
Multimodal underwater adhesion using self-assembled Dopa-bearing ABA triblock copolymer networks†
Xiaomin
Tang
 a and
Christopher J.
Bettinger
*ab
a and
Christopher J.
Bettinger
*ab
aDepartment of Materials Science and Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA. E-mail: cbetting@andrew.cmu.edu
bDepartment of Biomedical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA
First published on 21st November 2017
Abstract
Self-assembled mechanically robust Dopa-bearing triblock copolymer networks improve underwater adhesion through both energy dissipation and interfacial bonding. Polymer networks that incorporate energy dissipating motifs could improve the performance of high-performance wet adhesives that only use interfacial bonds.
Underwater adhesives have many applications ranging from functional materials in marine environments to biomedical implants and coatings.1–3 Bioinspired approaches are largely based on recapitulating the chemistry of mussel foot proteins (mfps) and have been very effective, to date.4–7 3,4-dihydroxyphenylalanine (Dopa) residues found in natural mfps increase the interfacial adhesion to many substrate materials by forming coordination bonds, hydrogen bonds and π–π stacking.8–11 More recently, cation–π interactions between Dopa residues and amine groups in flanking amino acid residues in mfps can also improve adhesion by enhancing interfacial bonding.7 Dopa residues and synergistic adhesion-promoting components can be incorporated as pendant groups or end groups in synthetic polymers to create underwater adhesives.12–16 Despite biomimetic chemistry, there are large gaps in performance between synthetic polymers and natural mfps in terms of interfacial adhesion and toughness.11,17,18 Dopa groups in mfps also form reconfigurable bonds that approximate the strength of covalent networks. Transient intra- and inter-molecular bonding can dissipate energy and improve the mechanical toughness in both natural17,19,20 and synthetic adhesives.21–24 Synthetic Dopa-bearing hydrogel networks with reconfigurable coordination bonds exhibit self-healing properties.25 However, there are few, if any, synthetic underwater adhesives that improve performance by using reconfigurable bonds.26–30
Here we describe the synthesis and characterization of Dopa-bearing ABA triblock copolymers that can self-assemble into mechanically robust networks. We posit that catechol groups in Dopa molecules confer adhesive properties by promoting interfacial bonding and increasing energy dissipation through several modes of intra- and inter-molecular interactions.
The synthesis of ABA triblock copolymers is shown in Fig. 1. Poly(ethylene glycol)-based (PEG) macroinitiators are first extended with an active esterified methacrylic acid (methacrylic acid N-hydroxysuccinimide ester; NHSMA) by atom transfer radical polymerization (ATRP) to obtain poly(NHSMA)-b-PEG-b-poly(NHSMA) (ABA-Dopa(−)). ABA-Dopa(−) triblock copolymers are then conjugated with dopamine to produce poly(NHSMA)-b-PEG-b-poly(NHSMA)-Dopa (ABA-Dopa(+)). The resulting triblock copolymers exhibit immiscible A and B blocks, which can produce physical crosslinked networks via microphase separation.31,32 Monodisperse macroinitiators combined with precise radical polymerization afforded by ATRP permit fine control over the degree of polymerization (DP) of network precursors (poly(NHSMA)70-b-PEG228-b-poly(NHSMA)70; A70B228A70). Furthermore, amine-reactive NHSMA can support a Dopa conjugation ratio approaching 70% on a NHSMA monomer basis as calculated by 1H NMR and supported by UV-vis spectroscopy (see Fig. S2, S4 and S5, ESI†). A70B228A70 triblocks with the nominal Dopa conjugation ratio of 70% were chosen for subsequent network formation studies because the role of Dopa groups could be best elucidated with the maximum concentration of Dopa in phases rich in A segments of the ABA triblock copolymers.
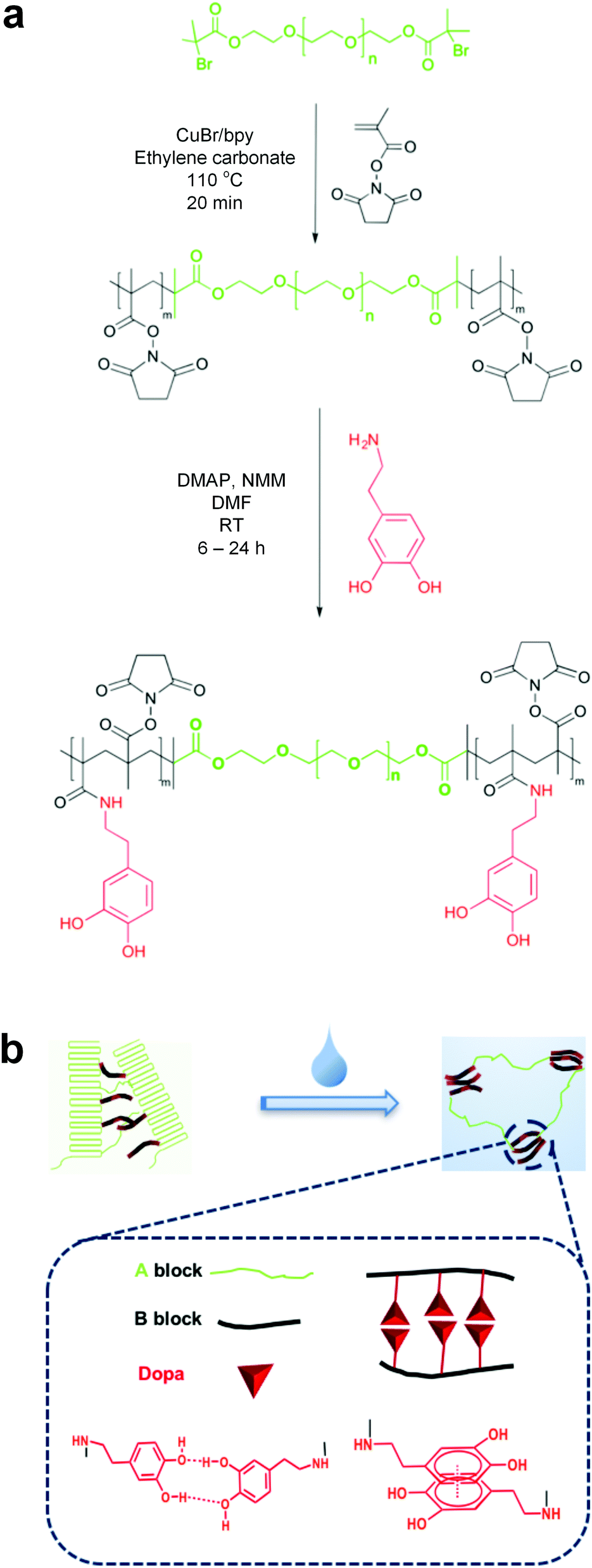 | ||
| Fig. 1 Design and processing of precursor. (a) Schematic showing the synthesis of poly(NHSMA)-b-PEG-b-poly(NHSMA) (ABA-Dopa(−)) and poly(NHSMA)-b-PEG-b-poly(NHSMA)-Dopa (ABA-Dopa(+)) triblock copolymers. (b) Schematic illustration of polymer processing to create the hydrated ABA-Dopa(+) network. | ||
Self-assembled polymer networks were prepared from both ABA-Dopa(−) and ABA-Dopa(+) polymer precursors. Concentrated polymer solutions in N,N-dimethylformamide (DMF) were first cast into films and then assembled into networks through phase separation by solvent exchange with excess water. The solvent exchange disrupts crystalline PEG domains as assessed by differential scanning calorimetry (DSC; Fig. S6, ESI†). We posit that the solvent exchange process generates phases rich in A blocks that serve as physical crosslinks and are joined together by PEG-based B blocks. Mechanically robust networks can be produced through solvent exchange using precursors with B blocks of MWPEG = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (DPPEG = 228). Precursors of B blocks of DPPEG = 450 and DPPEG = 92 create brittle networks and therefore the mechanical properties were not investigated further in this study. The former may be caused by excess PEG crystallization and the later may be attributed to excessive loop formation. The length of the B block is therefore critical in realizing mechanically robust adhesive polymer networks.
000 g mol−1 (DPPEG = 228). Precursors of B blocks of DPPEG = 450 and DPPEG = 92 create brittle networks and therefore the mechanical properties were not investigated further in this study. The former may be caused by excess PEG crystallization and the later may be attributed to excessive loop formation. The length of the B block is therefore critical in realizing mechanically robust adhesive polymer networks.
Dynamic mechanical analysis (DMA) was performed on both ABA-Dopa(−) and ABA-Dopa(+) films. Networks prepared from ABA-Dopa(+) triblock copolymers exhibit solid-like behavior with a storage modulus of E′ ∼ 107 Pa that monotonically increases for ω = 0.1–100 rad s−1. The storage modulus is orders of magnitude larger than other types of hydrated catechol-bearing networks (Table S5, ESI†).33,34 Strikingly, the loss modulus E′′ of ABA-Dopa(+) networks, which is 5 times larger than the E′′ of ABA-Dopa(−) networks, possesses the same order with E′. In addition, the markedly increased tan(δ) suggests that ABA-Dopa(+) networks have a larger viscosity compared to ABA-Dopa(−) films. The differential mechanical properties are attributed to dynamic interactions between Dopa motifs in phases rich in A blocks. The loss modulus of ABA-Dopa(+) networks increases from E′′ = 5 to 10 MPa over ω = 0.1–100 rad s−1 whereas the loss modulus of ABA-Dopa(−) networks is largely constant at E′′ = 1–2 MPa in the same frequency range. The value of E′′ in ABA-Dopa(+) networks increases monotonically with frequency, which suggests multiple dissipation processes are present and span time scales across several orders of magnitude. Rapid dissipation processes at high frequencies are present in ABA-Dopa(+) networks and typically associated with reconfigurable bonds.35,36 The solid-like, yet viscous behavior of this material is attributed to intra- and inter-molecular interactions of Dopa within phases rich in A blocks.
The viscoelastic properties of networks composed of ABA-Dopa(+) polymers were further studied using stress relaxation measurements and modeled using a Maxwell–Wiechert linear viscoelastic model (Fig. 2b). The best fit for the Maxwell–Wiechert model was achieved using the following four elements in parallel: three Maxwell elements plus an additional elastic element. The relaxation times of each corresponding Maxwell element are 0.76 ± 0.009, 9.87 ± 1.48, and 114.28 ± 11.91 s, respectively. These relaxation modes span three orders of magnitude and are associated with multiple dissipation processes from Dopa interactions concentrated in phases rich in A blocks and PEG bridges rich in B blocks. H-Bonding in polymer networks exhibits a characteristic relaxation time of 3–6 s.37 Based on the relation between relaxation time τ and bond energy Ea given by τ ∝ eEa/RT, the relaxation time of 0.76 s is consistent with dissipation due to π–π interactions. Relatively longer relaxation time scale could be related to the dissociation of H-bonding clusters or entanglements in PEG chains.38,39
 | ||
| Fig. 2 Viscoelastic properties of ABA-Dopa(+) networks comparison to linear viscoelastic models. (a) Storage (E′) and loss (E′′) modulus are plotted as a function of angular frequency obtained from tensile oscillatory sweeps (ε = 0.5%; ω = 0.1–100 rad s−1). (b) Plots of stress relaxation curves for both experimental data and fitting curve using Maxwell–Wiechert model (inset). | ||
Pendant Dopa residues improve the mechanical toughness and resilience of ABA triblock copolymer networks. The tensile properties of both ABA-Dopa(−) and ABA-Dopa(+) networks are shown in Fig. 3a. The Young's moduli of films composed of ABA-Dopa(+) and ABA-Dopa(−) polymers are EABA-Dopa(+) = 32 ± 4.7 MPa and EABA-Dopa(−) = 28 ± 2.8 MPa, respectively. Although the Young's moduli are comparable, the tensile toughness and maximum elongation are greatly increased in networks composed of ABA-Dopa(+) polymers (Table S2, ESI†). Networks prepared from ABA-Dopa(+) polymers exhibit rate-dependent tensile toughness, values for maximum elongation and breaking stress (Fig. 3b and Table S2, ESI†). This behavior has been observed previously in crosslinked networks that contain reconfigurable H-bonds and ionic cross-links.38,40 We anticipate that reconfigurable transient bonds in phases rich in A blocks produce a cycle-dependent toughness (Fig. 3c). The strain energy dissipated reduces larger than half from the first cycle to the second cycle. If a recovery time of 5 min is permitted, the strain energy dissipated in the second cycle is increased substantially (Fig. 3d and Table S3, ESI†). The observed time-dependent recovery of energy dissipation is attributed to the reformation of transient intra- and inter-molecular bonds between Dopa groups in physical crosslinks rich in A blocks.
 | ||
| Fig. 3 Mechanical properties of networks. (a) Tensile test results for networks prepared from ABA-Dopa(−) and ABA-Dopa(+) triblock copolymer precursors at a strain rate of 2 mm min−1 (see Experimental methods for details). (b) Stress–strain curves of networks prepared from ABA-Dopa(+) as a function of strain rate. (c) Cyclic tensile curves of films prepared from ABA-Dopa(+) at a nominal strain of ε = 80%: first loading–unloading cycle (n = 1, red); successive stretching curve (n = 2, orange); cycle after 5 min rest (after 5 min, n = 3, blue). (d) Work of extension of films at nominal strains of ε = 10%, 50%, and 80% for n = 1, 2, and 3 (after 5 min), respectively. | ||
Dopa groups increase underwater adhesion in polymers by enhancing interfacial bonding and increasing the energy dissipation capacity within the polymer network. Physically crosslinked polymer networks prepared from ABA-Dopa(−) polymers exhibit limited adhesion compared to polymer networks prepared from ABA-Dopa(+). These results suggest that catechol groups increase interfacial adhesion in hydrated conditions even if they are embedded in phases rich in A blocks (Fig. 4a). The work of adhesion in wet environments for ABA-Dopa(+) is also a strong function of the retraction rate of the probe (Fig. 4b and Table S4, ESI†), an observation that is consistent with the tensile rate-dependent toughness. ABA-Dopa(+) networks exhibit broad relaxation times ranging from 0.76 to 114 s. A force–distance measurement with a retraction velocity of 2 mm min−1 rate requires 450 s to complete. This time scale permits all observed dissipation modes to occur. Conversely, force–distance measurements using retraction velocities of 10 mm min−1 and 30 mm min−1 (requiring 90 and 30 s to complete, respectively) restrict some dissipation modes and therefore produce smaller values for the calculated work of adhesion. We infer that networks prepared from ABA-Dopa(+) triblock polymers enhance underwater adhesion by increasing the areal density of interfacial bonds and creating dissipative networks in physical crosslinks composed of phases rich in Dopa-bearing A blocks.
 | ||
| Fig. 4 Wet adhesion properties of ABA triblock copolymer networks. (a) Stress–strain curves extracted from force–distance curves between glass probes and substrates composed of networks prepared from ABA-Dopa(−) and ABA-Dopa(+) polymers. (b) Stress–strain curves for ABA-Dopa(+) networks performed at different retraction rates. | ||
We have designed and synthesized networks composed of Dopa-bearing self-assembled ABA triblocks with the following characteristic mechanical properties: a Young's modulus of E ∼ 28 MPa; an ultimate tensile stress of σb ∼ 1.1 MPa; and a maximum elongation at break of εmax ∼ 130% (Table S2, ESI†). ABA-Dopa(+) triblock polymers form mechanically resilient networks that exhibit adhesion in hydrated conditions. Compared to ABA-Dopa(−) films, Dopa-bearing polymer networks promote adhesion through mechanisms including interfacial bonding and strain energy dissipation through multiple types of intra- and inter-molecular bonds (Table 1). The synthesis strategy described herein affords molecular control over the composition of network precursors by using B blocks of monodisperse telechelic macroinitiators that can be extended to create A blocks with precise DP and degree of substitution of Dopa. In addition to H-bonding and π–π interactions, it may be possible to incorporate additional energy dissipation modes through reconfigurable coordination bonding between catechols and cations to further enhance underwater adhesion. The collective chemical and mechanical properties afforded by the unique molecular design suggest that ABA-Dopa(+) triblock polymers have potential applications as functional materials for marine environments, surgical adhesives, or substrate materials for ultracompliant hydrogel-based electronic devices.16,41
| Sample | Dopa ratioa (%) | Storage modulus, E′ (Pa) | Loss modulus, E′′ (Pa) | Toughness, We (MJ m−3) | Work of adhesion, Wad (J m−2) |
|---|---|---|---|---|---|
| a Dopa ratio (%) indicates the mole fraction of the Dopa conjugated A block in the total A block. b Below the detection limit of our instrumentation and methods. | |||||
| ABA-Dopa(+) | 70 | (0.7–8) × 107 | (0.55–1) × 107 | 1.55 ± 0.43 | 0.12 ± 0.01 |
| ABA-Dopa(−) | 0 | (2–4) × 107 | (0.1–0.2) × 106 | 0.025 ± 0.021 | 0b |
Author contributions
C. J. B. and X. M. T. designed the synthesis scheme, methodology for characterizing the material, and wrote the manuscript. X. M. T. performed the experiments.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
X. M. T. thanks to the following people for the help and discussion of synthesis, dynamic mechanical test and adhesion test. They are Liye Fu, Hangjun Ding, Yi Shi, Bill Pingitore, Sipei Li and Iksoo Kwon. The authors acknowledge financial support provided by the following organizations: National Institutes of Health (R21NS095250); the Defense Advanced Research Projects Agency (D14AP00040); the National Science Foundation (DMR1542196); the Carnegie Mellon University School of Engineering. The authors would also like to thank the CMU Thermomechanical Characterization Facility in the Department of Materials Science and Engineering.References
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS PubMed.
- N. Annabi, A. Tamayol, S. R. Shin, A. M. Ghaemmaghami, N. A. Peppas and A. Khademhosseini, Nano Today, 2014, 9, 574–589 CrossRef CAS PubMed.
- G. Kaplan, Plast. Reconstr. Surg., 1966, 37, 139–142 CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- D. G. Barrett, G. G. Bushnell and P. B. Messersmith, Adv. Healthcare Mater., 2013, 2, 745–755 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- Q. Zhao, D. W. Lee, B. K. Ahn, S. Seo, Y. Kaufman, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2016, 15, 407–412 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- T. Utzig, P. Stock and M. Valtiner, Angew. Chem., Int. Ed., 2016, 55, 9523–9527 CrossRef PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- H. G. Silverman and F. F. Roberto, Mar. Biotechnol., 2007, 9, 661–681 CrossRef CAS PubMed.
- C. R. Matos-Perez, J. D. White and J. J. Wilker, J. Am. Chem. Soc., 2012, 134, 9498–9505 CrossRef CAS PubMed.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CAS.
- C. E. Brubaker, H. Kissler, L. J. Wang, D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 420–427 CrossRef CAS PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- H. Wu, V. Sariola, C. Zhu, J. Zhao, M. Sitti and C. J. Bettinger, Adv. Mater., 2015, 27, 3398–3404 CrossRef CAS PubMed.
- J. J. Wilker, Science, 2015, 349, 582–583 CrossRef CAS PubMed.
- J. H. Waite, N. H. Andersen, S. Jewhurst and C. J. Sun, J. Adhes., 2005, 81, 297–317 CrossRef CAS.
- D. S. Hwang, M. J. Harrington, Q. Y. Lu, A. Masic, H. B. Zeng and J. H. Waite, J. Mater. Chem., 2012, 22, 15530–15533 RSC.
- S. Kim, A. Faghihnejad, Y. Lee, Y. Jho, H. Zeng and D. S. Hwang, J. Mater. Chem. B, 2015, 3, 738–743 RSC.
- X. H. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. B. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210–1216 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. DiMarco, L. H. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Leei, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- K. R. Shull and C. Creton, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 4023–4043 CrossRef CAS.
- A. Roos and C. Creton, Macromolecules, 2005, 38, 7807–7818 CrossRef CAS.
- N. Amouroux, J. Petit and L. Leger, Langmuir, 2001, 17, 6510–6517 CrossRef CAS.
- A. Lindner, B. Lestriez, S. Mariot, C. Creton, T. Maevis, B. Luhmann and R. Brummer, J. Adhes., 2006, 82, 267–310 CrossRef CAS.
- J. Li, A. D. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. R. Seo, N. V. Vasilyev, J. J. Vlassak, Z. Suo and D. J. Mooney, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- C. C. Zhu and C. J. Bettinger, Macromol. Rapid Commun., 2013, 34, 1446–1451 CrossRef CAS PubMed.
- C. C. Zhu and C. J. Bettinger, Macromolecules, 2015, 48, 1563–1572 CrossRef CAS.
- Y. Tanaka, K. Fukao and Y. Miyamoto, Eur. Phys. J. E: Soft Matter Biol. Phys., 2000, 3, 395–401 CrossRef CAS.
- S. Naficy, H. R. Brown, J. M. Razal, G. M. Spinks and P. G. Whitten, Aust. J. Chem., 2011, 64, 1007–1025 CrossRef CAS.
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. Annable, R. Buscall, R. Ettelaie and D. Whittlestone, J. Rheol., 1993, 37, 695–726 CrossRef CAS.
- X. B. Hu, J. Zhou, W. F. M. Daniel, M. Vatankhah-Varnoosfaderani, A. V. Dobrynin and S. S. Sheiko, Macromolecules, 2017, 50, 652–659 CrossRef CAS.
- X. B. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. X. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. A. Bao, Nat. Chem., 2016, 8, 619–625 Search PubMed.
- H. Wu, V. Sariola, J. Zhao, H. Ding, M. Sitti and C. Bettinger, Polym. Int., 2016, 65, 1355–1359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02371e |
| This journal is © The Royal Society of Chemistry 2018 |