
Highly selective, red emitting BODIPY-based fluorescent indicators for intracellular Mg2+ imaging†

Abstract
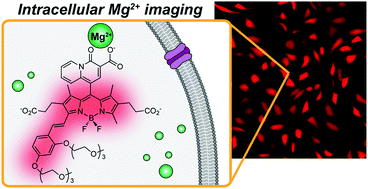
Most fluorescent indicators for Mg2+ suffer from poor selectivity against other divalent cations, especially Ca2+, thus do not provide reliable information on cellular Mg2+ concentrations in processes in which such metals are involved. We report a new set of highly selective fluorescent indicators based on alkoxystyryl-functionalized BODIPY fluorophores decorated with a 4-oxo-4H-quinolizine-3-carboxylic acid metal binding moiety. The new sensors, MagQ1 and MagQ2, display absorption and emission maxima above 600 nm, with a 29-fold fluorescence enhancement and good quantum yields (Φ > 0.3) upon coordination of Mg2+ in aqueous buffer. Fluorescence response to Mg2+ is not affected by the presence of competing divalent cations typically present in the cellular milieu, and displays minimal pH dependence in the physiologically relevant range. The choice of alkoxy groups decorating the styryl BODIPY core does not influence the basic photophysical and metal binding properties of the compounds, but has a marked effect on their intracellular retention and thus in their applicability for detection of cellular Mg2+ by fluorescence imaging. In particular, we demonstrate the utility of a triethyleneglycol (TEG) functionalization tactic that endows MagQ2 with superior cellular retention in live cells by reducing active extrusion through organic anion transporters, which are thought to cause fast leakage of typical anionic dyes. With enhanced retention and excellent photophysical properties, MagQ2 can be applied in the detection of cellular Mg2+ influx without interference of high concentrations of Ca2+ akin to those involved in signaling.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

