
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing conjugated microporous polymers for visible light-promoted photocatalytic carbon–carbon double bond cleavage in aqueous medium†
Cyrine
Ayed
ab,
Lucas
Caire da Silva
a,
Di
Wang
a and
Kai A. I.
Zhang
 *a
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
bGraduate School for Excellence Materials Science in Mainz, Johannes Gutenberg University Mainz, Staudingerweg 9, 55128 Mainz, Germany
First published on 8th August 2018
Abstract
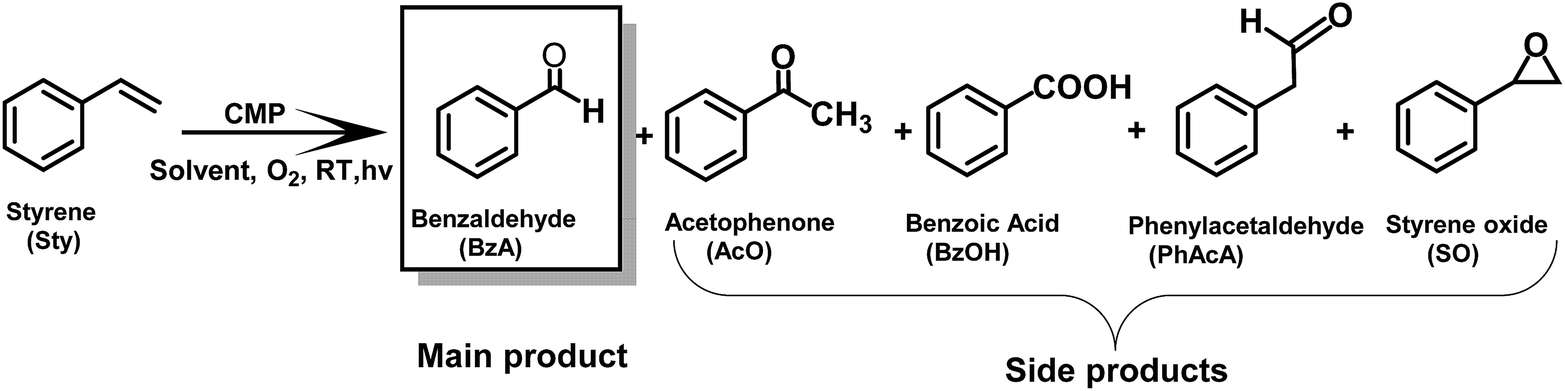
The production of valuable carbonyl compounds directly from alkenes via carbon–carbon double (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) bond cleavage is a highly important reaction. As a promising alternative to traditional metal-catalyzed thermal reaction conditions, photocatalysis offers an environmentally friendly and sustainable synthetic strategy. However, most photocatalysts used so far were homogeneous, molecular and transition metal-based catalytic systems. Here, we report a structural design strategy of conjugated microporous polymers (CMPs) as a class of metal-free, heterogeneous, stable and reusable photocatalysts for visible light-promoted CC bond cleavage reaction. Via different electron donor and acceptor combinations in the CMP backbone structure, we systematically investigated the structural influence of the CMPs on their optical and electrochemical properties, thereby revealing the structure and performance relationship in the photocatalytic CC bond cleavage reaction. As a result, the CMP with a specific electron donor–acceptor combination containing benzothiazole as a strong electron withdrawing unit and phenyl as a weak electron donating unit was the most efficient photocatalyst. The CC bond in various alkenes, mostly styrene derivatives, could be successfully cleaved, forming aldehydes as a product with high conversion and moderate to high selectivity. The green aspect of the work is demonstrated by the utilization of visible-light, molecular oxygen as a cheap oxidant and water-based reaction medium.
C) bond cleavage is a highly important reaction. As a promising alternative to traditional metal-catalyzed thermal reaction conditions, photocatalysis offers an environmentally friendly and sustainable synthetic strategy. However, most photocatalysts used so far were homogeneous, molecular and transition metal-based catalytic systems. Here, we report a structural design strategy of conjugated microporous polymers (CMPs) as a class of metal-free, heterogeneous, stable and reusable photocatalysts for visible light-promoted CC bond cleavage reaction. Via different electron donor and acceptor combinations in the CMP backbone structure, we systematically investigated the structural influence of the CMPs on their optical and electrochemical properties, thereby revealing the structure and performance relationship in the photocatalytic CC bond cleavage reaction. As a result, the CMP with a specific electron donor–acceptor combination containing benzothiazole as a strong electron withdrawing unit and phenyl as a weak electron donating unit was the most efficient photocatalyst. The CC bond in various alkenes, mostly styrene derivatives, could be successfully cleaved, forming aldehydes as a product with high conversion and moderate to high selectivity. The green aspect of the work is demonstrated by the utilization of visible-light, molecular oxygen as a cheap oxidant and water-based reaction medium.
Introduction
The synthesis of valuable carbonyl compounds such as aldehydes, acetic acid, acetaldehyde and ketone derivatives via carbon–carbon double (CC) bond cleavage of alkenes is a highly important reaction in the chemical and pharmaceutical industries. These products have wide applications as important molecular building blocks and intermediates for the manufacture of perfumes, dyestuffs, agrochemicals and pharmaceuticals.1–5 Various studies have reported the thermal catalytic oxidation of alkenes, notably styrene, for the desired carbonyl products. Important examples of metal catalysts are iron-based systems such as iron(II) chloride,6 Fe(NO3)·9H2O,7etc.8 Alternatively, Feng et al. have employed a water-soluble palladium(II) complex for the selective aerobic oxidation of styrene in water, obtaining benzaldehyde with high selectivity.9 Zhang et al. have also observed the CC bond cleavage and the formation of acetophenone with moderate selectivity over ZrO2-supported palladium nanoparticles.10 Recently, Liu et al. have reported the use of gold nanoparticle mobilized carbon as a catalyst for the selective oxidation of styrene.2 The researchers pointed out that the strong Au–carbon interaction could enhance the catalytic efficiency.
In comparison to traditional thermal reaction conditions, photocatalysis offers an environmentally friendly and sustainable synthetic strategy. Nevertheless, there are only few photochemical reports on the photo-oxidative cleavage of the CC bonds of styrene. For instance, Guo et al. have reported the use of Au–Ti-decorated mesoporous silica for the photo-epoxidation reaction of styrene with moderate selectivity.11 More recently, Shen et al. have developed a chiral manganese catalyst to trigger the photocatalytic enantioselective epoxidation of terminal olefins, using water as an oxygen source, yielding epoxides with relatively high enantioselectivities.12 So far, the photocatalytic systems reported were mainly based on transition metals. Very few molecular organic photocatalysts have been developed, such as eosin Y, which was used as an organophotoredox to mediate the oxidative cleavage of the CC bond.13 However, despite the progress made so far in this field, considerable drawbacks are still associated with those metal based or organic molecular systems. For example, the toxicity of heavy metals, the instability and the lack of reusability of the molecular photocatalysts lead to the need for the development of a more stable, reusable, metal-free and heterogeneous photocatalytic system.
Conjugated microporous polymers (CMPs), combining a visible light-active π-backbone and microporous properties, have recently emerged as a new class of efficient, stable and heterogeneous photocatalysts for chemical transformations.14–16 Recent research activities demonstrated their wide application range in various photocatalytic reactions such as molecular oxygen activation,14,17,18 selective oxidation of organic sulfides,19 C–C or C–N bond formation,20–22 dehalogenation,23,24 oxidative hydroxylation of arylboronic acids,25 visible light-initiated free radical and cationic polymerization,26,27 cycloaddition,28 oxidative homocoupling of amines,29 C-3 functionalization of indoles,30 and hydrogen evolution.31–34 So far, the photocatalytic oxidative CC bond cleavage using conjugated microporous polymers has not been reported.
Herein, we report the design of CMPs bearing different electron donor and acceptor-building blocks as heterogeneous photocatalysts for the CC bond cleavage of alkenes, mostly styrene derivatives. We investigated the structural influence of the CMPs on their photocatalytic efficiency. It was shown that the photocatalytic reaction could be conducted with almost quantitative conversion and selectivity using the CMP containing benzothiazole as a strong electron withdrawing unit, together with a phenyl group as an electron donating unit in the polymer backbone. Additionally, the use of aqueous reaction medium demonstrated its green and sustainable reaction nature. The mechanistic insight of the photoredox reaction was also investigated.
Results and discussion
In this work, for the structural design of the CMPs, we choose different building blocks according to their electron affinity. As displayed in Scheme 1, thiophene (Th) as a strong electron donor and benzothiodiazole (BT) as a strong electron acceptor were polymerized via the palladium-catalyzed Sonogashira cross-coupling reaction35 with triethynylbenzene (B) in the corresponding CMP backbone structures, referred to as BTh and BBT, respectively. A mixed CMP was also synthesized containing 50% BT unit and 50% Th unit in the same manner, referred to as BThBT. The CMPs were obtained as amorphous solids as shown by the X-ray diffraction patterns (Fig. S1†). The detailed synthetic pathway with detailed characterization methods of the polymers are described in the Experimental section.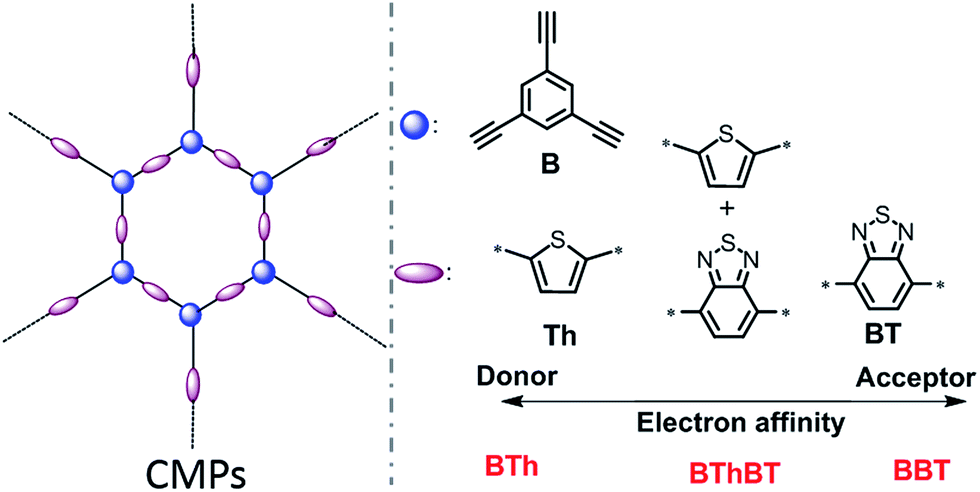 | ||
| Scheme 1 Structural design of a series of conjugated microporous polymers (CMPs) via combination of different electron donor and acceptor building blocks. | ||
As displayed in Fig. 1, the scanning electron microscopy (SEM) images of the CMPs showed a variety of shapes. BTh was obtained as irregularly shaped particles with a size ranging between 40 and 180 nm, whereas, BBT showed a fibrous structure with a diameter of ca. 200 nm. BThBT appeared as fused large particles and fibers.
 | ||
| Fig. 1 SEM images of (a) BTh, (b) BThBT and (c) BBT. | ||
The Brunauer–Emmett–Teller (BET) surface areas of BTh, BThBT and BBT were measured to be 806 m2 g−1, 445 m2 g−1 and 129 m2 g−1, with pore sizes of 1.7 nm, 1.41 nm and 1.7 nm, respectively. The nitrogen gas sorption isotherms and pore size distributions are displayed in Fig. S2.† Their physical properties are listed in Table 1.
| CMP | S BET (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Band gap (eV) |
|---|---|---|---|---|
| BTh | 806 | 0.620 | 1.7 | 2.36 |
| BThBT | 445 | 0.314 | 1.41 | 2.00 |
| BBT | 129 | 0.631 | 1.7 | 2.12 |
The solid state 13C CP/MAS NMR spectra of all CMPs are displayed in Fig. 2. The typical chemical shifts between 110 and 150 ppm shown for all CMPs can be assigned to the aromatic rings in the polymer backbone. The signals from 80 to 100 ppm can be assigned to the carbon of the triple bond. A resonance peak at 155 ppm could be observed, which can be assigned to the adjacent carbon next to nitrogen in the BT unit. The thiophene (Th) unit can be characterized by the signal at about 124 ppm, which is assigned to the C–S single bond.36–38
 | ||
| Fig. 2 Solid state 13C CP/MAS NMR spectra of the CMPs. | ||
The Fourier transform infrared (FTIR) spectra (Fig. 3a) showed a typical signal at 2300 cm−1 which could be assigned to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretching mode.39 The signals at 1570 cm−1 and 1480 cm−1 are characteristic of the N–S– and the –CN– stretching modes in the benzothiadiazole moiety.35 The vibrational peak at 790 cm−1 is attributed to the thiophene unit in the BTh network.37 The BThBT spectra contain signals corresponding to both the BT and Th units.40 Thermogravimetric analysis (TGA) indicated that BBT was thermally the most stable up to 300 °C, whereas BTh and BThBT showed lower initial decomposition temperatures with BTh exhibiting the lowest weight loss among all of the CMPs (Fig. 3b).
C– stretching mode.39 The signals at 1570 cm−1 and 1480 cm−1 are characteristic of the N–S– and the –CN– stretching modes in the benzothiadiazole moiety.35 The vibrational peak at 790 cm−1 is attributed to the thiophene unit in the BTh network.37 The BThBT spectra contain signals corresponding to both the BT and Th units.40 Thermogravimetric analysis (TGA) indicated that BBT was thermally the most stable up to 300 °C, whereas BTh and BThBT showed lower initial decomposition temperatures with BTh exhibiting the lowest weight loss among all of the CMPs (Fig. 3b).
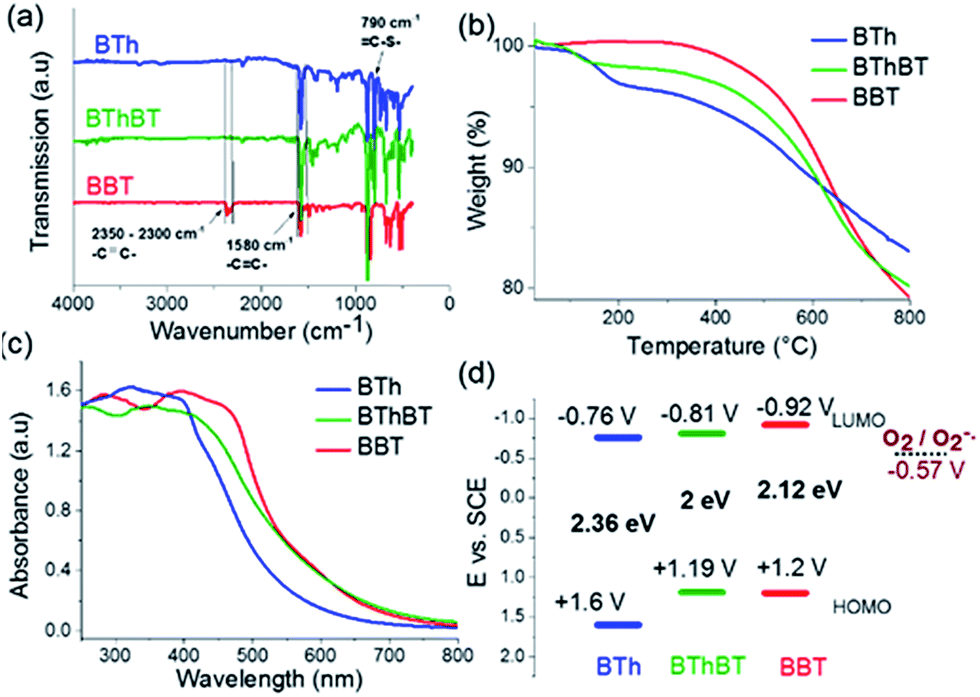 | ||
| Fig. 3 (a) FTIR and (b) TGA spectra of the materials under a N2 atmosphere with a heating rate of 10 °C min−1; (c) UV/vis DR spectra and (d) energy band structures of the as synthesized CMPs and the O2/O2˙− couple. | ||
Fig. 3c illustrates the UV/vis diffuse reflectance (DR) spectra of the CMPs. All three polymers showed a similar absorption range with maximal absorption between 300 and 450 nm and gradual levelling off until ca. 800 nm. Additionally, a clear extension effect of the absorption range of the BT unit was revealed. The strong donor-based CMP, i.e.BTh, absorbs mainly in the blue light range. In comparison, the spectra of BThBT as well as those of BBT exhibited a gradual bathochromic shift up to ca. 60 nm.
To gain further insight into the electronic properties, cyclic voltammetry (CV) measurements were conducted to reveal the energy band structure of the designed materials (Fig. S3†). The electronic band positions of the CMPs are displayed in Fig. 3d. BBT exhibited the highest LUMO level at −0.92 V vs. SCE among the CMPs, indicating its strong ability to activate molecular oxygen to its active states such as superoxide oxygen and singlet oxygen. The reduction potential of the O2/O2˙− lies at −0.57 V vs. SCE.41,42 The results strongly suggest the possible superior photocatalytic activity of BBT for oxidative reactions.
To investigate the photocatalytic performance of the designed CMPs, we tested the oxidative cleavage of the CC bond using styrene as a model substrate under visible light irradiation. The experiments were first carried out at room temperature using BBT as the photocatalyst due to its highest reduction potential and molecular oxygen as an oxidant in various solvents. The results are listed in Table 2. The conversion of styrene catalyzed by BBT was found to be strongly dependent on the solvent polarity. Benzaldehyde was mainly observed in various solvents, with relatively high selectivity (entries 1 to 8, Fig. S4†). For instance, in an apolar solvent such as hexane (entry 1 in Table 2), a high selectivity of around 81% to benzaldehyde with a total styrene conversion of 7% was observed after 18 h. Increasing the polarity of the solvent (ethanol, THF, nitromethane and acetonitrile) led to an increase of the conversion of styrene up to 44% with benzaldehyde yields ranging from 61% to 92% (entries 2, 3, 4 and 5 in Table 2).
C bond cleavage using various CMPs with various reaction conditions
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Photocatalyst | Solvent | O2 | Light | Additives | Conversionb (%) | Selectivity of BzAb (%) |
| a Standard reaction conditions: 0.1 mmol of styrene, 10 mg of CMP in 1.5 mL of solvent under the irradiation of a blue LED lamp (460 nm, 0.16 W cm−2), 1 atm O2 room temperature, 18 h. b Conversion determined by GC-MS. c BBT, no light, 1 atm O2, MeCN/H2O: v/v = 1/24. d With BBT, under light, no O2, MeCN/H2O: v/v = 1/24. e Without catalyst, under light, 1 atm O2, MeCN/H2O v/v = 1/24. f Without catalyst, under light, 1 atm O2, acetonitrile. g KI as a hole scavenger. h NaN3 as a singlet oxygen scavenger. i Isopropanol as a hydroxyl radical scavenger. j Benzoquinone (BQ) as a superoxide scavenger. | |||||||
| 1 | BBT | Hexane | + | + | — | 7 | 81 |
| 2 | BBT | Ethanol | + | + | — | 15 | 85 |
| 3 | BBT | THF | + | + | — | 23 | 92 |
| 4 | BBT | MeNO2 | + | + | — | 43 | 62 |
| 5 | BBT | MeCN | + | + | — | 44 | 60 |
| 6 | BBT | MeCN/H2O (v/v = 24/1) | + | + | — | 59 | 57 |
| 7 | BBT | MeCN/H2O (v/v = 1/1) | + | + | — | 77 | 51 |
| 8 | BBT | MeCN/H2O (v/v = 1/24) | + | + | — | 91 | 85 |
| 9 | BTh | MeCN/H2O (v/v = 1/24) | + | + | — | 16 | 69 |
| 10 | BThBT | MeCN/H2O (v/v = 1/24) | + | + | — | 58 | 43 |
| 11c | BBT | MeCN/H2O (v/v = 1/24) | + | − | — | n.d | n.d |
| 12d | BBT | MeCN/H2O (v/v = 1/24) | − | + | — | n.d | n.d |
| 13e | — | MeCN/H2O (v/v = 1/24) | + | + | — | 48 | 79 |
| 14f | — | MeCN | + | + | — | 30 | 85 |
| 15g | BBT | MeCN/H2O (v/v = 1/24) | + | + | KI as a hole scavenger | 2 | 100 |
| 16h | BBT | MeCN/H2O (v/v = 1/24) | + | + | NaN3 as a singlet oxygen scavenger | 17 | 69 |
| 17i | BBT | MeCN/H2O (v/v = 1/24) | + | + | Isopropanol as a hydroxyl radical scavenger | 88 | 45 |
| 18j | BBT | MeCN/H2O (v/v = 1/24) | + | + | Benzoquinone as a superoxide scavenger | 1 | 100 |
When further increasing the solvent polarity by mixing water with acetonitrile, it was found that the photooxidation of styrene was noticeably enhanced upon addition of H2O to acetonitrile under blue light irradiation (entries 6,7 and 8 in Table 2). For example, the styrene conversion was increased by more than 2-fold from 44% to 91%, with a high benzaldehyde yield of about 86%, when an optimized amount of water to acetonitrile (v/v = 1/24) was added (entry 8 in Table 2). These results shed light on the solvent-polarity-dependent-conversion on one hand and the effect of water on the acceleration of the photooxygenation of styrene by O2 on the other hand.43
The involvement of water in this reaction was further confirmed by the detection of 1-phenyl-1,2-ethanediol by GC-MS during the reaction, which is possibly due to the attack of water on the intermediate in the reaction process (Fig. S9†).
Taking into consideration the optimized reaction conditions above, the photocatalytic performance of the other designed CMPs was investigated under the same conditions using water-based-biphasic systems (MeCN/H2O: v/v = 1/24). As listed in Table 2 (Fig. S5†), the reaction catalyzed by the BThBT reached a lower conversion of about 58% with only 43% yield to benzaldehyde, whereas BTh was not efficient for this reaction exhibiting only 16% of conversion with formation of benzaldehyde as the main product (entries 9 and 10). The higher photocatalytic efficiencies of the BT-containing CMPs, BBT and BThBT could be explained by a more efficient photogenerated charge separation within the polymer networks due to the donor–acceptor combinations. In comparison, BTh only contains electron donor units, which inhibit an efficient charge separation and transfer inside the CMP. Indeed, Fig. 4a reveals the most intense photocurrent signal of BBT among the three CMPs under visible light, indicating that the photogenerated charge separation in BBT is more efficient than that in BThBT and BTh.
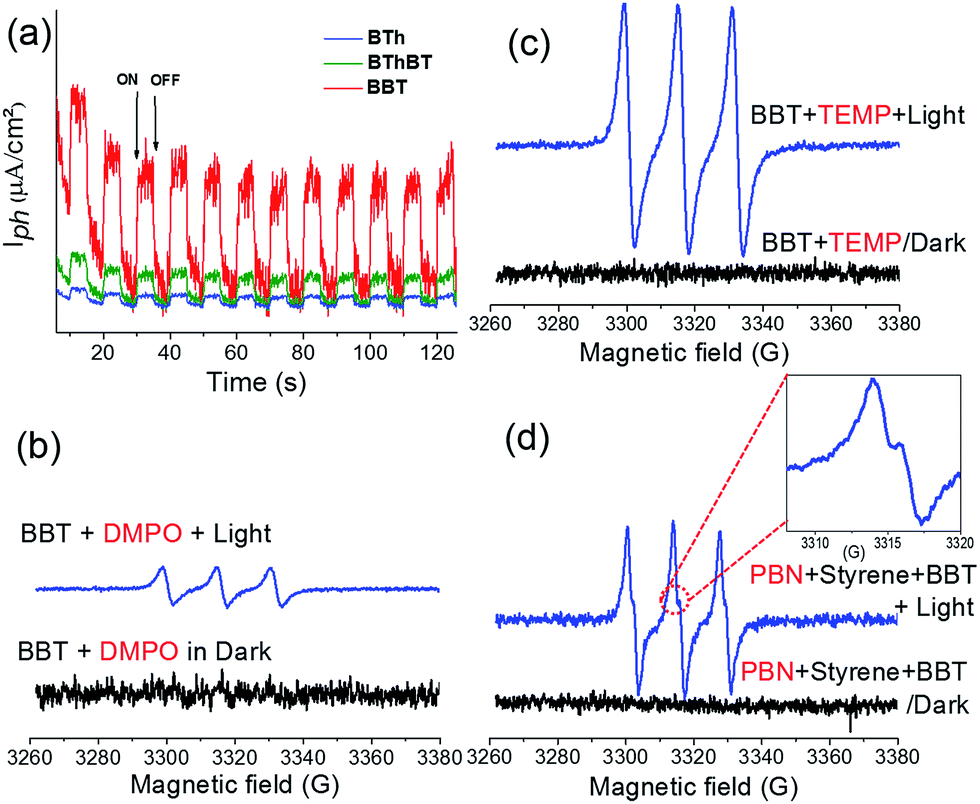 | ||
| Fig. 4 (a) Photocurrent responses of the CMPs under the illumination of visible light with a wavelength ≥ 420 nm with a light ON/OFF model. EPR spectra of (b) DMPO-O2− (g = 2.00639) and (c) TEMP-1O2 adducts (g = 2.00645) using BBT as the photocatalyst in the dark and under light irradiation (λ = 460 nm, 0.065 W cm−2). (d) EPR spectra using PBN as a radical trapping agent for the radical intermediate of styrene, with BBT, under O2 and light irradiation (g = 2.00667). | ||
To further investigate the reaction mechanism and reveal the specific role of the photogenerated electron–hole pairs during the photocatalytic process, we conducted a number of control experiments, using BBT as the photocatalyst. The results are also listed in Table 2 (Fig. S6†). In the absence of light and molecular oxygen, no reaction conversion was determined (entries 11 and 12 in Table 2). Without using a photocatalyst, only a conversion of 30–42% was determined (entries 13 and 14 in Table 2). The results indicate that all three components are indispensable. The effect of specific scavengers was also studied.
A conversion of 2% was obtained in the presence of KI as a hole scavenger (entry 15 in Table 2). A conversion of 17% was reached in the presence of NaN3 as a singlet oxygen scavenger (entry 16 in Table 2). Use of isopropanol as a hydroxyl scavenger led to a conversion of 88% (entry 17 in Table 2). Significantly, a conversion of only 1% was determined in the presence of benzoquinone as a superoxide scavenger (entry 18 in Table 2). These results indicate that the activated oxygen species (superoxide and singlet oxygen) and photogenerated hole all participate during the oxidative CC bond cleavage reaction. To precisely analyze the possible active oxygen species (superoxide and singlet oxygen) and their exact contribution during the photocatalytic reaction, we further conducted electron spin resonance (EPR) experiments using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) as superoxide and singlet oxygen trapping agents, respectively. As shown in Fig. 4b and c, both active oxygen species could be determined, confirming their active roles in the reaction.
Based on the observations drawn from the experiments described above, we suggest a modified reaction mechanism for the oxidative CC bond cleavage and aldehyde formation inspired from the literature13,43 (Fig. 5). Under visible light irradiation, the charge separation occurs within the CMP based photocatalyst. The photogenerated hole of BBT oxidizes styrene 1 into its cationic radical intermediate 1a. Further proof of the formation of the radical intermediate could be observed using N-tert-butyl-α-phenylnitrone (PNB) as a radical trapping agent (Fig. 4d). A typical pattern for the PBN trapped radical with g value equal to 2.00667 has been recorded.
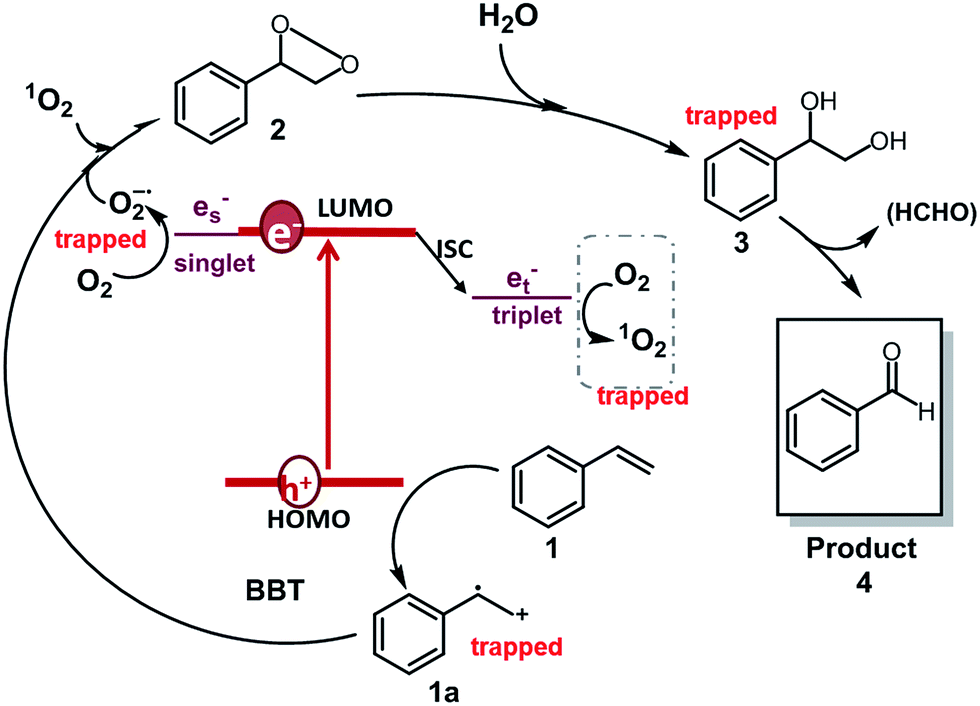 | ||
| Fig. 5 Suggested reaction mechanism for the aerobic CC double bond cleavage of styrene using BBT as the photocatalyst. | ||
In the same time, the electron from the LUMO of the CMP assisted the reductive activation of molecular oxygen into superoxide (O2−), which undergoes [2 + 2] cycloaddition with the cationic radical of styrene to generate the 3-phenyl-1,2-dioxetane 2. Note that, singlet oxygen (1O2) also took part in the oxidation step. 1-Phenyl-1,2-ethanediol 3 is then formed due to the attack of the water on the dioxetane intermediate, which ultimately is cleaved into benzaldehyde 4, releasing formaldehyde as the possible side product. High-performance liquid chromatography (HPLC) did not give a confirmation of the existence of formaldehyde. Nevertheless, the 1H NMR spectra showed traces of possible formaldehyde formation (Fig. S7†). The results indicate that the formed formaldehyde as the side product could be directly oxidized. Additionally, the overoxidation of the aldehyde may result in the formation of carboxylic acids, depending on the nature of the substrate (see the scope of substrates below).
To further demonstrate the general applicability of BBT as a photocatalyst, screening experiments with various styrene derivatives have been carried out under the same conditions and the results are illustrated in Fig. 6. High conversion was obtained in most cases under the same conditions. However, both electron-withdrawing substitution groups on the phenyl rings of the substrates such as –F (5a) or –Cl (5b) or –Br (5c and 5d) and electron-donating substitution groups such as methyl (5e and 5f) and methoxy (5j) led to lower benzaldehyde yield than the model reaction. Nevertheless, it appears that the variation of the position of the substituent on the phenyl ring does not have considerable effect on the conversion and on the yield of the corresponding aldehyde, either.
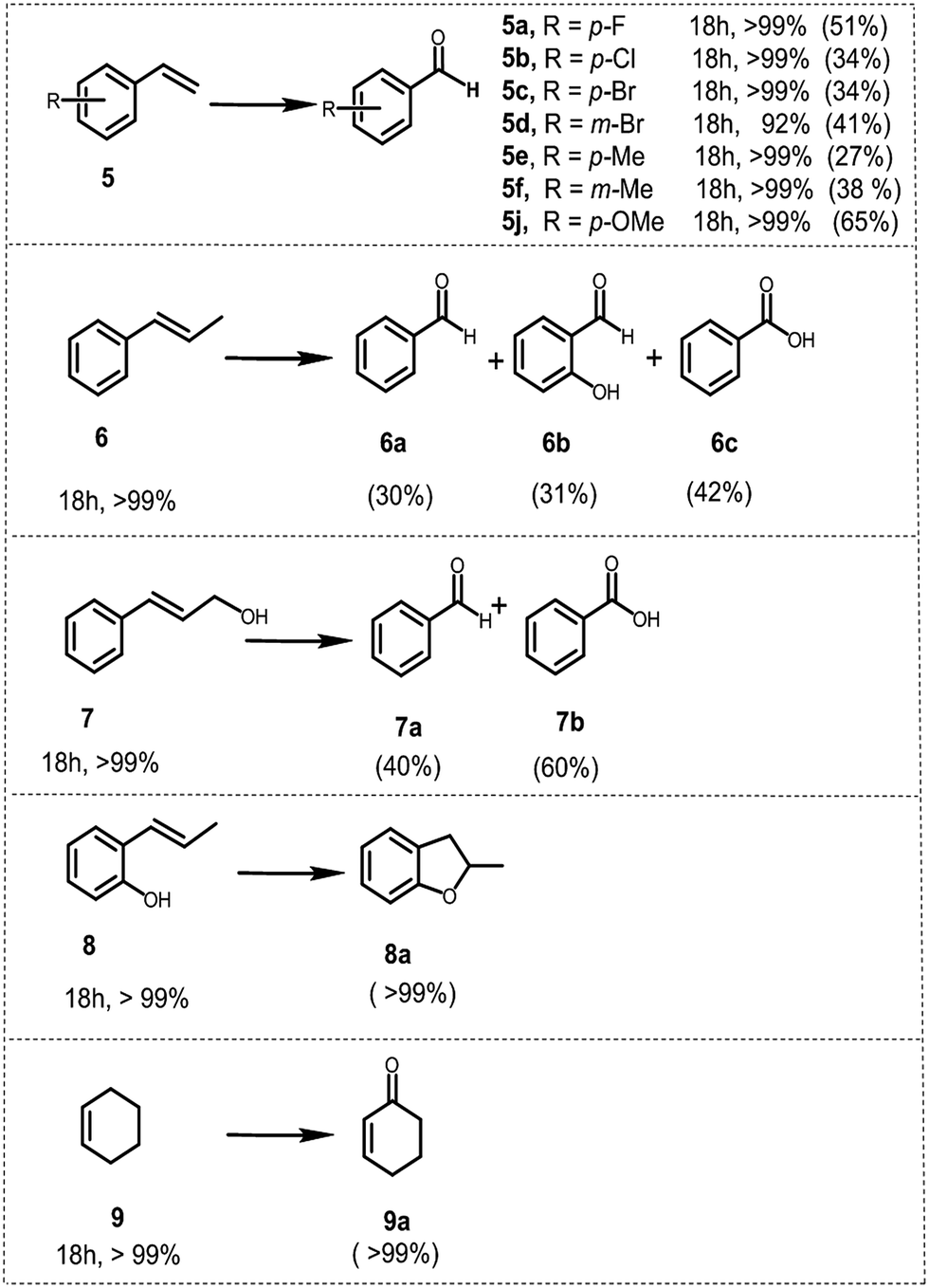 | ||
| Fig. 6 Scope of alkenes for the photocatalytic CC bond cleavage using BBT as the photocatalyst. | ||
Following the same procedure, trans-β-methyl styrene 6 and cinnamyl alcohol 7 were utilized as substrates to test secondary olefins under identical conditions. Both 6 and 7 were oxidized to the corresponding carbonyl compounds, offering benzoic acids as the main product with a yield of 40% (6c) and 60% (7b) respectively, which implied that BBT was also suitable for the secondary olefins. Additionally, BBT could be efficiently used for the synthesis of benzofuran 8a from 2-allylphenol 8 and 2-cyclohexen-1-one 9a from cyclohexene 9, both in excellent yields.
The stability and reusability of BBT were investigated via 5 additional repeat experiments under the same reaction conditions. As shown in Fig. S8,†BBT could catalyze the reaction for 5 additional cycles without significantly affecting its catalytic efficiency. However, the yield to benzaldehyde decreased in favor of the formation of 1-phenyl-1,2-ethanediol (compound 3 in Fig. 5). This indicates a slowing-down of the photocatalytic reaction after five cycles of use. FT-IR and UV/vis DR spectra (Fig. S9†) remain unchanged after the repeat experiments, which further confirm the high stability of the photocatalyst.
Conclusions
In summary, we demonstrated the design strategy of conjugated microporous polymers (CMPs) as heterogeneous, visible-light active and stable photocatalysts for the photocatalytic CC bond cleavage reaction of alkenes. By combining specific electron donor and acceptor building blocks with the CMP backbone structure, we found out that the CMP containing benzothiodiazole as a strong electron acceptor and phenyl as a weak electron donor was the most efficient photocatalyst. The photocatalytic cleavage reaction of the CC bond was found to be strongly dependent on the solvent polarity, with the aqueous reaction mixture being the ideal reaction medium with benzaldehydes as main products with high efficiency and selectivity.
Experimental procedure
Materials
Copper(I) iodide (99%), tetrakis(triphenylphosphine)palladium(0) (99%), p-benzoquinone (98%), sodium azide (99%), potassium iodide (>99.9%), triethylamine (99%) and 2,5-dibromothiophene (96%), were purchased from Sigma Aldrich. 1,3,5-Triethynylbenzene (>98%) and 4,4-dibromobiphenyl (98%) were purchased from Alfa Aesar (Massachusetts, USA). 4,7-Dibromo-2,1,3-benzothiadiazole (97%) (Br-BT) was purchased from COMBI-BLOCKS. Isopropanol (99.5%) and anhydrous N,N-dimethylformamide (99.8%) were purchased from Acros organics. Acetonitrile was purchased from Fischer Chemical. Styrene and styrene derivatives were purified by elution through neutral aluminium oxide (50–200 μm) and anhydrous CaCl2 (w/w, 95/5) in order to remove peroxides and other impurities. Chemicals and solvents purchased from commercial sources were used as received unless otherwise noted.Methods
X-ray diffraction (XRD) patterns were obtained using a Philips PW1050 diffractometer with CuKα radiation at k = 1.5418 Å. UV/vis absorption spectra were measured using a Perkin-Elmer Lambda 25 UV-Vis spectrometer. FTIR spectra were obtained using a FTIR spectrophotometer TENSOR II Bruker equipped with a platinum ATR. Solid state 13C CP/MAS NMR measurements were performed with a standard 4 mm magic angle spinning MAS double resonance probe head at 300 MHz Larmor frequency. TGA measurements were carried out on a Mettler Toledo TGA-851 system. The temperature was increased from 25 °C to 800 °C (10 °C min−1) under a nitrogen atmosphere. Electron paramagnetic resonance (EPR) spectroscopy was performed using a Magnettech MS 200 spectrometer. The nitrogen sorption data were measured to determine the surface area and the pore size distribution; the data were recorded at 77.3 K on an Autosorb 1 using QuadraWin Software (Quantchrome instruments). CV and photocurrent measurements were conducted using an Autolab Potentiostat PGSTAT204 (Metrohm). A glassy carbon working electrode, a Pt counter electrode and a SCE reference electrode were used. The SEM images were collected using a LEO Gemini 1530 (Carl Zeiss AG, Germany) with an in-lens SE detector.Synthesis of BTh, BThBT and BBT
The synthesis of BBT, BThBT and BTh was performed via palladium catalyzed Sonogashira–Hagihara cross coupling reactions. The cross-linker 1,3,5-triethynylbenzene (150 mg, 1 mmol, 1 equiv.), tetrakis(triphenylphosphine)palladium(0) (35 mg, 0.03 mmol, 0.03 equiv.) and copper(I) iodide (5.7 mg, 0.03 mmol, 0.03 equiv.) were dissolved in 20 mL of DMF/triethylamine mixture with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then, the co-monomer 4,7-dibromo-2,1,3-benzothiadiazole (294 mg, 1 mmol, 1 equiv.) was added for BBT, and the co-monomer 2,5-dibromothiophene (119 μL, 1 mmol, 1 equiv.) was added for BTh. As for the BThBT, both co-monomers (4,7-dibromo-2,1,3-benzothiadiazole and 2,5-dibromothiophene) were added in an equal ratio. The reaction mixtures were degassed with N2 for 10 min. After the degassing process, the reactive mixtures were heated at 80 °C for 48 h. After cooling, the precipitated powders were washed with methanol, acetone and water several times. The crude products were then extracted in a Soxhlet extractor for 24 h using a mixture of dichloromethane and methanol. Finally, the products were dried overnight at 60 °C under vacuum. Yield (BTh) = 52%, yield (BThBT) = 64% and yield (BBT) = 55%.
:1. Then, the co-monomer 4,7-dibromo-2,1,3-benzothiadiazole (294 mg, 1 mmol, 1 equiv.) was added for BBT, and the co-monomer 2,5-dibromothiophene (119 μL, 1 mmol, 1 equiv.) was added for BTh. As for the BThBT, both co-monomers (4,7-dibromo-2,1,3-benzothiadiazole and 2,5-dibromothiophene) were added in an equal ratio. The reaction mixtures were degassed with N2 for 10 min. After the degassing process, the reactive mixtures were heated at 80 °C for 48 h. After cooling, the precipitated powders were washed with methanol, acetone and water several times. The crude products were then extracted in a Soxhlet extractor for 24 h using a mixture of dichloromethane and methanol. Finally, the products were dried overnight at 60 °C under vacuum. Yield (BTh) = 52%, yield (BThBT) = 64% and yield (BBT) = 55%.
General procedure for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond cleavage using CMPs as photocatalysts
C bond cleavage using CMPs as photocatalysts
0.1 mmol of styrene or its derivatives or alkenes and 10 mg of the photocatalyst were added to 1.5 mL of solvent in a glass vial equipped with an oxygen balloon. A blue LED lamp (460 nm, 0.065 W cm−2, OSA Opto Light GmbH) was used as the light source. The reaction mixture was stirred at room temperature and under irradiation for 18 h. The conversion and selectivity were determined via GC-MS. The GCMS spectra are displayed in the ESI. For the repeat experiments, the CMP was filtered and washed several times with methanol, CH2Cl2 and acetone, and then dried under vacuum before being used for the repeat experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. I. Z. acknowledges the Max Planck Society for financial support. C. A. is a recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) of the Graduate School of Excellence “MAINZ” (Materials Science in Mainz). Open Access funding was provided by the Max Planck Society.Notes and references
- V. Kogan, M. M. Quintal and R. Neumann, Org. Lett., 2005, 7, 5039–5042 CrossRef PubMed.
- B. Liu, P. Wang, A. Lopes, L. Jin, W. Zhong, Y. Pei, S. L. Suib and J. He, ACS Catal., 2017, 7, 3483–3488 CrossRef.
- A. P. S. Oliveira, I. S. Gomes, A. S. B. Neto, A. C. Oliveira, J. M. Filho, G. D. Saraiva, J. M. Soares and S. Tehuacanero-Cuapa, Mol. Catal., 2017, 436, 29–42 CrossRef.
- A. Rubinstein, P. Jiménez-Lozanao, J. J. Carbó, J. M. Poblet and R. Neumann, J. Am. Chem. Soc., 2014, 136, 10941–10948 CrossRef PubMed.
- B. Wang, J. Zhang, X. Zou, H. Dong and P. Yao, Chem. Eng. J., 2015, 260, 172–177 CrossRef.
- B. Liu, F. Jin, T. Wang, X. Yuan and W. Han, Angew. Chem., Int. Ed., 2017, 56, 12712–12717 CrossRef PubMed.
- A. B. Khemnar and B. M. Bhanage, RSC Adv., 2014, 4, 8939–8942 RSC.
- A. P. S. Oliveira, I. S. Gomes, A. C. Oliveira, J. M. Filho, G. D. Saraiva, J. M. Soares, F. F. de Sousa and A. Campos, Catalysts, 2017, 7, 323 CrossRef.
- B. Feng, Z. Hou, X. Wang, Y. Hu, H. Li and Y. Qiao, Green Chem., 2009, 11, 1446–1452 RSC.
- Z. Zhang, Y. Kumamoto, T. Hashiguchi, T. Mamba, H. Murayama, E. Yamamoto, T. Ishida, T. Honma and M. Tokunaga, ChemSusChem, 2017, 10, 3482–3489 CrossRef PubMed.
- Y. Guo, L. Zhengwang, W. Guangjian, Y. Huang and F. Kang, Appl. Surf. Sci., 2011, 258, 1082–1090 CrossRef.
- D. Shen, C. Saracini, Y.-M. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 15857–15860 CrossRef PubMed.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2015, 56, 653–656 CrossRef.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef PubMed.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- S. Ghasimi, S. A. Bretschneider, W. Huang, K. Landfester and K. A. I. Zhang, Adv. Sci., 2017, 4, 1700101 CrossRef PubMed.
- Y. Li, M. Liu and L. Chen, J. Mater. Chem. A, 2017, 5, 13757–13762 RSC.
- X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536–6539 CrossRef PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef PubMed.
- N. Kang, J. H. Park, K. C. Ko, J. Chun, E. Kim, H. W. Shin, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, Angew. Chem., Int. Ed., 2013, 52, 6228–6232 CrossRef PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720–18724 RSC.
- W. Huang, B. C. Ma, D. Wang, Z. J. Wang, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2017, 5, 3792–3797 RSC.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef.
- Z. J. Wang, K. Landfester and K. A. I. Zhang, Polym. Chem., 2014, 5, 3559–3562 RSC.
- S. Dadashi-Silab, H. Bildirir, R. Dawson, A. Thomas and Y. Yagci, Macromolecules, 2014, 47, 4607–4614 CrossRef.
- R. Li, B. C. Ma, W. Huang, L. Wang, D. Wang, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 3097–3101 CrossRef.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, ACS Catal., 2018, 8, 6751–6759 CrossRef.
- W. Zhang, J. Tang, W. Yu, Q. Huang, Y. Fu, G.-C. Kuang, C. Pan and G. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef PubMed.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- C. Ayed, W. Huang, R. Li, L. C. da Silva, D. Wang, O. Suraeva, W. Najjar and K. A. I. Zhang, Part. Part. Syst. Charact., 2018, 35, 1700234 CrossRef.
- B. C. Ma, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. B, 2016, 4, 5112–5118 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. I. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- R. Li, B. C. Ma, W. Huang, L. Wang, D. Wang, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 3097–3101 CrossRef.
- Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 8, 3459–3464 CrossRef PubMed.
- R.-M. Latonen, C. Kvarnström and A. Ivaska, J. Electroanal. Chem., 2001, 512, 36–48 CrossRef.
- S. Ghosh, N. A. Kouame, S. Remita, L. Ramos, F. Goubard, P.-H. Aubert, A. Dazzi, A. Deniset-Besseau and H. Remita, Sci. Rep., 2015, 5, 18002 CrossRef PubMed.
- R. Li, Z. J. Wang, L. Wang, B. C. Ma, S. Ghasimi, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2016, 6, 1113–1121 CrossRef.
- Y. Ren, Y. Che, W. Ma, X. Zhang, T. Shen and J. Zhao, New J. Chem., 2004, 28, 1464–1469 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta05772a |
| This journal is © The Royal Society of Chemistry 2018 |