
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully bio-based zwitterionic membranes with superior antifouling and antibacterial properties prepared via surface-initiated free-radical polymerization of poly(cysteine methacrylate)†
Luis
Valencia
a,
Sugam
Kumar
a,
Blanca
Jalvo
a,
Andreas
Mautner
 b,
German
Salazar-Alvarez
a and
Aji P.
Mathew
*a
b,
German
Salazar-Alvarez
a and
Aji P.
Mathew
*a
aDivision of Materials and Environmental Chemistry, Stockholm University, Frescativägen 8, 10691, Sweden. E-mail: aji.mathew@mmk.su.se
bPolymer and Composite Engineering (PaCE) Group, Institute of Materials Chemistry and Research, Faculty of Chemistry, University of Vienna, Währinger Str. 42, 1090 Wien, Austria
First published on 30th July 2018
Abstract
This article proposes a strategy to prepare membranes that combine the network characteristics of micro/nanocellulose with grafted zwitterionic poly(cysteine methacrylate) (PCysMA) to develop fully bio-based membranes with antifouling properties. The surface characteristics of the membranes were studied, together with static adsorption of bovine serum albumin (BSA) and S. aureus for evaluating the antifouling properties of the membranes. Experimental data revealed a homogeneous modification that resulted in excellent antifouling properties with a reduction of 85% in biofilm formation and enhanced antimicrobial activity. Moreover, we introduced a novel method to determine the pore size of membranes in the wet-state and assess the antifouling performance in situ by synchrotron-based SAXS. This allowed us to observe in real-time the decrease in pore size upon adsorption of BSA during filtration, and how this phenomenon is strongly suppressed by grafting of PCysMA. The importance of this work lies in introducing a simple method to yield cellulosic membranes with superior antifouling properties, which could significantly increase their potential for water treatment applications.
Introduction
The non-specific adsorption of proteins and unwanted adhesion of cells to surfaces of materials, commonly referred to as surface fouling, is a problematic phenomenon associated with medical implantation, biosensing, and membrane performance.1–3 For this reason, the development and optimization of new approaches for designing both biocompatible and non-fouling surfaces has attracted considerable academic and industrial interest during the past few decades, especially in the fields of biomedical and environmental engineering. Non-toxic functional polymer brushes are effective in prevention of fouling, due to their large energetic barrier, constituted by the balance between volume extrusion, conformational entropy, and segment interactions of the polymer brushes that must be overcome for proteins to be adsorbed.4–6 Moreover, polymer brushes can also provide mechanical and chemical robustness with long-term stability.The most widely used polymer brush for antifouling properties is poly(ethylene glycol) (PEG) and its derivatives, which offer high resistance to nonspecific protein adsorption and cell adhesion. However, these materials have limitations, especially in long-term use, because they can be oxidized under physiological conditions, which can lead to activation of complementary responses.7,8 Therefore, over the past decade, diverse research activities have been devoted to designing alternative non-fouling biomaterials. For example, zwitterionic polymers have emerged as a promising candidate because of their good chemical stability, low cost and excellent anti-fouling activity. These antifouling properties arise from their electrical neutrality with equivalent positively and negatively charged groups, and through a hydration layer formed by solvation of the charged groups, assisted by hydrogen bonding, which creates a physical and energetic barrier.8 Polymers incorporating zwitterionic molecules such as phosphorylcholine,9 sulfobetaine10,11 and carboxybetaine12 have been reported to be promising for anti-biofouling surfaces. However, to the best of our knowledge, relatively little work has been focused on polymers containing amino acid motifs as side chains, in particular those containing cysteine.
In addition to the antifouling behaviour, cysteine has been reported to be a potent metal chelator, presenting a unique affinity to metal cations.13 The primary group responsible for cation binding is the thiol group of the constituent cysteine, but it also occurs through complex formation between the amine and carboxylate moieties (N, O-chelation).13 On the other hand, the high reactivity of cysteine has also been proven to affect several enzymatic reactions in vitro and many microorganisms in various metabolic functions.14,15 In the present study, zwitterionic polymer brushes based on poly(cysteine methacrylate) (PCysMA) were grafted from nanocellulose-based membranes by means of surface-initiated free radical polymerization. Static adsorption tests with bovine serum albumin (BSA) and bacteria were performed to evaluate the antifouling properties of the modified membranes. Moreover, a novel method to determine the pore size of membranes in the wet-state and to assess the antifouling performance in situ is introduced by means of synchrotron-based SAXS. Data collected from the results indicate that PCysMA-grafted nanocellulose membranes are highly stable and possess excellent antifouling properties.
Experimental section
Materials
High-purity cellulose from softwood fibers (Norwegian spruce) with high cellulose content (95% cellulose, 4.5% hemicellulose and 0.1% lignin content as provided by Domsjö Fabriker AB, Sweden) was used as the starting material to produce cellulose nanofibers. L-Cysteine (97%), potassium persulfate (KPS), dimethyl sulfoxide (99%), fluorescein diacetate (FDA), dodecyl sulfate (SDS), Bovine Serum Albumin (BSA), phosphate buffered saline (BioUltra), dimethylphenyl phosphine (DMPP) (99.0%), 3-(acryloxy)-2-hydroxypropyl methacrylate (99%), 3-(trimethoxysilyl) propyl methacrylate (MPS), and propylamine (99%) were purchased from Sigma Aldrich and were used as received. SYTO was purchased from Thermo Fisher Scientific and used as received.A detailed description of the extraction of nanocellulose and synthesis of monomer cysteine methacrylate15 is given in the ESI (S1.1 and S1.2).†
Manufacturing of membranes
The membranes were prepared by vacuum filtration of a mixture containing 2 wt% suspension of cellulose and 1 wt% suspension of CNF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.12 ratio using a Buchner funnel setup having an area of 143 cm2. The membranes were dried at room temperature for 2 days under a load of 5 kg. The thickness of the membranes was determined using scanning electron microscopy to be around 200 μm.
:0.12 ratio using a Buchner funnel setup having an area of 143 cm2. The membranes were dried at room temperature for 2 days under a load of 5 kg. The thickness of the membranes was determined using scanning electron microscopy to be around 200 μm.
Grafting of PCysMA brushes on the nanocellulose membranes via SI-FRP
A two-step process was employed: covalent surface immobilization of the initiator and subsequent surface-initiated free-radical polymerization (SI-FRP). In a typical procedure, three membranes (around 0.15 g each) were submerged in 20 mL of cyclohexane contained in a 50 mL glass vessel covered by a rubber septum under a nitrogen atmosphere. 1 mL (4 μmol) MPS was injected into the reaction system, followed by 0.260 mL (3.1 μmol) of n-propylamine, and the mixture was stirred for 24 h at 60 °C. The membranes were washed extensively with cyclohexane, ethanol and water (3× each) to remove any trace of reagent and dried under reduced pressure at room temperature. For the surface initiated-polymer grafting, the modified membranes were submerged in a 50 mL aqueous solution containing cysteine methacrylate, inside a 50 mL glass vessel covered with a rubber septum under a nitrogen atmosphere. The temperature was raised to 70 °C and a solution of KPS (30 mg, 0.11 μmol) was injected to initiate the graft polymerization. After 2 h the membranes were removed, washed repeatedly with ethanol and water in a sonication bath, and dried overnight under reduced pressure at 50 °C. A schematic illustration of the functionalization of the membranes is shown in Fig. 1.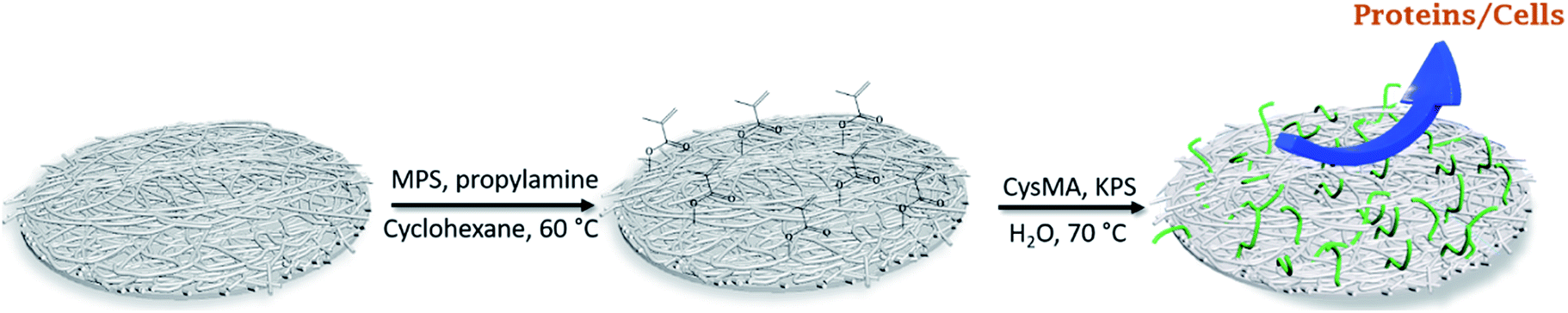 | ||
| Fig. 1 Schematic illustration of the surface-initiated polymer grafting of P(CysMA) on the nanocellulose-based membrane. | ||
Surface characterization of the membranes
Water permeability and flux
The water permeability of the membranes was measured in a dead-end cell (Sterlitech HP4750 stirred cell, USA). Prior to the measurements, membrane discs with a diameter of approx. 50 mm were cut from the membranes and placed in the dead-end cell on a stainless steel porous support disk and water was passed through the membranes at 23 °C at a head pressure of 0.5 MPa maintained by compressed air flow. The quantity of water that passed through the membrane for a defined time interval was measured and the flux was calculated (L h−1 m−2) for the active filtration area (14.6 cm2).Antibacterial and antifouling performance
The antibacterial properties of the membranes were assessed using the strain Staphylococcus aureus CECT 240. The antifouling capability of the modified and unmodified samples was studied in two different and complementary ways: on the one hand, biofilm formation of S. aureus and, on the other hand, protein adsorption using a solution of bovine serum albumin (BSA). Bacterial viability and biofilm assays were tested using different fluorescence techniques. For these tests, exponentially grown cultures of S. aureus on nutrient medium were diluted to an OD600 of 0.0138 (108 cells per mL). Diluted cultures were placed on the membranes inside polystyrene 24-well plates and incubated without stirring for 24 h at 37 °C. Afterwards, the membranes were carefully washed with distilled water to remove planktonic and loosely attached cells.Fluorescein diacetate (FDA), a fluorogenic substrate that permits the detection of enzymatic activity, was used for the relative quantification of the biofilm formation. The fluorescence was measured in a fluorometer/luminometer, Fluoroskan Ascent FL. 200 μL of FDA (0.02% (w/w) in DMSO) were spread over the entire surface of the samples. After 15 min of pre-incubation at 25 °C, FDA was excited at 485 nm and its emission recorded at 538 nm.
The visualization of the bacterial cells and biofilms was performed by confocal microscopy 24 h after inoculation using a Leica Microsystems Confocal SP5 fluorescence microscope (Leica Microsystems, Germany). Viable and non-viable bacteria were tracked using a Live/Dead BacLight Bacterial Viability Kit. For membrane staining, the surface of each specimen was covered with 30 μL of stain (a 0.5:1 mixture of SYTO 9 and PI in DMSO). For green fluorescence (SYTO 9, intact cells), excitation was performed at 488 nm and emission recorded at 500–575 nm. For red fluorescence (PI, dead cells), the excitation/emission wavelengths were 561 nm and 570–620 nm, respectively. Incubation was performed in the dark for 15–30 min at room temperature. For matrix visualization, the biofilms were stained with 200 μL of FilmTracer SYPRO Ruby per film, incubated in the dark for 30 min at room temperature, and rinsed with distilled water. For matrix staining, the excitation/emission wavelengths were 450 nm and 610 nm, respectively. Biofilm formation was also visualized by SEM. A process of dehydration and drying with ethanol at different concentrations was carried out to analyse samples in contact with microorganisms by SEM.
BSA adsorption was performed using PBS solution as a buffer. The proteins were dissolved in 0.01 M PBS solution at a concentration of 1 mg mL−1. The membranes were equilibrated with PBS overnight and then immersed in the protein solution for 2 h at 37 °C. After that, the membranes were rinsed with PBS solution three times. In one batch of samples, the adsorbed proteins were removed by immersing the membranes in 1 wt% sodium dodecyl sulfate (SDS) solution for 1 h at 37 °C under slight shaking conditions. In the other batch of samples, the proteins were kept on the membranes for their confocal microscopy visualization in order to study the antifouling properties of the membranes. For both characterizations, the proteins were quantified and visualized using a Qubit Protein Assay Kit in a fluorometer/luminometer, Fluoroskan Ascent FL, and with a Leica Microsystems Confocal SP5 fluorescence microscope, respectively. For BSA characterization, the excitation/emission wavelengths were 485 nm and 592 nm, respectively.
In situ SAXS measurements
To examine the evolution of the pore structures in both the unmodified and modified cellulose membranes in real time, synchrotron-based SAXS experiments were performed. The SAXS experiments were carried out at micro- and nano-focus X-ray scattering (MiNaXS) beamline P03 at synchrotron source PETRA III, DESY, Germany. The X-rays of wavelength (λ) 0.09 nm with a beam size of about 20 × 20 μm2 were made incident on the samples. The scattering patterns were recorded using a 2D-pixel detector (Pilatus 300 K), and the radial averaged intensity I(Q) was calculated for different wave vector transfers, Q (Q = (4πsin(θ))/λ, where 2θ is the scattering angle). The sample to detector distance was 1.5 m and the transmission of the X-rays across the membrane is monitored simultaneously by a PIN diode during the scanning experiment. The samples were scanned at five different sample positions and it was found that the samples were acceptably homogeneous. The antifouling properties of the modified and unmodified membranes were investigated by comparing the SAXS data of the two, measured in real time with the flow of the BSA protein solution (1 wt%). In SAXS, one measures the intensity of the scattered X-rays as a function of the wave vector transfer (Q). For a dilute system of monodisperse, identical scatterers (particles/pores), the scattering intensity can be given as16,17| I(Q) = ϕV(Δρ)2P(Q) | (1) |
The structural parameters are thus obtained by fitting the I(Q) using the P(Q) of different suitable models, employing a nonlinear least-squares fitting program. The expression of P(Q) for spherical scatterers of radius R can be given as18
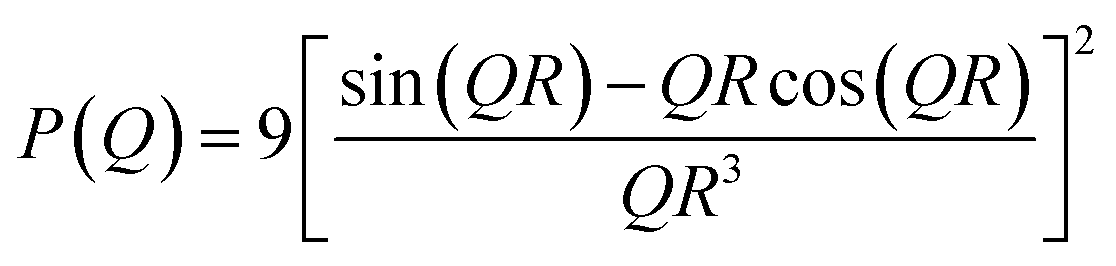 | (2) |
For a dilute ensemble of polydisperse particles, the intensity (eqn (1)) is modified as
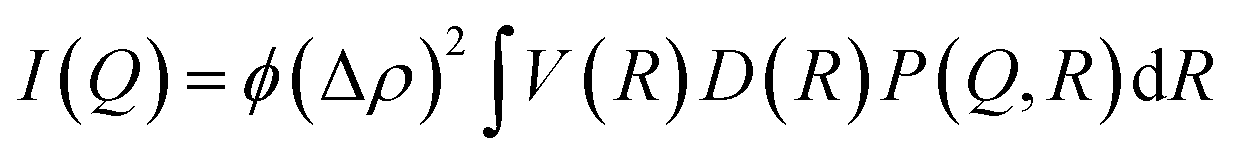 | (3) |
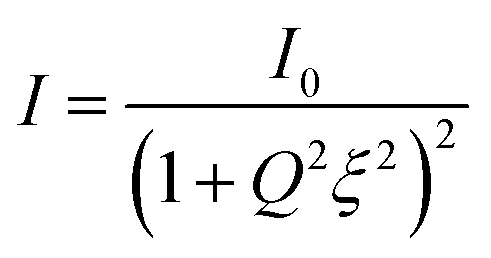 | (4) |
Results and discussion
Fig. 1 shows the processing route for zwitterionic membranes prepared by surface-initiated polymer grafting of poly(cysteine methacrylate) (PCysMA).Two different zwitterionic membranes were prepared (ZM-1 and ZM-2), with variation of the grafting yield, in order to investigate the influence of this factor on the structural properties of the membrane, and their antifouling performance. The main properties of the membranes are summarized in Table 1.
| Sample | [M]0a (mmol) | Surface areab (m2 g−1) | Pore diameterc (nm) | Permeate fluxd (Lmh bar−1) | Grafting densitye (mg cm−2) | Contact angle (°) |
|---|---|---|---|---|---|---|
| a Monomer concentration. b BET specific surface area. c BJH desorption pore diameter. d Measured at 1 bar pressure, using 200 mL of water. e Determined by the weight difference between the modified membrane and the virgin membrane divided by the surface area of the virgin membrane. | ||||||
| Unmodified | — | 2.40 | 4.74 | 11742 |
— | 28.3 |
| ZM-1 | 2.23 | 2.22 | 4.40 | 11228 |
0.91 ± 0.18 | 54.4 |
| ZM-2 | 4.47 | 2.10 | 4.23 | 10960 |
1.41 ± 0.13 | 66.2 |
A two-step functionalization strategy was followed: first, immobilization of a methacrylate group by means of a silylation reaction followed by surface-initiated free-radical polymerization of CysMA. The successful modification was confirmed by the intense change in the water contact angle of the membranes after each stage. After immobilizing the initiator, the hydrophilic surface of neat cellulose turned hydrophobic with a contact angle value of 128.1° (see Fig. S2†), which suggests the consumption of the surface hydroxyl groups. Then, upon polymer grafting of CysMA, the water contact angle decreased to 54. 5° and 66. 2° for ZM-1 and ZM-2, respectively (see Table 1), which suggests the uniform polymer grafting of hydrophilic PCysMA.
Surface characterization of the membranes
The chemical composition of the surface of the membranes was characterized by ATR-FTIR, and the resultant spectra are shown in Fig. 2a. Compared with the pristine cellulose membrane, the modified membranes showed an additional IR signal between 1700 and 1760 cm−1 that correlates with the characteristic stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from PCysMA. This indicates that the polymer brushes were successfully grafted from the cellulose membrane surface. Moreover, the higher peak intensity for ZM-2 reaffirmed the higher grafting yield. In addition, FT-IR mapping of ZM-2 was performed to provide information regarding the distribution of polymer brushes on the surface,20 and the results are displayed in Fig. 2b. ZM-2 was solely used for the analysis, chosen due to the higher degree of functionalization. At first sight, different regions, shown as different colours, were observed in the FT-IR mapping image; however, we found that they do not indicate a lack of effective functionalization, but rather indicate the surface roughness of the membranes. This was demonstrated by analysing the FT-IR spectra at each of the different spots, shown also in Fig. 2b, where the peak corresponding to the carboxyl group of PCysMA at 1700 cm−1 was found in all regions. These observations highlight the effectiveness of the grafting technique utilized here, which yields a rather homogeneous grafting density throughout the surface of the membrane, as well as the applicability of high-resolution FT-IR imaging to study polymer grafting. The homogeneity of the modification on the membranes was also corroborated by EDS spectroscopy (Fig. S4 and S5†).
O from PCysMA. This indicates that the polymer brushes were successfully grafted from the cellulose membrane surface. Moreover, the higher peak intensity for ZM-2 reaffirmed the higher grafting yield. In addition, FT-IR mapping of ZM-2 was performed to provide information regarding the distribution of polymer brushes on the surface,20 and the results are displayed in Fig. 2b. ZM-2 was solely used for the analysis, chosen due to the higher degree of functionalization. At first sight, different regions, shown as different colours, were observed in the FT-IR mapping image; however, we found that they do not indicate a lack of effective functionalization, but rather indicate the surface roughness of the membranes. This was demonstrated by analysing the FT-IR spectra at each of the different spots, shown also in Fig. 2b, where the peak corresponding to the carboxyl group of PCysMA at 1700 cm−1 was found in all regions. These observations highlight the effectiveness of the grafting technique utilized here, which yields a rather homogeneous grafting density throughout the surface of the membrane, as well as the applicability of high-resolution FT-IR imaging to study polymer grafting. The homogeneity of the modification on the membranes was also corroborated by EDS spectroscopy (Fig. S4 and S5†).
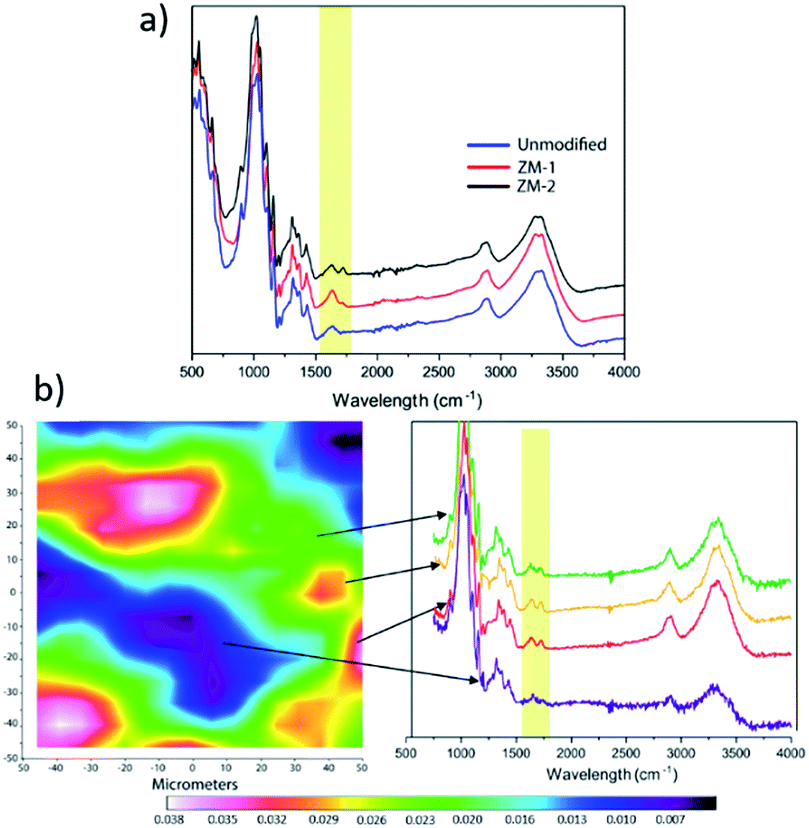 | ||
| Fig. 2 Chemical composition of the membranes: (a) FTIR spectra and (b) FTIR mapping. The mapped area corresponds to a surface of 32 × 32 μm. | ||
Zwitterionic membranes were characterized by XPS to reveal the chemical structure of the polymer grafts. High resolution C 1s and O 1s spectra of both unmodified and ZM-2 membranes were deconvoluted to confirm the proper polymer grafting on the surface. Furthermore, N 1s and S 2p and Si 2p XPS spectra were acquired from ZM-2. The results are shown in Fig. 3 and Table S1.†
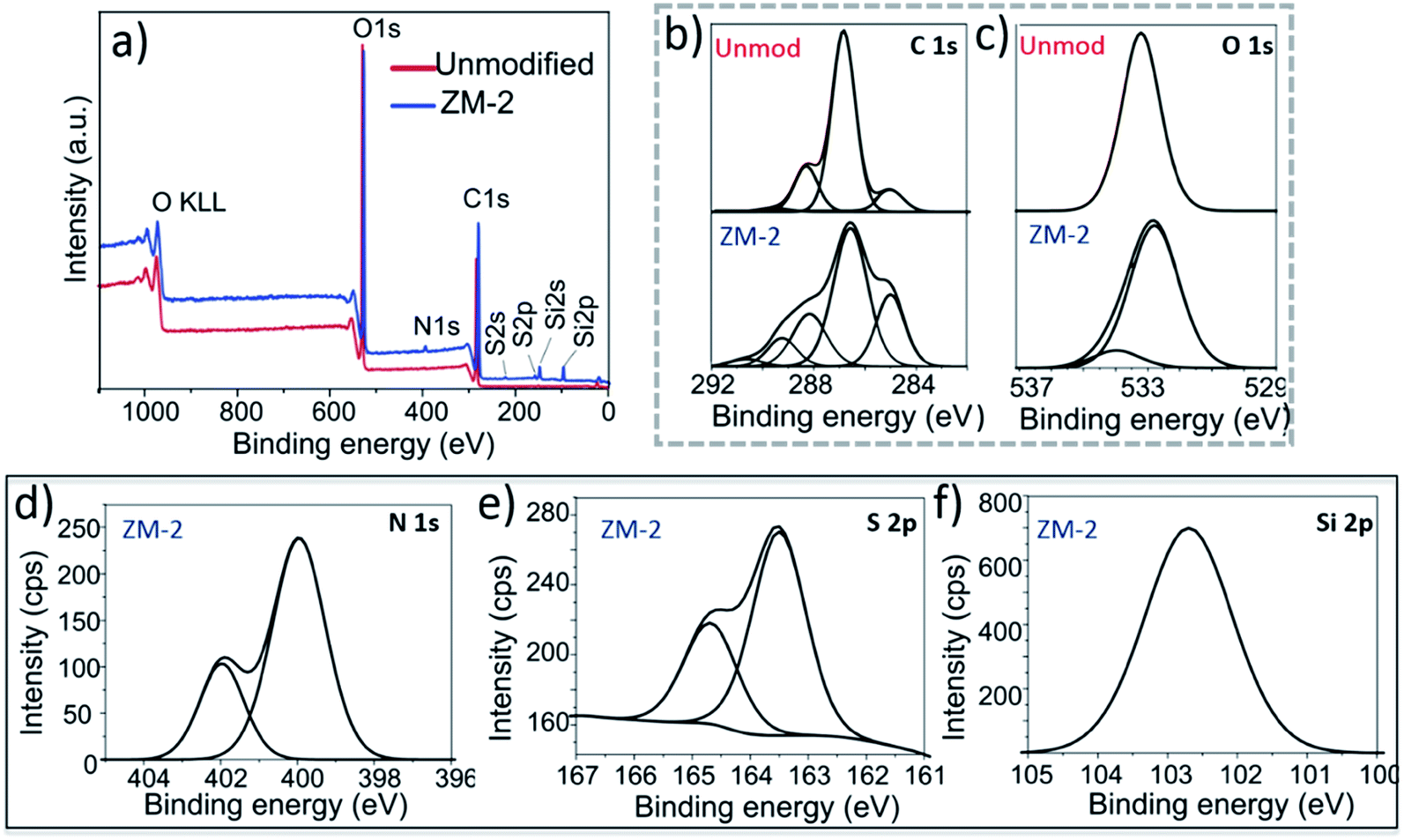 | ||
| Fig. 3 X-ray photoelectron spectra of the ZM-2 and unmodified membranes: (a) XPS survey spectra of the unmodified membrane and ZM-2. (b) C 1s high resolution XPS spectra and (c) O 1s high-resolution XPS spectra of the unmodified and ZM-2 membranes. (d) N 1s spectrum of ZM-2. (e) S 2p spectrum of ZM-2. (f) Si 2p spectrum of ZM-2. | ||
The C 1s high-resolution spectrum (Fig. 3b) of the unmodified membrane suggests three main chemical environments of carbon, as reported before for cellulosic materials: a major peak at 286.8 eV corresponding to C–O bonds, a distinguished peak at 285 eV corresponding to C–C bonds and the third one at 288.3 eV corresponding to O–C–O bonds.18 Furthermore, a fourth small population at 289.6 eV was also observed, corresponding to the carboxyl groups present readily in the cellulose fibres prior to modification. In the case of ZM-2, we fitted the XPS high-resolution C 1s spectra using five Gaussian distributions, introducing another population at 291 eV, corresponding to the methacrylate group (OC–O) of PCysMA. Furthermore, a large increase in the integral area of the peak at 286.4 eV was observed, which is due to the superposition of additional peaks corresponding to C–NH3+ and C–COOH, which are in that region as well.
Two types of O 1s photoemission signals at 532.6 and 534.3 eV, respectively, were observed for ZM-2 (Fig. 3c), in contrast to the unmodified membrane, which had a monomodal distribution at 533.2 eV. This also confirmed the presence of the carboxylate form derived from PCysMA at a binding energy of around 534 eV. Moreover, the N 1s spectrum (Fig. 3d) of ZM-2 was fitted using two components centered at 400.0 and 402.0 eV, which revealed C–NH2 and C–NH3+ species, respectively.15 With this, we estimated that about 1/3 of the surface primary amine groups were protonated at pH 7, at which the sample was prior to drying. On the other hand, the high-resolution S 2p spectrum (Fig. 3e) was deconvoluted into two components, S 2p3/2 at 163.5 and S 2p1/2 at 164.5 eV. The relative intensities of these components were approximately 2:1, as expected. In addition, the BE of the Si 2p (Fig. 3f) signal at around 102 eV is indicative of Si–O species, found in siloxanes, Si (–R) O, where R is a hydrocarbon chain, proving the presence of the immobilized siloxane initiator.15
The pH dependence of the surface charge of the PCysMA-grafted membranes was evaluated and the results are shown in Fig. 4. We observed that at acidic pH the amines on the surface were protonated and ionized yielding positive ζ-potential, while a negatively charged surface was present at basic pH values.
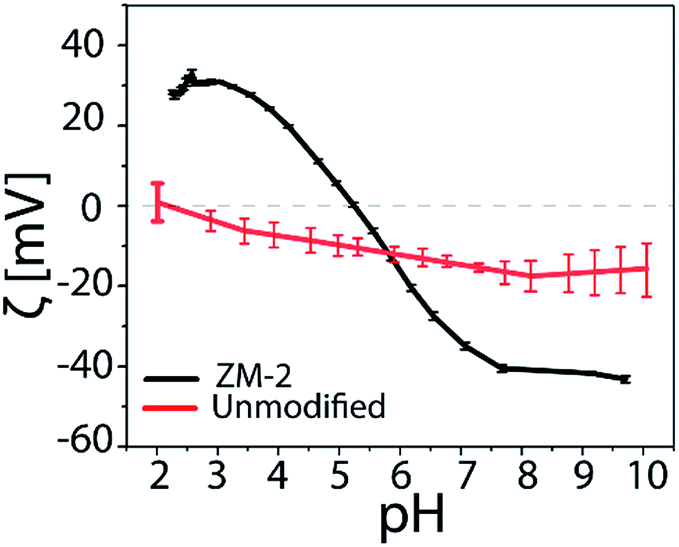 | ||
| Fig. 4 ζ-potential = f(pH) × l of the PCysMA-grafted zwitterionic membranes. | ||
Both types of functional groups were ionized at pH 5.3, where the zeta potential value was zero (isoelectric point, iep), providing the most non-fouling behavior resulting from the zwitterionic character. On the other hand, the unmodified cellulosic membrane exhibited a slight negative charge, characteristic of cellulose surfaces. The isoelectric point of the PCysMA-grafted membranes was very close to the value reported for cysteine (5.07), and it is worth highlighting that the iep at pH 5.3 almost matches the pH of the skin (about 5.5), which could suggest their high potential in areas such as wound dressing applications. Moreover, the topography of the membranes was examined using tapping AFM and the 3D micrographs are shown in Fig. 5. A significant change in the topography of the membranes was observed upon grafting of PCysMA brushes, compared to the unmodified membrane which exhibited a rather smooth surface, indicating that the entire surface of zwitterionic membranes was densely and uniformly covered by polymer brushes. This also demonstrated the potential of AFM to characterize the polymer brushes polymerized from the surface. The 2D micrographs are presented in Fig. S9.†
 | ||
| Fig. 5 3D AFM micrographs of the zwitterionic membranes: (a) unmodified; (b) ZM-1; and (c) ZM-2. | ||
Antifouling performance of the PCysMA-grafted zwitterionic membranes
As protein adsorption is believed to be the first event in many bio-related responses such as biofilm formation and cell adhesion, static protein adsorption is considered as a primary method to evaluate the antifouling properties of the surfaces of materials.21 The primary mechanisms for the resistance to protein adsorption from zwitterionic polymer brushes arise from several factors including solvation effect, conformational entropy and electrical neutrality.21,22 In addition, because of their practical importance, during the past few years, microbiological applications of zwitterionic polymers and their derivatives have been studied in depth, resulting in new mechanistic understanding.23–25Fig. 6j–l show the confocal microscopy images of BSA adsorption on the unmodified and PCysMA-grafted membranes using the Qubit Protein Assay Kit for protein quantification. It can be observed that the amount of proteins adsorbed on the surface was significantly reduced by surface grafting of PCysMA. More specifically, the relative fluorescence decreased from 70.27 in the case of the unmodified membrane to 59.71 and 11.17 for ZM-1 and ZM-2, respectively. These results indicate that the protein adsorption was significantly suppressed upon grafting of PCysMA, especially in the case of ZM-2, where the adsorption decreased by 84%, which also indicates the great influence of the grafting yield on the antifouling performance of the membranes.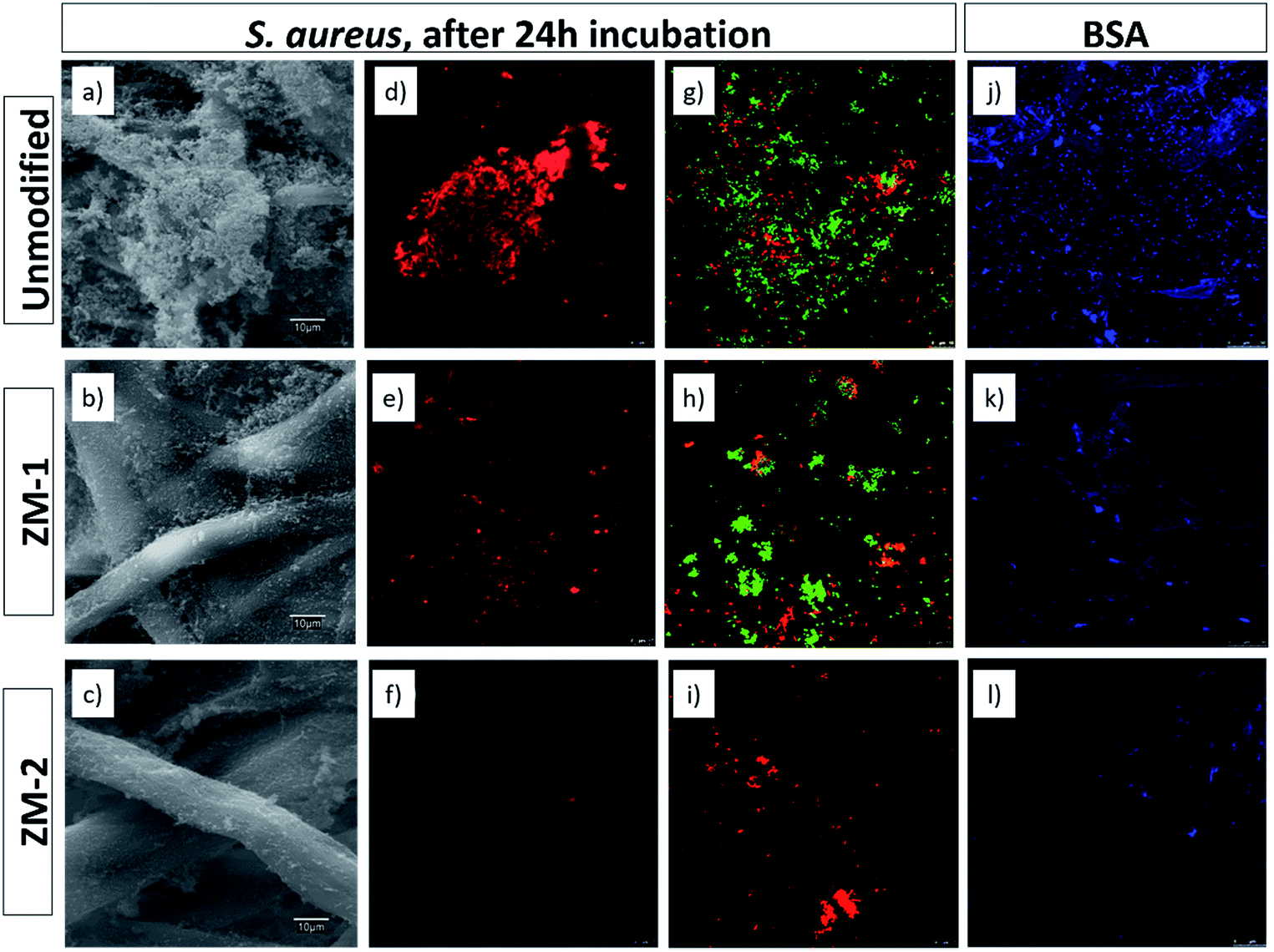 | ||
| Fig. 6 SEM images (a–c) and confocal microscopy micrographs showing the biofilm formation of S. aureus after 24 h of bacterial incubation (d–i), and BSA adsorption (j–l) on the surface of the unmodified and PCysMA-grafted membranes studied with the Qubit Protein Assay Kit. (d–f) correspond to FilmTracer SYPRO Ruby biofilm matrix staining. (g–i) correspond to live/dead double staining cell viability staining. Live cells were green stained by SYTO 9 and dead cells were red stained by PI. | ||
Similarly, we studied the inhibition of biofouling on the surface of the membranes upon grafting of PCysMA. Fig. 6a–i show the SEM and confocal microscopy micrographs of the growth, adhesion and viability of S. aureus on the surface of the membranes. The results from SEM microscopy were in agreement with those obtained with FilmTracer SYPRO Ruby biofilm matrix staining, where in both cases it could be seen that the bacterial adhesion and, consequently, biofilm formation was significantly suppressed upon grafting of PCysMA. FilmTracer SYPRO Ruby preferentially binds proteins, which are the components providing structural stability to biofilms.
This reduction in the formation of a protein matrix in the biofilm reaffirms the antifouling activity of the modified membranes, previously observed for BSA adsorption. Moreover, the reduction in bacterial adhesion was also supported by a reduction in the metabolic activity of the cells, according to the data obtained with the FDA staining. The relative quantification of S. aureus biofilm formation based on its metabolic activity was reduced by 86.92% in ZM-2, compared to the unmodified one (Table S2†). In addition, these membranes also decreased the viability of the cells adhering to the surface (Fig. 6g–i) as noted by the reduction of the number of cells and by the fact that the few remaining ones were clearly PI-marked as non-viable ones (in red), compromising their survival within the biofilm matrix.
This antibacterial activity observed for the PCysMa-grafted membranes could be mainly attributed to the presence of NH3+ groups, which remain at pH 7 at a 1:3 ratio, as confirmed by XPS analysis (Fig. 3d). These protonated groups interact with the negatively charged residues of carbohydrates, lipids and proteins located on the cell surface of bacteria, explaining their role in bacterial impairment. Previous studies support the model in which the interaction was mediated by the electrostatic forces between protonated –NH3+ groups and the negative residues, presumably by competing with Ca2+ for electronegative sites on the membrane surface. This electrostatic interaction results in a two-fold interference: (i) by promoting changes in the properties of the cell membrane wall permeability, inducing internal osmotic imbalances and the inhibition of microbial growth and (ii) by the hydrolysis of the peptidoglycans in the microorganism wall, leading to the leakage of intracellular electrolytes such as potassium ions and other low molecular weight proteinaceous constituents.26–28 On the other hand, several studies have been performed on the antibacterial activity of cysteine, establishing the mechanism by which the thiol groups induce growth inhibition.14,29 According to these studies, the main mechanism against K. pneumoniae and S. aureus is the occurrence of changes in metabolic activity of these bacteria due to a small membrane depolarization that leads to bacterial membrane permeabilization, which could result in bacterial cell death. The results obtained in the present work agree with these observations previously described. In addition, the ZM-2 surface was more active in preventing abiotic fouling and biofouling, which suggests the significant influence of the high grafting density in ZM-2 samples.23–25,30,31
In situ SAXS measurements
To examine the real time evolution of the pore structures, an in situ study of the antifouling performance in both the unmodified and modified (ZM-2) membranes, involving synchrotron SAXS experiments, was performed during the filtration of water and BSA solution.For the measurements, a homemade low vacuum filtering cell (Fig. 7a) was used, where water or the BSA suspension was vacuum filtered (at an approximate permeate flux of 5 Lmh) through a membrane (50 mm diameter), which was hermetically sealed by O-rings while shooting the X-rays. A schematic representation of the experimental setup and the measured data for the modified and unmodified membranes are shown in Fig. 7.
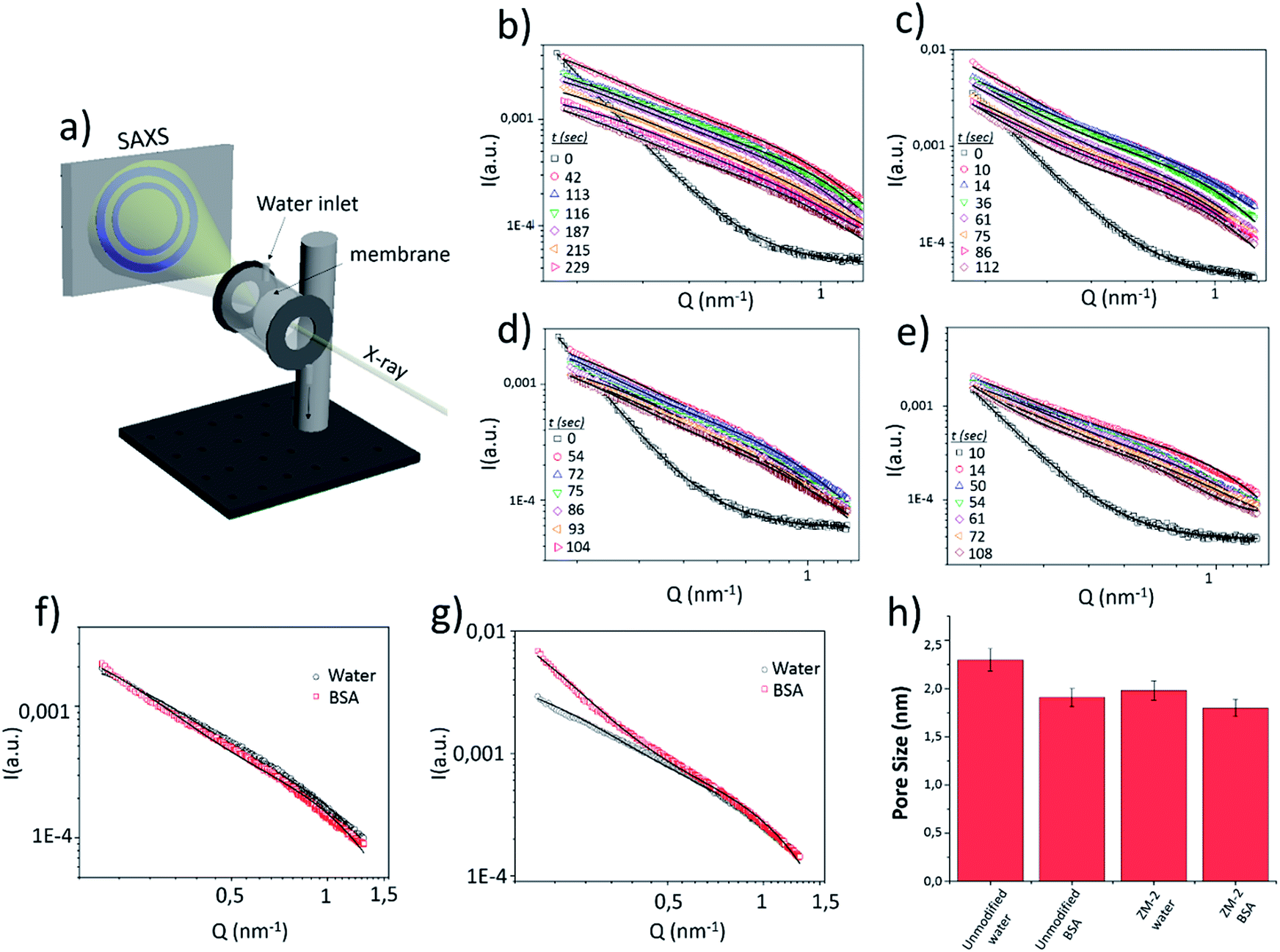 | ||
| Fig. 7 (a) Schematic drawing of the experimental setup for in situ SAXS measurements. SAXS data of the unmodified membrane with (b) water and (c) BSA solution flow and ZM-2 with (d) water and (e) BSA solution flow. Comparison of the SAXS data for water and BSA flow for (f) the modified and (g) unmodified membranes after 60 seconds of filtration. (h) Pore size variation upon filtration of both water and BSA solution. | ||
The SAXS data of the dry membranes (unmodified and ZM-2) were analyzed using the Debye–Anderson–Brumberger (DAB) model, which is usually used for a randomly distributed two-phase system, similar to our highly dense membranes. The system in this model is characterized by a single length scale, the correlation length (ξ), which is a measure of the average spacing between regions of phase 1 and phase 2. This model considers smooth interfaces between the phases and hence the scattering pattern exhibits Porod behaviour [I(Q)αQ−4 ]. This is also consistent with the observed Q−4 dependence in the low Q data of the dry cellulose membranes in both the cases (unmodified and ZM-2) [Fig. 7b–e, time = 0]. The correlation lengths in the dry state were found to be 10 and 12 nm, respectively.
The addition of water leads to the swelling of the cellulose fibers (both unmodified and ZM-2). These swollen fibers form the matrix of the system and the pores embedded in it scatters the X-rays. Any increase in the porosity may increase the number density (volume fraction) of the pores and/or the pore size distribution in the system where the former only scales up the intensity while the latter modifies the scattering profile (as described in eqn (1)). On addition of water to the dry membranes, we observe an increase in the scattering intensity (mainly in the intermediate Q region) as well as changes in the scattering pattern (Fig. 7b–e). This is attributed to the evolution of the pore distribution in the matrix of the swollen cellulose fibers. However, further incorporation of water only reduces the scattering contrast in the membrane, leading to a systematic decrease in the scattering intensity without any significant modifications in the Q dependence of the data (Fig. 7b–e). The scattering contrast is expected to decrease as more and more pores are filled with water, while the fact that there is almost no change in the scattering profile suggests that the pore size remains almost constant. In the presence of water, the SAXS data were analyzed by summing the two scattering contributions from the correlated structure (DAB model) and that from the pores embedded in the cellulose matrix. The contribution from the correlated structure decreases systematically as more and more water penetrates the membrane. The pores were considered spherical (fitted using a form factor as given in eqn (2)) in shape with a size distribution modelled by a log-normal distribution.
The variations in the mean pore radius for both membranes (unmodified and ZM-2) under the flow of water and BSA solution are plotted in a histogram [Fig. 7h]. The pore radius was found to be slightly smaller in the case of ZM-2, which is also in good agreement with the results of BET measurements (see Table 1). This can be interpreted in a way that the grafted polymer brushes, whose scattering contrast is comparable to that of cellulose, overshadow some of the pores. Moreover, the pore radius in both membranes decreased upon addition of the BSA solution with respect to that obtained for the water flow; however, the reduction in pore size was much smaller in the case of ZM-2, compared to the unmodified membrane. The decrease of the pore radius upon addition of BSA is obviously a consequence of larger pores being filled with adsorbed BSA and hence the mean shifts towards smaller values in both the cases, while the smaller reduction in the case of the modified membrane can be attributed to decreased BSA adsorption.
The SAXS data of both membranes with the flow of water and BSA solution were also compared at a specific time (after 60 seconds of filtration) and the results are displayed in Fig. 7f and g. The scattering profiles [Fig. 7g] of the unmodified membrane with water and BSA solution were remarkably different, particularly in the low and intermediate Q range. On the other hand, for ZM-2, the scattering data were similar for both cases (water and BSA solution). This clearly suggests that the BSA adsorption was significantly suppressed upon grafting of PCysMA.
Conclusions
In this work, novel cellulose-based membranes with antifouling and antimicrobial properties were prepared via surface-initiated free radical polymerization of cysteine methacrylate. Successful and homogeneous modification was demonstrated by FT-IR mapping and EDS spectroscopy, while XPS revealed the chemical structure of the polymer grafts. Furthermore, streaming zeta potential measurements demonstrated the zwitterionic behaviour of the PCysMA-g-cellulose membrane and an isoelectric point at pH 5.3. Static adsorption tests with bovine serum albumin (BSA), biofilm formation and S. aureus viability helped us to elucidate the antifouling performance of the modified membranes, together with enhanced antimicrobial activity. Moreover, a novel method to examine the evolution of the pore structures in real time using synchrotron-based SAXS for both unmodified and modified membranes and to assess the antifouling performance in situ was successfully introduced.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the MULTIMAT project (H2020-MSCA-ITN-2014, Grant No. 676045) for research funding and Dr Shun Yu for valuable scientific discussions. LV and AM thank Eva-Malmstrom and team, KTH for support with FTIR mapping. LV, SK and AM thank DESY, Hamburg, Germany for the beam time I-20170358 EC and beamline scientist Wiebke Ohm for the support.Notes and references
- T. Mohammadi, S. S. Madaeni and M. K. Moghadam, Desalination, 2003, 153, 155–160 CrossRef.
- J. Nilsson and B. Hallström, J. Membr. Sci., 1992, 67, 177–189 CrossRef.
- C. Jönsson and A.-S. Jönsson, J. Membr. Sci., 1995, 108, 79–87 CrossRef.
- J. J. Keating, J. Imbrogno and G. Belfort, ACS Appl. Mater. Interfaces, 2016, 8, 28383–28399 CrossRef PubMed.
- K. H. A. Lau, C. Ren, T. Sileika, S. H. Park, I. Szleifera and P. B. Messersmith, Langmuir, 2012, 28, 16099–16107 CrossRef PubMed.
- W. Zhao, Q. Ye, H. Hu, X. Wang and F. Zhou, J. Mater. Chem. B, 2014, 2, 5352–5357 RSC.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef PubMed.
- H. J. Kwon, Y. Lee, L. T. Phuong, G. M. Seon, E. Kim, J. C. Park, H. Yoon and K. D. Park, Acta Biomater., 2017, 61, 169–179 CrossRef PubMed.
- P. Bengani-Lutz, E. Converse, P. Cebe and A. Asatekin, ACS Appl. Mater. Interfaces, 2017, 9, 20859–20872 CrossRef PubMed.
- Y. Chang, W. Chang, Y. Shih, T. Wei and G. Hsiue, ACS Appl. Mater. Interfaces, 2011, 3, 1228–1237 CrossRef PubMed.
- H. S. Sundaram, X. Han, A. K. Nowinski, J. R. Ella-Menye, C. Wimbish, P. Marek, K. Senecal and S. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 6664–6671 CrossRef PubMed.
- X. Shen, Y. Gao, Y. He, Y. Zhao and L. Chen, Sep. Sci. Technol., 2016, 51, 1189–1198 CrossRef.
- H. Akbulut, S. Yamada and T. Endo, RSC Adv., 2016, 6, 108689–108696 RSC.
- E. Caldeira, E. Piskin, L. Granadeiro, F. Silva and I. C. Gouveia, J. Biotechnol., 2013, 168, 426–435 CrossRef PubMed.
- A. M. Alswieleh, N. Cheng, I. Canton, B. Ustbas, X. Xue, V. Ladmiral, S. Xia, R. E. Ducker, O. El Zubir, M. L. Cartron, C. N. Hunter, G. J. Leggett and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 9404–9413 CrossRef PubMed.
- D. I. Svergun and M. H. J. Koch, Rep. Prog. Phys., 2003, 66, 1735–1782 CrossRef.
- B. Chu and B. S. Hsiao, Chem. Rev., 2001, 101, 1727–1762 CrossRef PubMed.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef.
- P. Debye, H. R. Anderson and H. Brumberger, J. Appl. Phys., 1957, 28, 679–683 CrossRef.
- S. Hansson, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmström, Polym. Chem., 2012, 3, 307–309 RSC.
- P. Wang, J. Meng, M. Xu, T. Yuan, N. Yang, T. Sun, Y. Zhang, X. Feng and B. Cheng, J. Membr. Sci., 2015, 492, 547–558 CrossRef.
- B. Te Li, Y. X. Wu, H. Cheng and W. H. Liu, Polymer, 2012, 53, 3726–3734 CrossRef.
- L. Mi and S. Jiang, Angew. Chem., Int. Ed., 2014, 53, 1746–1754 CrossRef PubMed.
- S. Chen and S. Jiang, Adv. Mater., 2008, 98195, 335–338 CrossRef.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef PubMed.
- B. Jalvo, A. P. Mathew and R. Rosal, J. Membr. Sci., 2017, 544, 261–271 CrossRef.
- Y. Andres, L. Giraud, C. Gerente and P. Le Cloirec, Environ. Technol., 2007, 28, 1357–1363 CrossRef PubMed.
- L. A. Goetz, B. Jalvo, R. Rosal and A. P. Mathew, J. Membr. Sci., 2016, 510, 238–248 CrossRef.
- I. C. Gouveia, D. Sa and M. Henriques, J. Appl. Polym. Sci., 2011, 124, 1352–1358 CrossRef.
- Z. Zhu, J. Jiang, X. Wang, X. Huo, Y. Xu, Q. Li and L. Wang, Chem. Eng. J., 2017, 314, 266–276 CrossRef.
- G. Cheng, G. Li, H. Xue, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2009, 30, 5234–5240 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details about the extraction of nanocellulose, synthesis of cysteine methacrylate and manufacturing of the membranes; EDS spectroscopy, SEM and AFM micrographs of the membranes; and relative quantification of S. aureus biofilm formation and BSA adsorption on the membranes. See DOI: 10.1039/c8ta06095a |
| This journal is © The Royal Society of Chemistry 2018 |