
Metallic P3C monolayer as anode for sodium-ion batteries†
Ziyuan
Zhao‡
,
Tong
Yu‡
,
Shoutao
Zhang
 ,
Haiyang
Xu
*,
Guochun
Yang
* and
Yichun
Liu
,
Haiyang
Xu
*,
Guochun
Yang
* and
Yichun
Liu
Centre for Advanced Optoelectronic Functional Materials Research, Laboratory for UV Light-Emitting Materials and Technology of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: hyxu@nenu.edu.cn; yanggc468@nenu.edu.cn
First published on 28th November 2018
Abstract
Sodium-ion batteries (SIBs) have become one of the most promising energy storage devices due to the high abundance and safety of sodium. On the other hand, most of the well-developed technologies in lithium-ion batteries (LIBs) can be migrated to SIBs. However, unfortunately, the known anode materials are not suitable in SIBs as a result of the larger atomic radius of sodium compared with lithium. Phosphorus demonstrates an unprecedented high theoretical capacity in SIBs, but its inherent low electrical conductivity and huge volume expansion upon sodiation greatly restrict the cycle stability. Two-dimensional materials as electrodes have shown unique advantages in contrast to their bulk counterparts. In this work, we identify a hitherto unknown P3C monolayer with a puckered honeycomb structure through first-principles swarm-intelligence structure calculations. More intriguingly, the predicted theoretical capacity of P3C as an anode for SIBs is up to 1022 mA g h−1, corresponding to a stoichiometry of P3CNa4. Metallic P3C and P3CNa4 can provide good electrical conductivity during the battery cycle. The acceptable barrier energy and lower open-circuit voltage ensure good rate capacity and safety in practical applications. Its outstanding performance is attributed to the unique arrangement of two kinds of hexagon rings (i.e. P6 and P4C2), producing high cohesive energy and thermodynamic stability. These desirable properties render P3C monolayers as a promising two-dimensional material for application in SIBs.
Introduction
Sodium (Na) is more abundant and evenly distributed in the earth than lithium. Moreover, the mature technologies of lithium-ion batteries (LIBs) can be migrated to sodium-ion batteries (SIBs) owing to the similar chemical properties of lithium (Li) and Na.1 Therefore, SIBs are deemed to be promising energy storage devices and attractive alternatives to LIBs, so alleviating reliance on increasingly depleted lithium sources.2,3 A very tricky scientific issue in SIBs is the performance of anode materials, because the well-developed ones in LIBs are unsuitable for SIBs as a result of the larger atomic radius of Na compared with Li.4–6 For example, commercial graphite in LIBs hardly accommodates Na ions. To solve this problem, various kinds of materials (e.g. carbonaceous compounds,7,8 phosphorus (P) allotropes,9–12 phosphides,13–15 sulfides16,17 and metal alloys18–24) have been tried in SIBs. Unfortunately, their performance is still far from satisfactory. Therefore, design and preparation of anode materials with desirable properties is indispensable to develop high-performance SIBs.25 To accelerate the development of SIBs, many theoretical efforts have been made through structure screening,26 prediction,27,28 and structural modification29,30 to search for ideal anode materials. Moreover, theoretical calculations play an important role in understanding the charge/discharge mechanisms and electronic properties of electrode materials.31,32Among various anode material candidates for SIBs, black P has received much attention due to its remarkable large theoretical capacity (i.e. 2596 mA g h−1) and high stability.33 However, its large volume expansion (∼400%) upon sodiation leads to rapid capacity fading thus impeding battery performance. On the other hand, the low electrical conductivity of black P decreases the reversibility of the cyclic and coulombic efficiency of the battery.34 After extensive efforts, P/C composite materials have been found not only to have high mechanical stability, but also to improve electrochemical performance with respect to black P.35–38 This is attributed to formation of P–C bonds and the good electrical conductivity of C.30
Two-dimensional (2D) materials as anodes exhibit unique advantages, in contrast to their bulk counterparts, such as large surface–volume ratio, broad electrochemical window, fascinating chemical activities, and excellent mechanical strength.39–42 Some 2D materials (e.g. graphene,43 transition metal oxides,44 and transition metal dichalcogenides45) have exhibited excellent performance in LIBs. Phosphorene, stripped from black P, demonstrates intriguing electron properties.46 Phosphorene as anode in SIBs shows acceptable theoretical capacity, however, its inherent non-metallic nature affects electrochemical performance.12,47 Once again, phosphorene–graphene hybrid material shows high performance, in which graphene serves as a mechanical backbone and an electrical highway.38,48 By all appearances, the combination P and C is an effective way to enhance performance of the anode. A natural and immediate thought is therefore to examine whether P and C could form stable two-dimensional compounds with high performance in SIBs.
Considering the high capacity of P as anode, we mainly focus on P-rich P–C stoichiometric monolayers. In this work, an extensive structural search was carried out with various stoichiometries (PxCy, x = 1–8 and y = 1) through first-principles swarm structural search calculations. As expected, a stable and metallic P3C monolayer with a buckled hexagonal structure was identified. More importantly, the P3C monolayer shows high adsorption capacity for sodium (i.e. P3CNa4), corresponding to a theoretical capacity of 1022 mA g h−1. The intrinsic metallicity of P3C and P3CNa4 ensures a good rate performance. The Na diffusion barrier of 0.19 eV in the P3C monolayer is comparable to that of the well-known Ti2C39 and Sb.49 Its high cohesive energy and thermodynamic stability provide good opportunities for experimental synthesis.
Computational methods
A structure search has been carried out by employing a particle swarm optimization algorithm as implemented in the CALYPSO code,50 which is a leading structure prediction method in the field. It can efficiently find ground or metastable structures depending on just the given chemical compositions and its validity has been confirmed by the application to both bulk51–54 and two-dimensional materials.55–59 The structure optimizations and property calculations were performed by using the density functional theory method60 within the generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE)61 as implemented in the VASP code.62 The ion–electron interaction was described by the projector augmented wave (PAW) technique,63 and van der Waals interactions were taken into account.64 To eliminate interactions between adjacent layers, a large vacuum distance of ∼20 Å along the c-axis was used and a plane-wave cutoff energy of 700 eV was employed. Phonon dispersion calculations were based on a supercell approach, as used in the Phonopy code,65 in order to determine the dynamical stability of the predicted structure. The thermal stability was analyzed through first-principles molecular dynamics (MD) simulations.66 Detailed descriptions of the computational method can be found in the ESI.† The cohesive energy of the P-rich monolayer was calculated by using the following formula:| Ecoh = (xEP + EC − EPxC)/(x + 1) | (1) |
The adsorption energy of the Na atom on the P3C monolayer can be obtained by:
| Ead = (EP3C + nENa − EP3CNan)/n | (2) |
Here, the positive adsorption energy indicates that the P3C monolayer has the intrinsic ability to adsorb Na ions. The average open-circuit voltage (OCV) is evaluated by the equation expressed as:
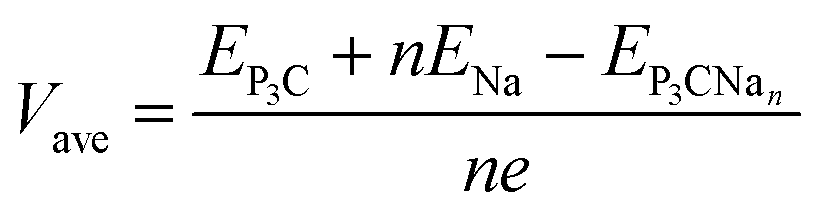 | (3) |
Results and discussion
A stable P3C monolayer with C2/m symmetry was identified through comprehensive structure search calculations. Its basic building blocks are P6 and P4C2 rings. The alternative arrangement of the two kinds of hexagon rings makes the P3C monolayer stabilize in a puckered single-layered structure akin to the structural configuration of layer-structured GeP3,67,68 InP3,69 and SnP3 (ref. 70) (see Fig. 1a). In more detail, each atom is three-fold coordinated with the same or other atoms, having P–P and P–C distances of 2.28 Å and 1.78 Å, which are comparable to that in black P (2.22 Å)71 and almost the same as that in β0-PC (1.77 Å).72 Their covalent bonding characters are evidenced by the presence of localized electrons between two nearest-neighbor atoms, as clearly shown in the plot of electron localization function (ELF, Fig. 1b). On the other hand, the slight torsion of two hexagon rings causes the P3C monolayer to have a thickness of 1.31 Å, which is much thinner than phosphorene (thickness of 2.10 Å)12 and GeP3 (thickness of 2.44 Å).73 Considering these characteristics, P3C monolayers have a large surface–volume ratio,74,75 facilitating intercalation of Na, as discussed later.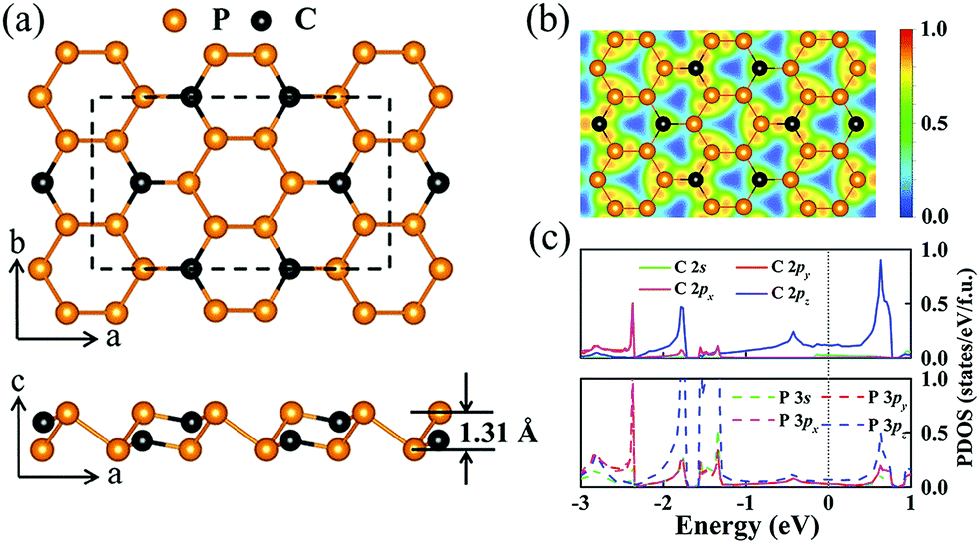 | ||
| Fig. 1 (a) Optimized structure of P3C monolayer with C2/m symmetry. (b) Electron localization function (ELF) of P3C monolayer. (c) Decomposed PDOS of P3C monolayer, projected on four orbitals (s and px,y,z) of P and C atom. | ||
The feasibility of experimental synthesis of the predicted two-dimensional materials correlates closely with the amount of cohesive energy.59,76 The larger the cohesive energy, the easier is experimental synthesis. The resultant cohesive energy of 4.18 eV per atom is significantly higher than that of synthesized phosphorene (3.48 eV per atom)77 or GeP3 (3.34 eV per atom),73 and is comparable to that of the 1T-NiSe2 monolayer (4.66 eV per atom)78 and the 2H–CoTe2 monolayer (4.48 eV per atom).79,80 Notably, a layer-structured GeP3/C nanocomposite has been synthesized showing excellent Li storage performance.81 Besides, the absence of the imaginary frequency mode in the whole Brillouin zone indicates the dynamical stability of the P3C monolayer (Fig. S1a and b†). As shown in projected phonon density of states (PHDOS), the appearance of extensive overlapping between P and C vibrations confirms a strong interaction in the P–C bond. To examine the temperature-dependent stability, we performed MD simulations at 1000 K. The framework of the P3C monolayer maintains its original configuration (Fig. S1c and d†) after simulation. However, the interlayer interaction caused by unique structural characters (i.e. P lone pair electrons, C dangling bond) might hinder synthesis of the P3C monolayer. This inspired us to probe the feasibility of synthesizing the P3C monolayer. According to the symmetry of P3C, five kinds of bilayer stacking patterns can be considered to investigate the exfoliation energy (Fig. S2†). After full structural relaxation, including van der Waals interaction, a stable P3C bilayer is obtained. The interlayer distance of 4.05 Å (Fig. S2d†) is much larger than for graphite (3.23 Å)82 and black phosphorus (3.10 Å).83 Moreover, the exfoliation energy of the P3C bilayer is 25.2 meV Å−2, which is smaller than hexagonal BN (28 meV Å−2) and slightly larger than graphene (21 meV Å−2) and phosphorene (22 meV Å−2).84 These results suggest that our predicted P3C has outstanding thermal and dynamical stability and favors experimental synthesis.
To better understand the nature of the chemical bonding and electron properties of the P3C monolayer, we calculated its electronic band structure (Fig. S3†) and projected density of states (PDOS) (Fig. 1c). As can be seen in Fig. 1c, there is a large overlap of P and C orbitals below the Fermi level, indicating the formation of covalent bonds between P–P and P–C, which is consistent with the structural analysis given above. More intriguingly, the P3C monolayer is metallic because of several bands across the Fermi level, which is in sharp contrast with the GeP3 monolayer with semiconducting character,73 having similar atoms arrangement and electronic configuration between P3C and GeP3. However, in contrast to C, the large atomic radius of Ge leads to a dihedral angle of 38.64° in the GeP3 unit, which is much bigger than the 10.67° in the P3C unit. With replacement of the Ge atoms in the GeP3 monolayer with C atoms, the GeP3-structured P3C monolayer is a semiconductor with a band gap of 0.16 eV (Fig. S4†). In the P3C monolayer, one C atom with the other three P atoms forms quasi-sp2 hybridization, leaving one C 2pz free electron. Each P atom adopts an sp3 hybridization, in which one of the hybrid orbitals is filled with a lone electron pair (3pz), and the other three hybrid orbitals form covalent bonds with the neighboring C and P atoms. However, the dihedral angles in P3C are 106.20° and 88.58°, which is distant from the typical sp3 hybridization of 109.47°. The imperfect sp3 and sp2 hybridization of P and C provides delocalized electrons, which might be responsible for the metallic behavior. This point can also be found from the PDOS (e.g. the main contribution of PDOS around the Fermi energy level coming from C 2pz and P 3pz orbitals, Fig. 1c). To confirm this, we performed hydrogenation on the P3C monolayer (Fig. S5†). The added H atoms can immobilize the delocalized electrons donated by C or P.85 Both P3CH and HP3C are semiconducting (Fig. S5c and d†). A similar phenomenon has been observed in the β0-PC monolayer.72,85
The P3C monolayer is expected to be a potential anode material in SIBs in view of its favorable stability, structural character, and inherent metallicity. For anode materials, spontaneously adsorbing Na atom is one of the necessary conditions. Considering the symmetry of the P3C monolayer, nine possible adsorption sites were considered (Fig. S6†). After full geometrical relaxations, there are two favorable adsorption sites (A1 and A2), localizing at the top of the C atoms (A1) and the P6 ring (A2), as shown in Fig. 2a and b. The resultant adsorption energies at A1 and A2 sites are 1.08 eV and 0.90 eV, respectively, indicating that our predicted monolayer can adsorb Na ions. The larger adsorption energy at the A1 site can be attributed to the delocalized electrons on the C surface providing a suitable habitat for Na ions. As can be seen in the ELF (Fig. S7†), the interaction type between Na and C is ionic with a Na–C distance of 2.49 Å. This is further supported by the Bader charge analysis: each Na atom transfers 0.84 electrons to the C atom.
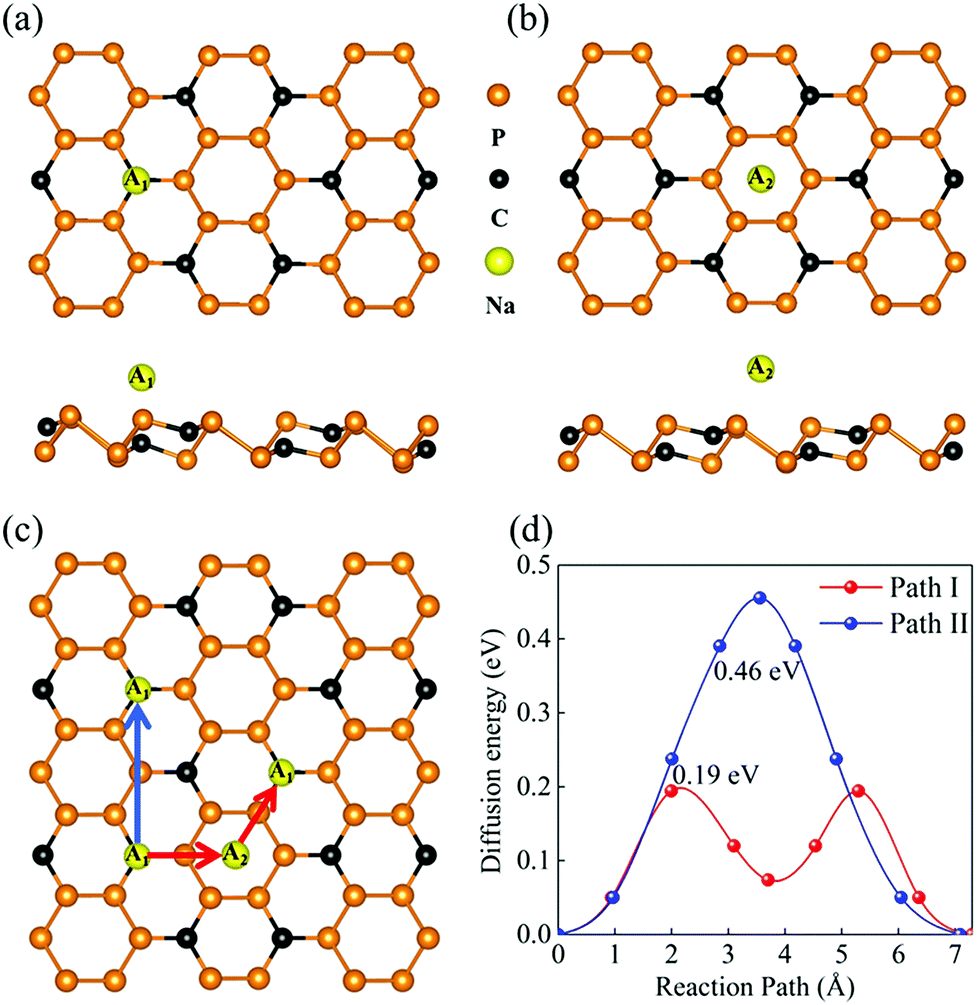 | ||
| Fig. 2 (a) Na adsorbs at top of the lower C atom, named A1. (b) Na sits at the center of a P6 ring, as A2. (c) The considered migration paths of Na diffusion on the P3C monolayer. (d) The corresponding diffusion energy barrier profiles of Path I and Path II. | ||
The Na ion diffusion energy barrier has a great effect on the charging and circuit rate capacity of SIBs. Based on the above results, there are two different diffusion paths for Na ion (I and II, Fig. 2c) between the two nearest-neighbor low-energy adsorption sites. The calculated diffusion energy barriers are shown in Fig. 2d. Path I (i.e. A1 → A2 → A1) has the lowest diffusion energy barrier, of 0.19 eV, whereas the diffusion energy barrier of path II, which is directly from an A1 site to another nearest A1 site, is up to 0.46 eV. Based on the above analysis, the P6 ring plays a key role in accelerating Na-ion migration. Notably, the diffusion energy barrier is much lower than that of traditional MoS2 (0.28 eV)86 and comparable with that of Ti2C (0.18 eV)39 and Sb (0.21 eV).49 To clarify the migration paths I and II, the movement trails of adsorbed Na ion crossing the energy barriers are shown in Fig. S8.† Notably, the diffusion length of 7.30 Å in Path I is similar to 7.09 Å in Path II.
To determine the specific capacity of the P3C monolayer, we explored concentration-dependent Na ion adsorption behavior on the P3C monolayer. Four different Na concentrations (P3CNan, n = 1–4) have been used to simulate the adsorption process. All the most stable configurations of the four considered stoichiometries are shown in Fig. 3. The resultant adsorption energies (i.e. 0.97, 0.53, 0.48, and 0.35 eV for P3CNa1, P3CNa2, P3CNa3, and P3CNa4) indicate that the P3C monolayer favorably holds Na ions in these Na ion concentrations. Determining the relative stabilities of P3CNan is important to understand which P3CNan has a more favored formation in the sodiation process. The calculated formation energies of each P3CNan with respect to the P3C monolayer and body-centered cubic Na solid can be used to build the convex hull (Fig. S9†). In general, a structure whose formation energy lies on the convex hull (i.e. the solid line) is deemed stable with respect to decomposition into other compounds or elemental solids, whereas a structure whose formation energy sits above the convex hull is metastable. In our case, P3CNa and P3CNa3 sit on the solid line, but P3CNa4 lies slightly above the solid line. This means that P3CNa4 has a greater possibility of forming during the dynamic charge/discharge processes.87,88 For P3CNa4, the P3C monolayer spontaneously adsorbs two-layer Na atoms (Fig. 3d) on its sides, forming P3CNa4. This can be understood by the unique distribution of the dispersive electron cloud (Fig. 4a and b) acting as anion,89,90 which plays a dominant role in stabilizing the second-layer Na ions. In addition, electron clouds distributed around Na ions can effectively alleviate repulsive interactions between Na ions.91
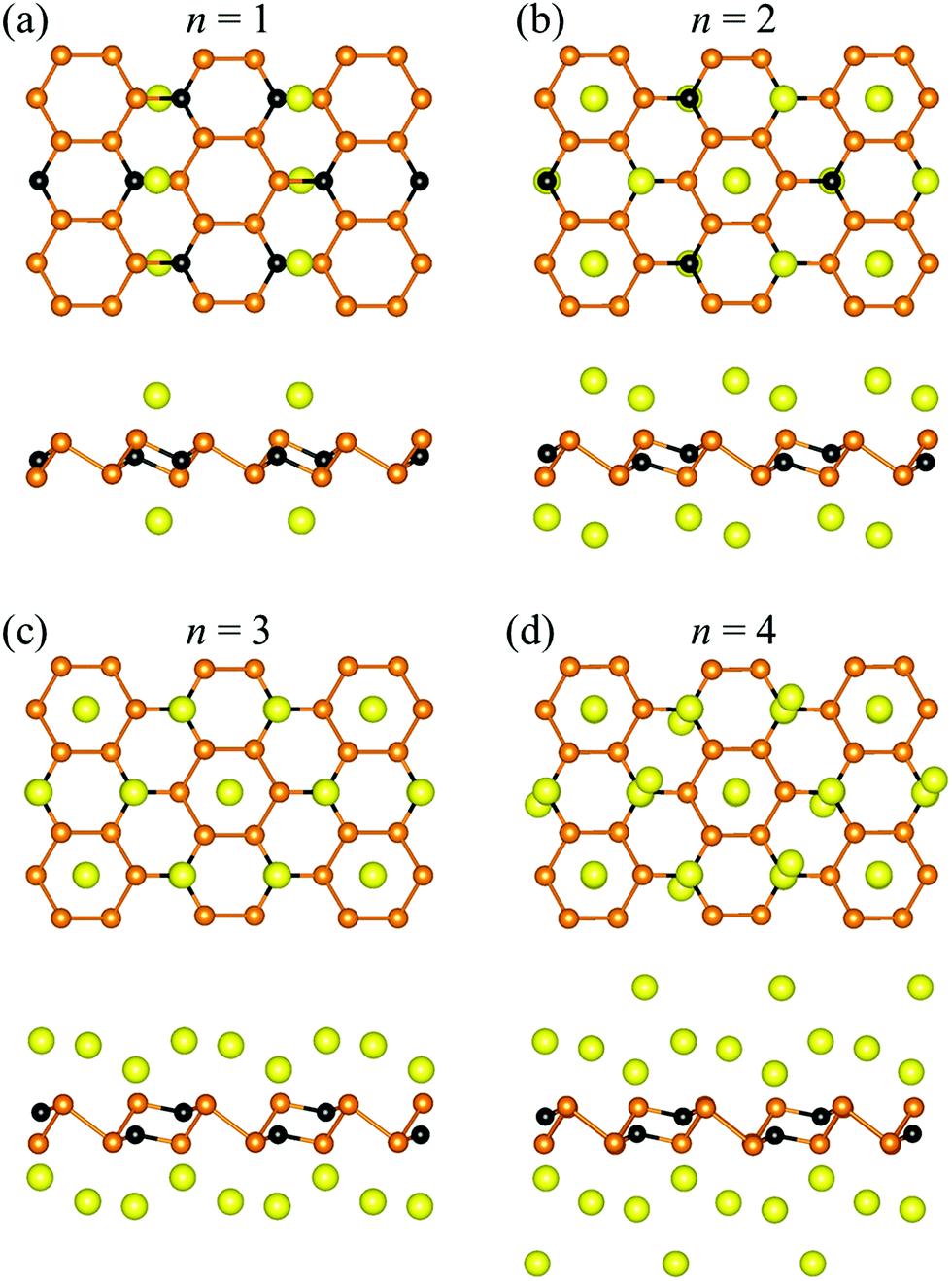 | ||
| Fig. 3 The top and side views of optimized adsorption configuration for the four different Na concentrations (i.e. (a) P3CNa, (b) P3CNa2, (c) P3CNa3 and (d) P3CNa4). | ||
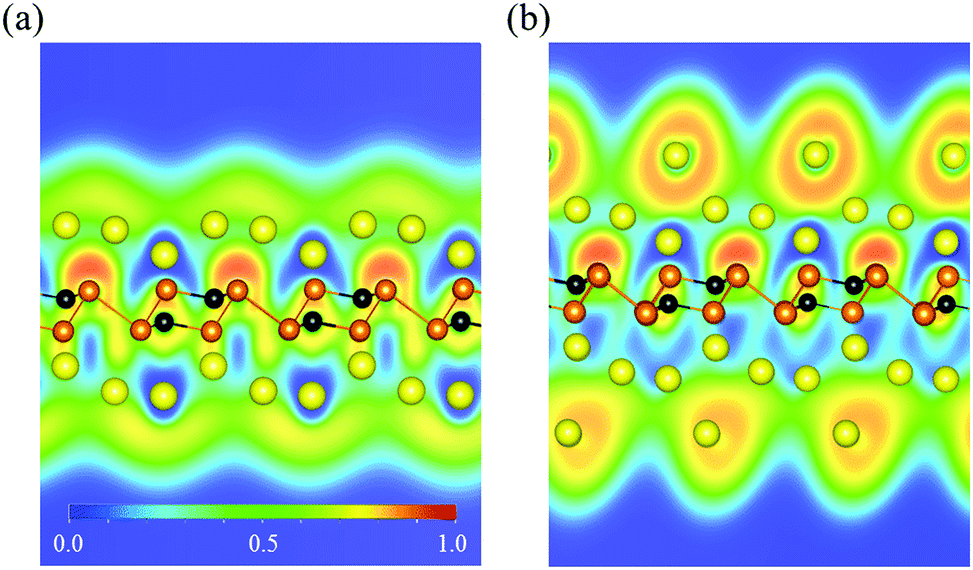 | ||
| Fig. 4 ELF map of P3C with (a) one-layer and (b) two-layer Na atoms. | ||
Finally, we studied the anode performance of the P3C monolayer. Besides the good thermodynamic stability mentioned above, P3C in P3CNan (n = 1–4) only shows slight structural distortion. As we expected, the stoichiometry P3CNa4 corresponds to a theoretical capacity of 1022 mA g h−1, which is much higher than that of known anode materials (e.g. graphite92 is 284 mA g h−1, TiO2 (ref. 93) is 193 mA g h−1, and MoS2 (ref. 94) is 350 mA g h−1). Even if the final product of sodiation could not reach P3CNa4, the stable P3CNa3 stoichiometry can also achieve a theoretical capacity up to 766.5 mA g h−1. The OCV values for different Na concentrations (e.g. 0.97, 0.53, 0.48, and 0.35 V) are in the range of 0–1.0 V, which can efficiently prevent dendrite formation of alkali metals during the discharge/charge process.95,96 The inherent metallicity of the P3C monolayer is beneficial for rate capacity. More intriguingly, except for P3CNa, the other three adsorption stoichiometries (i.e. P3CNa2, P3CNa3, and P3CNa4) are still metallic (Fig. S10†). P3CNa shows a band gap of 0.77 eV, which is much smaller than the reported sodiation products of black P (e.g. Na3P11 is ∼1.81 eV, Na3P7 is ∼1.94 eV, and Na3P is ∼1.67 eV).33
Under experimental conditions, anode materials could consist of multiple monolayers. This inspired us to explore the intercalation of Na within layers and the resultant volume expansion, which closely correlates with practical applications. Here, we mainly focus on the most stable P3C bilayer, as discussed above. As shown in Fig. S11,† the P3C bilayer can adsorb two-layer Na atoms within the layer with an adsorption energy of 0.63 eV, which is larger than 0.48 eV for P3CNa3 and 0.35 eV for P3CNa4 with Na atoms on both sides of P3C monolayer. The interlayer distance of sodiation for the P3C bilayer is 9.51 Å, corresponding to a volume expansion of ∼135% with respect to the P3C bilayer.33,97 This is much smaller than the volume expansion of black phosphorus (∼400%).33 Finally, we investigated whether the sodiation P3C bilayer can be restored to its original configuration. After removing all the adsorbed Na atoms, the relaxed P3C bilayer recovered its initial structure.
Conclusion
In this work, a novel P3C monolayer with a puckered honeycomb structure has been found through swarm-intelligence structural search calculations, with large cohesive energy and good thermodynamic stability. The P3C monolayer can spontaneously adsorb Na ions with an unexpected stoichiometry of P3CNa4, leading to a remarkably large theoretical capacity of 1022 mA g h−1. Its Na ion diffusion barrier is as low as 0.19 eV, ensuring a rapid charge/discharge rate capacity for SIBs. The inherent metallicity of the P3C monolayer provides good electron conductivity. Its structural integrity can be well maintained in the sodiation process. By all appearances, our predicted P3C monolayer is a promising anode material for SIBs, and awaits experimental confirmation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Natural Science Foundation of China under No. 21573037, 21873017, 11704062, and 51732003, the Postdoctoral Science Foundation of China under grant 2013M541283, and the Natural Science Foundation of Jilin Province (20150101042JC), and the Fundamental Research Funds for the Central Universities (2412017QD006).References
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed. Engl., 2018, 57, 102–120 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2012, 23, 947–958 CrossRef.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- Q. Bai, H. Chen, L. Yang and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS.
- J. Yang, X. Zhou, D. Wu, X. Zhao and Z. Zhou, Adv. Mater., 2017, 29, 1604108 CrossRef.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., 2013, 125, 4731–4734 CrossRef.
- N. Yabuuchi, Y. Matsuura, T. Ishikawa, S. Kuze, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2014, 1, 580–589 CrossRef.
- T. Ramireddy, T. Xing, M. M. Rahman, Y. Chen, Q. Dutercq, D. Gunzelmann and A. M. Glushenkov, J. Mater. Chem. A, 2015, 3, 5572–5584 RSC.
- A. Sibari, A. El Marjaoui, M. Lakhal, Z. Kerrami, A. Kara, M. Benaissa, A. Ennaoui, M. Hamedoun, A. Benyoussef and O. Mounkachi, Sol. Energy Mater. Sol. Cells, 2018, 180, 153–157 CrossRef.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139–4144 CrossRef CAS.
- Q. Xia, W. Li, Z. Miao, S. Chou and H. Liu, Nano Res., 2017, 10, 4055–4081 CrossRef CAS.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS.
- K. Zhang, M. Park, L. Zhou, G. H. Lee, J. Shin, Z. Hu, S. L. Chou, J. Chen and Y. M. Kang, Angew. Chem., Int. Ed., 2016, 55, 12822–12826 CrossRef CAS PubMed.
- X. Song, X. Li, Z. Bai, B. Yan, D. Li and X. Sun, Nano Energy, 2016, 26, 533–540 CrossRef CAS.
- X. Xu, L. Si, X. Zhou, F. Tu, X. Zhu and J. Bao, J. Power Sources, 2017, 349, 37–44 CrossRef CAS.
- J. Liu, Z. Yang, J. Wang, L. Gu, J. Maier and Y. Yu, Nano Energy, 2015, 16, 389–398 CrossRef CAS.
- M. He, M. Walter, K. V. Kravchyk, R. Erni, R. Widmer and M. V. Kovalenko, Nanoscale, 2015, 7, 455–459 RSC.
- J. M. Stratford, M. Mayo, P. K. Allan, O. Pecher, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, C. J. Pickard, A. J. Morris and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 7273–7286 CrossRef CAS PubMed.
- L. Baggetto, J. K. Keum, J. F. Browning and G. M. Veith, Electrochem. Commun., 2013, 34, 41–44 CrossRef CAS.
- D. Chao, C. Zhu, P. Yang, X. Xia, J. Liu, J. Wang, X. Fan, S. V. Savilov, J. Lin and H. J. Fan, Nat. Commun., 2016, 7, 12122 CrossRef CAS.
- N. Zhang, Y. Liu, Y. Lu, X. Han, F. Cheng and J. Chen, Nano Res., 2015, 8, 3384–3393 CrossRef CAS.
- T. Wang, D. Su, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Electrochemical Energy Reviews, 2018, 1, 200–237 CrossRef.
- X. Zhang, Z. Zhang, S. Yao, A. Chen, X. Zhao and Z. Zhou, npj Comput. Mater., 2016, 28, 2011–2021 Search PubMed.
- T. Yu, Z. Zhao, L. Liu, S. Zhang, H. Xu and G. Yang, J. Am. Chem. Soc., 2018, 140, 5962–5968 CrossRef CAS PubMed.
- Q. Sun, Y. Dai, Y. Ma, T. Jing, W. Wei and B. Huang, J. Phys. Chem. Lett., 2016, 7, 937–943 CrossRef CAS PubMed.
- P. Li, Z. Li and J. Yang, J. Phys. Chem. Lett., 2018, 9, 4852–4856 CrossRef CAS PubMed.
- J. Liu, C.-S. Liu, X.-J. Ye and X.-H. Yan, J. Mater. Chem. A, 2018, 6, 3634–3641 RSC.
- X. Zhao, X. Zhang, D. Wu, H. Zhang, F. Ding and Z. Zhou, J. Mater. Chem. A, 2016, 4, 16606–16611 RSC.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- M. Mayo, K. J. Griffith, C. J. Pickard and A. J. Morris, Chem. Mater., 2016, 28, 2011–2021 CrossRef CAS.
- L. E. Marbella, M. L. Evans, M. F. Groh, J. Nelson, K. J. Griffith, A. J. Morris and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 7994–8004 CrossRef CAS.
- Z. Yu, J. Song, D. Wang and D. Wang, Nano Energy, 2017, 40, 550–558 CrossRef CAS.
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS.
- X. Ma, L. Chen, X. Ren, G. Hou, L. Chen, L. Zhang, B. Liu, Q. Ai, L. Zhang and P. Si, J. Mater. Chem. A, 2018, 6, 1574–1581 RSC.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed.
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef CAS PubMed.
- X. Xie, Z. Ao, D. Su, J. Zhang and G. Wang, Adv. Funct. Mater., 2015, 25, 1393–1403 CrossRef CAS.
- D. Sun, D. Ye, P. Liu, Y. Tang, J. Guo, L. Wang and H. Wang, Adv. Energy Mater., 2017, 8, 1702383 CrossRef.
- F. Zhang, C. Xia, J. Zhu, B. Ahmed, H. Liang, D. B. Velusamy, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601188 CrossRef.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- A. A. AbdelHamid, Y. Yu, J. Yang and J. Y. Ying, Adv. Mater., 2017, 29, 1701427 CrossRef PubMed.
- Z. Wu, B. Li, Y. Xue, J. Li, Y. Zhang and F. Gao, J. Mater. Chem. A, 2015, 3, 19445–19454 RSC.
- E. S. Reich, Nature, 2014, 506, 19 CrossRef PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- G.-C. Guo, D. Wang, X.-L. Wei, Q. Zhang, H. Liu, W.-M. Lau and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 5002–5008 CrossRef CAS PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. Li, J. Hao, H. Liu, Y. Li and Y. Ma, J. Chem. Phys., 2014, 140, 174712 CrossRef.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644 CrossRef CAS.
- G. Yang, Y. Wang, F. Peng, A. Bergara and Y. Ma, J. Am. Chem. Soc., 2016, 138, 4046–4052 CrossRef CAS PubMed.
- S. Zhang, Z. Zhao, L. Liu and G. Yang, J. Power Sources, 2017, 365, 155–161 CrossRef CAS.
- H. Zhang, Y. Li, J. Hou, A. Du and Z. Chen, Nano Lett., 2016, 16, 6124–6129 CrossRef CAS PubMed.
- T. Yu, S. Zhang, F. Li, Z. Zhao, L. Liu, H. Xu and G. Yang, J. Mater. Chem. A, 2017, 5, 18698–18706 RSC.
- Y. Wang, F. Li, Y. Li and Z. Chen, Nat. Commun., 2016, 7, 11488 CrossRef CAS PubMed.
- W. Ming, M. Yoon, M.-H. Du, K. Lee and S. W. Kim, J. Am. Chem. Soc., 2016, 138, 15336–15344 CrossRef CAS PubMed.
- H. Zhang, Y. Li, J. Hou, K. Tu and Z. Chen, J. Am. Chem. Soc., 2016, 138, 5644–5651 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- J. Gullman and O. Olofsson, J. Solid State Chem., 1972, 5, 441–445 CrossRef CAS.
- P. C. Donohue and H. S. Young, J. Solid State Chem., 1970, 1, 143–149 CrossRef CAS.
- N. Kinomura, K. Terao, S. Kikkawa, H. Horiuchi, M. Koizumi and H. Setoguchi, Mater. Res. Bull., 1983, 18, 53–57 CrossRef CAS.
- C.-S. Liu, X.-L. Yang, J. Liu and X.-J. Ye, ACS Appl. Energy Mater., 2018, 1, 3850–3859 CrossRef CAS.
- X. Ling, H. Wang, S. Huang, F. Xia and M. S. Dresselhaus, ACS Appl. Energy Mater., 2015, 112, 4523–4530 CAS.
- J. Guan, D. Liu, Z. Zhu and D. Tománek, Nano Lett., 2016, 16, 3247–3252 CrossRef CAS PubMed.
- Y. Jing, Y. Ma, Y. Li and T. Heine, Nano Lett., 2017, 17, 1833–1838 CrossRef CAS PubMed.
- J. Zhang, J. Jiang, H. Li and X. S. Zhao, Energy Environ. Sci., 2011, 4, 4009–4015 RSC.
- H. R. Jiang, Z. Lu, M. C. Wu, F. Ciucci and T. S. Zhao, Nano Energy, 2016, 23, 97–104 CrossRef CAS.
- L.-M. Yang, V. Bačić, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS PubMed.
- Y. Zhang, Z.-F. Wu, P.-F. Gao, D.-Q. Fang, E.-H. Zhang and S.-L. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 2245–2251 RSC.
- H. Sun, Z. Liang, K. Shen, M. Luo, J. Hu, H. Huang, Z. Zhu, Z. Li, Z. Jiang and F. Song, Appl. Surf. Sci., 2018, 428, 623–629 CrossRef CAS.
- Q. Gao, C.-Q. Huang, Y.-M. Ju, M.-R. Gao, J.-W. Liu, D. An, C.-H. Cui, Y.-R. Zheng, W.-X. Li and S.-H. Yu, Angew. Chem., 2017, 129, 7877–7881 CrossRef.
- B. Xu, H. Xiang, J. Yin, Y. Xia and Z. Liu, Nanoscale, 2018, 10, 215–221 RSC.
- W. Qi, H. Zhao, Y. Wu, H. Zeng, T. Tao, C. Chen, C. Kuang, S. Zhou and Y. Huang, Sci. Rep., 2017, 7, 43582 CrossRef PubMed.
- X. Chen, F. Tian, C. Persson, W. Duan and N.-x. Chen, Sci. Rep., 2013, 3, 3046 CrossRef PubMed.
- W. H. Han, S. Kim, I. H. Lee and K. J. Chang, J. Phys. Chem. Lett., 2017, 8, 4627–4632 CrossRef CAS PubMed.
- J. H. Jung, C. H. Park and J. Ihm, Nano Lett., 2018, 18, 2759–2765 CrossRef CAS PubMed.
- B. Rajbanshi and P. Sarkar, J. Phys. Chem. Lett., 2017, 8, 747–754 CrossRef CAS PubMed.
- M. Mortazavi, C. Wang, J. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125416 CrossRef.
- H. Liu, F. C. Strobridge, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Science, 2014, 344, 1252817 CrossRef PubMed.
- Y. Ma, M. Eremets, A. R. Oganov, Y. Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle and V. Prakapenka, Nature, 2009, 458, 182 CrossRef CAS PubMed.
- M.-s. Miao and R. Hoffmann, J. Am. Chem. Soc., 2015, 137, 3631–3637 CrossRef CAS PubMed.
- T. Zhao, S. Zhang, Y. Guo and Q. Wang, Nanoscale, 2016, 8, 233–242 RSC.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CrossRef CAS PubMed.
- K.-T. Kim, G. Ali, K. Y. Chung, C. S. Yoon, H. Yashiro, Y.-K. Sun, J. Lu, K. Amine and S.-T. Myung, Nano Lett., 2014, 14, 416–422 CrossRef CAS PubMed.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- E. Yang, H. Ji, J. Kim, H. Kim and Y. Jung, Phys. Chem. Chem. Phys., 2015, 17, 5000–5005 RSC.
- C. Eames and M. S. Islam, J. Am. Chem. Soc., 2014, 136, 16270–16276 CrossRef CAS PubMed.
- A. Agrawal, K. Biswas, S. K. Srivastava and S. Ghosh, J. Solid State Electrochem., 2018, 11, 3443–3455 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description of the computational method and structural prediction, main structural parameters, relative stability, electronic energy band structure, PDOS, average voltages, phonon dispersion curves, and temperature-dependent molecular dynamical simulations. See DOI: 10.1039/c8ta09155b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |