
Structural effects of phosphate groups on apatite formation in a copolymer modified with Ca2+ in a simulated body fluid
Ryo
Hamai
a,
Hirotaka
Maeda
b,
Hikaru
Sawai
cd,
Yuki
Shirosaki
 e,
Toshihiro
Kasuga
b and
Toshiki
Miyazaki
*a
e,
Toshihiro
Kasuga
b and
Toshiki
Miyazaki
*a
aGraduate School of Life Science and Systems Engineering, Kyushu Institute of Technology, 2-4, Hibikino, Wakamatsu-ku, Kitakyushu, 808-0196, Japan. E-mail: tmiya@life.kyutech.ac.jp; Fax: +81-93-695-6025; Tel: +81-93-695-6025
bDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Japan
cFukushima Prefectural Centre for Environmental Creation, Japan
dInstitute of Environmental Radioactivity, Fukushima University, Japan
eGraduate School of Engineering, Kyushu Institute of Technology, Japan
First published on 5th December 2017
Abstract
Organic–inorganic composites are novel bone substitutes that can ameliorate the mismatch of Young's moduli between natural bone and implanted ceramics. Phosphate groups contribute to the formation of apatite in a simulated body fluid (SBF) and the adhesion of osteoblast-like cells. Therefore, modification of a polymer with these functional groups is expected to enhance the ability of the organic–inorganic composite to bond with bone. Two phosphate groups have been used, phosphonic acid (–C–PO3H2) and phosphoric acid (–O–PO3H2). However, the effects of structural differences between these phosphate groups have not been clarified. In this study, the apatite formation of copolymers modified with Ca2+ and either –C–PO3H2 or –O–PO3H2 was examined. The mechanism of apatite formation is discussed based on analytical and computational approaches. The copolymers containing –O–PO3H2, but not those containing –C–PO3H2, formed apatite in the SBF, although both released similar amounts of Ca2+ into the SBF. Adsorption of HPO42− from –O–PO3H2 in the SBF following Ca2+ adsorption was confirmed by zeta-potential measurement and X-ray photoelectron spectroscopy. The measurement of the complex formation constant revealed that the –O–PO32−⋯Ca2+ complex was thermodynamically unstable enough to convert into CaHPO4, which was not the case with –C–PO32−⋯Ca2+. The formation of CaHPO4-based clusters was found to be a key factor for apatite nucleation. In conclusion, this study revealed that modification with –O–PO3H2 was more effective for enhancing apatite formation compared with –C–PO3H2.
1. Introduction
The ability to bond to existing bone is one of the essential properties for permanent implanted bone substitutes used to repair defects. Bioactive ceramics, such as Bioglass,1 the glass-ceramic A-W,2 and sintered hydroxyapatite3 can bond to living bone without encapsulation by fibrous tissue. They are able to form bone-like apatite with low levels of crystallinity on their surfaces in physiological environments. Although these materials are used clinically as bone substitutes, implantation may cause stress-shielding and subsequent bone absorption due to their higher Young's moduli compared with natural bone. Natural bone is an organic–inorganic composite consisting of collagen fibers and apatite. Here, the fabrication of artificial bioactive organic–inorganic composites is used to solve this mismatch problem. However, specific functional groups, such as Si–OH,4 Ti–OH,5 carboxyl (–COOH),6 and sulfonate (–SO3H)7 can contribute to the formation of a bone-like apatite layer in simulated body fluid (SBF). SBF has inorganic ion concentrations similar to human blood plasma.8,9 These groups interact with Ca2+ ions in the SBF, and induce heterogeneous nucleation of apatite. Thus, modifications to these functional groups is an important method for preparing composites possessing bone-bonding ability.Phosphate groups (–PO3H2) have also attracted a great deal of attention for improving the biocompatibility of implants, and their ability to bond to bone. Tanahashi et al. found that phosphate groups immobilized on gold in self-assembled monolayers exhibited better apatite-forming ability compared with carboxyl groups as a result of stronger interactions with Ca2+.6 Datta et al. prepared nanofibers and films of poly(vinyl alcohol) modified with phosphate groups via phosphorylation with phosphoric acid.7 They reported that such modification led to the formation of calcium phosphate in SBF, and the enhancement of the proliferation of osteoblast-like MG63 cells.10 Moreover, López-Pérez et al. showed that the number of SaOs-2 osteoblast-like cells attached on a poly(ε-polycaprolactone) scaffold increased after surface modification using poly(vinylphosphonic acid).11
We previously examined the effect of phosphate group content on the apatite-forming ability of copolymers based on vinylphosphonic acid treated with a CaCl2 solution.12 As a result, an increase in the phosphate group content actually inhibited apatite formation in SBF, despite the fact that the copolymer released Ca2+ into the SBF. These results indicate that the incorporation of phosphate groups using vinylphosphonic acid has an adverse effect on apatite formation in SBF.
To clarify these different phenomena, we focused on the molecular structures of the phosphate groups. They have two types of structure, phosphonic acid (–C–PO3H2) and phosphoric acid (–O–PO3H2). Tanahashi et al. and Datta et al. used –O–PO3H2, while we used –C–PO3H2. These structural differences are expected to affect the interactions with Ca2+ ions, and subsequent apatite formation, because the stability of the ionized phosphate groups is dependent on element substitution as a result of the inductive effect.13 However, this hypothesis has not been previously examined.
In this study, we prepared copolymers containing Ca2+ and either –C–PO3H2 or –O–PO3H2 under identical synthetic conditions and examined the apatite formation on their surfaces in SBF. The mechanism of apatite formation on the copolymers is discussed using chemical analytical and computational approaches.
2. Materials and methods
2.1 Preparation of copolymers modified with Ca2+ and different phosphate groups
Vinylphosphonic acid (VPA, 95%, Tokyo Chemical Industry Co., Ltd, Tokyo, Japan) and methacryloyloxyethyl phosphate (MOEP, 90%, Phosmer™ M, Yuni Chemical Co., Ltd, Nara, Japan) were used to incorporate the –C–PO3H2 and –O–PO3H2 phosphate groups, respectively, in the copolymers. 2-Hydroxyethyl methacrylate (HEMA, 95%, Wako Pure Chemical Industries, Ltd, Osaka, Japan) and triethylene glycol dimethacrylate (TEGDMA, 90%, Wako Pure Chemical Industries, Ltd) were mixed with VPA or MOEP. The molar ratio of the phosphate monomers, HEMA and TEGDMA was VPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HEMA:TEGDMA = 10:85:5 or MOEP:HEMA:TEGDMA = 7:88:5, and the total weight of the monomers was 10 g. Then, 0.5 wt% sodium p-toluene sulfinate (p-TSS, 98%, Tokyo Chemical Industry Co., Ltd) and 2 wt% of N,N′-dimethyl-p-toluidine (97%, Wako Pure Chemical Industries, Ltd) were added to the monomers. Then, 1 mol% of (±)-camphorquinone (97%, Wako Pure Chemical Industries, Ltd) was added, and the mixture was stirred for 1 hour with light shielding.
:HEMA:TEGDMA = 10:85:5 or MOEP:HEMA:TEGDMA = 7:88:5, and the total weight of the monomers was 10 g. Then, 0.5 wt% sodium p-toluene sulfinate (p-TSS, 98%, Tokyo Chemical Industry Co., Ltd) and 2 wt% of N,N′-dimethyl-p-toluidine (97%, Wako Pure Chemical Industries, Ltd) were added to the monomers. Then, 1 mol% of (±)-camphorquinone (97%, Wako Pure Chemical Industries, Ltd) was added, and the mixture was stirred for 1 hour with light shielding.
Then, the mixture was polymerized for 1 hour using irradiation with blue light (λ = 460 nm) and the obtained copolymers were dried at 60 °C for 1 day. The copolymers were cut into rectangles and polished with #1000 SiC waterproof abrasive paper. To remove the unreacted regents, the copolymers were soaked in ultra-pure water for one day at room temperature, and dried at 60 °C for 1 day. The final size of the copolymers was 10 × 10 × 1 mm (weight = 0.1 g). The copolymers were soaked in 30 cm3 of 0, 0.1, or 1 kmol m−3 CaCl2 solution at 36.5 °C for 1 day. The specimens prepared from VPA and MOEP were denoted VPA(C)xCa and MOEP(O)xCa, respectively, and x gives the molarity of the CaCl2 solution used.
2.2 Soaking specimens in SBF, 1.5CaSBF, or Tris–NaCl buffer
The SBF (Na+ 142.0, K+ 5.0, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3− 4.2, HPO42− 1.0, SO42− 0.5 mol m−3) at pH 7.40 was prepared as follows: NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, and Na2SO4 (Nacalai Tesque, Inc., Kyoto, Japan) were dissolved in ultrapure water, and the pH was adjusted with the addition of 1 kmol m−3 HCl and tris(hydroxymethyl) aminomethane (Nacalai Tesque, Inc.). In addition, a solution with a Ca2+ concentration 1.5 times that of the final SBF (1.5CaSBF) with a pH 7.25 was also prepared by the same method as the SBF.14 The specimens were soaked in 30 cm3 of SBF or 1.5CaSBF at 36.5 °C for various time intervals.The Tris–NaCl buffer (NaCl 142, tris(hydroxymethyl) aminomethane 50 mol m−3) at pH 7.40 was prepared by dissolving NaCl and tris(hydroxymethyl) aminomethane in ultrapure water. Then, the pH of the resulting solution was adjusted with the addition of 1 kmol m−3 HCl. The specimens were soaked in 30 cm3 of Tris–NaCl buffer at 36.5 °C for various time intervals to measure the amount of released Ca2+.
2.3 Characterization of the specimens
To measure the phosphate group content and acid dissociation constant (pKa) of the specimens, 30 cm3 of a 0.16 kmol m−3 NaCl solution, after soaking a VPA(C)0Ca or MOEP(O)0Ca sample in it, was titrated using 0.01 kmol m−3 NaOH solution at 36.5 °C. During the titration, the pH value of the solution was measured with a pH meter (F-23IIC, Horiba Ltd, Kyoto, Japan).The Ca content of the specimens before soaking in the SBF was measured by an energy-dispersive X-ray (EDX) analysis system (EMAX Energy, Horiba Ltd). The surfaces of the specimens after soaking in SBF or 1.5CaSBF were analyzed by thin-film X-ray diffraction (TF-XRD; MXP3V, Mac Science, Co., Yokohama, Japan), X-ray photoelectron spectroscopy (XPS; KRATOS Nova, KRATOS Analytical Ltd, Manchester, UK), and scanning electron microscopy (SEM; S-3500N, Hitachi Co., Tokyo, Japan). For the TF-XRD analysis, the angle of the incident X-rays (CuKα) was fixed at 1° with respect to the surface of the specimens. In the XPS analysis, AlKα (hν = 1486.6 eV) was used as the X-ray source, and the spectra were measured with a high-resolution scan at 20 eV of pass energy. The binding energies of these elemental spectra were calibrated with the binding energy of a methylene group (284.6 eV). The observed spectra was subjected to peak fitting using a blend function consisting of 70% Gaussian and 30% Lorentzian curves.
The zeta-potentials of the surfaces of the specimens were measured with a zeta-potential analyzer (Otsuka Electronics Co., Osaka, Japan) connected to a box-like quartz cell. After being soaked in SBF for various periods, the specimens were washed with ultrapure water, and then placed in a quartz cell. Fresh SBF mixed with polyethylene latex particles (Otsuka Electronics Co.) was introduced into the cell, and the electrophoretic mobilities of the particles were measured using the laser Doppler method to find the zeta-potentials.
The Ca and P concentrations, along with the pH of the SBF and Tris–NaCl buffers after soaking the copolymer specimens with Ca2+, were measured with an inductively coupled plasma atomic emission spectrometer (ICP-AES; ICPE-9820, SHIMADZU Co., Kyoto, Japan) and pH meter, respectively.
2.4 Evaluation of the complex formation constant
The stability constant of the polymer–Ca2+ complex was determined in accordance with the method reported by Szabadka et al.15 A 0.1 g sample of a VPA(C)0Ca or MOEP(O)0Ca copolymer was soaked in 30 cm3 of a 10 mol m−3 CaCl2 solution for 5 days at 36.5 °C. Then, they were soaked in 30 cm3 of the Tris–NaCl buffer, simulating physiological conditions (ion strength: I = 0.16, pH 7.40, 36.5 °C), for 5 days. The common logarithm of the stability constant (logβ) of the complex in the buffer was calculated using eqn (1):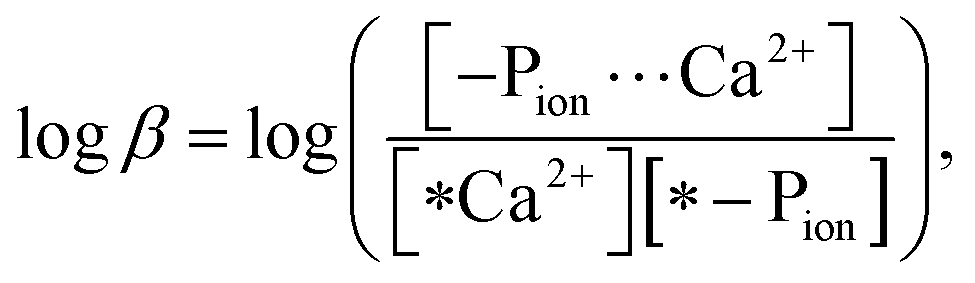 | (1) |
2.5 Simulation of the dipole moments and molecular orbitals
The structure with the lowest energy using each of the two phosphate groups was found using the restricted local density approximation and the Perdew–Wang correlational (LDA/PWC) method® with double-numerical basis sets, as implemented in the DMol3 software package®.16,17 The ground-state geometries were calculated with the phosphate groups bound to Ca as structural models to evaluate their electron density states by DMOl3. The dipole moments of the species were determined using the Hirshefeld approach.3. Results
Table 1 shows the phosphate group content in the copolymers prepared from different monomers, along with their acid dissociation constants at 36.5 °C measured via neutralizing titration. The VPA(C) and MOEP(O) copolymers contained nearly equivalent amounts of the phosphate groups. The first and second proton dissociation constants for MOEP(O) were lower than those of VPA(C).| VPA(C) | MOEP(O) | |
|---|---|---|
| Phosphate group/mmol g−1 | 0.24 ± 0.02 | 0.24 ± 0.002 |
| pKa1 | 3.63 | 3.48 |
| pKa2 | 6.11 | 5.96 |
Fig. 1(A) and (B) show the Ca/P molar ratios of the specimens after treatment with different concentrations of the CaCl2 solution, and the Ca concentration in the Tris–NaCl buffer after soaking the specimens. The ratios for the VPA(C) and MOEP(O) samples were very similar with regard to CaCl2 concentration. The Ca concentration in the Tris–NaCl buffer increased and became almost constant after 7 days for all the specimens. The Ca concentrations after 14 days satisfied MOEP(O)1Ca > VPA(C)1Ca ≈ MOEP(O)01Ca > VPA(C)01Ca.
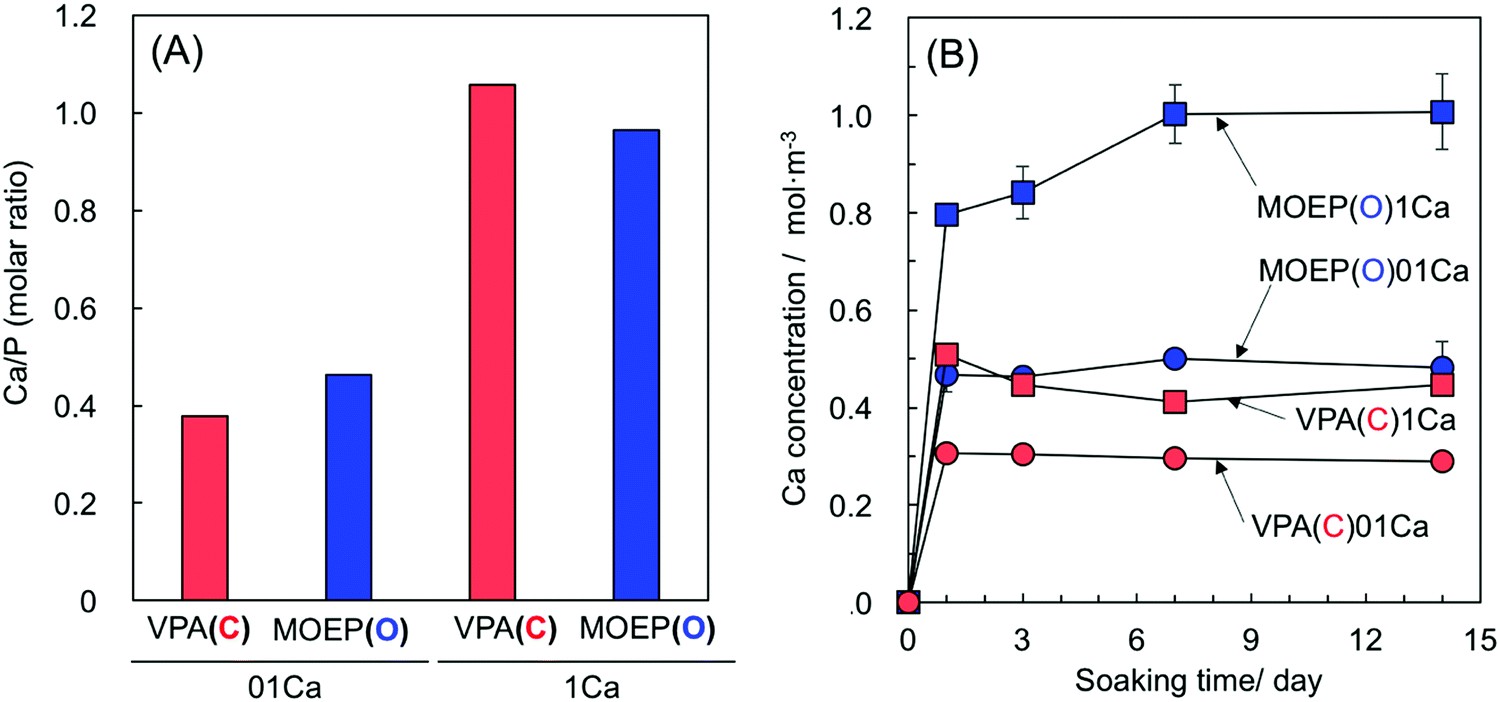 | ||
| Fig. 1 Ca/P molar ratio of the specimens after soaking in 0.1 or 1 kmol m−3 of CaCl2 solution (A) and Ca concentration in Tris–NaCl buffer following soaking of the different specimens (n = 3) (B). | ||
Fig. 2 shows the TF-XRD results of the specimens after soaking in SBF for various time intervals. The two broad peaks at 2θ = 26° and 32° corresponding to apatite (JCPDS #09-0432) were observed in MOEP(O)01Ca and MOEP(O)1Ca after 7 days and 1 day, respectively. Conversely, these peaks were not observed in VPA(C)01Ca or VPA(C)1Ca even after 14 days.
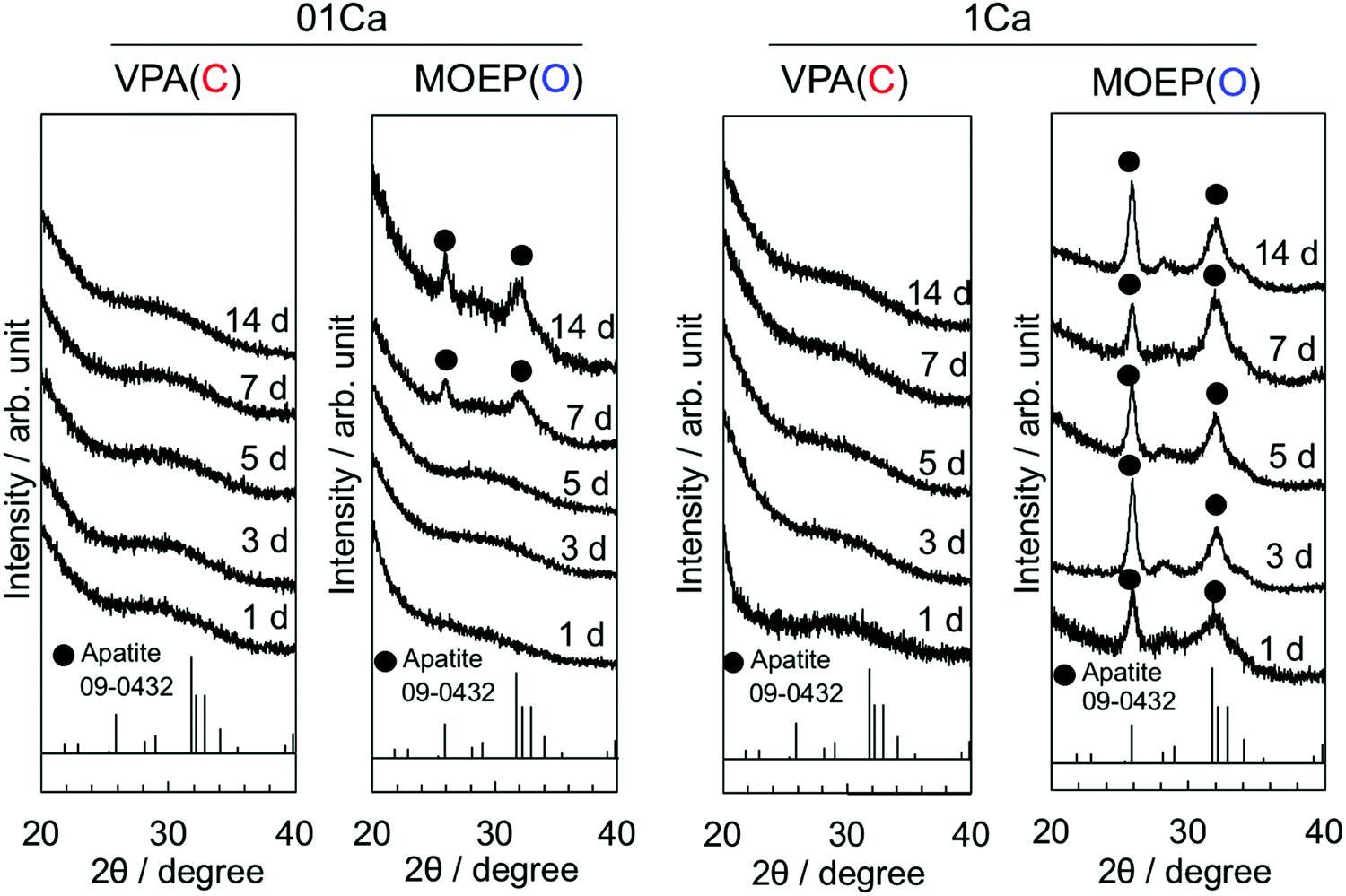 | ||
| Fig. 2 TF-XRD patterns of the specimens following soaking in SBF for various periods. | ||
Fig. 3 shows surface SEM images of different specimens that were soaked in SBF for various time intervals. Deposition was observed on the MOEP(O)01Ca and MOEP(O)1Ca samples after 7 days and 1 day, respectively, but not on the VPA(C)01Ca or VPA(C)1Ca surfaces. The scale-like morphology of the primary particles was similar to the apatite formed in the SBF.7
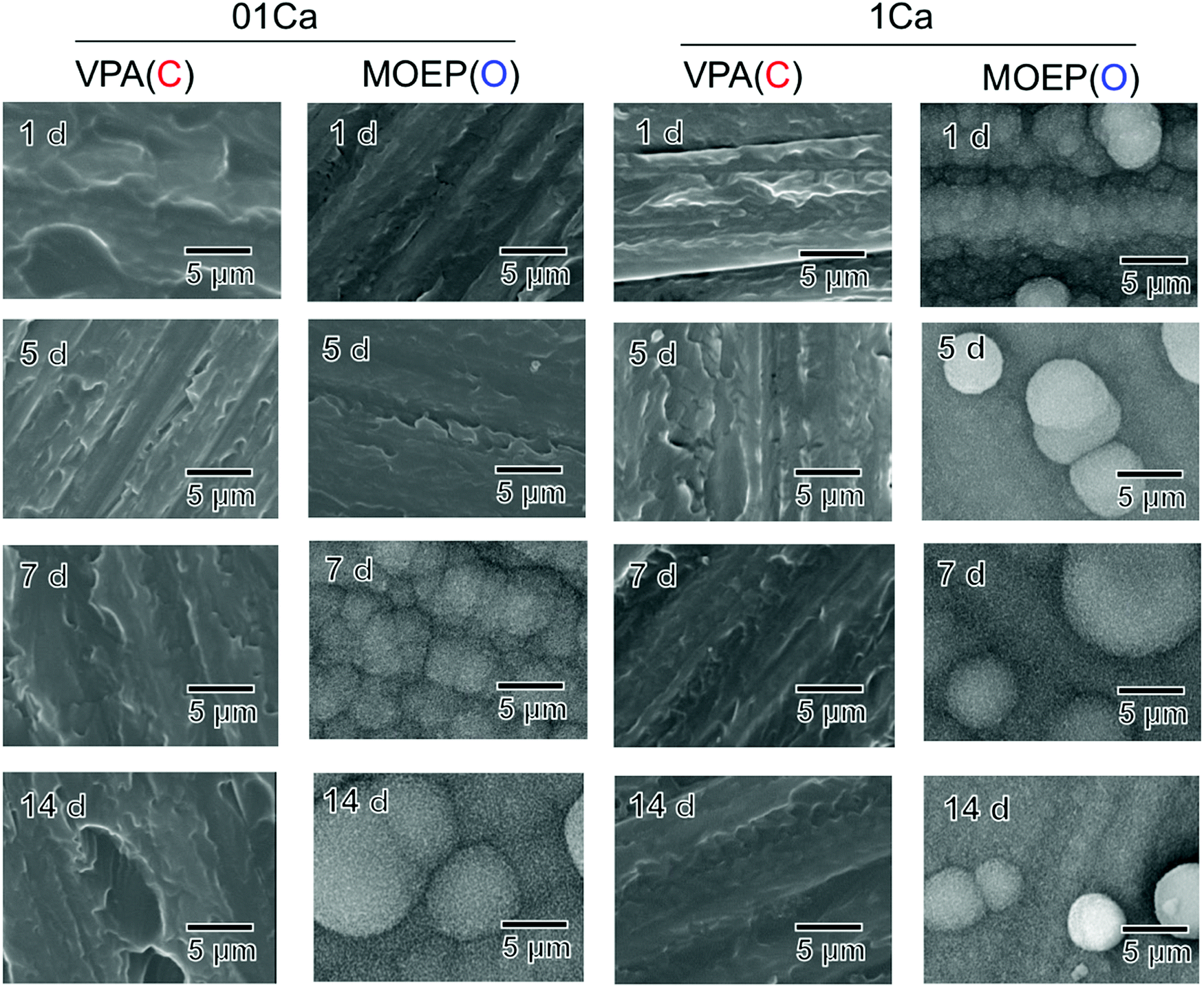 | ||
| Fig. 3 SEM images of the surfaces of the specimens following soaking in SBF for various periods. | ||
Fig. 4(A) shows the XPS spectra of the surfaces of VPA(C)1Ca and MOEP(O)01Ca after soaking in SBF for various time intervals. Peaks attributed to –C–PO3H2 and –C–PO3Ca were observed in VPA(C)1Ca at 133.2 and 132.7 eV, respectively.18 The peak position did not shift regardless of the soaking time. In contrast, the peaks attributed to –O–PO3H2 and –O–PO3Ca were observed in MOEP(O)01Ca at 134.5 and 133.8 eV, respectively.19 These peaks decreased after 3 hours, while those corresponding to CaHPO420 and octacalcium phosphate (OCP)21 became visible at 133.4 and 133.2 eV, respectively. Then, the peak corresponding to apatite was observed at 133.0 eV after 1 day.22Fig. 4(B) shows the change in the rate for each component of calcium phosphate formed on the MOEP(O)01Ca surface calculated from the area of the fitted peaks in the P2p spectra. The ratio of CaHPO4 to OCP initially increased during the first 3 hours, and then decreased with soaking time. Conversely, the signal from apatite monotonically increased, and the calcium phosphate phase was almost entirely apatite after 7 days.
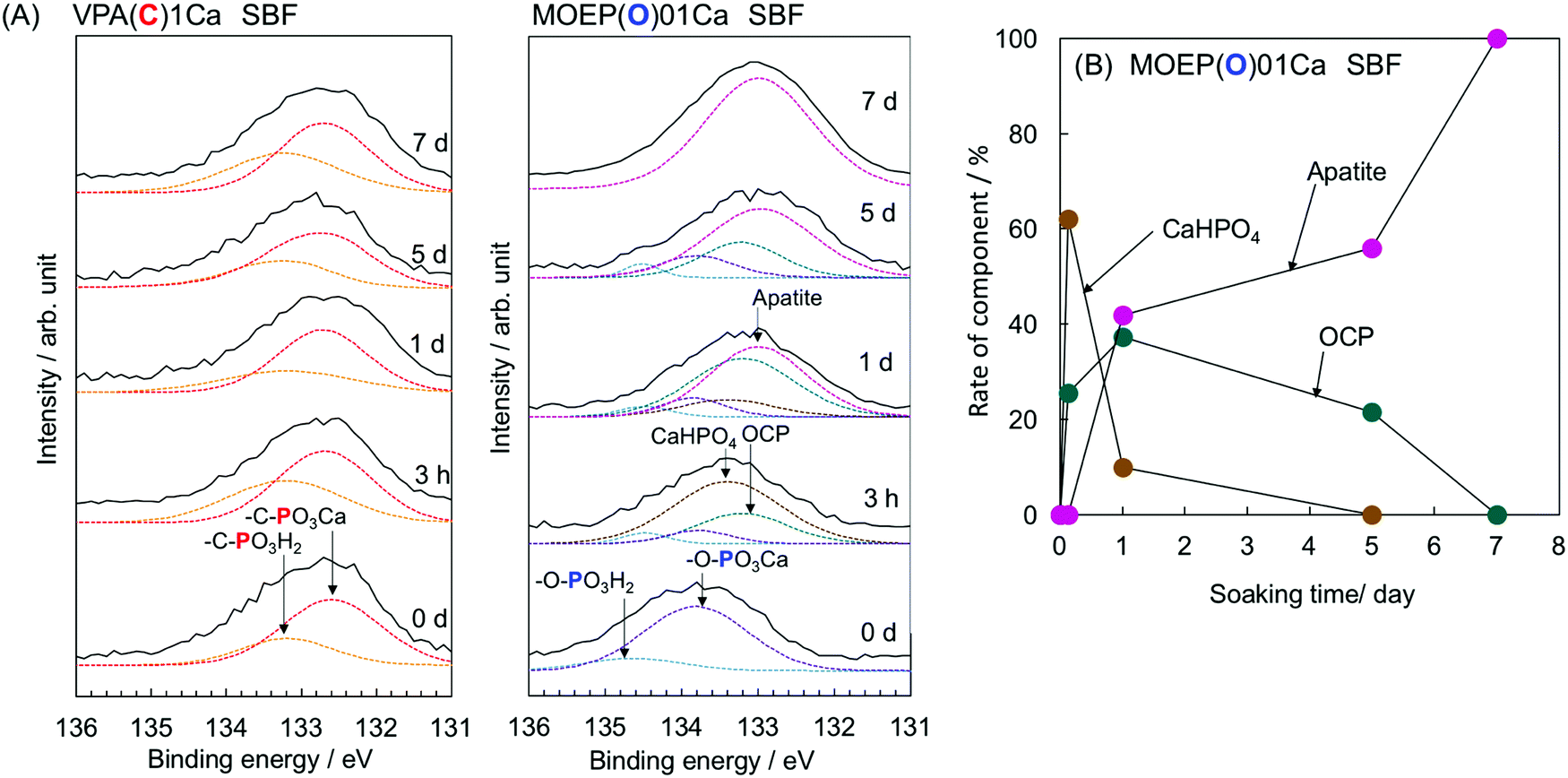 | ||
| Fig. 4 P2p XPS spectra of VPA(C)1Ca and MOEP(O)01Ca (A) and change in rate of component of calcium phosphate formed on the MOEP(O)01Ca (B) following soaking in SBF for various periods. | ||
Fig. 5 shows the changes in the surface zeta-potential of the VPA(C)1Ca and MOEP(O)01Ca samples after soaking in the SBF. The potential of both specimens changed from negative to positive after 5 days. Afterwards, the potential of VPA(C)1Ca was almost constant, while that of MOEP(O)01Ca decreased and became negative after 7 days.
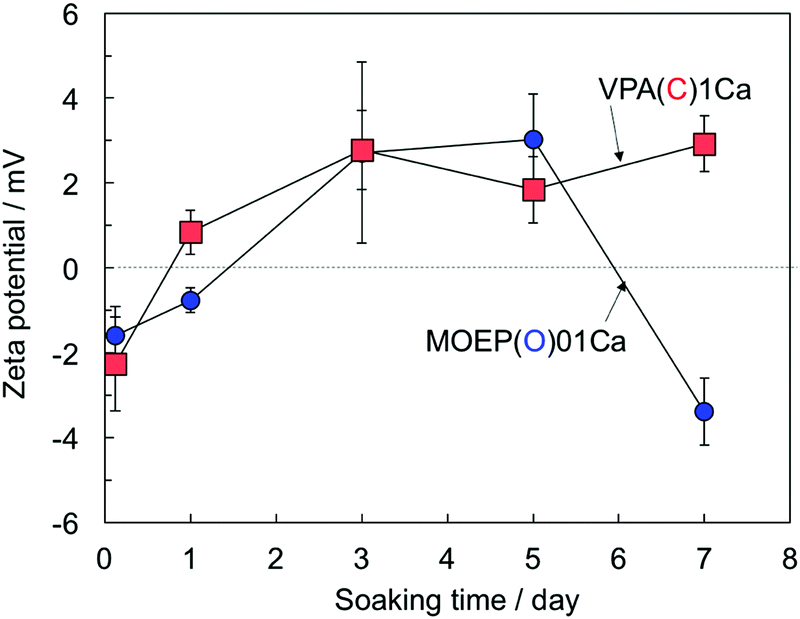 | ||
| Fig. 5 Change in surface zeta potential of VPA(C)1Ca and MOEP(O)01Ca following soaking in SBF for various periods (n = 3). | ||
Fig. 6 shows the changes in the Ca and P concentrations, along with the pH value after soaking the specimens in SBF for various time periods. The Ca concentration after 1 day decreased according to VPA(C)1Ca > MOEP(O)1Ca ≈ MOEP(O)01Ca > VPA(C)01Ca. Then, the Ca concentration gradually decreased in all specimens. The P concentration significantly decreased in MOEP(O)01Ca and MOEP(O)1Ca, while it was almost constant in VPA(C)01Ca and VPA(C)1Ca. The pH value of the SBF following soaking for all the specimens increased after 1 day, and then gradually decreased.
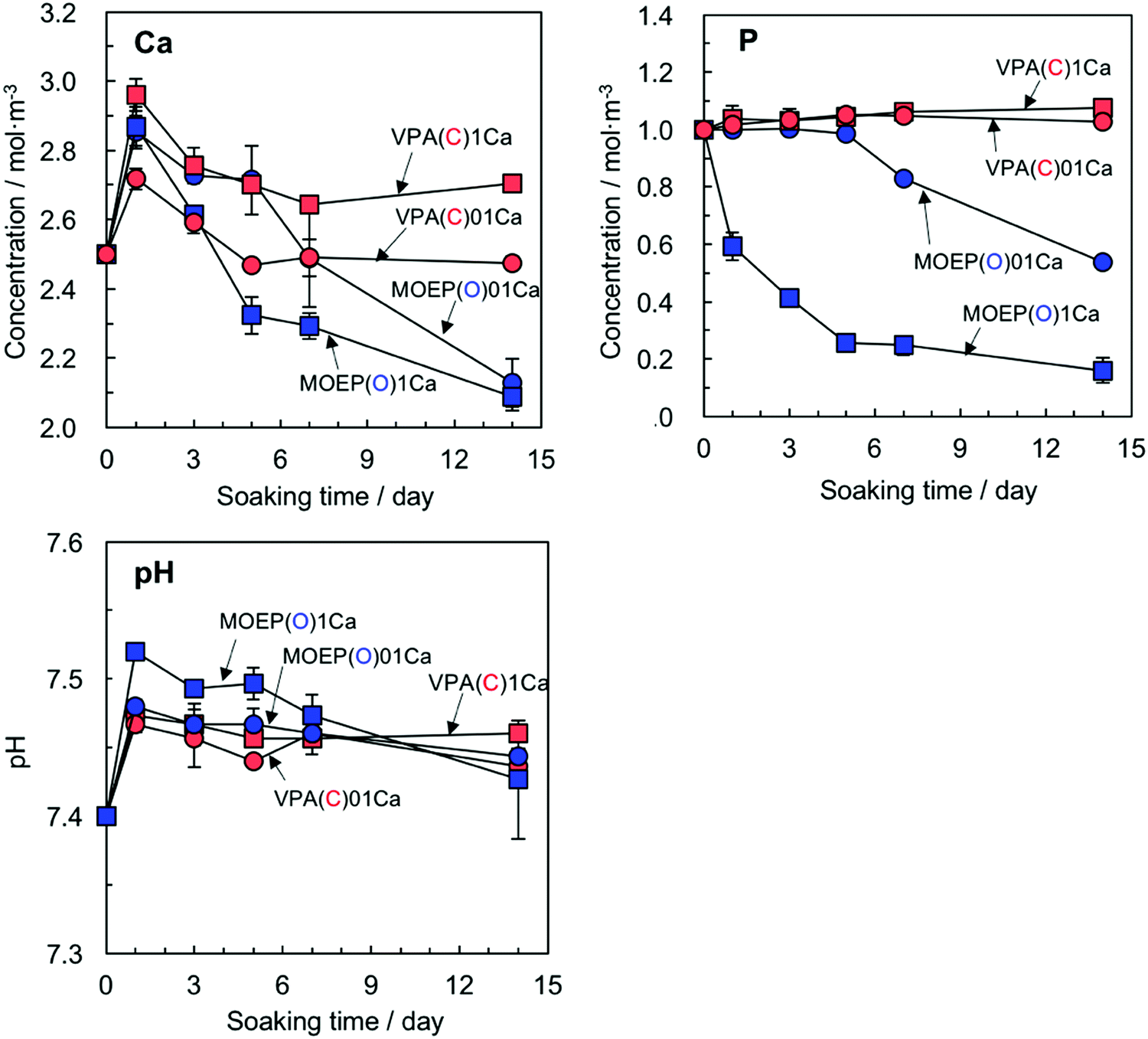 | ||
| Fig. 6 Change in Ca and P concentrations and pH in SBF following soaking of different specimens (n = 3). | ||
Fig. 7 shows the P2p XPS spectra of the VPA(C)0Ca and MOEP(O)0Ca samples after being soaked in 1.5CaSBF for various time periods. The peaks attributed to –C–PO3H2 and –C–PO3Ca were observed in VPA(C)0Ca, and remained constant regardless of the soaking time. On the other hand, the peaks attributed to CaHPO4 and OCP, as well as those from –O–PO3H2 and –O–PO3Ca, were observed in MOEP(O)0Ca after 3 hours. The peaks from CaHPO4 and OCP were almost replaced by those of apatite after 1 day. This indicates that the initial processes of calcium phosphate formation were similar, irrespective of previous Ca2+ incorporation (see Fig. 4).
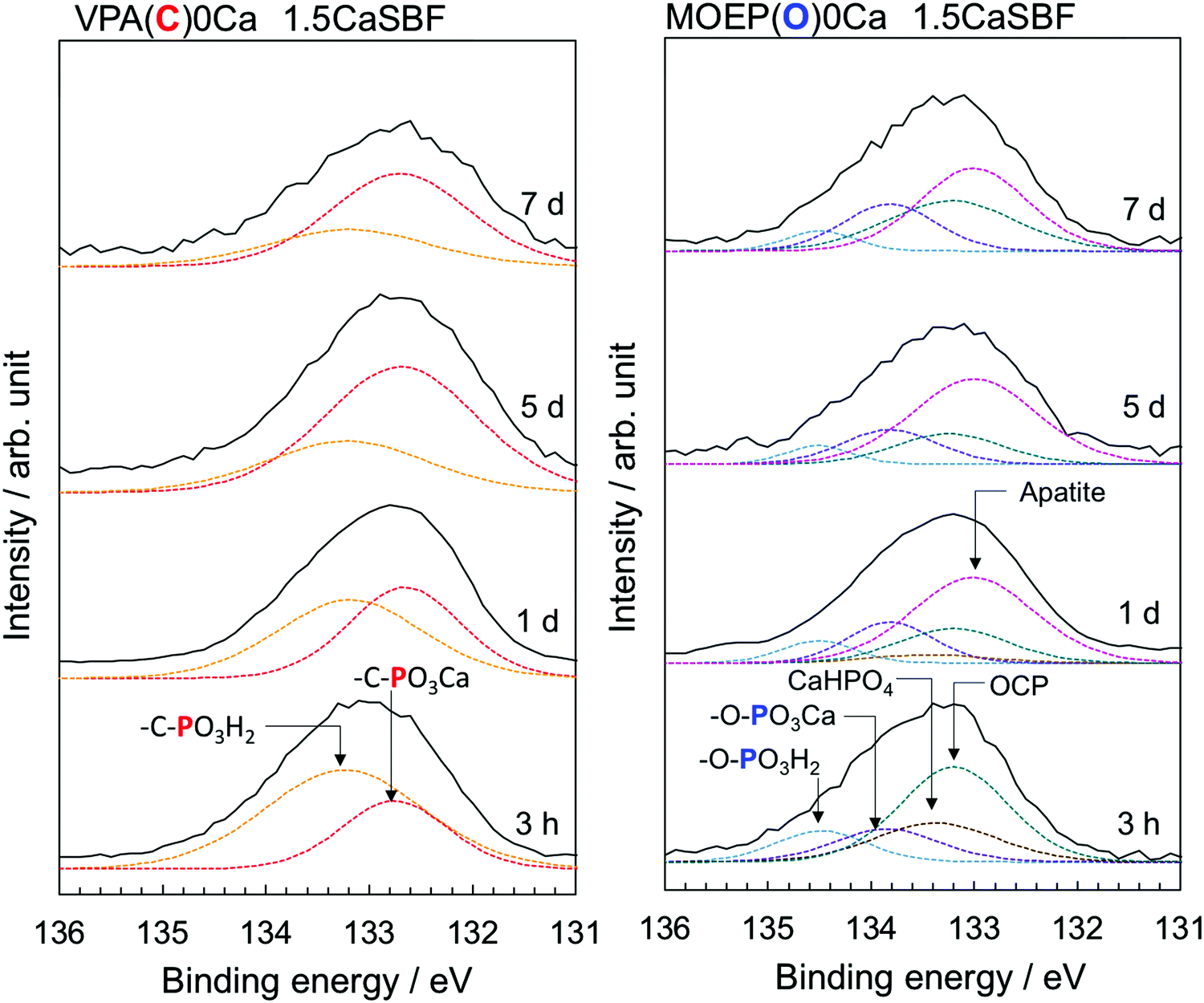 | ||
| Fig. 7 P2p XPS spectra of VPA(C)0Ca and MOEP(O)0Ca following soaking in 1.5CaSBF for various periods. | ||
Table 2 shows the stability constants of the polymer–Ca2+ and CaHPO4 (aq.) complex under physiological conditions. The constant for the CaHPO4 (aq.) complex under these conditions was calculated using the ion strength I = 0.02 at 37 °C reported by Chughtai et al.23 The constant increased according to MOEP(O) < HPO42− < VPA(C).
| VPA(C) | CaHPO4(aq.) | MOEP(O) | |
|---|---|---|---|
| logβ |
4.44 ± 0.07 | 3.41 | 3.08 ± 0.13 |
Fig. 8 shows the structures with the lowest energy from the molecular models of CH3–CH2–PO3Ca and CH3–O–PO3Ca with –C–PO3H2 and –O–PO3H2, respectively. The O atom forms a double bond with P close to the Ca atom in the CH3–CH2–PO3Ca. On the other hand, this was located in the reverse direction from Ca in CH3–O–PO3Ca. The calculated highest occupied molecular orbital (HOMO) was distributed around the O atoms, while the lowest unoccupied molecular orbital (LUMO) was widely distributed around the Ca atom in both of the phosphate groups. The energy levels of the HOMO and LUMO for CH3–CH2–PO3Ca were −0.156 and −0.118, and for CH3–O–PO3Ca they were −0.186 and −0.146 eV, respectively. The value of the dipole moment of CH3–CH2–PO3Ca was 9.35 Debye, and for CH3–O–PO3Ca it was 15.71 Debye. This suggests that the structural differences between –C–PO3Ca and –O–PO3Ca were not at the molecular orbital level, but rather a result of the most stable structures and polarizability.
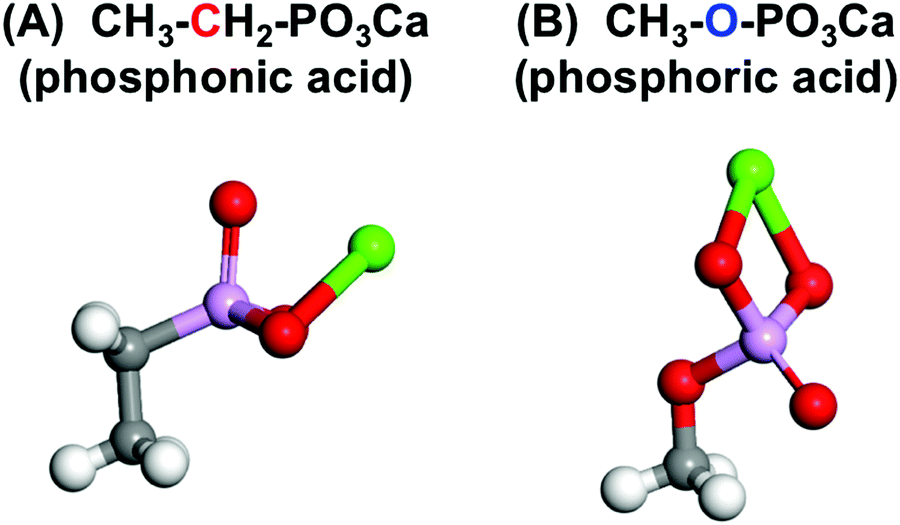 | ||
| Fig. 8 The structure of the molecular model of (A) CH3–CH2–PO3Ca and (B) CH3–O–PO3Ca with the lowest energy for each type of phosphate group (white: H, gray: C, red: O, purple: P, green: Ca atom). | ||
4. Discussion
Copolymers containing equivalent amounts of the phosphate group and Ca2+ were prepared by radical polymerization (see Table 1 and Fig. 1(A)). It was found that the copolymers modified with –O–PO3H2 had much higher apatite-forming ability in SBF than those modified with –C–PO3H2 when treated with the CaCl2 solution (see Fig. 2 and 3). This means that modification with –O–PO3H2 was more effective for apatite formation on the copolymers modified with Ca2+, when compared with –C–PO3H2.The formation of embryos, which are pre-nucleation clusters, larger than the critical size r* is important for the spontaneous nucleation of apatite on the substrate. The activation free energy (ΔG*) for heterogeneous nucleation is given by eqn (2):24
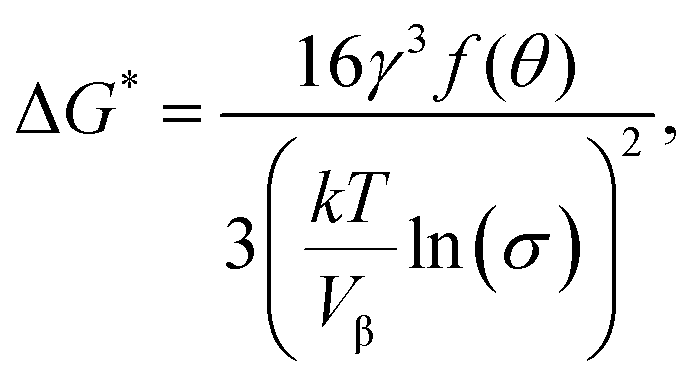 | (2) |
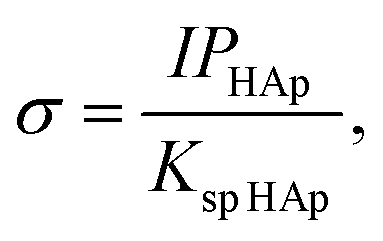 | (3) |
HAp are the ion activity and solubility product with respect to apatite, respectively. Moreover, IPHAp can be calculated from the pH and concentrations of Ca and P using eqn (4):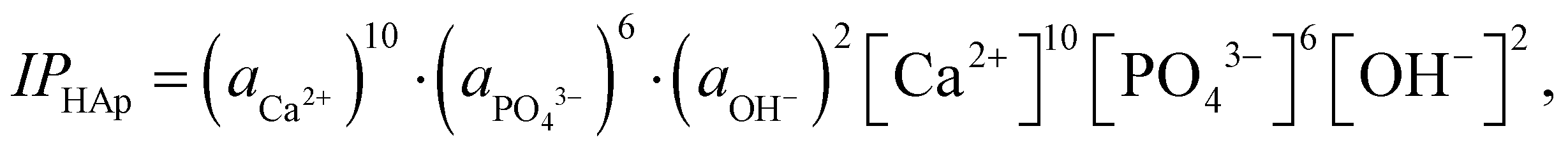 | (4) |
HAp is the solubility product for hydroxyapatite (5.5 × 10−118).25,26 Therefore, the heterogeneous apatite nucleation in this study was governed by the supersaturation degree and the surface condition of the specimens.
The supersaturation degree with respect to apatite in the SBF can be increased by the release of Ca2+ from the substrate.27Fig. 9 shows the changes in the logarithm of σ in the SBF after soaking the specimens for various time periods. The supersaturation degree decreased as VPA(C)1Ca ≈ MOEP(O)01Ca > VPA(C)01Ca > MOEP(O)1Ca. Although the largest amount of Ca2+ was released from MOEP(O)1Ca in the Tris–NaCl buffer (see Fig. 1(B)), the supersaturation degree was low for all specimens. It was speculated that apatite formation on MOEP(O)1Ca occurred rapidly, within 1 day (see Fig. 2 and 3), and that the supersaturation degree rapidly decreased due to ion consumption in the SBF after that time. Although the supersaturation degree values in MOEP(O)01Ca and VPA(C)1Ca were similar, the former formed apatite, but not the latter. These results indicate that structural differences in the phosphate groups affected the surface conditions related to the heterogeneous nucleation of apatite.
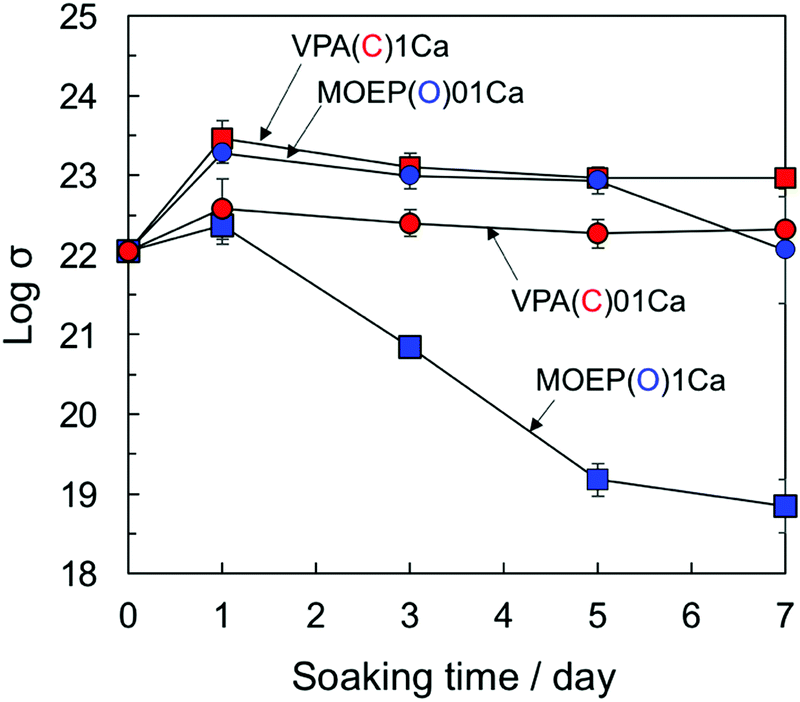 | ||
| Fig. 9 Change in supersaturation degree with respect to the apatite in SBF following soaking of different specimens (n = 3). | ||
It was confirmed by XPS that the surface reaction process was significantly different in –C–PO3H2 and –O–PO3H2 (see Fig. 4(A)). VPA(C)1Ca only adsorbs Ca2+ to form the –C–PO32−⋯Ca2+ complex, but not HPO42− in SBF. This is supported by the fact that the P concentration in SBF was almost constant (see Fig. 6). On the other hand, MOEP(O)01Ca formed CaHPO4 and OCP at the initial stage, and subsequently the OCP that formed was transformed into apatite (see Fig. 4(B)). A similar transformation from OCP to apatite on a plasma-treated polymer in SBF was discussed by Oyane et al.28 These results indicate that only MOEP(O)01Ca adsorbs HPO42− following Ca2+ adsorption to form calcium phosphate as the precursor of apatite. In addition, MOEP(O)0Ca formed calcium phosphate in 1.5CaSBF even without a release of Ca2+ (see Fig. 7). Thus, these results suggest that the specific structures of the phosphate groups provide the surface conditions favorable for apatite nucleation via the adsorption of HPO42− in SBF.
It has been reported that the surface zeta-potential is one of the parameters that governs apatite formation on bioactive materials in SBF.29,30 In the case of titanium metal bound to bone prepared by treatment with an alkaline solution, the negatively charged surfaces becomes positive due to Ca2+ adsorption in SBF, then becomes negative again via the subsequent adsorption of phosphate, ultimately forming apatite.29 In contrast, VPA(C)1Ca did not demonstrate such behavior, even though the zeta-potential of both specimens changed from negative to positive (see Fig. 5). Based on the results of XPS and the zeta-potential measurement, differences in apatite formation cannot be explained in terms of surface charge alone.
With regard to the initial reaction process for each phosphate group on the surface of specimens with Ca2+ and HPO42− in the SBF, we consider two viewpoints: (1) the interaction energies between the reactive chemical species in the transition state, and (2) the thermodynamic stability of the products.
With viewpoint (1), it is assumed that a transition state –PO3Ca⋯HPO42− is formed for all the specimens. The interaction energy is obtained as the sum of the frontier orbital interaction and the Coulomb interaction.31,32 The LUMO around the Ca atom in the both models (see Fig. 8) was expected to accept the HOMO electron of HPO42−. When comparing the energy levels of the orbitals, a significant difference is observed for the LUMO in both models (−0.146 and −0.118 eV) and the HOMO level of O atoms in HPO42− (−8.9 eV) estimated from the vertical ionization energy.33 This means that the Coulomb interaction contributes more to the formation of the transition state than the frontier orbital. A computational simulation of the polarizability indicated that the Coulomb interaction of the –O–PO32−⋯Ca2+ complex with HPO42− was stronger than with –C–PO32−⋯Ca2+. Therefore, the transition state in MOEP(O) was predicted to be more stable than VPA(C), leading to a decrease in activation energy for the formation of CaHPO4 in the former.
Using viewpoint (2), the thermodynamic stability values between CaHPO4 (aq.) and the complex composed of Ca2+ and one of the phosphate groups were evaluated. The magnitude of the complex formation constants in Table 2 indicates that Ca2+ bound with MOEP(O) has a higher ability to bond to HPO42−via a ion–dipole interaction, and subsequently become converted into CaHPO4, because the stability of the –O–PO32−⋯Ca2+ complex was lowest.
The Ca2+⋯HPO42− interaction on the surface of the MOEP(O) sample had an important role for apatite deposition. It was recently reported that heterogeneous apatite nucleation on a titanium substrate treated with H2O2 in SBF progresses via the formation of a pre-embryo consisting of Ca2+, HPO42−, and OH−.34,35 Moreover, self-assembled monolayers containing carboxyl groups formed clusters by accumulation of pre-nucleation complexes of [Ca(HPO4)3]4−.36,37
Summarizing the mechanism described above, apatite formation on the surface of MOEP(O) is assumed to progress as follows: an increase in [Ca(HPO4)3]4− concentration results in the formation of many clusters larger than r*. Subsequently, the clusters transformed into OCP, the apatite precursor, via ion adsorption in the SBF. In fact, Habraken et al. demonstrated that amorphous calcium phosphate with the formula [Ca2(HPO4)3]2− grew into Ca-deficient OCP via the aggregation of the pre-nucleation complex and ions in a solution supersaturated with respect to the apatite.37
Finally, the results of this study reveal that differences in the molecular structures of the phosphate groups govern the apatite-forming ability of the copolymers modified with Ca2+, because the low thermodynamic stability of the phosphate⋯Ca2+ complex on the specimen enhances calcium phosphate cluster formation that is favorable for apatite nucleation. It is also expected that such structural differences affect the ability of the composites to bond with bone. Recently, Huang et al. reported that modification of –O–PO3H2 promoted the differentiation of mesenchymal stem cells to provide mineralization on the polymer.38 However, it has never been revealed whether structural differences affect the behavior of calcification induced by the cells. To clarify this point in detail, the adsorption behavior of proteins, and the response of the cells, should be investigated in future work.
5. Conclusion
The structural effects of phosphate groups on apatite formation on the surface of a copolymer modified with Ca2+ in SBF were examined. The copolymers modified with phosphoric acid (–O–PO3H2) induced heterogeneous nucleation of apatite, but not those modified with phosphonic acid (–C–PO3H2), because the amount of adsorbed HPO42− in the SBF was insufficient for apatite nucleation. The reason for this phenomenon was that the thermodynamic stability of the ionized –O–PO32−⋯Ca2+ complex was lower than that of –C–PO32−⋯Ca2+. It was found that the reaction with HPO42− that allowed the calcium phosphate clusters to become precursors of apatite was a key issue that significantly affected apatite formation on the copolymer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Grant-in-Aid for JSPS Research Fellow Grant Number 17J02000. We thank Louis R. Nemzer, PhD, from Edanz Group for editing a draft of this manuscript.References
- L. L. Hench, J. Am. Ceram. Soc., 1998, 81, 1707–1728 Search PubMed.
- T. Kokubo, H. M. Kim and M. Kawashita, Biomaterials, 2003, 24, 2161–2175 CrossRef CAS PubMed.
- M. Jarcho, C. H. Bolen, M. B. Thomas, J. Bobick, J. F. Kay and R. H. Doremus, J. Mater. Sci., 1976, 11, 2161–2175 CrossRef.
- P. Li, C. Ohtsuki, T. Kokubo, K. Nakanishi, N. Soga and K. de Groot, J. Biomed. Mater. Res., 1994, 28, 7–15 CrossRef CAS PubMed.
- M. Uchida, H. M. Kim, T. Fujibayashi and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 64, 164–170 CrossRef PubMed.
- M. Tanahashi and T. Matsuda, J. Biomed. Mater. Res., 1997, 34, 305–315 CrossRef CAS PubMed.
- T. Kawai, C. Ohtsuki, M. Kamitakahara, T. Miyazaki, M. Tanihara, Y. Sakaguchi and S. Konagaya, Biomaterials, 2004, 25, 4529–4534 CrossRef CAS PubMed.
- S. B. Cho, K. Nakanishi, T. Kokubo, N. Soga, C. Ohtsuki, T. Nakamura, T. Kitsugi and T. Yamamuro, J. Am. Ceram. Soc., 1995, 78, 1769–1774 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- P. Datta, J. Chatterjee and S. Dhara, Colloids Surf., B, 2012, 94, 177–183 CrossRef CAS PubMed.
- P. M. López-Pérez, R. M. P. Silva, R. A. Sousa, I. Pashkuleva and R. L. Reis, Acta Biomater., 2010, 6, 3704–3712 CrossRef PubMed.
- R. Hamai, Y. Shirosaki and T. Miyazaki, J. Mater. Sci.: Mater. Med., 2016, 27, 152 CrossRef PubMed.
- O. Exner, J. Phys. Org. Chem., 1999, 12, 265–274 CrossRef CAS.
- S. B. Cho, F. Miyazi, T. Kokubo, K. Nakanishi, N. Soga and T. Nakamura, J. Ceram. Soc. Jpn., 1996, 104, 399–404 CrossRef CAS.
- Ö. Szabadka, E. Varga and L. Nagy, Talanta, 2003, 59, 1081–1088 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron gad correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- B. Delly, From molecules to solids with the DMOl3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef.
- X. Zhou, S. H. Goh and S. Y. Lee, Polymer, 1997, 38, 5333–5338 CrossRef CAS.
- I. F. Amaral, P. L. Granja and M. A. Barbosa, J. Biomater. Sci., Polym. Ed., 2005, 16, 1575–1593 CrossRef CAS PubMed.
- T. Hanawa and M. Ota, Biomaterials, 1991, 12, 767–774 CrossRef CAS PubMed.
- M. J. Van Stiodonk, D. R. Justes and E. A. Schweikert, Anal. Chem., 1999, 71, 149–153 CrossRef PubMed.
- L. Radev, M. H. V. Fernandes, I. M. Salvado and D. Kovacheva, Cent. Eur. J. Chem., 2009, 7, 721–730 CAS.
- A. Chughtai, R. Marshall and G. H. Nancollas, J. Phys. Chem., 1968, 72, 208–211 CrossRef CAS PubMed.
- T. Kawai, C. Ohtsuki, M. Kamitakahara, M. Tanihara, T. Miyazaki, Y. Sakaguchi and S. Konagaya, J. Ceram. Soc. Jpn, 2005, 113, 588–592 CrossRef CAS.
- W. Neuman and M. Neuman, The Chemical Dynamics of Bone Mineral, University of Chicago, Chicago, 1958, p. 3 Search PubMed.
- H. McDowell, T. M. Gregory and W. E. Brown, J. Res. Natl. Bur. Stand., Sect. A, 1977, 81, 273–281 CrossRef.
- C. Ohtsuki, T. Kokubo and T. Yamamuro, J. Non-Cryst. Solids, 1992, 43, 84–92 CrossRef.
- A. Oyane, M. Uchida, Y. Yokogawa, C. Choong, J. Triffitt and A. Ito, J. Biomed. Mater. Res., Part A, 2005, 75, 138–145 CrossRef PubMed.
- H.-M. Kim, T. Himeno, M. Kawashita, J.-H. Lee, T. Kokubo and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 67, 1305–1309 CrossRef PubMed.
- H.-M. Kim, T. Himeno, T. Kokubo and T. Nakamura, Biomaterials, 2005, 21, 4366–4373 CrossRef PubMed.
- Chemical Reactivity and Reaction Paths ed. G. Klopman, Wiley-Interscience, New York, 1974 Search PubMed.
- G. Klopman, J. Am. Chem. Soc., 1968, 90, 223–234 CrossRef CAS.
- E. Pluhařová, M. Ončák, R. Seidel, C. Schroeder, W. Schroeder, B. Winter, S. E. Bradforth, P. Jungwirth and P. Slavíček, J. Phys. Chem. B, 2012, 116, 13254–13264 CrossRef PubMed.
- S. Hayakawa, K. Tsuru, K. Uetsuki, K. Akasaka, Y. Shirosaki and A. Osaka, J. Mater. Sci.: Mater. Med., 2015, 26, 222 CrossRef PubMed.
- K. Uetsuki, S. Nakai, Y. Shirosaki, S. Hayakawa and A. Osaka, J. Biomed. Mater. Res., Part A, 2013, 101, 712–719 CrossRef PubMed.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. Schoot, B. Aichmayer, G. With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 CrossRef PubMed.
- P. Huang, X. Bi, J. Gao, L. Sun, S. Wang, S. Chen, X. Fan, Z. You and Y. Wang, J. Mater. Chem. B, 2016, 4, 2090–2101 RSC.
| This journal is © The Royal Society of Chemistry 2018 |