
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic liquid containing electron-rich, porous polyphosphazene nanoreactors catalyze the transformation of CO2 to carbonates†
Zhangjun
Huang
ab,
Jorge G.
Uranga
a,
Shiliu
Zhou
b,
Haiyan
Jia
ab,
Zhaofu
Fei
 *a,
Yefeng
Wang
a,
Felix D.
Bobbink
a,
Qinghua
Lu
*b and
Paul J.
Dyson
*a
*a,
Yefeng
Wang
a,
Felix D.
Bobbink
a,
Qinghua
Lu
*b and
Paul J.
Dyson
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: zhaofu.fei@epfl.ch; paul.dyson@epfl.ch
bSchool of Chemistry and Chemical Engineering, The State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai, P. R. China. E-mail: qhlu@sjtu.edu.cn
First published on 17th October 2018
Abstract
We show that ionic liquids (ILs) interact with electron-rich, porous polyphosphazene (PPZ), to form hybrid PPZ-IL nanoreactors able to simultaneously capture and transform CO2 into carbonates. The PPZ nanospheres swell in organic solvents and effectively absorb IL cations by virtue of the electron-rich sites, while leaving the anions exposed and increasing their nucleophilicity. This leads to considerably higher catalytic activity compared to the IL alone in the cycloaddition reaction of CO2 to epoxides. The cation shielding effect is dependent on the structure of the IL cation and, hence, the catalytic activity can be tuned by varying the structure of the cation in the IL and DFT calculations were used to rationalize the experimentally observed differences in catalytic activity. These studies indicate that PPZ nanospheres could find widespread uses in catalysis, acting as active nanosupports for homogeneous catalysts, not only for the transformations of CO2, but also for other substrates.
Introduction
Porous polymers are formed from the reaction of rigid organic building blocks with complementary steric and geometric features.1 They have been evaluated in numerous applications including gas adsorption and storage,2 separations,3 sensors,4 opto/electronic devices,5 drug delivery and therapeutics,6 energy storage,7 and catalysis.8 However, the preparation of highly porous polymers requires reversible bond-forming conditions.9 In certain systems the pore size is adjustable and swelling of porous materials, induced by solvents, can facilitate the transport of compounds into the porous network under actual reaction conditions.10Porous polymers have recently been evaluated in CO2 separation and conversion processes. For example, a flexible porous polymer exhibits gate opening-type abrupt adsorption for C2H2, but not for CO2, leading to an appreciable separation for CO2 from CO2/C2H2 mixtures at near ambient temperature (273 K).11 A porous polymer containing an embedded ionic-polymer was used to catalyze the cycloaddition reaction of CO2 to epoxides under ambient temperature,12 although Cu(OAc)2 was required as a co-catalyst to achieve high efficiency under ambient conditions. Cross-linked ionic polymers based on poly(styrene) and encompassing imidazolium cations catalyze this reaction in the absence of a co-catalyst and under mild conditions.13
Porous polyphosphazenes (PPZs) are prepared from the polycondensation of compounds containing o-dihydroxybenzene/o-phenylenediamine groups with hexachlorocyclophosphazene (HCCP).14 PPZs possess unique frameworks with each aromatic plane linked in a perpendicular fashion to the plane of the cyclophosphazene ring, leading to large gaps between the layers of the polymer and preventing the formation of π–π stacking or Lewis acid–base interactions, which leads to flexible amorphous structures that self-assemble to form spheroids.
Herein, we describe the preparation of a new electron-rich PPZ material that forms nanospheres which exhibit solvent-dependent size and porosity. The PPZ nanospheres readily absorb a range of ionic liquid (IL) cations, leaving the anions largely exposed. The resulting hybrid nanospheres catalyze the reaction of CO2 and epoxides to form cyclic carbonates considerably more effectively than the pure IL. This reaction has been extensively investigated and is even conducted on an industrial scale.15
Results and discussion
The PPZ nanospheres were prepared and optimized from the polycondensation reaction between 9,10-dimethyl-9,10-ethano-9,10-dihydro-2,3,6,7-tetrahydroxy-anthracene (ATC) and HCCP in the presence of triethylamine in acetonitrile (Scheme 1 and Fig. S2†). The reaction of the phenol hydroxyl group (3280 cm−1) and HCCP (1209 cm−1) and the formation of P–O–Ar bonds (921 cm−1), is apparent from the FT-IR spectrum (Fig. S3†), confirming the formation of the PPZ nanospheres.16 | ||
| Scheme 1 Synthetic route used to prepare the PPZ material. | ||
The 13C solid state NMR spectrum of the PPZ nanospheres (Fig. 1a) shows signals centered at 9, 20, 40, 112, 140, 219 ppm, all of which are consistent with the presence of the ATC building block, providing further evidence of the incorporation of ATC into the PPZ nanospheres. The 31P solid state NMR spectrum shows one broad singlet at −1 ppm characteristic of P(V) in a single environment (Fig. 1b).14b
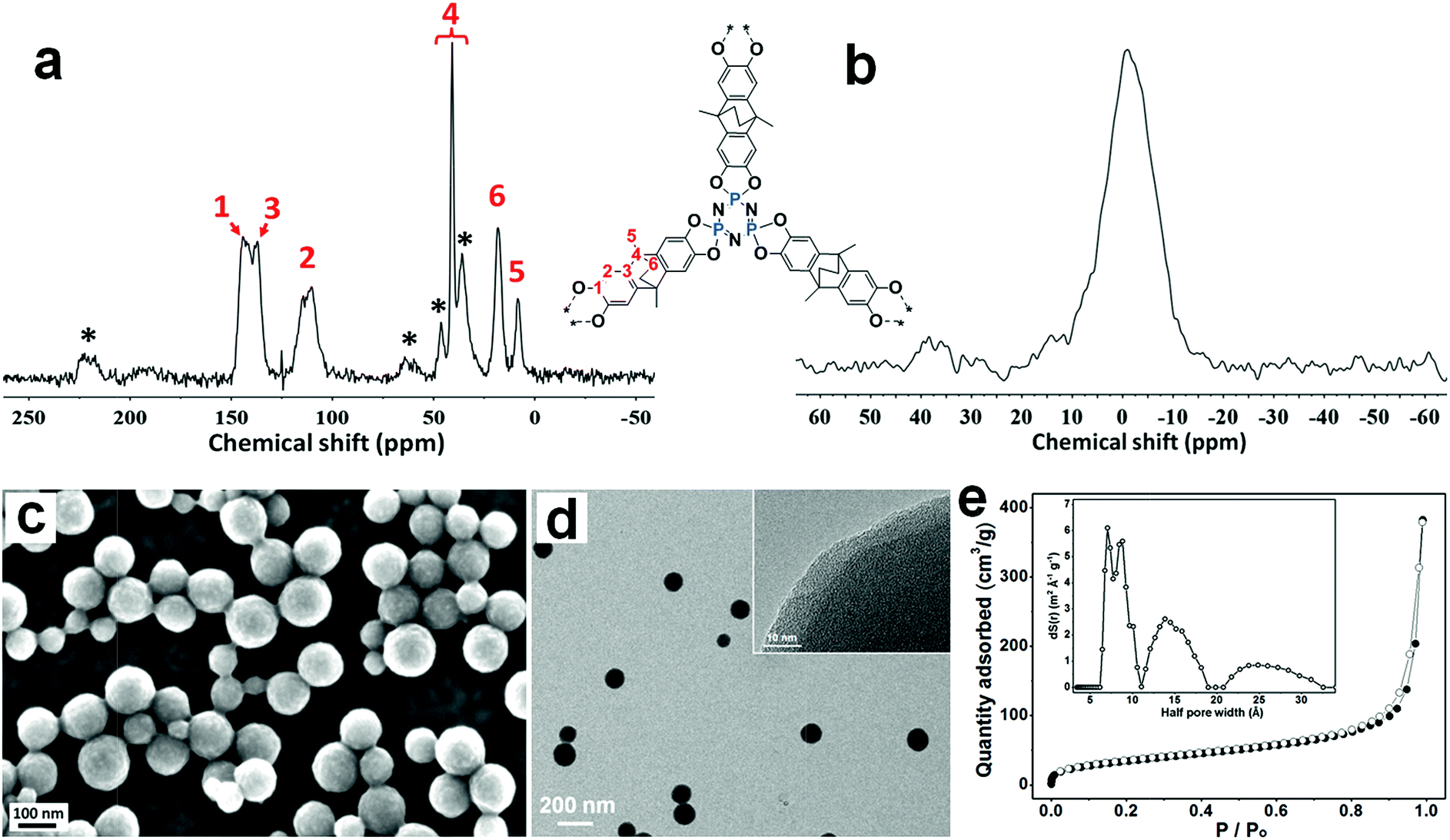 | ||
| Fig. 1 (a) 13C solid state NMR spectrum and (b) 31P solid state NMR spectrum of the PPZ nanospheres. Side bands are indicated with stars (*). (c) SEM and (d) TEM image of the PPZ nanospheres. Inset shows the high resolution image. (e) N2 adsorption–desorption isotherms of the PPZ nanospheres recorded at 77 K. Inset shows the DFT estimated pore size distribution. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) shows that the PPZ material comprises nanospheres (Fig. 1c and d) that are free-standing and well dispersed. Statistical analysis indicates that the PPZ nanospheres have a mean diameter of 121 nm and a reasonably narrow size distribution (Fig. S4†). The expected lack of long-range order is apparent from the powder X-ray diffraction (PXRD) analysis (Fig. S5†), which shows three amorphous broad peaks around 16, 39 and 41°. The amorphous character of PPZ material is caused by the interspersal of domains with eclipsed ordering and domains with staggered ordering,17 and the peak around 16° reveals some ordering in the range 4.9 to 6.5 Å, presumably corresponding to shoulder-to-shoulder packing (4.9 Å, staggered ordering, Fig. S6a†) and head-to-head packing (6.5 Å, eclipsed ordering, Fig. S6b†) of the layers. Considering the PXRD signal is relatively weak, most of the layers should pack obliquely, which leads to the macroscopic spherical shape.
The porosity of the PPZ nanospheres was established using Brunauer–Emmett–Teller (BET) surface area analysis. The N2 adsorption–desorption isotherms of the PPZ nanospheres (Fig. 1e) provide a surface area value of 128.8 m2 g−1 at 77 K, with a hierarchical pore size distribution of 7, 15 and 25 Å for half pore widths (inset of Fig. 1e). Smaller pores, i.e. those with a half pore width < 10 Å, are expected to correspond to the holes created by the stacking of the frames,18 and should open if the PPZ nanospheres swell. The pores with half pore widths of 15 and 25 Å presumably correspond to the intrinsic size of the frameworks and the gap between the random packed frameworks, respectively. CO2 adsorption–desorption isotherms of the PPZ nanospheres conducted at 273 K (Fig. S7a†) reveal that CO2 uptake reaches a value of 1.5 mmol g−1 at 1 atm. A broad adsorption/desorption hysteresis loop indicates the presence of intra-pore CO2–PPZ interactions, presumably due to polarization of CO2 by the electron-rich material leading to dipole–dipole interactions that inhibits the release of CO2 at low pressures. Notably, such dipole–dipole interactions are unfavorable with CH4 which is much less polarizable (Fig. S7b†).19
The ability of the PPZ nanospheres to capture CO2, observed previously with a related material,14a should be advantageous in reactions employing CO2 as a substrate. Since the PPZ nanospheres are electron-rich, they can potentially interact strongly with the cations of ILs, although the dense nature of the PPZ nanospheres and relatively low porosity could reduce their accessibility. However, when the PPZ nanospheres are dispersed in solvents, the polymeric layers separate and the nanospheres swell. AFM images of the PPZ nanospheres show that the overall size of the nanospheres swells from ca. 120 nm (Fig. 2a and c) in dry state to ca. 236 nm (Fig. 2b and d) in styrene oxide (SO). Further swelling of the PPZ nanospheres is accompanied by an increase in porosity, allowing interactions with solvent molecules and solvates. Thus, the PPZ nanospheres and ILs combine in situ to afford PPZ-IL nanoreactors.
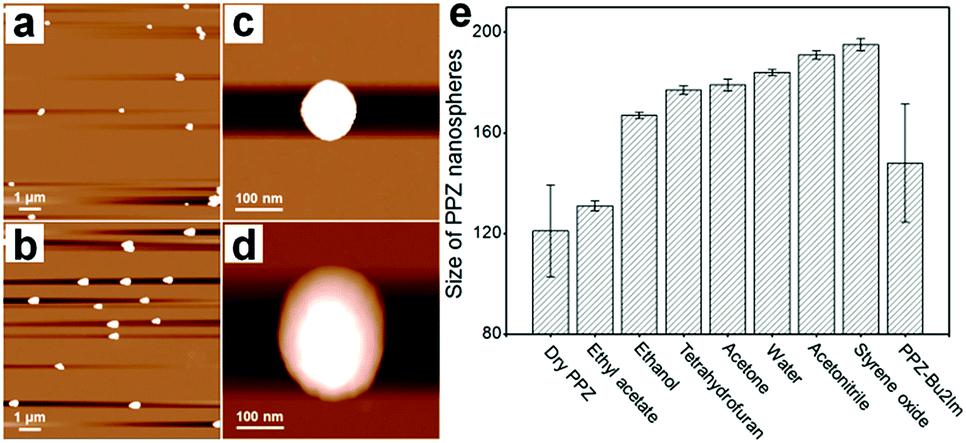 | ||
| Fig. 2 AFM images of the PPZ nanospheres (a) in a dry state and (b) after swelling in SO. (c) and (d) are single particle images corresponding to (a) and (b), respectively. The size of the nanospheres was determined at 25 °C at a concentration of 0.1 mg mL−1. (e) Data for the average size and standard derivation of the PPZ nanospheres when dry (determined from SEM) and when swelled in solvents (determined from DLS, Fig. S8†). | ||
The cycloaddition of CO2 to epoxides was selected as an ideal test reaction20 as the PPZ nanospheres interact with CO2, and appear to polarize it, and therefore the absorbed CO2 could be somewhat activated. Moreover, the cycloaddition of CO2 to epoxides is of industrial importance,21 and ILs are good catalysts for this reaction,13a with highest activities obtained for catalysts with highly nucleophilic dynamic light scattering (DLS) shows that the extent of swelling of the PPZ nanospheres is solvent dependent (Fig. 2e). Swelling of the PPZ nanospheres in styrene oxide (SO) doubled their size, which was accompanied by an increase in porosity and catalytic activity. Due to the electron rich nature of the PPZ nanospheres, IL cations should interact strongly with them, increasing the nucleophilicity of the anions, and potentially enhancing catalytic activity. In this respect, the rate-determining step of the reaction catalyzed by imidazolium salts involves ring-opening of the epoxide by the anion.22 In this respect, the rate-determining step of the reaction catalyzed by allowing interactions with solvent molecules and solvates. Thus, the PPZ nanospheres and ILs selected as solvent and substrate for cycloaddition with CO2. Although many catalytic systems have been established for this highly atom-economic reaction,15 certain amorphous polymers have advantages including excellent stability, high efficiency under mild conditions, and they are readily recyclable and reusable.23 A series of ILs (Fig. 3a) with different structures were combined with the PPZ nanospheres and a schematic of a PPZ-IL interaction is shown in Fig. 3b. The PPZ nanospheres do not catalyze the cycloaddition of CO2 to SO in the absence of IL (Table 1, entry 1). In contrast, ILs that dissolve in SO under the given conditions, i.e. 57 °C and 1 atm CO2, catalyze the reaction to afford styrene carbonate (SC) in moderate yields, with the activity being comparable to structurally related ILs.24 The PPZ-IL nanoreactors, which form in situ, catalyze the reaction considerably more efficiently, which is as expected due to the enhanced nucleophilicity of the IL anion. Notably, the magnitude of the enhancement in activity of the PPZ-IL nanoreactors relative to the pure IL strongly depends on the structure of the cation, with the catalytic efficiency of less sterically encumbered ILs increasing by over 100% (Table 1, entries 2/3, 5/6 and 11/12), and over 80% (Table 1, entries 7/8, 9/10 and 13/14). In contrast, those with bulky substituents, e.g. PhIm, increase by only ca. <10% (Table 1, entries 15/16) and a decrease in activity is observed for the extremely bulky cation in 4PhIm (Table 1, entries 19/20). These differences in the magnitude of the reaction enhancement may be attributed to the strength of the interactions between the PPZ nanospheres and the IL, i.e. the least bulky IL cations interact strongly with the PPZ leaving the IL anion more exposed and reactive. The largest increase in activity was observed for the non-hindered bis-imidazolium salt, Bu2Im, with a –(CH2)4– linker connecting the rings, i.e. PPZ-Bu2Im led to near quantitative yields (Table 1, entry 22) whereas Bu2Im alone resulted in a yield of 14% (Table 1, entry 21). This represents an increase in activity of 7-fold. It has previously been shown that hydroxyl groups can enhance the catalytic efficiency of the cycloaddition reaction of CO2 to epoxides,25 however, in the PPZ nanoparticles the majority of hydroxyl groups in the ATC starting material are consumed, as demonstrated by the IR spectrum of the product (Fig. S3†), Moreover, the ATC starting material combined with Bu2Im is not a particularly active catalyst for the reaction (Table 1, entry 24), implying that the cation shielding effect provided by the electron-rich PPZ material is responsible for the enhancements in catalytic activity. While the ATC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCCP substrate ratios of PPZ was 3:1 (PPZ3) and 1:1 (PPZ1), the conversion efficiency of corresponding nanoreactors decreased (entries 26/27), due to the interfere of defects to the interaction between Bu2Im and PPZ.
:HCCP substrate ratios of PPZ was 3:1 (PPZ3) and 1:1 (PPZ1), the conversion efficiency of corresponding nanoreactors decreased (entries 26/27), due to the interfere of defects to the interaction between Bu2Im and PPZ.
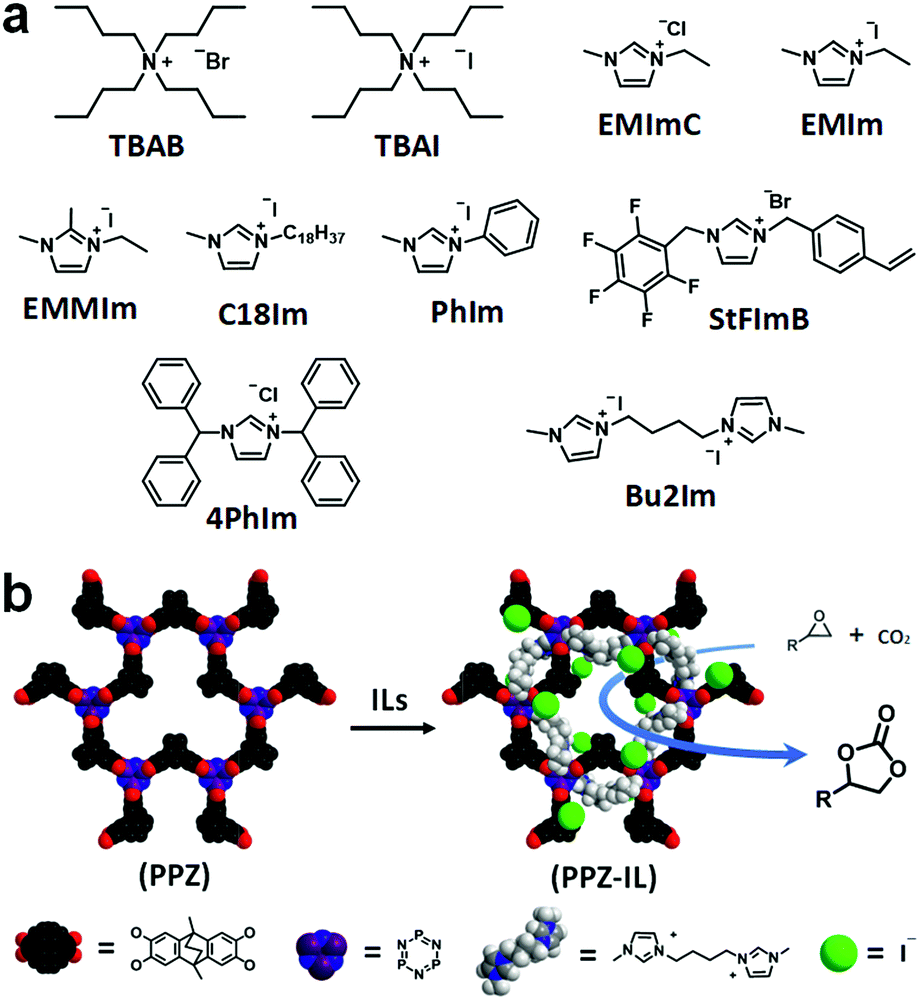 | ||
| Fig. 3 (a) Structures of the ILs used to prepare PPZ-IL nanoreactors. (b) The proposed representation of interactions of the IL in the PPZ-Bu2Im system. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yield (%) | Entry | Catalyst | Yield (%) |
|
a Reaction conditions: SO (480 mg, 4.00 mmol), CO2 (1 atm, using a balloon), 57 °C and 20 h. ILs (2.5 mol% halide), PPZ (19.2 mg, containing 2.5 mol% N). Yield determined by 1H NMR spectroscopy. The selectivity of the reaction is all >99% (determined by GC-MS). Entry 22 is shown as an example in Fig. S9 ESI.
b Using PPZ-TBAB in acetone.
c Using PPZ-Bu2Im in ethanol.
d PPZ3-Bu2Im and PPZ1-Bu2Im.
e Were synthesized from with ATC:HCCP substrate ratios of 3:1 and 1:1, respectively.
|
|||||
| 1 | PPZ | 0 | 15 | PhIm | 58 |
| 2 | TBAB | 44 | 16 | PPZ-PhIm | 63 |
| 3 | PPZ-TBAB | 89 | 17 | StFImB | 13 |
| 4 | PPZ-TBABb | 89 | 18 | PPZ-StFImB | 34 |
| 5 | TBAI | 37 | 19 | 4PhIm | 48 |
| 6 | PPZ-TBAI | 88 | 20 | PPZ-4PhIm | 45 |
| 7 | EMImC | 66 | 21 | Bu2Im | 14 |
| 8 | PPZ-EMImC | 81 | 22 | PPZ-Bu2Im | 99 |
| 9 | EMIm | 73 | 23 | PPZ-Bu2Imc | 98 |
| 10 | PPZ-EMIm | 84 | 24 | ATC + Bu2Im | 32 |
| 11 | EMMIm | 41 | 25 | HCCP + Bu2Im | 14 |
| 12 | PPZ-EMMIm | 89 | 26 | PPZ3-Bu2Imd | 63 |
| 13 | C18Im | 67 | 27 | PPZ1-Bu2Ime | 73 |
| 14 | PPZ-C18Im | 88 | |||
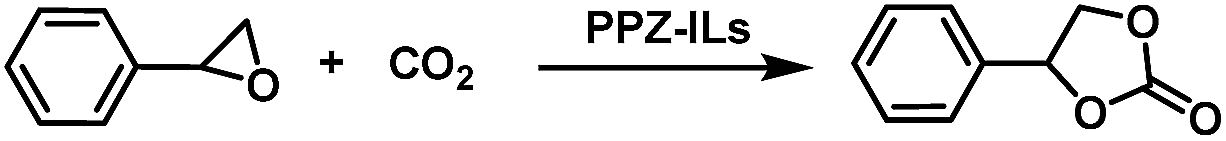
In order to better understand the differences in catalytic activity, DFT calculations were performed on three ILs as representative examples, including the systems with the best and worst enhancements in activity when combined in PPZ-IL nanoreactors, i.e. PPZ-Bu2Im, PPZ-EMIm and PPZ-4PhIm.
First, the three ILs, Bu2Im, EMIm and 4PhIm, were independently optimized, and the most stable conformation showed hydrogen bonds (HBs) as the main interaction. Only in the case of EMIm the conformation with “on top” electrostatic I−–imidazolium+ interaction was also important, but for comparative purpose our analysis only considered the HB conformation. For Bu2Im and EMIm, the iodide interacts with the C2–H bond from the imidazolium ring, whereas, 4PhIm exhibits a HB network between the chloride anion and the four aromatic C–H bonds at the ortho position with an average HB distance of 2.44 Å. Optimized geometries of the ILs are presented in Fig. 4a, with HBs shown in yellow and the optimized structures of the adducts, i.e. PPZ-Bu2Im, PPZ-EMIm and PPZ-4PhIm are illustrated in Fig. 4c, with HBs shown in green and yellow. The PPZ moiety is represented by means of electrostatic potential map (see Fig. 4b) and, as expected, the phosphazene and dihydroxyphenyl rings are more electron rich than other regions. Hence, the phosphazene ring interacts preferentially with the IL cations.
 | ||
| Fig. 4 (a) Optimized geometries for ILs Bu2Im, EMIm and 4PhIm. H-bonding interactions shown as yellow with their corresponding distances. (b) Schematic representation of the optimized PPZ (represented as an electrostatic potential map according to the adjacent scale), and (c) PPZ-IL adducts for: PPZ-Bu2Im (adducts A and B), PPZ-4PhIm (adduct C) and PPZ-EMIm (adducts D and E). In addition, the main H-bonds and their corresponding distances are indicated using green lines for PPZ-cation interactions and using yellow lines for cation–anion interactions. The main H-bonds were identified by changes in C–H vibrations and only the H-bonds with distances <3 Å are shown. Two constraints (fixed distances) were included in the optimization of the PPZ structure (shown as purple lines). | ||
Once the PPZ-Bu2Im adduct is formed, the stronger HB interactions are between the imidazolium cation and the phosphazene ring. Moreover, the orientation of imidazolium ring is not parallel to any of the π systems in the PPZ structure, i.e. π–π stacking interactions are not observed (see adducts A and B, Fig. 4c). Instead, HBs (marked in green in Fig. 4c) appear to be responsible of adduct formation. In contrast, the smaller IL (EMIm) interacts forms π–π stacking interactions between the imidazolium and PZ ring (see adduct D) or phenyl ring in the polymer (see adduct E) with an average distance between the rings of 3.50 Å for adduct D, and 3.55 Å for adduct E. In the case of the more sterically hindered 4PhIm system, the main interactions between PPZ and IL cation comprise HBs with the aromatic and benzylic C–H bonds (see green lines in adduct C). This leads to a more compact adduct in which the 4PhIm occupies most of the free space inside the PPZ framework. Note that due to the high volume of 4PhIm only one stable conformation was found.
A binding energy (BE) of 34.7 kcal mol−1 was calculated for the most stable conformer of PPZ-Bu2Im in the gas phase (adduct A in Table 2). Adduct B, is slightly less stable than adduct A (binding energy = 33.7 kcal mol−1). Lower binding energies were computed for PPZ-EMIm, with values of 21.1 kcal mol−1 for adduct D and 25.1 kcal mol−1 for adduct E. Intermediate stability is predicted for PPZ-4PhIm.
| Structure | Binding energya | rCsp2H⋯Xb/Δr rCsp2H⋯Xc | νCsp2–He/ΔνCsp2–Hf |
|---|---|---|---|
| a Calculated binding energies in kcal mol−1. b H⋯Y distances in Å. c Computed changes in CH⋯X distance in Å. d Average (Cortho)H⋯X distance in Å. e Calculated C2–H or Cortho–H stretching frequencies in cm−1. f Computed changes in above mentioned frequencies. g Asymmetric Cortho–H stretching. h Symmetric Cortho–H stretching. (ν1) vibrational mode for the imidazolium ring 1 and (ν2) vibrational mode for ring 2. | |||
| Isolated Bu2Im | (I1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) I2) 2.51 I2) 2.51 |
3036 | |
| PPZ-Bu2Im (A) | 34.7 | (I1) 2.73; (I2) 2.71, Δr1 = 0.22; Δr2 = 0.20 | (ν1) 3222; (ν2) 3249, Δν1 = 186; Δν2 = 213 |
| PPZ-Bu2Im (B) | 33.7 | (I1) 2.64; (I2) 2.73, Δr1 = 0.13; Δr2 = 0.28 | (ν1) 3213; (ν2) 3195, Δν1 = 178; Δν2 = 159 |
| Isolated 4PhIm | 2.44d | 3164g, 3173h | |
| PPZ-4PhIm (C) | 25.8 | 2.46d, Δr = 0.02 | 3163g, 3169h, Δνg = −1; Δνh = −4 |
| Isolated EMIm | 2.46 | 2987 | |
| PPZ-EMIm (D) | 21.1 | 2.58, Δr = 0.12 | 3150, Δν = 163 |
| PPZ-EMIm (E) | 25.1 | 2.56, ΔI = 0.10 | 3110, Δν = 123 |
HBs in ILs are evidenced by changes in the Csp2–H stretching frequencies in their IR spectra.26 Notably, the Csp2–H stretching frequency moves to lower energy as the strength of the HB increases and the distance between the CH group and the halide shortens.27 Taking these properties into account, our analysis was carried out considering the geometric and vibrational changes on the IL structure before and after adduct formation, as a descriptor of HB strength between the IL fragments. The frequency shift was calculated according to the formula Δν = νCHPPZ-IL − νCHIL, where νCHPPZ-IL represents the stretching frequency of the Csp2–H bonds in the adduct, and νCHIL the corresponding stretching frequency in the isolated ionic liquid. Accordingly, the analyzed frequency shifts were derived from the calculated νCHIL for the isolated ILs as reference (see Table 2), i.e. 3036 cm−1 for Bu2Im, 2987 cm−1 for EMIm, and 3166 and 3173 cm−1 for the asymmetric and symmetric stretching modes in 4PhIm. In addition, the changes in CH⋯X distances were estimated from the initial values of isolated ILs, i.e. 2.51 Å for PPZ-Bu2Im, 2.46 Å for PPZ-EMIm and 2.44 Å for 4PhIm (see Table 2).
In the case of adduct A, which involves a double imidazolium iodide structure, there are two different C(2)–H⋯I1 and C(2)–H⋯I2 moieties in which a reduction of the C–H stretching frequencies from the initial value was calculated, 186 cm−1 for C(2)–H⋯I1 and 213 cm−1 for C(2)–H⋯I2 (Table 2, adduct A). In parallel the C(2)–H⋯I1 HB is elongated by 0.22 Å and the C(2)–H⋯I2 HB is elongated by 0.20 Å relative to the initial value of 2.51 Å (Table 2, Bu2Im). In adduct B, frequency shifts relative to those in the isolated IL of 159 cm−1 and 177 cm−1 for C(2)–H⋯I1 and C(2)–H⋯I2 are calculated, respectively, with corresponding H⋯I elongations of 0.13 and 0.18 Å. In the case of adduct D, frequency shift of 163 cm−1 and H⋯I distance increase of 0.12 Å were obtained for the corresponding C(2)–H⋯I moiety. Similarly with adduct E Δν and Δr were 123 cm−1 and 0.10 Å respectively, upon adduct formation. These changes indicate that formation of the PPZ-EMIm adduct has a weaker effect on the ion–pair association compared to the PPZ-Bu2Im system (adducts A and B). Thus, HB strength calculations may be used to rationalize the changes in reactivity of systems before and after adduct formation with the PPZ material, in that the anion interacts less strongly with the cation in the PPZ-Bu2Im than in the PPZ-EMIm systems, and is therefore more nucleophilic and hence more reactive.
For PPZ-4PhIm, no important geometric and vibrational changes in IL structure were observed before and after adduct formation. For example, the average C–H⋯Cl distances (H-bonding network) in the adduct PPZ-4PhIm are comparable with those observed in isolated 4PhIm. The same was calculated for the Cortho–H bonds for which stretching frequency were reduced by only 1 and 4 cm−1. This suggests that the interaction between IL ion pairs is still significant and the chloride anion is less nucleophilic. Indeed, the chloride anion shown to be encapsulated within the HB network of the cation and in part by the surrounding PPZ material. As a result, it is less accessible and thus less reactive.
According to the computational results, the structure of the cation in ILs is strongly influenced by the interaction with the PPZ support and, consequently, determines the degree of activation of the anion which could be estimated from the strength of the H-bonds.
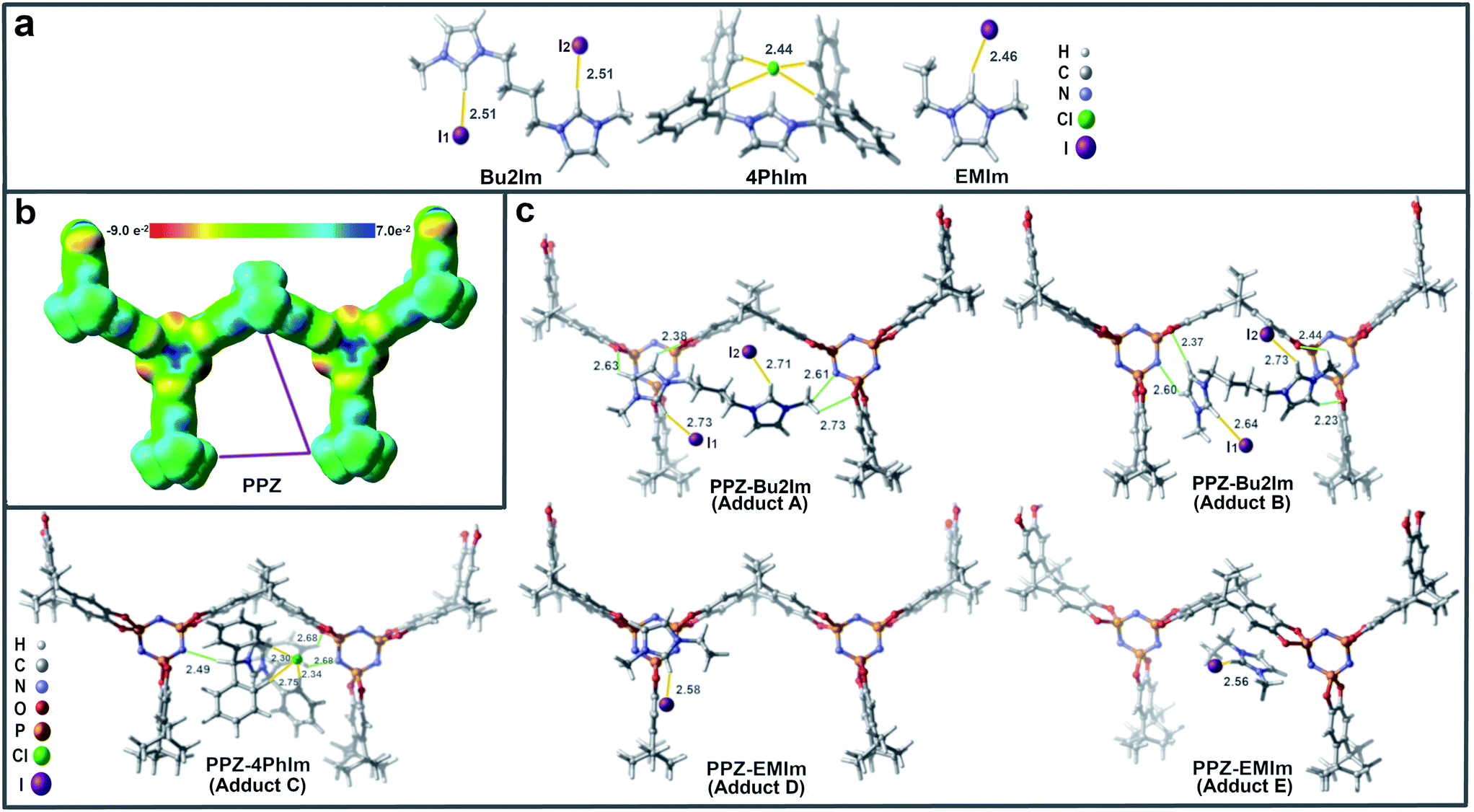
The kinetic profiles for the PPZ-Bu2Im nanoreactors and the Bu2Im IL are compared in Fig. S10,† showing that the former is considerably more active. These differences between the PPZ-Bu2Im nanoreactors and the other PPZ-IL nanoreactors may be attributed to the superior interaction of the Bu2Im cation with the PPZ nanospheres (see computational results above and Fig. 4c). The PPZ-IL nanoreactors form in various solvents such as acetone or ethanol and exhibit similar catalytic activities to those formed in situ in SO (Table 1, cf. entries 4 and 23 with entries 3 and 22). The PPZ-Bu2Im nanoreactors were recycled and reused 5 times with the yield of SC remaining above 95% (Fig. S11†). The slight decrease in activity may be attributed to ca. 2% loss of the Bu2Im IL after each reaction. No changes to the catalyst were detected by solid-state 31P NMR spectroscopy (Fig. S12†). The SEM image of the PPZ-Bu2Im nanoreactors after catalysis (Fig. 5a) shows them to be coated by a smooth 14 nm thick layer (Fig. 5b), corresponding to the precipitated Bu2Im IL in ethyl acetate (poor solvent). After removing the excess Bu2Im coating by washing with anhydrous ethanol and ethyl acetate, the PPZ nanomaterial presented a bigger diameter of ca. 150 nm (Fig. 5c) than the that of the freshly prepared PPZ (ca. 121 nm), due to the residual Bu2Im inside (elemental mapping of EDS), Fig. 5d, e and S13,† which has a detection depth of c.a. 100 nm.28 The amount of the residual IL was estimated to be 24% from thermogravimetric analysis (Fig. S14†).
 | ||
| Fig. 5 (a) SEM image of the recovered PPZ-Bu2Im nanoreactors with an average size of 174 nm. (b) Circular fitting on the enlarged area shows that the thickness of Bu2Im is 14 nm. (c) Scanning TEM (STEM) image, (d) P and (e) I of EDS elemental mapping of the PPZ-Bu2Im nanoreactors after washing. The scale bar is 100 nm. | ||
The scope of the PPZ-Bu2Im nanoreactors in the cycloaddition of CO2 to various epoxides was studied at atmospheric pressure (Table 3), with the corresponding carbonates obtained in excellent yields.
|
|
||||
|---|---|---|---|---|
| Entry | Epoxide | Product | Time (h) | Yield (%) |
| a Reaction conditions: epoxide (4.00 mmol), CO2 (entry 1, 10 bar CO2 in an autoclave; others, 1 atm using a CO2-filled balloon), 57 °C. Bu2Im (23.7 mg, 1.25 mol%), PPZ (19.2 mg, containing 2.5 mol% N). Yield was determined by 1H NMR spectroscopy. The selectivity of the products is all above 99% by GC-MS. | ||||
| 1a |
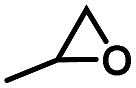
|
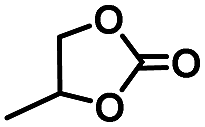
|
3 | 99 |
| 2 |
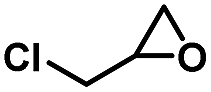
|
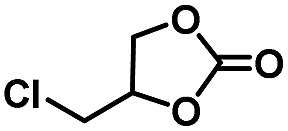
|
3 | 99 |
| 3 |
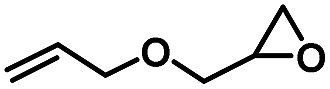
|
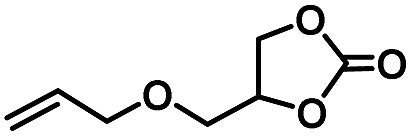
|
4 | 99 |
| 4 |
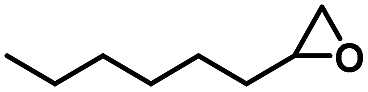
|
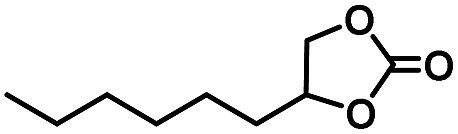
|
20 | 99 |
| 5 |
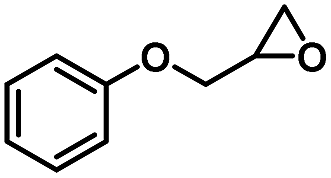
|
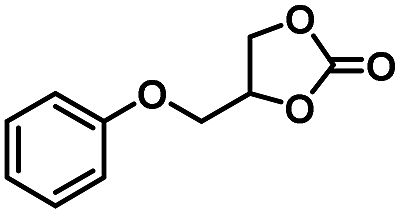
|
4 | 99 |

Conclusions
Electron-rich, semi-porous and swellable PPZ nanospheres were prepared using a facile polycondensation reaction. The PPZ nanospheres are thermally stable and can absorb CO2 under ambient conditions with weak interactions formed with the framework. The PPZ material interacts with IL cations, in particular the bis-imidazolium Bu2Im cation, to form PPZ-IL nanoreactors, which catalyze the cycloaddition of CO2 to epoxides to form cyclic carbonates under mild reaction conditions. Highest activities are obtained when the IL fits intimately into the PPZ host material, as demonstrated by DFT calculations, which reduces interactions between the cation and anion of the IL, leading to more nucleophilic anions and higher activities with catalytic efficiency exceeding 7-fold. Importantly, the PPZ-Bu2Im nanoreactors are stable, tolerant to a range of substrates, and can be recycled and reused multiple times. Such activity is essential for this industrially important reaction and it is likely that this new class of nanoreactor can be adapted to many other transformations.Experimental
Materials and methods
All starting materials and solvents were obtained from commercial sources and used as received. The following compounds were prepared using literature procedures: 3-ethyl-1,2-dimethylimidazolium iodide (EtMMIm), 3,3′-(butane-1,4-diyl)bis(1-methylimidazolium)iodide (Bu2Im), 3-methyl-1-octadecylimidazolium iodide (C18Im), 1,3-dibenzhydrylimidazolium chloride (4PhIm), 3-methyl-1-phenylimidazolium iodide (PhMIm), 1-((perfluorophenyl)methyl)-3-(4-vinylbenzyl)-1H-imidazol-3-ium bromide (StFImB), and 9,10-dimethyl-9,10-ethano-9,10-dihydro-2,3,6,7-tetrahydroxy-anthracene (ATC).29Synthesis of EtMMIm
Iodoethane (15.60 g, 100 mmol) was slowly added to a dry THF solution (200 mL) of 1,2-dimethylimidazole (9.61 g, 100 mmol). The mixture was stirred at reflux under a nitrogen atmosphere for 2 d. The solid obtained was removed by filtration and washed with dry ethyl ether, dissolved in methanol, and recrystallized by addition of dry ethyl ether at −20 °C. The white solid was obtained by filtration and dried under vacuum. Yield: 81%. FT-IR (neat): 3121, 3094, 2970, 1739, 1639, 1579, 1534, 1452, 1415, 1347, 1283, 1228, 1201, 1132, 1091, 1041, 855, 808, 772, 726, 667, 630 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 7.67 (s, 2H), 4.15 (s, 2H), 3.75 (s, 3H), 2.59 (s, 3H), 1.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 144.51, 122.82, 120.77, 43.25, 35.16, 15.34, 9.63. HR-ESI-MS: 125.1073 (C7H13N2+; calc. 125.1083). Anal. calcd. For C7H13N2I: C, 33.35; H, 5.20; N, 11.11; I, 50.34. Found: C, 33.51; H, 5.60; N, 11.40; I, 50.14.Synthesis of Bu2Im
1,4-Diiodobutane (30.99 g, 100 mmol) was gradually added to an acetonitrile solution (60 mL) of 1-methylimidazole (16.42 g, 200 mmol). The mixture was stirred at reflux under a nitrogen atmosphere for 3 d. The solution was then condensed by evaporation, and was washed by diethyl ether. The salt was precipitated as a brown powder, which was recrystallized from ethanol/diethyl ether for several times, and dried under vacuum. The purified product was finally obtained as a white solid. Yield: 76%. FT-IR (neat): 3132, 3072, 2979, 2939, 2911, 1628, 1556, 1451, 1436, 1339, 1311, 1230, 1158, 1106, 816, 780, 756, 696, 636, 615, 595 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (s, 2H), 7.75 (m, 4H), 4.21 (s, 4H), 3.86 (s, 6H), 1.78 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 137.04, 124.16, 122.70, 48.48, 36.36, 26.56.Synthesis of C18Im
1-Iodooctadecane (38.04 g, 100 mmol) was added to an acetonitrile solution (50 mL) of 1-methylimidazole (8.21 g, 100 mmol). The solution was stirred under reflux for 2 d, and the resulting yellow solution was concentrated and precipitated in diethyl ether. The solid precipitation was collected by filtration, and dried under vacuum. Yield: 79%. FT-IR (neat): 3092, 2915, 2848, 1564, 1468, 1377, 1166, 1080, 1020, 826, 735, 721, 645, 617 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (d, J = 1.7 Hz, 1H), 7.73 (dt, J = 26.7, 1.8 Hz, 2H), 4.14 (t, J = 7.2 Hz, 2H), 3.84 (s, 3H), 1.77 (p, J = 7.4 Hz, 2H), 1.23 (s, 30H), 0.95–0.77 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 136.94, 124.07, 122.73, 49.23, 36.24, 31.76, 29.85, 29.50, 29.46, 29.42, 29.29, 29.17, 28.85, 25.96, 22.56, 14.42. Anal. calc. for C22H43IN2 (462.50): C 57.13, H 9.37, N 6.06; found C 56.98, H 9.32, N 5.89.Synthesis of 4PhIm
A mixture of (trimethylsilyl)imidazole (14.026 g, 0.100 mol) and chlorodiphenylmethane (40.5 g, 0.2 mol) in acetonitrile (200 mL) was refluxed for 24 h. After removal of the solvents, the resulting solid was filtered, washed with diethyl ether (3 × 30 mL), and dried under vacuum for 24 h. Yield: 98%. FT-IR (neat): 3166, 3069, 2977, 1536, 1492, 1452, 1183, 1138, 1078, 1030, 854, 838, 749, 737, 693, 661, 637, 592, 508 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.34 (t, J = 1.7 Hz, 1H), 7.83 (d, J = 1.7 Hz, 2H), 7.58–7.24 (m, 20H), 7.20 (s, 2H), 3.32 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 137.52, 129.63, 129.43, 128.59, 123.34, 66.50.Synthesis of PhMIm
A mixture of methyl iodide (17.03 g, 120 mmol) and phenylimidazole (14.41 g, 100 mmol) in acetonitrile (200 mL) was refluxed for 48 h under N2. The solvent was removed under reduced pressure, the crude product was recrystallized from acetone and ether, and the product was obtained as white solid. Yield: 80%. FT-IR (neat): 3171, 3120, 1584, 1557, 1451, 1428, 1189, 1156, 1110, 1028, 821, 733, 651, 618, 554 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 9.80 (d, J = 1.7 Hz, 1H), 8.31 (t, J = 1.9 Hz, 1H), 7.99 (t, J = 1.8 Hz, 1H), 7.86–7.51 (m, 5H), 3.97 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 136.44, 135.20, 130.69, 130.22, 124.92, 122.30, 121.42, 36.74.Synthesis of StFImB
4-Vinylbenzylimidazole (1.0 g, 5.43 mmol) was gradually added to an acetonitrile solution (5 mL) of bromopentafluorobenzene (1.20 g, 4.86 mmol). The mixture was stirred and heated to 60 °C under a nitrogen atmosphere for 2 h. The solution was then condensed by evaporation, and was washed by ethyl acetate. The salt was precipitated as a white powder, which was sonicated for 1 h and collected by filtration, washed with diethyl ether and dried. Yield: 95%. 1H-NMR (400 MHz, DMSO-d6): 9.52 (s, 1H); 7.93 (d, J = 14.8, 2H); 7.61 (d, J = 8.2, 2H); 7.48 (d, J = 7.9, 2H); 6.83 (s, 1H); 5.96 (d, J = 17.7, 1H); 5.73 (s, 2H); 5.48 (s, 2H); 5.39 (d, J = 11.0, 1H). 13C-NMR (101 MHz, DMSO-d6): 139.0; 138.1; 137.4; 136.4; 134.5; 129.2; 127.1; 123.6; 123.3; 115.8; 52.3. HR-ESI-MS: 365.1081 (C19H14F5N2+, M+; calc. 365.1072). Anal. calc. for C19H14BrF5N2 (445.22): C 51.26, H 3.17, N 6.29; found: C 51.05, H 3.30, N 6.04.Synthesis of ATC
Powdered pyrocatechol (11.01 g, 100 mmol) was added to ice-cooled sulfuric acid (220 mL, 70%) to give a colorless suspension. 2,5-Hexanedione (5.71 g, 50 mmol) was added drop-wise to form a green mixture. After 1 hour of stirring, the ice bath was removed and the color of the mixture turned to reddish-brown and stirring was continued for 7 d at room temperature. The precipitate was removed from the acid by filtration with a glass frit and washed with water. The dark red crude product was recrystallized twice from ethyl acetate, then filtrated and dried under vacuum to form light gray powder. Yield: 71%. Mp 265 °C. FT-IR (neat): 3497, 3295, 2945, 1617, 1445, 1297, 1220, 1139, 992, 879, 813, 802 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 4H), 6.61 (s, 4H), 1.68 (s, 6H), 1.41 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 142.2, 138.1, 109.2, 36.7, 19.0.Synthesis of PPZ nanospheres
HCCP (300 mg, 0.862 mmol) and ATC (385 mg, 1.293 mmol) were mixed in acetonitrile (120 mL) and triethylamine (TEA) (522 mg, 5.17 mmol) was added. The reaction mixture was stirred in an ultrasonic bath (100 W, 80 kHz) at room temperature for 5 h. The resulting precipitate was collected by centrifugation at 6000 rpm for 6 min and then washed three times with tetrahydrofuran and water. The collected solid was dried under vacuum to yield light brown powder (435 mg, yield 88%, based on HCCP used).Synthesis of the PPZ-TBAB and PPZ-Bu2Im nanoreactors
TBAB (33 mg, 2.5 mol%) or Bu2Im (24.3 mg, 1.25 mol%) was dissolved in acetone (300 μL), and PPZ (19.2 mg, containing 2.5 mol% N) was added and the solution heated at 40 °C for 6 h under N2. The solvent was evaporated and dried under vacuum to afford the desired material.Computational details
Gas phase calculations were carried out at the M062X30/6-31G* level using the Gaussian 09 suite of programs,31 and DGDZVP basis set was employed for iodide,32 as it has been used to describe iodide anions in related systems.33 M06-2X functional has demonstrated to be suitable for describing non-bonding interaction34 including ionic hydrogen bonds.35 The compounds were optimized and all structures were confirmed as minima by Hessian matrix calculations. Two constraints were included in order to avoid deformations of the PPZ cavity which are not representative of the system considering the effect of the surrounding polymer and the multiple layer nature of this material. Relevant vibrational frequencies were calculated at the same level of theory showing acceptable accuracy to describe the more relevant vibrational modes in the isolated ILs (see ESI†). Whereas, binding energies were estimated at M062X/6-311G*//M062X/6-31G* level, as a difference between energy of a given adduct and the sum of energies of isolated components constituting it, and including zero-point vibrational energies. It is worth to mention that Coulomb interactions were not considered due to the size of the system and the required computational cost in order to avoid self-interaction errors.36Characterization in the solid-state
31P and 13C NMR spectra were recorded on a Bruker AvanceIIIHD 400 spectrometer operating at 100.6 MHz for 13C and 162.0 MHz for 31P. All experiments were performed at ambient probe temperature using hydrogen high-power decoupling. Cross-polarization with magic angle spinning (CP/MAS) was adopted. Liquid-state 1H and 13C NMR spectra were recorded on a Bruker 400 MHz instrument. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Vertex 80 instrument. Mass spectra were recorded on a Bruker MALDI-TOF AutoFlex speed machine. Field emission SEM images were obtained using a Merlin instrument at an activation voltage of 2 kV under high resolution analysis mode. TEM and STEM microphotographs were recorded on a Tecnai Osiris instrument at an activation voltage of 200 kV. The number-average diameter (Dn) and the standard deviation (SD) were determined with Image Tool software by counting 1000 individual particles from TEM microphotographs. Brunauer–Emmett–Teller (BET) experiments were performed on a Quantachrome Autosorb-IQ/MP-XR with N2 (77 K), CO2 (273 K) and CH4 (273 K) as analyte gases. Atomic force microscopy (AFM) images were collected on a Cypher S AFM (Asylum Research) in tapping mode under ambient conditions. The dry PPZ sample was prepared by dropping an acetone suspension (1 mg mL−1) on a silicon wafer, then dried with N2 and kept under vacuum for 3 d. The swelled PPZ sample was prepared by dropping the styrene oxide suspension (1 mg mL−1) on a silicon wafer, dried with N2 until there was no apparent solvent stain. Powder X-ray diffraction (PXRD) patterns were collected on a powder diffractometer (D/max-2200/PC, Rigaku, Japan) using Cu-K irradiation (40 kV, 20 mA). Diffraction patterns were collected from 8° to 80° at a speed of 5° min−1. Thermal degradation of the crosslinked microspheres was examined with a thermogravimetric analyzer (TGA) Perkin Elmer TGA-7 with a heating rate of 10 °C min−1 under nitrogen condition. Dynamic light scattering (DLS) measurements were carried out on a Zeta sizer Nano ZS from Malvern Instruments with a laser at 633 nm.Catalytic studies
Typical procedure for reactions at an atmospheric pressure of CO2: a mixture of styrene oxide (480 mg, 4 mmol) and the TBAB (33 mg, 2.5 mol%) with or without PPZ (19.2 mg, containing 2.5 mol% N) was heated at 57 °C for 20 h under a CO2 atmosphere (1 atm, using a balloon). After the reaction, the system was cooled to room temperature and the products were analyzed by 1H NMR spectroscopy. For the bis-imidazolium salts 1.25 mol% were used.Typical procedure for reactions under high CO2 pressures: to a 100 mL stainless steel autoclave equipped with a glass vial and a magnetic stirrer, propylene oxide (232 mg, 4 mmol), Bu2Im (23.7 mg, 1.25 mol%) and PPZ (19.2 mg, containing 2.5 mol% N) were added. The autoclave was sealed and purged three times with CO2 and then set to 10 bar. The autoclave was heated in a 57 °C oil bath. After reaction, the autoclave was cooled in an ice bath and the yield of the product was determined by 1H NMR spectroscopy.
Kinetic study
A mixture of styrene oxide (1920 mg, 16 mmol) and Bu2Im (94.8 mg, 1.25 mol%) with or without PPZ (76.8 mg, containing 2.5 mol% N) were heated at 57 °C under a CO2 atmosphere (1 atm, using a balloon). After the appropriate time (1–20 h), one drop of the reaction mixture was removed and the sample was diluted with CDCl3 (0.5 mL) and analyzed by 1H NMR spectroscopy.Recycling experiments
The experiments were conducted according to the typical procedure at atmospheric pressure. In addition, the PPZ-Bu2Im catalyst was further washed with ethyl acetate and dried before reuse.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Sino Swiss Science and Technology Cooperation (SSSTC) Program. We are grateful to Dr Kurt Schenk for helpful discussions, Baudat Emilie for recording the solid-state NMR spectra, Xavier Adrian Jeanbourquin Vasilyev for AFM, and José Bila for BET.References
- (a) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed; (b) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- (a) D. Yuan, W. Lu, D. Zhao and H. C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed; (b) C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- (a) H. Oh, S. B. Kalidindi, Y. Um, S. Bureekaew, R. Schmid, R. A. Fischer and M. Hirscher, Angew. Chem., Int. Ed., 2013, 52, 13219–13222 CrossRef CAS PubMed; (b) S. Keskin, J. Phys. Chem. C, 2012, 116, 1772–1779 CrossRef CAS; (c) H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773–9775 RSC.
- (a) S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed; (b) S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed; (c) G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- (a) G. H. Bertrand, V. K. Michaelis, T. C. Ong, R. G. Griffin and M. Dincă, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4923–4928 CrossRef CAS PubMed; (b) D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, ACS Nano, 2017, 11, 2706–2713 CrossRef CAS PubMed; (c) M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann and D. Trauner, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed; (d) L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- (a) A. Larrañaga, M. Lomora, J. Sarasua, C. Palivan and A. Pandit, Prog. Mater. Sci., 2017, 90, 325–357 CrossRef; (b) Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed; (c) V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef CAS PubMed.
- (a) F. Wu, E. Zhao, D. Gordon, Y. Xiao, C. Hu and G. Yushin, Adv. Mater., 2016, 28, 6365–6371 CrossRef CAS PubMed; (b) C. R. DeBlase, K. H. Burgos, K. E. Silberstein, G. G. R. Calero, R. P. Bisbey, H. D. Abruna and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed; (c) C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed; (d) H. Liao, H. Wang, H. Ding, X. Meng, H. Xu, B. Wang, X. Ai and C. Wang, J. Mater. Chem. A, 2016, 4, 7416–7421 RSC; (e) C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed; (f) F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814–6818 CrossRef CAS PubMed.
- (a) H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS; (b) S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed; (c) Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 53, 2878–2882 CrossRef CAS PubMed; (d) S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed; (e) H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed; (f) X. Wang, X. Han, J. Zhang, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332–12335 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- O. R. Evans, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2001, 123, 10395–10396 CrossRef CAS PubMed.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato and Y. Kubota, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed.
- (a) W. Zhong, F. D. Bobbink, Z. Fei and P. J. Dyson, ChemSusChem, 2017, 10, 2728–2735 CrossRef CAS PubMed; (b) F. D. Bobbink, A. P. Van Muyden, A. Gopakumar, Z. Fei and P. J. Dyson, ChemPlusChem, 2017, 82, 144–151 CrossRef CAS.
- (a) P. Mohanty, L. D. Kull and K. Landskron, Nat. Commun., 2011, 2, 401 CrossRef PubMed; (b) H. R. Allcock, Accounts Chem. Res., 1978, 11, 81–87 CrossRef CAS.
- (a) W. Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615–2619 CrossRef CAS PubMed; (b) J. Dong, P. Cui, P. F. Shi, P. Cheng and B. Zhao, J. Am. Chem. Soc., 2015, 137, 15988–15991 CrossRef CAS PubMed; (c) M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed; (d) P. Z. Li, X. J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed; (e) Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS PubMed; (f) S. Kaneko and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 2836–2840 CrossRef CAS.
- (a) L. Zhu, Y. Xu, W. Yuan, J. Xi, X. Huang, X. Tang and S. Zheng, Adv. Mater., 2006, 18, 2997–3000 CrossRef CAS; (b) J. Zhou, L. Meng, X. Feng, X. Zhang and Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 8476–8479 CrossRef CAS PubMed.
- (a) H. Liu, S. Li, H. Yang, S. Liu, L. Chen, Z. Tang, R. Fu and D. Wu, Adv. Mater., 2017, 29, 1700723 CrossRef PubMed; (b) J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky and Y. Z. Khimyak, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS PubMed.
- (a) L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2012, 135, 562–565 CrossRef PubMed; (b) S. Yang, X. Lin, W. Lewis, M. Suyetin, E. Bichoutskaia, J. E. Parker, C. C. Tang, D. R. Allan, P. J. Rizkallah and P. Hubberstey, Nat. Mater., 2012, 11, 710–716 CrossRef CAS PubMed.
- (a) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52–61 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- H. Sun and D. Zhang, J. Phys. Chem. A, 2007, 111, 8036–8043 CrossRef CAS PubMed.
- J. Liang, Y. B. Huang and R. Cao, Coord. Chem. Rev., 2017 DOI:10.1016/j.ccr.2017.11.013.
- A. Mirabaud, J. C. Mulatier, A. Martinez, J. P. Dutasta and V. R. Dufaud, ACS Catal., 2015, 5, 6748–6752 CrossRef CAS.
- (a) D.-H. Lan, Y.-X. Gong, N.-Y. Tan, S.-S. Wu, J. Shen, K.-C. Yao, B. Yi, C.-T. Au and S.-F. Yin, Carbon, 2018, 127, 245–254 CrossRef CAS; (b) D.-H. Lan, H.-T. Wang, L. Chen, C.-T. Au and S.-F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS; (c) D.-H. Lan, L. Chen, C.-T. Au and S.-F. Yin, Carbon, 2015, 93, 22–31 CrossRef CAS; (d) D. H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C. T. Au and S. F. Yin, Chin. J. Catal., 2016, 37, 826–845 CrossRef CAS; (e) D. H. Lan, F. M. Yang, S. L. Luo, C. T. Au and S. F. Yin, Carbon, 2014, 73, 351–360 CrossRef CAS.
- (a) S. A. Katsyuba, M. V. Vener, E. E. Zvereva, Z. Fei, R. Scopelliti, G. Laurenczy, N. Yan, E. Paunescu and P. J. Dyson, J. Phys. Chem. B, 2013, 117, 9094–9105 CrossRef CAS PubMed; (b) S. A. Katsyuba, M. V. Vener, E. E. Zvereva, Z. Fei, R. Scopelliti, G. Laurenczy, J. G. Brandenburg, S. Siankevich and P. J. Dyson, J. Phys. Chem. Lett., 2015, 6, 4431–4436 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, et al., Definition of the Hydrogen Bond (IUPAC Recommendations 2011), Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- D. E. Newbury and N. W. Ritchie, J. Anal. At. Spectrom., 2013, 28, 973–988 RSC.
- (a) D. Peixoto, M. Figueiredo, M. B. Gawande, M. C. Corvo, G. Vanhoenacker, C. A. Afonso, L. M. Ferreira and P. S. Branco, J. Org. Chem., 2017, 82, 6223–6231 CrossRef PubMed; (b) T. V. Goncharova, L. V. Zatonskaya and A. S. Potapov, Procedia Chem., 2014, 10, 485–489 CrossRef CAS; (c) J. Arcau, V. Andermark, M. Rodrigues, I. Giannicchi, L. Pérez Garcia, I. Ott and L. Rodríguez, Eur. J. Inorg. Chem., 2014, 2014, 6117–6125 CrossRef CAS; (d) S. Li, J. Tang, Y. Zhao, R. Jiang, T. Wang, G. Gao and J. You, Chem. Commun., 2017, 53, 3489–3492 RSC; (e) F. D. Bobbink, Z. Fei, R. Scopelliti, S. Das and P. J. Dyson, Helv. Chim. Acta, 2016, 99, 821–829 CrossRef CAS; (f) D. Fritsch, G. Bengtson, M. Carta and N. B. McKeown, Macromol. Chem. Phys., 2011, 212, 1137–1146 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, et al.,Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- M. Aono, H. Miyazaki, T. Takekiyo, S. Tsuzuki and H. Abe, Phys. Chem. Chem. Phys., 2018, 20, 5780–5784 RSC.
- N. Mardirossian and M. Head-Gordon, J. Chem. Theory Comput., 2016, 12, 4303–4325 CrossRef CAS PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and Ca. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- (a) M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed; (b) I. Lage-Estebanez, A. Ruzanov, J. M. García de la Vega, M. V. Fedorov and V. B. Ivaništšev, Phys. Chem. Chem. Phys., 2016, 18, 2175–2182 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta08856j |
| This journal is © The Royal Society of Chemistry 2018 |