
Thick and freestanding MXene/PANI pseudocapacitive electrodes with ultrahigh specific capacitance†

Abstract
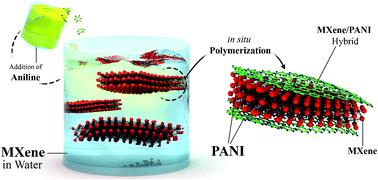
Two-dimensional (2D) titanium carbide MXene (Ti3C2Tx) has shown great promise as a high-performance electrode material for electrochemical capacitors (ECs). However, similar to other 2D materials, processing MXenes into freestanding films results in their restacking, thus decreasing the ion transport inside the electrodes. This problem significantly hinders the specific capacitance and rate capability of freestanding electrodes, particularly for those with thicknesses higher than a few microns. Here, we demonstrate a strategy based on surface modification of MXene sheets to fabricate electrodes with highly accessible structure and improved electrochemical performance even at very high electrode thicknesses. 2D Ti3C2Tx and polyaniline (PANI) hybrid materials were synthesized through oxidant-free in situ polymerization of PANI on the surface of MXene sheets and were assembled into freestanding films with various thicknesses. Thin MXene/PANI hybrid electrodes delivered outstanding gravimetric and volumetric capacitances as high as 503 F g−1 and 1682 F cm−3, respectively. As the electrode thicknesses and mass loadings were increased, the hybrid electrodes still showed high electrochemical performance. For example, an electrode with a thickness of 90 μm and a mass loading of 23.82 mg cm−2 could deliver a specific capacitance of about 336 F g−1 (∼888 F cm−3 volumetric capacitance). The hybrid electrodes also showed a high cycle lifetime with a capacitance retention of 98.3% after 10 000 cycles. This paper explains a simple and fast approach for the fabrication of MXenes/conducting polymer hybrid electrodes with superior electrochemical performance.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

