchi, S. Yoshida, Y. Uraki and J. Ojima, Bull. Chem. Soc. Jpn., 1998, 71, 2229–2237 CrossRef.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods-Fundamentals and Applications, Wiley, New York, 1984 Search PubMed.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bassler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef PubMed.
- K. Asadi, F. Gholamrezaie, E. C. P. Smits, P. W. M. Blom and B. de Boer, J. Mater. Chem., 2007, 17, 1947–1953 RSC.
- A. Pivrikas, M. Ullah, H. Sitter and N. S. Sariciftci, Appl. Phys. Lett., 2011, 98, 092114 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The results of synthetic details, NMR measurements, X-ray information, OFET device information, and computational details. CCDC 1837054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc01802b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Masashi
Nitani
a,
Norimitsu
Tohnai
*abc,
Masashi
Nitani
a,
Norimitsu
Tohnai