
S-Nitrosothiols (SNO) as light-responsive molecular activators for post-synthesis fluorescence augmentation in fluorophore-loaded nanospheres†
Shu-Yi
Lin
a,
Meng-Ren
Wang
a,
Shih-Jiuan
Chiu
b,
Chien-Yu
Lin
c and
Teh-Min
Hu
 *ac
*ac
aSchool of Pharmacy, National Defense Medical Center, Taipei, Taiwan, Republic of China
bSchool of Pharmacy, Taipei Medical University, Taipei, Taiwan, Republic of China
cFaculty of Pharmacy, School of Pharmaceutical Sciences, National Yang-Ming University, Taipei, Taiwan, Republic of China. E-mail: tehmin@ym.edu.tw
First published on 4th December 2017
Abstract
Dye-loaded, fluorescent nanoparticles (FNPs) have been extensively studied as promising imaging or probing agents for therapeutic and biomedical applications, because of improved photostability and reduced cytotoxicity (compared with free dyes). The synthesis of FNPs often involves entrapment of fluorescent dye molecules into nanostructures, which, however, would encounter a problem associated with the formation of molecular aggregates within the confined matrix space. The formation of nonfluorescent aggregates seems unavoidable for some conventional fluorescent dyes, thereby leading to fluorescent quenching. The problem is well-recognized, but frequently ignored; and FNPs are usually applied as prepared without addressing it either during or after the synthesis of FNPs. The ignorance may be due to the difficulty in altering post-synthetically the intraparticulate molecular arrangements and interactions. Herein, we describe how light-responsive S-nitrosothiol (SNO) can be engineered into fluorophore-loaded silica nanoparticles for efficient dye-loading and post-synthesis fluorescence augmentation. Silica nanoparticles loaded with various fluorescent molecules were prepared using one-pot, simultaneous, acid-catalyzed sol–gel condensation and nitrosation of a single mercaptosilane source, 3-mercaptopropyl trimethoxysilane (MPTMS), followed by nanoprecipitation. We first show how doxorubicin-loaded silica NPs respond to stepwise visible-light exposure and exhibit ON/OFF fluorescent response. We then demonstrate that rhodamine 6G (R6G) can be stably incorporated into SNO-enriched silica nanostructure with negligible payload leakage (<0.2%) and ∼1000-fold reduction in cytotoxicity (compared with free R6G). Remarkably, visible light irradiation leads to ∼100-fold increase in fluorescent intensity. Thus, for the first time, SNO is proposed as a light-responsive entity for post-synthesis fluorescence intensification in FNPs.
Introduction
Fluorescent nanomaterials have been extensively studied because of their broad applications.1 In biomedical and many other applications, conventional fluorescent molecules have several disadvantages, including chemical and/or photo-instability, high toxicity, and fluorescence quenching at an aggregate state.2 The incorporation of fluorescent molecules into a nanostructure (or nanocarriers) thus represents a promising strategy for preparing more stable and less toxic fluorescent bio-materials.3–9 Moreover, such fluorescent nanocarrier systems can be further engineered into multifunctional theranostic platforms.5,10,11Silica nanoparticles (silica NPs) are promising drug delivery systems.12–17 Silica species is detectable in human plasma, and daily intake of silica from foods can be quantified.18 Therefore, silica is generally biocompatible and biodegradable.6,9,17 Recently, the first fluorescent silica NPs have been tested first-time in human for cancer diagnosis and treatment.19 To prepare silica-based, fluorescent dye-loaded materials, two distinctly different strategies are commonly employed: one is physisorption (i.e. the physical route) and the other is through the formation of chemical bonds (i.e. the chemical route).20 The physical route involves doping of fluorescent molecules in pre-formed bare silica matrix (post-doping).20 Alternatively, fluorescent molecules can be encapsulated in silica structures during sol–gel condensation or microemulsion, and before particle formation (pre-doping).21–28 In either way, fluorescent molecules interact with silica structures via non-covalent supramolecular interactions. The physical route is straightforward, but it may have suboptimal loading efficiency and significant payload leakage. In some cases, even for materials prepared by the pre-doping method, the leaching can be as high as 50%.29 In contrast, the chemical route requires chemical conjugation of fluorescent molecules either to silica precursors or to pre-formed silica particles.20,30–34 Thus, the dye molecules are covalently encapsulated in silica matrix. The chemical route allows for more stable and versatile loading, but it is more complicated than the physical route and requires chemical modification of the dye molecules. Although both physical and chemical routes are frequently used, a common problem associated with these conventional routes is fluorescence quenching,35–38 which is due to molecular aggregation within the confined matrix space (also known as “cage effect”20). The phenomenon has been well known and its underlying mechanism extensively studied;28,39–42 however, the strategies to overcome fluorescence quenching remain limited.
The photophysical properties of fluorophores embedded in confined matrix are complex; the outcome may be fluorescence quenching or enhancement, depending on types of fluorophores, loading concentrations, preparation methods, dye-silica interactions, and spatial distribution of dye molecules.25,43–49 For example, by controlling molecular distribution (e.g. condensed vs. expanded or homogeneous) of a fluorescent probe within silica nanoparticles, Wilsner and Webb et al.43,45 demonstrated how precise control of particle architecture could lead to significant fluorescence enhancement. To control particle architecture (e.g. core–shell), dye precursor was synthesized before a sol–gel condensation reaction.43 Thus, the dye was covalently encapsulated. For the simpler physical loading approach, however, precise control of dye location within particles would be difficult, if not impossible. The distribution of fluorophores is predetermined by the physicochemical properties (e.g. molecular charges and propensities for hydrogen bonding and hydrophobic interactions) of both dye and scaffold molecules.29,46 Depending on dye molecules, fluorescence quenching may occur if fluorescent molecules aggregate at certain loading concentrations.28,37,46 Moreover, the microenvironment within confined cages or pores may affect the molecular orientation and aggregation states.29,37,50,51 It has been reported that the aggregation states may be altered by thermal treatment or by adding surfactants.37,52,53 However, they are not further studied as an effective post-synthetic measure for fluorescence enhancement.
In our previous study, we have successfully developed a silica-based nanoplatform for simultaneous cellular delivery of nitric oxide (NO) and drugs/theranostics.54 The method is based on one-pot S-nitrosation and polycondensation of 3-mercaptopropyl trimethoxysilane (MPTMS), a thiol-functionalized organosilane, followed by a nanoprecipitation process.55–58 Drugs and fluorophores can be simultanesouly entrapped in the polycondensed silica species with high efficiency during the solvent-shifting process.54 The particles formed are in the nanoscale range (ca. 100 nm), carrying both the S-nitrosothiol (SNO) functionality for sustained release of NO and a chemotherapeutic agent, doxorubicin (DOX).54 In this paper, we propose a novel application of SNO chemistry in the fabrication of dye-loaded NPs: i.e. SNO as a photoresponsive unit for post-synthesis augmentation of fluorescence intenstiy (Scheme 1). Herein, we first describe how DOX fluorescence in SNO–DOX silica NPs responds to visible-light irradiation. We then report the extended findings on another fluorophore, rhodamine 6G (R6G), and demonstrate that light irradiation resulted in >100-fold enhancement in fluorescence intensity. SNO–R6G silica NPs had extremely low leaching of R6G (<0.2%) and were about 1000-fold less toxic than free R6G. The potential role of light-triggered NO release played in the observed fluorescence enhancement was investigated and discussed.
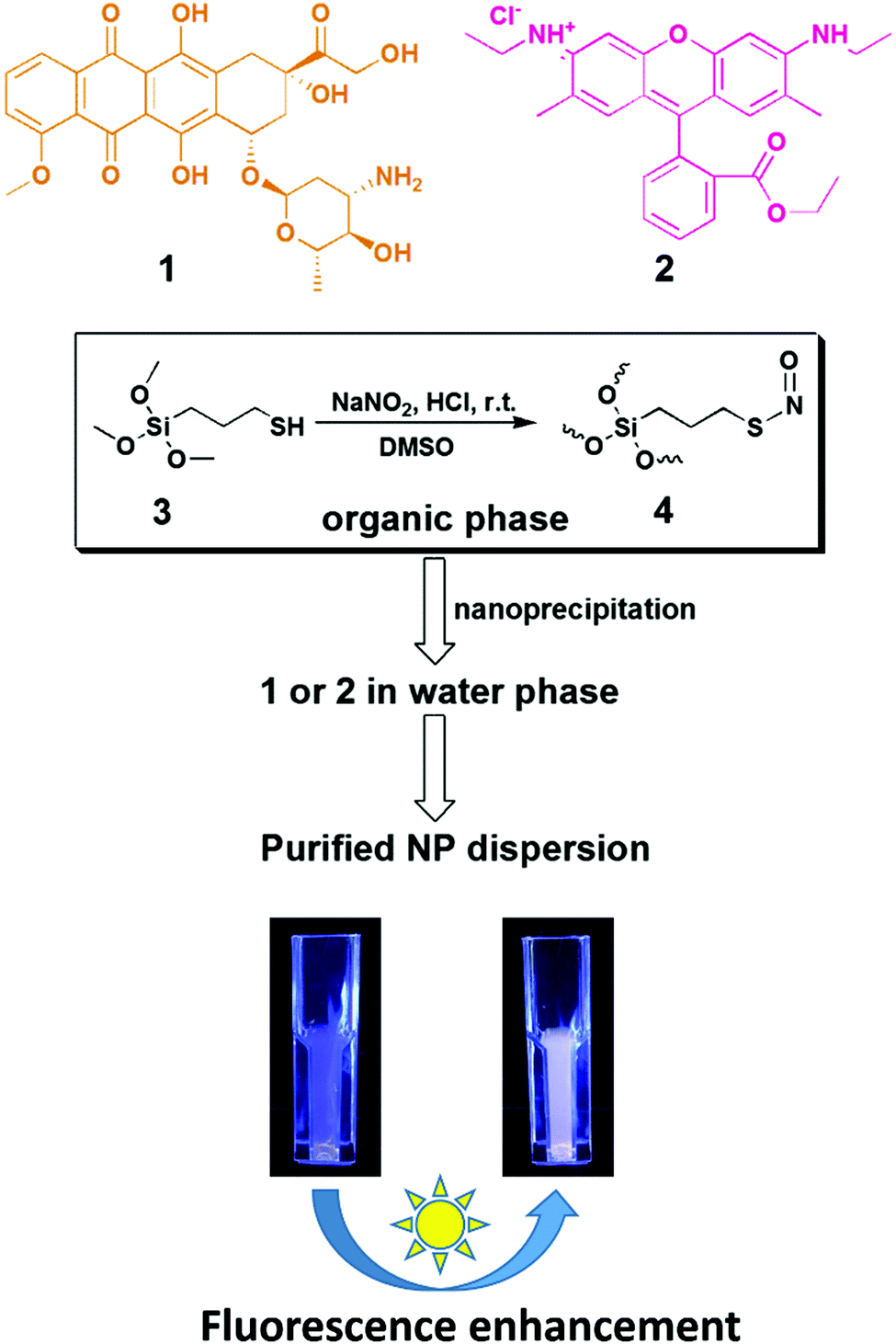 | ||
| Scheme 1 Schematic illustration of S-nitrosothiol (SNO)-based synthesis of fluorophore-loaded SiNPs and post-synthesis, light-triggered augmentation of the fluorescence intensity. (1) Doxorubicin, (2) rhodamine 6G, (3) 3-mercaptopropyl trimethoxysilane, (4) S-nitroso-polysilsesquioxane oligomeric species. | ||
Results
Photoresponsive fluorescence enhancement of doxorubicin (DOX)-loaded SNO–silica nanoparticles (SNO–DOX)
SNO–DOX silica NPs were prepared using a solvent-shifting procedure, in which SNO-functionalized silica oligomers self-assembled into nanoparticles with simultaneous DOX encapsulation.54 Initially, we attempted to shine light on SNO–DOX to facilitate NO release, because SNO is photolabile.59 Surprisingly, when freshly prepared SNO–DOX was exposed to light, we observed continuous increase in fluorescence spectra of DOX over time; and the fluorescence intensity increased about 10-fold at 24 h (Fig. 1a). For comparison, SNO–DOX exhibited insignificant spectral change when kept in the dark for 24 h (Fig. 1b). The light responsiveness of the system was further realized by conducting a cyclic ON/OFF experiment: the particle dispersion of SNO–DOX was irradiated for 5 min (“ON” phase) and then subjected to fluorescence measurement for another 5 min (in the dark chamber of the fluorometer, “OFF” phase). The result in Fig. 1c shows a step-like fluorescence time profile for DOX that captures the essence of light responsiveness of the system. Remarkably, the fluorescence signal for NO release (i.e. NAT fluorescence) also exhibited ON/OFF characteristics (Fig. 1d).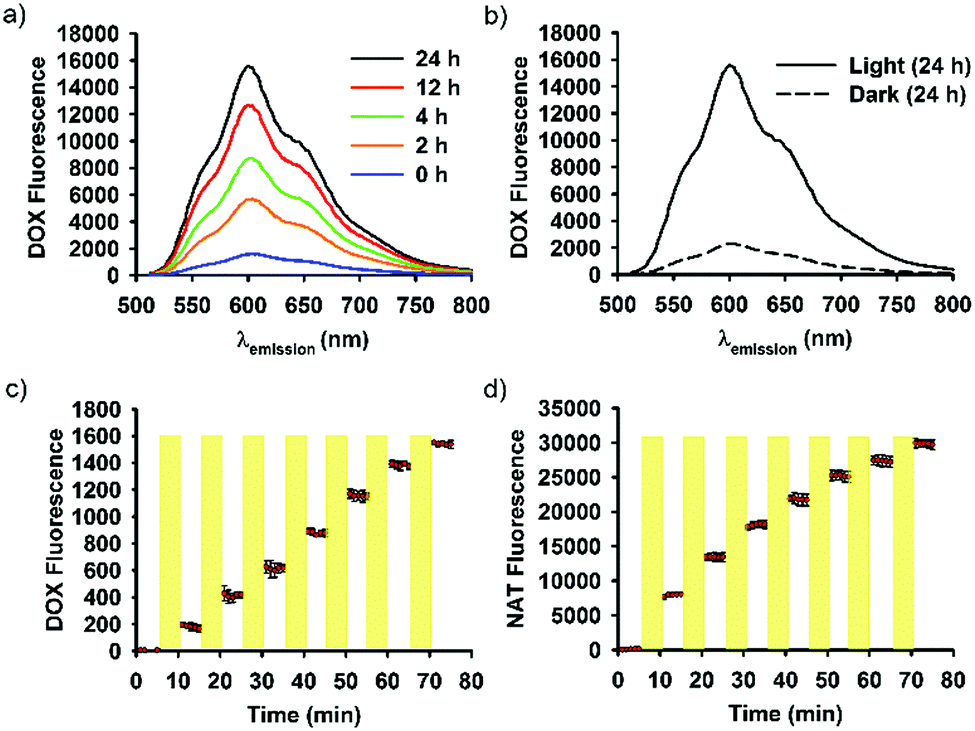 | ||
| Fig. 1 Visible-light mediated fluorescence response of SNO–DOX NPs. (a) Emission spectra of an aqueous SNO–DOX dispersion under continuous light irradiation up to 24 h. (b) Comparison of emission spectra between light-treated and control SNO–DOX (in the dark) at 24 h. (c) and (d) ON/OFF fluorescence response of DOX and an NO probe, respectively, in an aqueous dispersion of SNO–DOX. The yellow stripes indicate the ON period where the sample was irradiated with light. | ||
DOX has been extensively studied as a model drug in advanced drug delivery systems, partly because it can be easily detected by utilizing its fluorescent property. The encapsulation or conjugation of DOX into a nanocarrier may lead to fluorescence quenching. However, DOX would regain its fluorescence after releasing in a free form (“dequenching”), which is the rationale for fluorescence-based release study.60–63 It is therefore important to investigate whether unloaded or released, free DOX may contribute to observed fluorescence enhancement. First, the supernatant of the light-irradiated particle dispersion remained at low fluorescence intensity during light irradiation (Fig. 2a), indicating that light-triggered fluorescence enhancement was mainly from loaded DOX, but not from free DOX. Second, the release study in a dialysis bag revealed identical DOX-release profiles for the light-treated particles and the control particles (Fig. 2b); however, within the dialysis bag, light-treated particles remained highly fluorescent, compared with low fluorescence intensity for the control particles (Fig. 2b, inset). The results further suggest that light treatment did not lead to alteration of DOX release, and that the increment of fluorescence came from within the nanoparticle. Third, particles remained intact and spherical after 12 h exposure to light (Fig. 2c), and particle sizes were not significantly changed by light (86 ± 25 nm (dark) vs. 95 ± 25 nm (light); Fig. 2d). Thus, the macrostructure of SNO–DOX seemed not to be modified by photo-irradiation. Taken together, light-mediated fluorescence augmentation was not associated with DOX release or unloaded DOX. Instead, some intraparticle mechanisms may operate (discussed later).
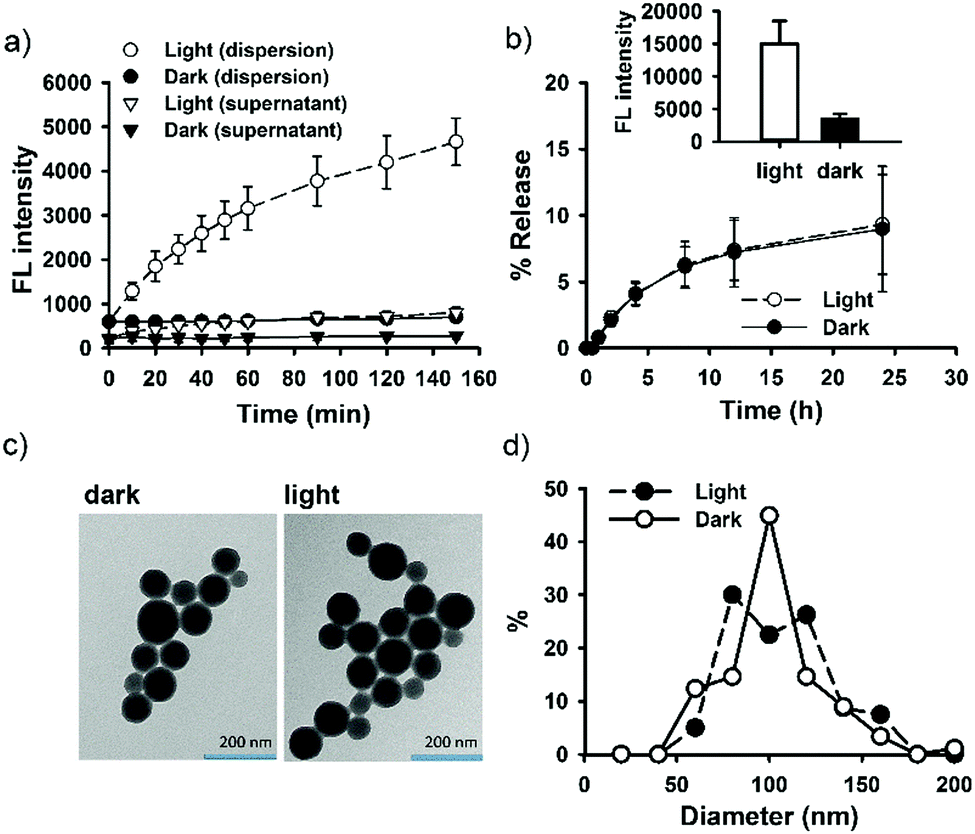 | ||
| Fig. 2 Light-treated SNO–DOX NPs. (a) Kinetic fluorescence measurements for SNO–DOX (aqueous dispersion and isolated supernatant) with or without light irradiation. (b) Release of DOX from SNO–DOX with or without light pretreatment for 4 h (i.e. light vs. dark). The release setup was placed in the dark and protected from light during the release study. Inset: Fluorescence intensities of particle dispersions in the dialysis bag at the end of release study. (c) The TEM images of light treated SNO–DOX (12 h irradiation) and of control particles (without light pretreatment). (d) Comparison of particle size between particles with or without light treatment (12 h). | ||
Light-mediated fluorescence turn-on of rhodamine 6G-loaded SNO–silica nanoparticles (SNO–R6G)
The interesting finding on SNO–DOX prompted us to further utilize SNO chemistry in the synthesis of fluorophores-loaded silica nanoparticles. We propose that SNO plays two roles (Scheme 1). First, it allows for facile nanoprecipitation-based preparation of silica nanoparticles with high payload-loading efficiency. Second, it can act as a light-responsive unit for post-synthetic manipulation of fluorescent properties. In the following study, we used R6G as a model fluorophore. R6G has been frequently used as a fluorescent probe for biological detection, because of its long absorption and emission wavelengths (500–600 nm) that can avoid autofluorescence interference from biomolecules (400–500 nm).38 Besides, R6G has high fluorescent intensities and laser tunability at 550–600 nm.38 However, the toxicity of R6G may limit its use in the biological system or in a laser device. It is therefore important to stably incorporate R6G in a carrier material with minimal leaching. In so doing, potential fluorescent quenching should be alleviated.In the present study, SNO–R6G was synthesized according to the method used for the synthesis of SNO–DOX.54 The entire preparation procedure was conducted in the dark, to avoid light. Similar to SNO–DOX, the maximum entrapment efficiency of R6G was above 80% (81.6 ± 2.2%). The formed particles have hydrodynamic sizes of about 240 nm with a polydispersity index of 0.21. Furthermore, under TEM, the particles are sub-100 nm spheres (Fig. S1, ESI†). Fig. 3a shows that light exposure caused continuous increase in fluorescence intensity of the particle dispersion over time, with a lag time of about 20 min. Remarkably, after 150 min, the increase reached about 100-fold. Fig. 3c shows contrasting images of light-treated SNO–R6G (left panel) vs. light-protected SNO–R6G (right panel). Obviously, light has “turned on” the fluorescence of SNO–R6G. To rule out the possibility that unloaded or free R6G may contribute to increased fluorescence, the supernatant of the SNO–R6G dispersion was separated from particles and its fluorescence intensity measured. The result clearly indicates that the fluorescence of the SNO–R6G dispersion was mainly originated from R6G-bound particles, but not from dissociated and released R6G (Fig. 3b). Moreover, less than 0.2% of R6G was released over 24 h in a release study performed in a dialysis bag (Fig. 3d), indicating that R6G is firmly entrapped within the nanostructure of SNO–R6G. This is an intended and favorable property for fluorescent silica NPs. Indeed, the benefit of low leakage can be realized in the photobleaching test showing no bleaching for R6G–silica NPs (Fig. S2, ESI†). Furthermore, the results of cell cytotoxicity assays confirms that free R6G is very toxic, especially when it is exposed to light (Fig. 3b). The data further demonstrate that SNO–R6G is about 100–1000-fold less toxic than free R6G in two cell lines, MDA-MB-231 (Fig. 3e) and A2058 (Fig. S3, ESI†). Finally, the advantage of the light-treated particles is further demonstrated in a cell labeling study using flow cytometry, where light-irradiated NPs also exhibit much higher cellular fluorescence intensities than light-protected NPs (Fig. 3f and g).
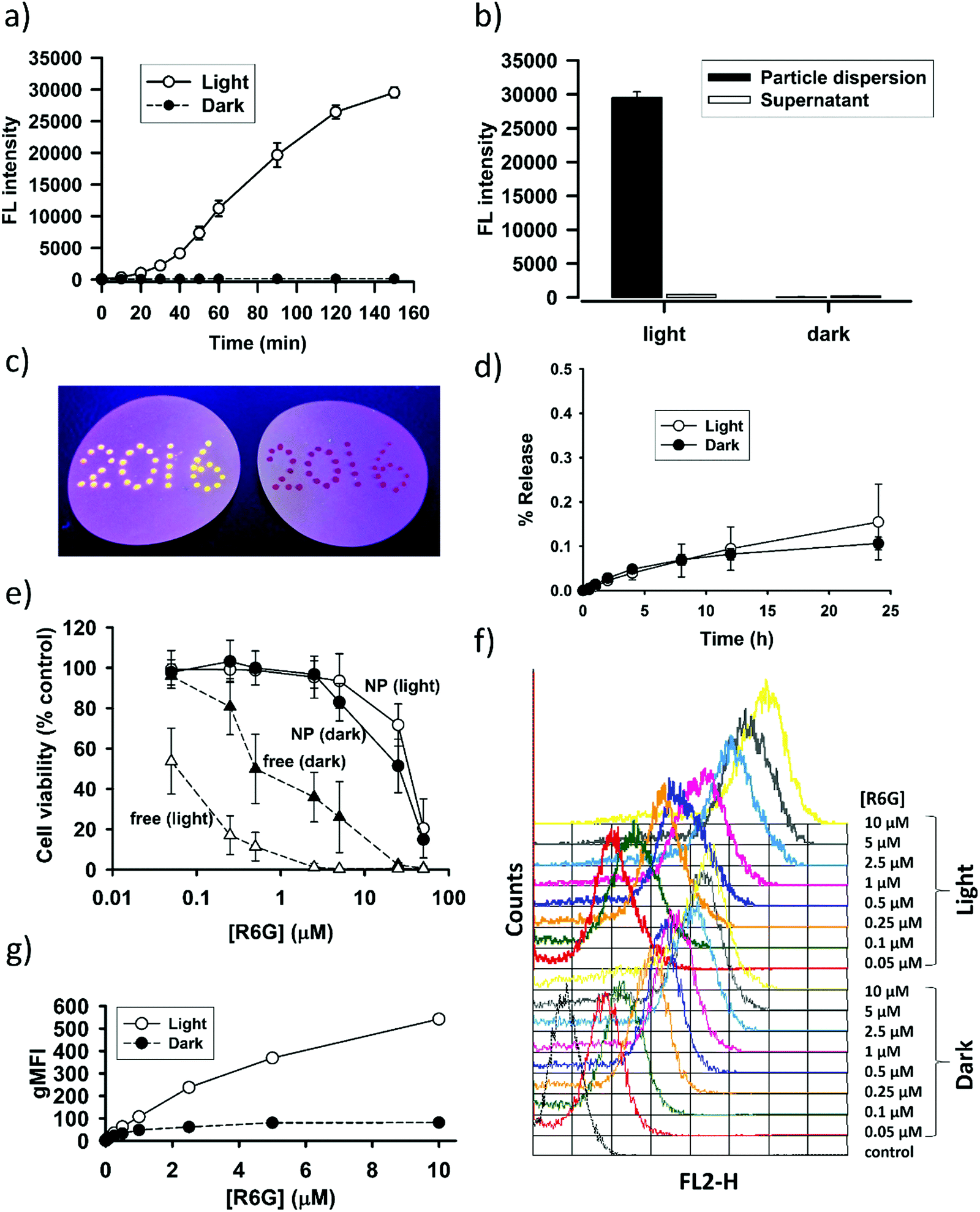 | ||
| Fig. 3 Light-treated SNO–R6G. (a) Kinetic fluorescence measurements for SNO–R6G with or without light irradiation. (b) Comparisons of fluorescence intensities between SNO–R6G dispersion and its supernatant for both light and dark samples. (c) Contrasting images obtained by dotting the droplets of SNO–R6G dispersion (light-treated, left vs. dark control, right) on a hydrophobic surface and viewed under UV light. (d) Release of R6G from SNO–R6G with or without light pretreatment for 4 h (i.e. light vs. dark). The release setup was placed in the dark and protected from light during the release study. (e) Cytotoxicity of SNO–R6G and R6G in MDA-MB-231 cells with or without light exposure during incubation. (f) Fluorescence histograms obtained from the flow cytometric analysis of MDA-MB-231 cells after cellular uptake of various concentrations of SNO–R6G (light vs. dark). (g) Geometric mean fluorescence intensities (gMFI) estimated from the flow cytometric analysis. | ||
Emission spectra of SNO–DOX and SNO–R6G with and without light treatment
Fig. 4 shows the emission spectra recorded for SNO–DOX and SNO–R6G in a dispersed state (in water) and in an extraction solvent (DMSO). The molecular solution of DOX in water exhibited maximum emission at 596 nm with two shoulders at 560 nm and 630 nm, respectively (Fig. 4a). Encapsulation of DOX in SNO–DOX resulted in spectral red-shift (from 596 nm to 608 nm) with a depressed shoulder at 560 nm (Fig. 4a, NPs in the dark). The result suggests that DOX was encapsulated in a hydrophobic environment.64 Remarkably, red-shifting was diminished for light-irradiated SNO–DOX (from 596 nm to 602 nm). In Fig. 4b, encapsulation of R6G in SNO–R6G also exhibited red-shifted spectrum (peak wavelength for R6G, 556 nm, and for SNO–R6G, 562 nm); however, light irradiation has shifted the spectrum of SNO–R6G further to the right (peaked at 570 nm). The difference between SNO–DOX and SNO–R6G may be attributed to different photophysical nature of fluorophores. For R6G, it has been reported that the formation of different molecular aggregates within the confined silica cage may result in spectral red-shifting.44Fig. 4c and d show superimposed spectra for DMSO extracted fluorophores and authentic fluorophores, suggesting that photo-irradiation seemed not to modify the structure of the fluorophore molecules.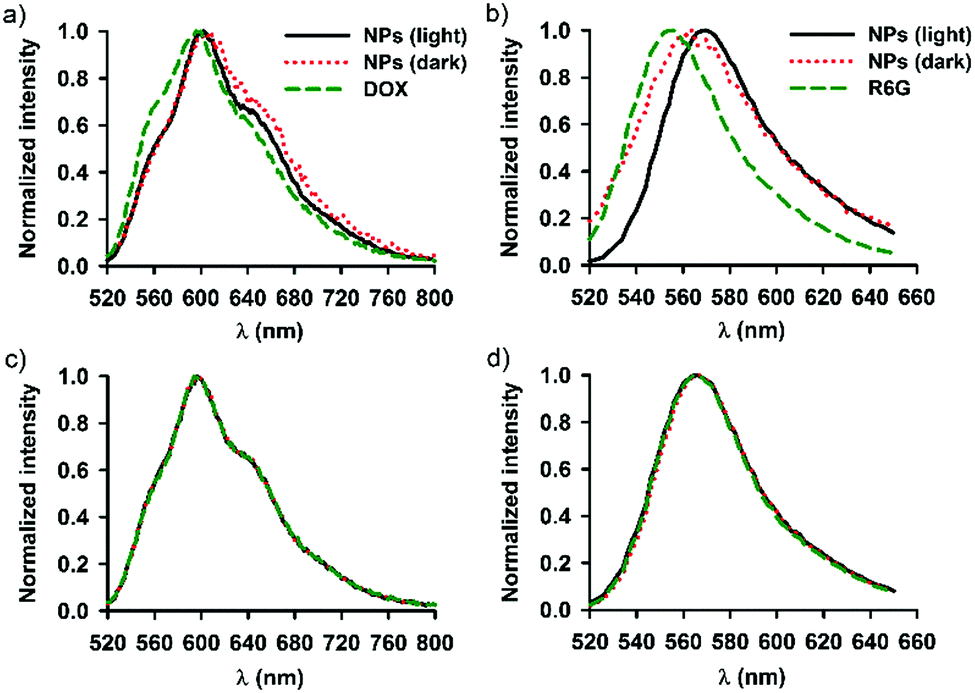 | ||
| Fig. 4 Emission spectra of DOX (excitation at 479 nm) and R6G (excitation at 480 nm). (a) DOX (1 μM) and SNO–DOX NPs (with or without 3 h light-pretreatment) in water. (b) R6G (1 μM) and SNO–R6G NPs (with or without 3 h light-pretreatment) in water. (c) Superimposed emission spectra for authentic DOX dissolved in DMSO and for extracted DOX (in DMSO from light and dark NPs). (d) Superimposed emission spectra for authentic R6G dissolved in DMSO and for extracted R6G. All curves are normalized by the respective peak values. | ||
Triggered NO release and fluorescence enhancement
As described above, light-triggered fluorescence enhancement seemed to correlate with light-induced NO release (Fig. 1c and d). It is therefore hypothesized that SNO played an important role in the observed fluorescent property, because SNO is photolabile.59 To provide further evidence, continuous fluorescence measurements for both fluorophores and a NO-releasing probe were performed for nanoparticle dispersions under continuous light irradiation or in the dark. The dark experiment lasted for 9 days to ensure complete NO release. The results are shown in Fig. 5 for SNO–DOX (left panel, a–c) and SNO–R6G (right panel, d–f), respectively. For SNO–DOX, its fluorescence increased immediately upon exposure to light with a kinetic profile that is almost in parallel with that for the fluorescent NO probe (Fig. 5a). However, in the dark the fluorescence of SNO–DOX only increased slowly with much lower intensities at an equivalent amount of NO release (Fig. 5b), compared with light-irradiated particles. Fig. 5c shows the correlation between fluorescence intensities (i.e. SNO–DOX vs. the NO probe). The correlation plot indeed reveals a sharp difference between light-irradiated and light-protected particles. It indicates that fluorescence intensities of light-irradiated NPs corresponded sensitively to the fluorescence signal of NO release. In contrast, light-protected nanoparticles did not have similar acute responsiveness. Similar observations could be made for SNO–R6G, although the onset of fluorescent response is somewhat delayed, with a lag time of about 30 min (Fig. 5d–f). Together, the findings for both SNO–DOX and SNO–R6G suggest that NO release is not sufficient to produce significant augmentation of particle fluorescence. It seemed that a large amount of NO has to be released within a short time, in an augmented manner (e.g. triggered by light), so that particle fluorescence can be much enhanced. To further test this premise, fluorescence measurements were conducted in the presence of mercuric ions, based on the labile nature of SNO towards mercuric ions, causing facilitated SNO breakdown and subsequent NO release.65,66 The experiment was initially performed in the dark to avoid light interference. Fig. 6a shows mercuric-ion-dependent increase in fluorescence intensity of SNO–DOX over time (in the dark), where the fluorescence-enhancing effect is saturable at high concentrations of HgCl2 (Fig. 6b). Moreover, mercuric ion had little effect on R6G release (Fig. 6c), and as in the light experiments described above, particle-encapsulated R6G, but not the free R6G, is the major source of fluorescence. Remarkably, when the particle samples were further subjected to visible-light irradiation, further fluorescence increase was observed for particles that were initially treated with low amounts of mercuric ions (Fig. 6d); and after prolonged irradiation all samples exhibited comparable fluorescence intensities. Similar results were obtained for SNO–R6G (Fig. S4, ESI†). The finding suggests that light and mercuric ions may operate under a similar mechanism, where both stimuli triggered a burst release of NO, thereby resulting in potential intraparticle perturbation.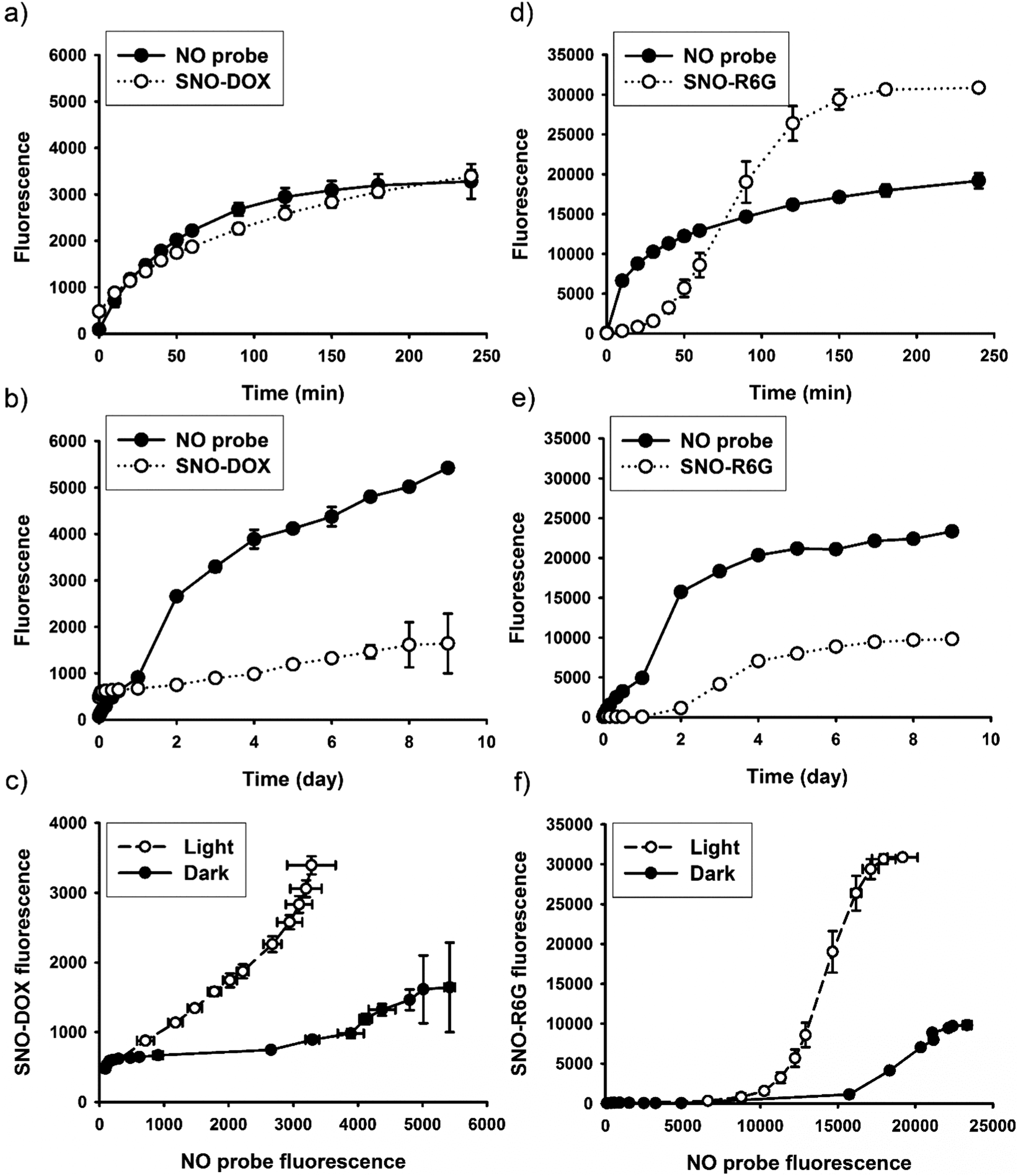 | ||
| Fig. 5 Fluorescent response under light-triggered NO release vs. slow NO release in the dark. Left panel: SNO–DOX, (a) kinetic profiles of fluorescence intensities for SNO–DOX and the NO probe under light irradiation; (b) kinetic profiles of fluorescence intensities for SNO–DOX and the NO probe in the dark; (c) the correlation between SNO–DOX and the NO probe (fluorescence). Right panel: SNO–R6G, (d) kinetic profiles of fluorescence intensities for SNO–R6G and the NO probe under light irradiation; (e) kinetic profiles of fluorescence intensities for SNO–R6G and the NO probe in the dark; (f) the correlation between SNO–R6G and the NO probe (fluorescence). | ||
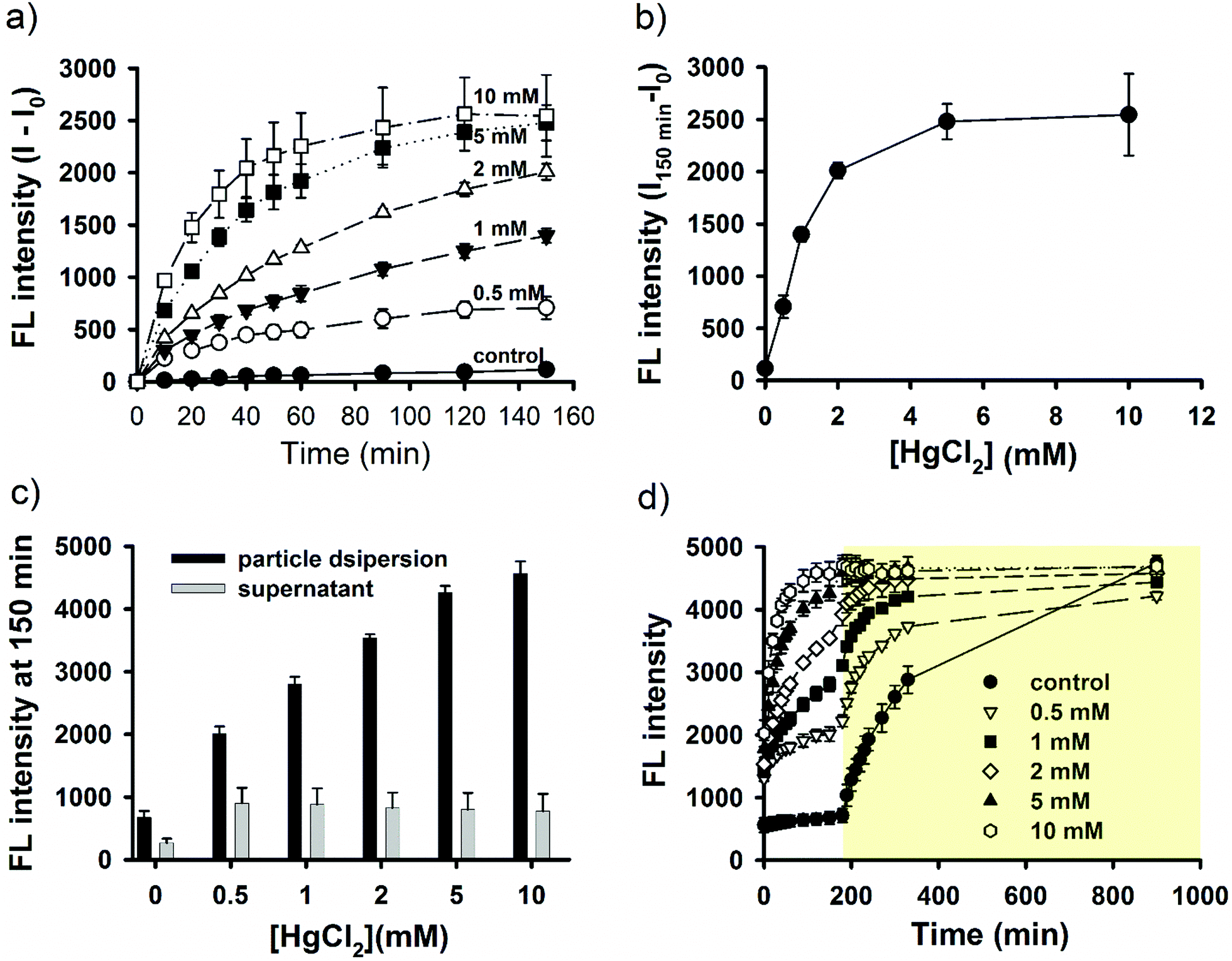 | ||
| Fig. 6 Mercuric ion-triggered fluorescent response. (a) Kinetic fluorescent profiles for SNO–DOX dispersions in the presence of various concentrations (0.5–10 mM) of mercuric chloride. (b) The plot of maximum fluorescence increment vs. the concentration of mercuric chloride. (c) The comparison of absolute fluorescent intensities between particle dispersion and supernatant at various concentrations of mercuric chloride. (d) Mercuric ion-triggered fluorescent response before and after (yellow shaded area) light exposure. Symbols represent different concentrations of mercuric ions added. | ||
Since S-nitrosothiols are also thermally labile, a logical next step is to demonstrate the thermal effect. We first measured the kinetic profile of fluorescence intensity over time for aqueous SNO–R6G dispersions incubated at 60 °C with and without light irradiation. The result, as shown in Fig. S5 (ESI†), is somewhat surprising. In the dark, fluorescence is not increased both at room temperature and at 60 °C up to 150 min. However, when the sample was incubated at 60 °C and under light irradiation, a synergistic effect between light and temperature could be observed (Fig. S5a, ESI†). In the following study, the incubation time was extended to 6 h at various temperatures and in the dark. The result indeed shows that fluorescence intensities increased with increasing temperature after extended period of time (Fig. S5b and c, ESI†). Remarkably, marked fluorescence response similar to the light effect is only observable at 80 °C. Overall, while the additional experiment confirms the temperature effect, it indicates that temperature is less efficient than light in triggering the fluorescence response. The finding may be attributed to the low thermal conductivity property of nano-porous silica materials, which are frequently studied for thermal insulation.67–69 Indeed, our previous study showed that SNO breakdown is much faster under light exposure than at elevated temperature.70
Discussion
S-Nitrosothiols (SNO) are a class of nitric oxide (NO)-donating groups. In the biological system, it has been considered as NO carriers that can store and carry NO functionality. Recently, SNO chemistry has been extensively used for fabricating NO-releasing materials for biomedical applications.54,55,71–75 In the present study, we describe a potential novel application of SNO chemistry in the preparation of fluorophore-loaded nanoparticles (Scheme 1). For the first time, we demonstrate that light-responsive SNO can be turned on post-synthetically for fluorescence augmentation. The finding is partly attributable to versatile synthesis of silica nanoparticles with SNO and fluorophores co-loading, based on our single-silane-based nanoprecipitation approach.54 Upon shining visible light, the as-synthesized SNO–DOX or SNO–R6G NPs exhibited much enhanced fluorescence. Importantly, we ruled out the possibility that the observation is due to fluorescence dequenching as a result of fluorophore release. Moreover, slow, gentle breakdown of SNO in the dark seemed not to generate the same degree of fluorescence enhancement seen in light-irradiated NPs (or when in the presence of mercuric ions). Therefore, we envision that SNO can be engineered into fluorophore-based nanomaterials for light-mediated modulation of the emission behavior of entrapped fluorophores (i.e. “remote control” of fluorescence intensity).In this paper, we provide multiple evidence supporting the role of SNO breakdown in mediating fluorescence response. First, in Fig. 1 and 5, we have shown the correlation between NO release and fluorescence activation, although correlation does not imply causation. Second, in Fig. 6 and Fig. S4 (ESI†), we show that fluorescence can also be activated by mercuric ions (because SNO is also labile to mercuric ions); particularly, when the fluorescence activation reached maximum in the presence of high concentration of mercuric ions, the system did not respond to light further, suggesting that light–thiol interaction may not cause the phenomenon observed. Moreover, the role of SNO breakdown is further verified by the demonstration of the thermal effect (Fig. S5, ESI†). Finally, to further solidify our argument, we attempted to carry out the synthesis of control particles without the nitrosation step. In so doing, the procedure has to be modified since nitrosation was used intentionally to facilitate nanoparticle formation; without nitrosation, it is difficult to form nanoparticle using only MPTMS as a sole silane source and based on a solvent-shifting scheme.55–57 Eventually, we managed to obtain control R6G-loaded NPs without SNO groups. Then, a similar experiment was conducted to monitor the fluorescence kinetics over time with and without light irradiation on the control particles. The preparation and the result of the kinetic evaluation of control particles were appended (Fig. S6, ESI†). As can be seen, the fluorescence intensity of control particles was slowly decreased over time with and without light exposure; and instead of enhancing fluorescence, light irradiation seemed to facilitate the fluorescence bleaching of the control NPs. Taken together, SNO, but not thiol groups are responsible for light-triggered fluorescence enhancement.
Potential perturbations of inner microenvironment
The emission behavior of a fluorophore depends, in addition to its molecular structure, on various other factors—including, but not limited to, hydrophobicity/hydrophilicity of the environment that surrounds the fluorophore molecules and the formation of aggregation states.25,28,29,40,50 In SNO–DOX or SNO–R6G, the encapsulated fluorophores (DOX or R6G) are likely to be surrounded by siloxane (Si–O–Si) and silanol (Si–OH) groups, which are less polar than water. Since SNO is also hydrophobic, it is probable that the dye molecules are enclosed in a hydrophobic microenvironment. Therefore, how light-mediated SNO unloading may have changed the intraparticle microenvironment, thereby affecting fluorescence, is of great interest.Co-encapsulation of SNO and fluorophores in the same silica NPs may result in fluorescence quenching due to potential energy transfer. Under this premise, the removal of SNO may alleviate quenching, thereby boosting fluorescence. However, our data in Fig. 5 demonstrate that even after near complete decomposition of SNO (by placing aqueous NP dispersions in the dark for an extended period of time), the gain of NP fluorescence cannot match that observed in the experiment with light. The result suggests that light-mediated fluorescence enhancement cannot be simply interpreted as dequenching through SNO removal. It further implies that intraparticle perturbation due to vigorous, facilitated SNO breakdown and NO release may be involved. Therefore, in the follow-up experiments, we hypothesize that light irradiation may lead to changes in the interaction between fluorophore molecules and their surrounding silica structure. To test this hypothesis, we conducted a solvent extraction study, in which SNO–DOX NPs were irradiated for various lengths of time. Then, both the light-irradiated NPs and control NPs (without pre-exposure to light) were subjected to solvent extraction using DMSO. Remarkably, the result in Fig. 7a shows that the percentage of DOX extracted by 60% DMSO was progressively reduced as the irradiation time increased, whereas the extraction ratio remained unchanged for the control NPs (light-protected in the dark). The data suggest that light-triggered burst release of NO may render a less solvent-accessible intraparticle microenvironment. Besides, reduced solvent extractability may be due to increased interaction of the fluorophore molecule with the surrounding silica structures. Indeed, evidence from solid-state 29Si NMR points to a possibility that light-triggered burst release of NO may result in the cleavage of some siloxane (Si–O–Si) linkages (reducing T3 fraction in Fig. 7c as compared with Fig. 7b) and the resulting formation of new silanol (Si–OH) groups (corresponding to increasing T2 fraction in Fig. 7c) within the silica backbone. The increase of Si–OH linkages within the silica cavity may contribute to enhanced fluorophore interaction via increased propensity of H-bonding and electrostatic interaction (particularly between the cationic fluorophores and negative silanolate, Si–O−). Such an increase in molecular interaction may further immobilize the fluorophore, thereby leading to fluorescence enhancement.50,51 Notably, this conjecture is consistent with the finding in the solvent extraction study.
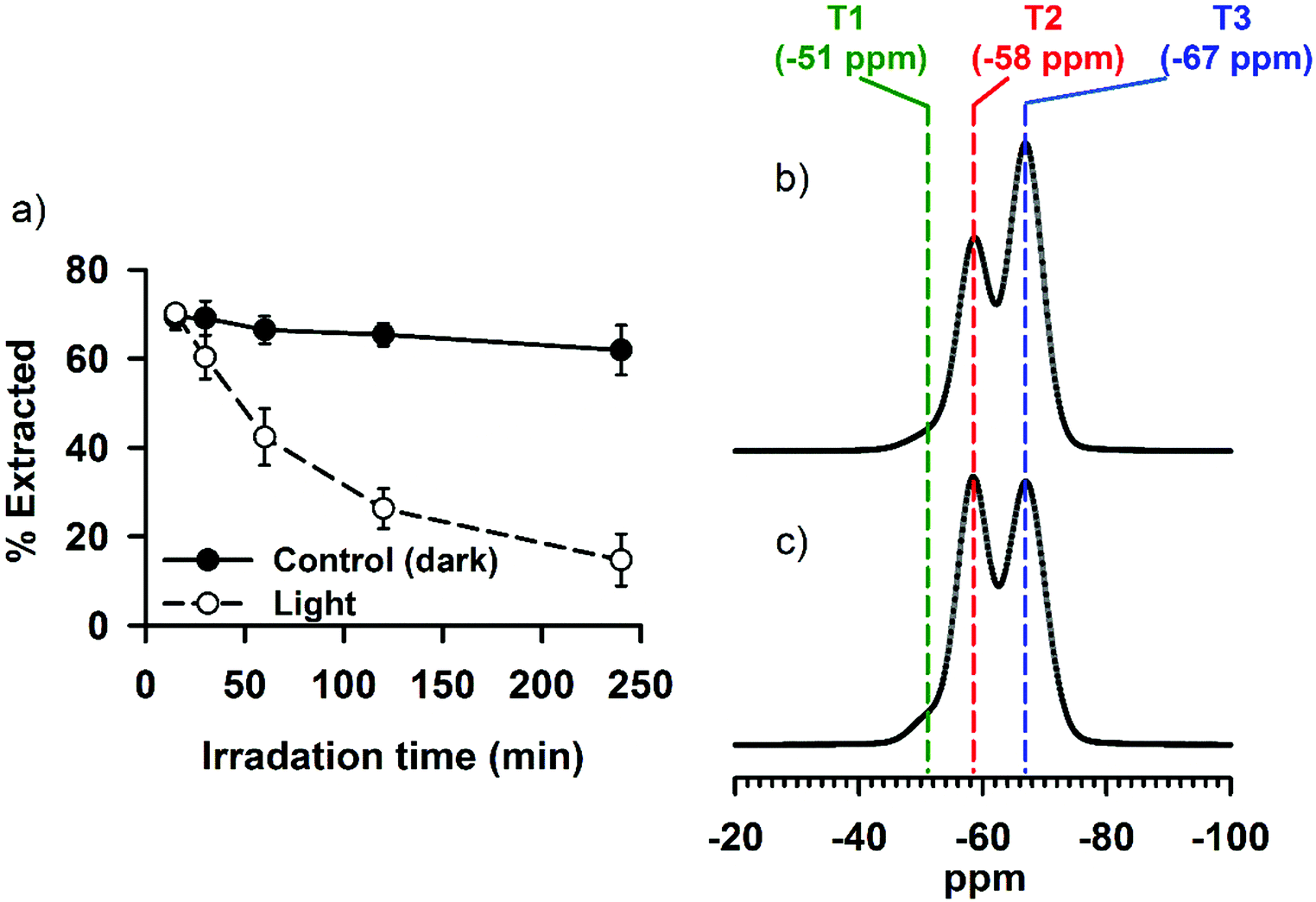 | ||
| Fig. 7 Solvent extractability and solid-state 29Si NMR spectra. (a) Differential extractability between light-treated and control (dark) SNO–DOX. For light-treated samples, particle dispersions were irradiated at various times, followed by solvent extraction using 60% DMSO. (b) NMR spectra for SNO–DOX prepared and stored in the dark. The fraction of T1, T2, and T3 is 4.9%, 36.1%, and 59.0%, respectively. (c) NMR spectra for light-treated (24 h) SNO–DOX. The fraction of T1, T2, and T3 is 4.2%, 43.5%, and 52.3%, respectively. | ||
Although, in Fig. 7, we offer two lines of evidence (Si-NMR and solvent extractability) to provide some inkling about the intraparticle mechanism potentially involved in the observed phenomenon, it is quite perplexing as to how photo-induced decomposition of S-nitrosothiols (i.e. through hemolytic S–N cleavage76) would have an effect on the breaking of siloxane bonds; to the best of our knowledge, this has not been reported. Indeed, it has been known that siloxane bonds are hydrolysable in water, which can be facilitated by several catalysts.77–80 The chemistry of S-nitrosothiol decomposition in the solution is itself complex, involving the formation of multiple free radicals including, NO, thiyl radical (RS˙) and other radicals.76,81–83 Besides, the decomposition reaction would also produce disulfide linkages and nitrite ions.83 Accordingly, within the confined matrix of particles one of the reaction intermediates or products may act as a catalyst for siloxane hydrolysis. Moreover, other than siloxane breaking, the formation of new intraparticle disulfide linkage may render silica nano-cavities more rigid and confined such that the movement of dye molecules is further hindered.
Can other fluorophores exhibit similar light response, when prepared using the same nanoprecipitation approach? The answer to this question may offer insights into the mechanism of the phenomenon recorded. Both DOX and R6G are cationic amine-containing fluorophores and they were stably encapsulated in the SNO–silica structure and were responsive to light. In this regard, we chose fluorescein (FLU), an anionic fluorophore, for comparison. Fig. S7a (ESI†) shows the fluorescent behavior of fluorescein-loaded SNO–silica nanoparticles (abbreviated as SNO–FLU) measured in an ON/OFF manner. As can be seen, the kinetic fluorescence profile is not a step-like curve as observed for SNO–DOX (Fig. 1), but more like a smooth curve with continuous increase of fluorescence in the dark condition. The result suggests that light has little effect on SNO–FLU. Indeed, when the particle dispersion was centrifuged to separate the supernatant from particle, one immediately sees that there is no difference in fluorescence intensity between particle dispersion and its supernatant (Fig. S7b, ESI†). The result indicates that unlike SNO–DOX or SNO–R6G, the payload of SNO–FLU was continuously released into the aqueous medium, which contributed to fluorescence increase over time and light unresponsiveness. The finding could be understood as the electrostatic repulsion between anionic fluorescein molecules and the anionic silanolate of the particle, given that silanol (Si–OH) groups are weakly acidic and prepared particles are negatively charged.54,55 In contrast, the anionic silica structure would favor the retention of cationic dyes within particles (Fig. 2, 3 and 6). Therefore, this stark contrast between cationic and anionic dyes in terms of payload retention further supports our surmise that intraparticle molecular interactions played a significant role in light-triggered, SNO-mediated fluorescence augmentation.
Implications and outlook
Thus far we have demonstrated that the fluorescence intensity of silicon-based NPs loaded with conventional organic dyes can be tuned on post-synthetically, via a novel S-nitrosothiol route. The major focus of this paper is to communicate this novel fluorescence activation phenomenon. One of the dyes presented herein is doxorubicin (DOX), a widely prescribed anticancer drug. Although DOX is generally not used for labeling or imaging purpose, it is the first example for post-loading fluorescence augmentation remotely controlled by external light stimuli. It remains to be tested whether the light-boosted NPs could have additional advantages such as improving the tracing or detection of the DOX-loaded NP during a therapeutic trial. Our second example is R6G, a widely studied fluorescent and laser dye. R6G is intrinsically toxic and has poor photostability; besides, it is an extremely hydrophobic molecule (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 6.36, Source: ALOGPS). A dilemma for R6G and other dyes with similar physicochemical properties is the potential tradeoff between the loading efficiency and the fluorescence intensity. By using a hydrophobic loading (i.e. loading based on hydrophobic interaction), as used in this study, R6G would be efficiently entrapped within NP; however, its fluorescence would be significantly quenched due to formation of dye aggregates.40,42 In the present study, we appear to resolve this issue by engineering SNO groups in the NP and by utilizing the light-sensitive SNO chemistry to possibly alter the inner particle environment. We show some improved properties of the light-treated SNO–R6G NP, such as enhanced fluorescence intensity and photostability, and much reduced cytotoxicity. The future studies should focus on applying the proposed approach to a wide variety of fluorophores, both conventional and emerging ones,84 in an attempt to increase the versatility of dye-loaded NPs,85 as well as match the performance of quantum dots (Q-dots), polymer dots (P-dots) or C-dots, in the in vitro cellular labeling and in vivo imaging.3,9,10
P = 6.36, Source: ALOGPS). A dilemma for R6G and other dyes with similar physicochemical properties is the potential tradeoff between the loading efficiency and the fluorescence intensity. By using a hydrophobic loading (i.e. loading based on hydrophobic interaction), as used in this study, R6G would be efficiently entrapped within NP; however, its fluorescence would be significantly quenched due to formation of dye aggregates.40,42 In the present study, we appear to resolve this issue by engineering SNO groups in the NP and by utilizing the light-sensitive SNO chemistry to possibly alter the inner particle environment. We show some improved properties of the light-treated SNO–R6G NP, such as enhanced fluorescence intensity and photostability, and much reduced cytotoxicity. The future studies should focus on applying the proposed approach to a wide variety of fluorophores, both conventional and emerging ones,84 in an attempt to increase the versatility of dye-loaded NPs,85 as well as match the performance of quantum dots (Q-dots), polymer dots (P-dots) or C-dots, in the in vitro cellular labeling and in vivo imaging.3,9,10
Conclusions
In the present study, fluorophore-loaded silica NPs were prepared using our previously published nanoprecipitation method.54,55 The use of one-pot, single-mercaptosilane-based approach allows us to synthesize SNO and fluorophores co-loading silica NPs with ease. We demonstrate that the fluorescence intensity of as-synthesized SNO–DOX or SNO–R6G can be significantly turned on by simply shining visible light on the NPs. Remarkably, the maximum fluorescence enhancement is about 100-fold for SNO–R6G. Both DOX and R6G are stably encapsulated in SNO–DOX and SNO–R6G with low payload release; especially the release of R6G is almost negligible. Besides, the observed photo-triggered emission behavior was not due to fluorophore release. Instead, we provide several lines of evidence showing that triggered NO release plays an important role in fluorescence augmentation. Thus, SNO is proposed as an intraparticle light-responsive unit for post-synthesis enhancement of NP fluorescence.SNO is known to be photolabile and frequently employed in the design and synthesis of NO donating compounds or materials. Here, a new concept is emerging: i.e. SNO-based post-synthesis fluorescence modulation of dye-doped nanomaterials (Scheme 1). First, SNO modification of silica species facilitates the assembly of silica NPs with high fluorophore loading efficiency. Second, after particle synthesis, vigorous NO release from light-mediated SNO breakdown results in a change of the microenvironment within the nanostructure. Such an intraparticle perturbation would alter the emission behavior of fluorophore molecules.
Although fluorophore-doped silica NPs can be prepared by various approaches, fluorescence quenching remains a challenge. In this regard, our current SNO-based approach represents a promising strategy for alleviating quenching. Besides, the method offers additional advantages, including the use of only a single silane to synthesize functionalized silica NPs, facile non-covalent loading with high loading efficiency and low payload loss (at least for some fluorophores), and the simplicity of the method. In the future, we envisage that the concept may be applied to modulate emission behaviors of emerging aggregation-induced emissive fluorogens (AIEgens). Moreover, we also envision that a more extended and generalizable use of SNO as a photo-responsive modulator of intraparticle microenvironment, e.g. applications even beyond fluorescence modulation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Ministry of Science and Technology of Taiwan (NSC 102-2320-B-016-003-MY3, MOST 105-2320-B-010-037-MY3 & MOST 106-2320-B-010-027). We thank Ms Huei-Min Chen (Core Facility Center at Taipei Medical University) for the technical support (TEM). We also thank the Instrumentation Center in the National Taiwan University for the solid-state 29Si NMR measurement. A2058 cells were kindly provided by Prof. Dr Shih-Ming Huang of National Defense Medical Center.Notes and references
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- M. H. Leung, T. Harada, S. Dai and T. W. Kee, Langmuir, 2015, 31, 11419–11427 CrossRef CAS PubMed.
- T. T. Morgan, H. S. Muddana, E. I. Altinoglu, S. M. Rouse, A. Tabakovic, T. Tabouillot, T. J. Russin, S. S. Shanmugavelandy, P. J. Butler and P. C. Eklund, Nano Lett., 2008, 8, 4108–4115 CrossRef CAS PubMed.
- A. A. Burns, J. Vider, H. Ow, E. Herz, O. Penate-Medina, M. Baumgart, S. M. Larson, U. Wiesner and M. Bradbury, Nano Lett., 2008, 9, 442–448 CrossRef PubMed.
- X. Wu and W. Zhu, Chem. Soc. Rev., 2015, 44, 4179–4184 RSC.
- B. Korzeniowska, R. Nooney, D. Wencel and C. McDonagh, Nanotechnology, 2013, 24, 442002 CrossRef CAS PubMed.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- C. Caltagirone, A. Bettoschi, A. Garau and R. Montis, Chem. Soc. Rev., 2015, 44, 4645–4671 RSC.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- S. W. Bae, W. Tan and J. I. Hong, Chem. Commun., 2012, 48, 2270–2282 RSC.
- B. Cohen, C. Martin, S. K. Iyer, U. Wiesner and A. Douhal, Chem. Mater., 2012, 24, 361–372 CrossRef CAS.
- Q. Huo, J. Liu, L. Q. Wang, Y. Jiang, T. N. Lambert and E. Fang, J. Am. Chem. Soc., 2006, 128, 6447–6453 CrossRef CAS PubMed.
- E. Rampazzo, S. Bonacchi, M. Montalti, L. Prodi and N. Zaccheroni, J. Am. Chem. Soc., 2007, 129, 14251–14256 CrossRef CAS PubMed.
- C. Caltagirone, A. Bettoschi, A. Garau and R. Montis, Chem. Soc. Rev., 2015, 44, 4645–4671 RSC.
- S. Sripanyakorn, R. Jugdaohsingh, W. Dissayabutr, S. H. Anderson, R. P. Thompson and J. J. Powell, Br. J. Nutr., 2009, 102, 825–834 CrossRef CAS PubMed.
- E. Phillips, O. Penate-Medina, P. B. Zanzonico, R. D. Carvajal, P. Mohan, Y. Ye, J. Humm, M. Gönen, H. Kalaigian and H. Schöder, Sci. Transl. Med., 2014, 6, 260ra149 CrossRef PubMed.
- A. Anedda, C. Carbonaro, F. Clemente, R. Corpino, S. Grandi, A. Magistris and P. Mustarelli, J. Non-Cryst. Solids, 2005, 351, 1850–1854 CrossRef CAS.
- D. R. Larson, H. Ow, H. D. Vishwasrao, A. A. Heikal, U. Wiesner and W. W. Webb, Chem. Mater., 2008, 20, 2677–2684 CrossRef CAS.
- X. Zhao, R. P. Bagwe and W. Tan, Adv. Mater., 2004, 16, 173–176 CrossRef CAS.
- S. Santra, P. Zhang, K. Wang, R. Tapec and W. Tan, Anal. Chem., 2001, 73, 4988–4993 CrossRef CAS PubMed.
- L. M. Rossi, L. Shi, F. H. Quina and Z. Rosenzweig, Langmuir, 2005, 21, 4277–4280 CrossRef CAS PubMed.
- D. Ma, A. J. Kell, S. Tan, Z. J. Jakubek and B. Simard, J. Phys. Chem. C, 2009, 113, 15974–15981 CAS.
- M. Ogawa, T. Nakamura, J.-I. Mori and K. Kuroda, J. Phys. Chem. B, 2000, 104, 8554–8556 CrossRef CAS.
- D. Avnir, D. Levy and R. Reisfeld, J. Phys. Chem., 1984, 88, 5956–5959 CrossRef CAS.
- A. Anedda, C. Carbonaro, R. Corpino, P. Ricci, S. Grandi and P. Mustarelli, J. Non-Cryst. Solids, 2007, 353, 481–485 CrossRef CAS.
- J. W. Gilliland, K. Yokoyama and W. T. Yip, Chem. Mater., 2005, 17, 6702–6712 CrossRef CAS.
- A. Van Blaaderen and A. Vrij, Langmuir, 1992, 8, 2921–2931 CrossRef CAS.
- R. Nyffenegger, C. Quellet and J. Ricka, J. Colloid Interface Sci., 1993, 159, 150–157 CrossRef CAS.
- L. Wang, B. Li, L. Zhang, L. Zhang and H. Zhao, Sens. Actuators, B, 2012, 171, 946–953 CrossRef.
- Y.-C. Wu and F. Nie, Sens. Actuators, B, 2015, 212, 120–126 CrossRef CAS.
- C. Graf, W. Schärtl, K. Fischer, N. Hugenberg and M. Schmidt, Langmuir, 1999, 15, 6170–6180 CrossRef CAS.
- A. Imhof, M. Megens, J. Engelberts, D. De Lang, R. Sprik and W. Vos, J. Phys. Chem. B, 1999, 103, 1408–1415 CrossRef CAS.
- P. J. Dewar, T. F. MacGillivray, S. M. Crispo and T. Smith-Palmer, J. Colloid Interface Sci., 2000, 228, 253–258 CrossRef CAS PubMed.
- P. Innocenzi, H. Kozuka and T. Yoko, J. Non-Cryst. Solids, 1996, 201, 26–36 CrossRef CAS.
- C. M. Carbonaro, J. Photochem. Photobiol., A, 2011, 222, 56–63 CrossRef CAS.
- B. Lasio, L. Malfatti and P. Innocenzi, J. Photochem. Photobiol., A, 2013, 271, 93–98 CrossRef CAS.
- J. Bujdák and N. Iyi, J. Phys. Chem. B, 2006, 110, 2180–2186 CrossRef PubMed.
- C. M. Carbonaro, F. Meinardi, P. C. Ricci, M. Salis and A. Anedda, J. Phys. Chem. B, 2009, 113, 5111–5116 CrossRef CAS PubMed.
- L. Malfatti, T. Kidchob, D. Aiello, R. Aiello, F. Testa and P. Innocenzi, J. Phys. Chem. C, 2008, 112, 16225–16230 CAS.
- D. R. Larson, H. Ow, H. D. Vishwasrao, A. A. Heikal, U. Wiesner and W. W. Webb, Chem. Mater., 2008, 20, 2677–2684 CrossRef CAS.
- F. Del Monte, J. D. Mackenzie and D. Levy, Langmuir, 2000, 16, 7377–7382 CrossRef CAS.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS PubMed.
- S. Liang, K. Shephard, D. T. Pierce and J. X. Zhao, Nanoscale, 2013, 5, 9365–9373 RSC.
- E. Rampazzo, S. Bonacchi, M. Montalti, L. Prodi and N. Zaccheroni, J. Am. Chem. Soc., 2007, 129, 14251–14256 CrossRef CAS PubMed.
- B. Cohen, F. Sanchez and A. Douhal, J. Am. Chem. Soc., 2010, 132, 5507–5514 CrossRef CAS PubMed.
- A. Auger, J. Samuel, O. Poncelet and O. Raccurt, Nanoscale Res. Lett., 2011, 6, 328 CrossRef PubMed.
- M. Xue and J. I. Zink, J. Phys. Chem. Lett., 2014, 5, 839–842 CrossRef CAS PubMed.
- W.-Y. Huang and J. I. Zink, J. Phys. Chem. C, 2016, 120, 23780–23787 CAS.
- J. R. Sánchez-Valencia, I. Blaszczyk-Lezak, J. P. Espinós, S. Hamad, A. N. R. González-Elipe and A. Barranco, Langmuir, 2009, 25, 9140–9148 CrossRef PubMed.
- T. Pirzada and S. S. Shah, Colloid Polym. Sci., 2015, 293, 3361 CAS.
- M. R. Wang, S. J. Chiu, H. C. Chou and T. M. Hu, Chem. Commun., 2015, 51, 15649–15652 RSC.
- H. C. Chou, S. J. Chiu, Y. L. Liu and T. M. Hu, Langmuir, 2014, 30, 812–822 CrossRef CAS PubMed.
- S. J. Chiu, S. Y. Wang, H. C. Chou, Y. L. Liu and T. M. Hu, Langmuir, 2014, 30, 7676–7686 CrossRef CAS PubMed.
- S. J. Chiu, C. Y. Lin, H. C. Chou and T. M. Hu, Langmuir, 2016, 32, 211–220 CrossRef CAS PubMed.
- H. C. Chou, S. J. Chiu and T. M. Hu, Biomacromolecules, 2015, 16, 2288–2295 CrossRef CAS PubMed.
- D. J. Sexton, A. Muruganandam, D. J. McKenney and B. Mutus, Photochem. Photobiol., 1994, 59, 463–467 CrossRef CAS PubMed.
- T. Ishida, M. Kirchmeier, E. Moase, S. Zalipsky and T. Allen, Biochim. Biophys. Acta, Biomembr., 2001, 1515, 144–158 CrossRef CAS.
- F. Wang, Y.-C. Wang, S. Dou, M.-H. Xiong, T.-M. Sun and J. Wang, ACS Nano, 2011, 5, 3679–3692 CrossRef CAS PubMed.
- A. Fritze, F. Hens, A. Kimpfler, R. Schubert and R. Peschka-Süss, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1633–1640 CrossRef CAS PubMed.
- J. Wang, J. Bhattacharyya, E. Mastria and A. Chilkoti, J. Controlled Release, 2017, 260, 100–110 CrossRef CAS PubMed.
- K. K. Karukstis, E. H. Thompson, J. A. Whiles and R. J. Rosenfeld, Biophys. Chem., 1998, 73, 249–263 CrossRef CAS PubMed.
- J. A. Cook, S. Y. Kim, D. Teague, M. C. Krishna, R. Pacelli, J. B. Mitchell, Y. Vodovotz, R. W. Nims, D. Christodoulou and A. M. Miles, Anal. Biochem., 1996, 238, 150–158 CrossRef CAS PubMed.
- D. Jourd’heuil, K. Hallén, M. Feelisch and M. B. Grisham, Free Radical Biol. Med., 2000, 28, 409–417 CrossRef.
- H. Abe, I. Abe, K. Sato and M. Naito, J. Am. Ceram. Soc., 2005, 88, 1359–1361 CrossRef CAS.
- M. Schmidt and F. Schwertfeger, J. Non-Cryst. Solids, 1998, 225, 364–368 CrossRef CAS.
- F. Meng, M. Elsahati, J. Liu and R. F. Richards, J. Appl. Phys., 2017, 121, 194302 CrossRef.
- C.-H. Lo and T.-M. Hu, Soft Matter, 2017, 13, 5950–5960 RSC.
- W.-L. Chang, K.-J. Peng, T.-M. Hu, S.-J. Chiu and Y.-L. Liu, Polymer, 2015, 57, 70–76 CrossRef CAS.
- J. Kim, G. Saravanakumar, H. W. Choi, D. Park and W. J. Kim, J. Mater. Chem. B, 2014, 2, 341–356 RSC.
- H. T. Duong, Z. M. Kamarudin, R. B. Erlich, Y. Li, M. W. Jones, M. Kavallaris, C. Boyer and T. P. Davis, Chem. Commun., 2013, 49, 4190–4192 RSC.
- D. A. Riccio, J. L. Nugent and M. H. Schoenfisch, Chem. Mater., 2011, 23, 1727–1735 CrossRef CAS PubMed.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, J. Controlled Release, 2015, 205, 190–205 CrossRef CAS PubMed.
- R. J. Singh, N. Hogg, J. Joseph and B. Kalyanaraman, FEBS Lett., 1995, 360, 47–51 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed.
- Y. Zuo, Z. Gou, J. Cao, Z. Yang, H. Lu and S. Feng, Chem. – Eur. J., 2015, 21, 10972–10977 CrossRef CAS PubMed.
- C. Rücker and K. Kümmerer, Chem. Rev., 2014, 115, 466–524 CrossRef PubMed.
- M. Cypryk and Y. Apeloig, Organometallics, 2002, 21, 2165–2175 CrossRef CAS.
- D. R. Arnelle and J. S. Stamler, Arch. Biochem. Biophys., 1995, 318, 279–285 CrossRef CAS PubMed.
- R. J. Singh, N. Hogg, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 1996, 271, 18596–18603 CrossRef CAS PubMed.
- P. D. Wood, B. Mutus and R. W. Redmond, Photochem. Photobiol., 1996, 64, 518–524 CrossRef CAS.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- I. Sokolov and S. Naik, Small, 2008, 4, 934–939 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary Fig. S1–S7. See DOI: 10.1039/c7tb02233f |
| This journal is © The Royal Society of Chemistry 2018 |