
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of oxygen with halide perovskites†
Alessandro
Senocrate
 a,
Tolga
Acartürk
a,
Gee Yeong
Kim
a,
Rotraut
Merkle
a,
Ulrich
Starke
a,
Michael
Grätzel
ab and
Joachim
Maier
*a
a,
Tolga
Acartürk
a,
Gee Yeong
Kim
a,
Rotraut
Merkle
a,
Ulrich
Starke
a,
Michael
Grätzel
ab and
Joachim
Maier
*a
aMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569, Stuttgart, Germany. E-mail: weiglein@fkf.mpg.de
bEcole Polytechnique Fédérale (EPFL) de Lausanne, Lausanne CH-1015, Switzerland
First published on 18th May 2018
Abstract
We systematically investigate the consequences of exposing halide perovskites to an oxygen-containing atmosphere, in particular methylammonium lead iodide, the archetypal compound used as photo-absorber in perovskite solar cells. For this purpose, we study oxygen solubility and global reaction with oxygen both in the dark and under light, and we refer to the kinetics in terms of surface reaction and bulk diffusion. As thermodynamics reveals, the material is unstable against oxygen, primarily because of the large driving force of water formation. While under light the material quickly degrades, in the dark the surface reaction kinetics – not the bulk transport – is very sluggish and keeps it metastable. For the same reason, oxygen incorporation into the lattice is negligible in the dark. On illumination, an accelerated oxygen in-diffusion occurs (as shown by 18O incorporation experiments) that severely modifies the electronic and ionic conductivities in the way that is expected for an acceptor dopant. Lastly, we investigate the impact of cation as well as anion mixing on the degradation and stress the necessity of using encapsulation during solar cell operation.
Mixed organic–inorganic halide perovskites (MOIHPs) have been extensively studied in recent years due to their prospective application as light harvesters in solar cells.1,2 This new class of devices, named perovskite solar cells, offers both simple processing and outstandingly high photo-conversion efficiencies. Unfortunately, the development and application of such devices is impeded by the poor stability of the halides. In particular, degradation processes in MOIHPs can be easily induced by temperature,3–5 water or high humidity6,7 and even high-intensity light.8,9 A major source of instability is exposure to oxygen.10–13 Notwithstanding the fact that oxygen is omnipresent during material synthesis as well as device operation, the interaction of O2 with MOIHPs has received only minor attention. Some of these reports show that oxygen can strongly and reversibly improve photoluminescence in MOIHPs.14–16 Other investigations deal with the formation of reactive superoxide species,10,17,18 attributed to charge transfer between O2 molecules and the photo-excited MOIHP.11,19,20 Notably, several commonly used organic hole- or electron-transport materials are known to degrade when exposed simultaneously to oxygen and light,21–25 making it crucial to distinguish the influence of O2 on the halide perovskite layer from the effect on the hole- or electron-transport materials used in perovskite solar cell devices. Surprisingly, there are no studies on oxygen solubility in MOIHPs, even though O-defects will act as dopants and severely influence not only their electronic26 but also ionic transport properties. For these reasons, we investigate reactivity and solubility of oxygen in the pure archetypical halide perovskite, i.e. methylammonium lead iodide (MAPbI3), comparing dark and illuminated conditions. We study both thermodynamics and kinetics of oxygen interaction, ranging from solubility to phase conversion (degradation). We observe that oxygen incorporation in MAPbI3 is modest in the dark, but strongly enhanced under illumination. By combining 18O chemical diffusion techniques with electrical characterizations and with UV-Vis and XRD degradation studies, as well as thermodynamic data, we obtain a consistent picture of the effects of O2 not only on transport properties, but also phase stability and degradation behavior of MAPbI3. Based on this, as a final point we also analyzed the stability of mixed halide (I, Br) and mixed cation (MA, FA) perovskite formulations, since such solid solutions represent the state of the art in terms of device applications.
Oxygen dissolution
Let us start with discussing the oxygen dissolution in MAPbI3 and its effect on transport properties (next section) under conditions where overall the perovskite phase remains intact. Owing to the configurational entropy involved, a certain solubility of oxygen in the MAPbI3 lattice must always occur. Considering the situation on the atomistic scale, atomic oxygen species can be introduced substitutionally, occupying an iodine site or an iodine vacancy and hereby forming a substitutional oxygen defect with a negative effective charge (denoted as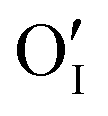 ). Alternatively, since the ionic radius of O2− is only 140 pm (I− is 210 pm), oxygen can be introduced interstitially, thus forming a doubly negatively charged defect (denoted as
). Alternatively, since the ionic radius of O2− is only 140 pm (I− is 210 pm), oxygen can be introduced interstitially, thus forming a doubly negatively charged defect (denoted as 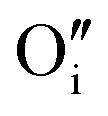 ). For the purpose of a direct experimental observation of oxygen dissolution in MAPbI3, we performed chemical diffusion experiments with a 18O tracer (Fig. 1). As shown in Fig. 1b (inset), in the dark we find incorporation of O2 in thin films to be poor and sluggish. This remains true even when elevated temperatures and long waiting times are involved, as shown for pellets (Fig. 1d). This is in striking contrast to the situation under illumination, where oxygen incorporation is substantially accelerated and enhanced (Fig. 1b). XRD studies show that the MAPbI3 phase was almost entirely preserved after these treatments (Fig. S3†). The detailed analysis of the diffusion profiles (Fig. 1c) reveals that the light-enhanced kinetics is due to an accelerated surface rate constant (kδ) while the impurity diffusion coefficient (Dδ) remains rather unchanged. While the effect on kδ is not unexpected for perovskites, the insensitivity of Dδ deserves some explanation, as our previous work showed a largely increased ionic carrier concentration under light.27 An independence of the impurity diffusion constant follows directly for the motion of interstitial oxygen defects (
). For the purpose of a direct experimental observation of oxygen dissolution in MAPbI3, we performed chemical diffusion experiments with a 18O tracer (Fig. 1). As shown in Fig. 1b (inset), in the dark we find incorporation of O2 in thin films to be poor and sluggish. This remains true even when elevated temperatures and long waiting times are involved, as shown for pellets (Fig. 1d). This is in striking contrast to the situation under illumination, where oxygen incorporation is substantially accelerated and enhanced (Fig. 1b). XRD studies show that the MAPbI3 phase was almost entirely preserved after these treatments (Fig. S3†). The detailed analysis of the diffusion profiles (Fig. 1c) reveals that the light-enhanced kinetics is due to an accelerated surface rate constant (kδ) while the impurity diffusion coefficient (Dδ) remains rather unchanged. While the effect on kδ is not unexpected for perovskites, the insensitivity of Dδ deserves some explanation, as our previous work showed a largely increased ionic carrier concentration under light.27 An independence of the impurity diffusion constant follows directly for the motion of interstitial oxygen defects (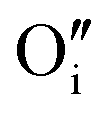 ), but it may also apply for the diffusion of substitutional defects (
), but it may also apply for the diffusion of substitutional defects (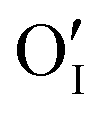 ), provided such species move strongly coupled with the induced iodine vacancy (such coupling is, in fact, expected for room temperature, as we detail in the ESI Section 5†). As far as the increased rate constant of the surface step under light is concerned, this may be attributed to an accelerated O2 reduction caused by the photo-generated electrons (ESI Section 3, eqn (S10)†), as in the case of SrTiO3.28 This is in also in agreement with previous studies showing how perovskite solar cells stability can be extended by improving the extraction of photo-generated carriers.11,17,20 These considerations also imply that in the dark oxygen incorporation is limited by the slow surface reaction. We note that the tracer profiles we observe here are typical of a bulk process, and also that the property changes discussed in detail below (e.g. doping effect) clearly show that the bulk of the material is affected by oxygen treatment. Nevertheless, even though we have no indications of comparatively fast grain boundary contribution (see ESI Section 13†) in our samples, we recognize that other preparation procedures could result in more open grain boundaries that would even enhance the incorporation rate.17,29,30
), provided such species move strongly coupled with the induced iodine vacancy (such coupling is, in fact, expected for room temperature, as we detail in the ESI Section 5†). As far as the increased rate constant of the surface step under light is concerned, this may be attributed to an accelerated O2 reduction caused by the photo-generated electrons (ESI Section 3, eqn (S10)†), as in the case of SrTiO3.28 This is in also in agreement with previous studies showing how perovskite solar cells stability can be extended by improving the extraction of photo-generated carriers.11,17,20 These considerations also imply that in the dark oxygen incorporation is limited by the slow surface reaction. We note that the tracer profiles we observe here are typical of a bulk process, and also that the property changes discussed in detail below (e.g. doping effect) clearly show that the bulk of the material is affected by oxygen treatment. Nevertheless, even though we have no indications of comparatively fast grain boundary contribution (see ESI Section 13†) in our samples, we recognize that other preparation procedures could result in more open grain boundaries that would even enhance the incorporation rate.17,29,30
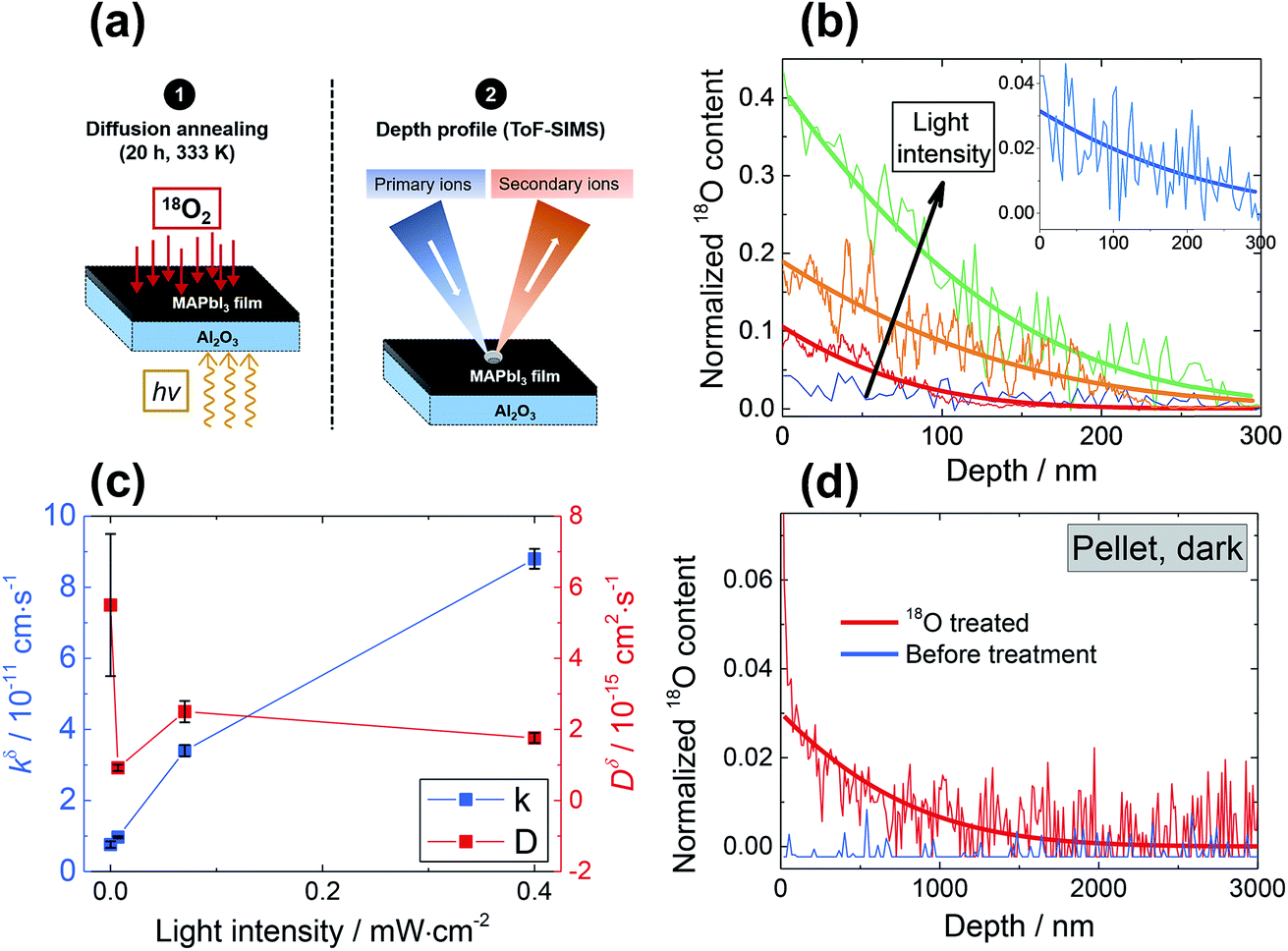 | ||
Fig. 1 (a) Schematic of the 18O incorporation experiment performed on MAPbI3 films deposited on Al2O3 substrates. First (1), the samples are annealed under oxygen; when necessary, light is simultaneously applied. After the treatment (2), the oxygen content is analyzed as a function of depth from the surface using Time of Flight Secondary Ion Mass Spectroscopy (ToF-SIMS). (b) ToF-SIMS depth profiles of four MAPbI3 thin films exposed to pure 18O2 at 333 K for 20 hours. One of the films was kept in dark ( blue), while the others illuminated with 1 ( blue), while the others illuminated with 1 ( green), 0.3 ( green), 0.3 ( orange) and 0.015 ( orange) and 0.015 ( red) mW cm−2 light. Insets show a magnification of the profile of the film kept under dark, where the profile is still visible and in agreement with the one observed for the pellet of panel (d). (c) Chemical diffusion coefficients (Dδ) and surface rate constants (kδ) extracted from the curves in panel (b) (at 333 K). (d) ToF-SIMS depth profiles of a MAPbI3 pellet, before and after exposure to 18O2 at 378 K for 46 hours under dark conditions ( red) mW cm−2 light. Insets show a magnification of the profile of the film kept under dark, where the profile is still visible and in agreement with the one observed for the pellet of panel (d). (c) Chemical diffusion coefficients (Dδ) and surface rate constants (kδ) extracted from the curves in panel (b) (at 333 K). (d) ToF-SIMS depth profiles of a MAPbI3 pellet, before and after exposure to 18O2 at 378 K for 46 hours under dark conditions (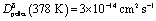 , , 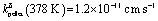 ). The amount of 18O found before treatment is too little to be properly detected, and appears lower than natural abundance, thus the normalized content lies below 0. ). The amount of 18O found before treatment is too little to be properly detected, and appears lower than natural abundance, thus the normalized content lies below 0. | ||
Dependence of transport properties on oxygen partial pressure
Upon oxygen incorporation, be it substitutional or interstitial, a negative dopant is generated (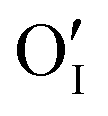 or
or 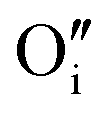 ), which gives rise to an electronic acceptor effect (increase of hole, decrease of excess electron concentration) as well as production of positively charged iodine vacancies (
), which gives rise to an electronic acceptor effect (increase of hole, decrease of excess electron concentration) as well as production of positively charged iodine vacancies (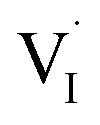 ), that we have identified as decisive ionic charge carriers in this material.31 For the effect of such doping to be significant, the incorporated oxygen content needs to exceed the intrinsic defect concentrations but may be still very small when compared with the number of regular lattice constituents. The quantitative consequences of oxygen doping on electronic and ionic carrier concentrations can be deduced from mass action considerations. The results of these considerations are reflected by the defect diagram shown in Fig. 2c and, more in detail, in the ESI (Section 3†). To analyze the oxygen effect quantitatively, we monitor the changes in electronic and ionic conductivities of a MAPbI3 film as a function of oxygen partial pressure, as reported in Fig. 2 (kinetics considerations are given in ESI Section 8†). As the comparison of the defect diagram of Fig. 2c with Fig. 2b (and Fig. S5†) demonstrates, the behavior under illumination well agrees with the predicted doping effect: At low oxygen pressures (P(O2) < 10−2 bar), the electronic conductivity increases with P(O2), while the ionic conduction appears invariant (this behavior is also observed in the dark, see below). At higher pressures (P(O2) > 10−2 bar), however, the situation changes drastically, in that here the ionic conductivity increases as well, while the electronic conductivity changes more steeply. We note that, in principle, also superoxide ions could cause such effect, but only if incorporated interstitially, which is thermodynamically not expected. If superoxide species would, as proposed, occupy iodine vacancies,17 no heterovalent doping effect would be achieved, in contrast with the experimental observations (see ESI Section 14† for more details). Having established that under illumination oxygen incorporation takes place and leads to the expected acceptor doping, we now turn to the conductivity effects caused by oxygen exposure in the dark, which appears puzzling at a first glance but fits nicely into the thermodynamic and kinetic picture given below. As the 18O incorporation experiments show, without illumination only little oxygen incorporation takes place (Fig. 1), hence one would not expect a significant effect on the transport properties of MAPbI3. We observe, however, that the samples (Fig. 2a) exhibit an increase in electronic conduction upon increasing P(O2), while the ionic conductivity stays rather constant. This behavior is not the one expected from O-doping, but is qualitatively comparable to what is observed upon exposure of MAPbI3 to an increasing iodine partial pressure.31 In fact the variations completely disappear when the outer iodine partial pressure is fixed (compare Fig. 2a and d). These results show that the O2 effect in the dark originates from the surface, where in simple terms (ESI Section 3†) oxygen converts I− to iodine, thus changing the iodine activity. Unlike for oxygen, the communication of the lattice with iodine is rather quick.31 Below we will show that such a surface conversion reaction consuming oxygen and producing iodine is indeed expected. Also under illumination, the simultaneous presence of iodine and oxygen severely weakens the O-influence on the transport properties (compare Fig. 2b and e), in agreement with a reduced iodine removal due to the fixed external I2 partial pressure (details in ESI Section 3†). A further interesting point is the observed reversibility (Fig. S6†), in spite of oxygen incorporation leading to iodine excorporation (ESI Section 3†); it follows that iodine is not excorporated into the gas phase, but rather it appears to be accommodated at higher dimensional defects, or even in the interstitial lattice.27
), that we have identified as decisive ionic charge carriers in this material.31 For the effect of such doping to be significant, the incorporated oxygen content needs to exceed the intrinsic defect concentrations but may be still very small when compared with the number of regular lattice constituents. The quantitative consequences of oxygen doping on electronic and ionic carrier concentrations can be deduced from mass action considerations. The results of these considerations are reflected by the defect diagram shown in Fig. 2c and, more in detail, in the ESI (Section 3†). To analyze the oxygen effect quantitatively, we monitor the changes in electronic and ionic conductivities of a MAPbI3 film as a function of oxygen partial pressure, as reported in Fig. 2 (kinetics considerations are given in ESI Section 8†). As the comparison of the defect diagram of Fig. 2c with Fig. 2b (and Fig. S5†) demonstrates, the behavior under illumination well agrees with the predicted doping effect: At low oxygen pressures (P(O2) < 10−2 bar), the electronic conductivity increases with P(O2), while the ionic conduction appears invariant (this behavior is also observed in the dark, see below). At higher pressures (P(O2) > 10−2 bar), however, the situation changes drastically, in that here the ionic conductivity increases as well, while the electronic conductivity changes more steeply. We note that, in principle, also superoxide ions could cause such effect, but only if incorporated interstitially, which is thermodynamically not expected. If superoxide species would, as proposed, occupy iodine vacancies,17 no heterovalent doping effect would be achieved, in contrast with the experimental observations (see ESI Section 14† for more details). Having established that under illumination oxygen incorporation takes place and leads to the expected acceptor doping, we now turn to the conductivity effects caused by oxygen exposure in the dark, which appears puzzling at a first glance but fits nicely into the thermodynamic and kinetic picture given below. As the 18O incorporation experiments show, without illumination only little oxygen incorporation takes place (Fig. 1), hence one would not expect a significant effect on the transport properties of MAPbI3. We observe, however, that the samples (Fig. 2a) exhibit an increase in electronic conduction upon increasing P(O2), while the ionic conductivity stays rather constant. This behavior is not the one expected from O-doping, but is qualitatively comparable to what is observed upon exposure of MAPbI3 to an increasing iodine partial pressure.31 In fact the variations completely disappear when the outer iodine partial pressure is fixed (compare Fig. 2a and d). These results show that the O2 effect in the dark originates from the surface, where in simple terms (ESI Section 3†) oxygen converts I− to iodine, thus changing the iodine activity. Unlike for oxygen, the communication of the lattice with iodine is rather quick.31 Below we will show that such a surface conversion reaction consuming oxygen and producing iodine is indeed expected. Also under illumination, the simultaneous presence of iodine and oxygen severely weakens the O-influence on the transport properties (compare Fig. 2b and e), in agreement with a reduced iodine removal due to the fixed external I2 partial pressure (details in ESI Section 3†). A further interesting point is the observed reversibility (Fig. S6†), in spite of oxygen incorporation leading to iodine excorporation (ESI Section 3†); it follows that iodine is not excorporated into the gas phase, but rather it appears to be accommodated at higher dimensional defects, or even in the interstitial lattice.27
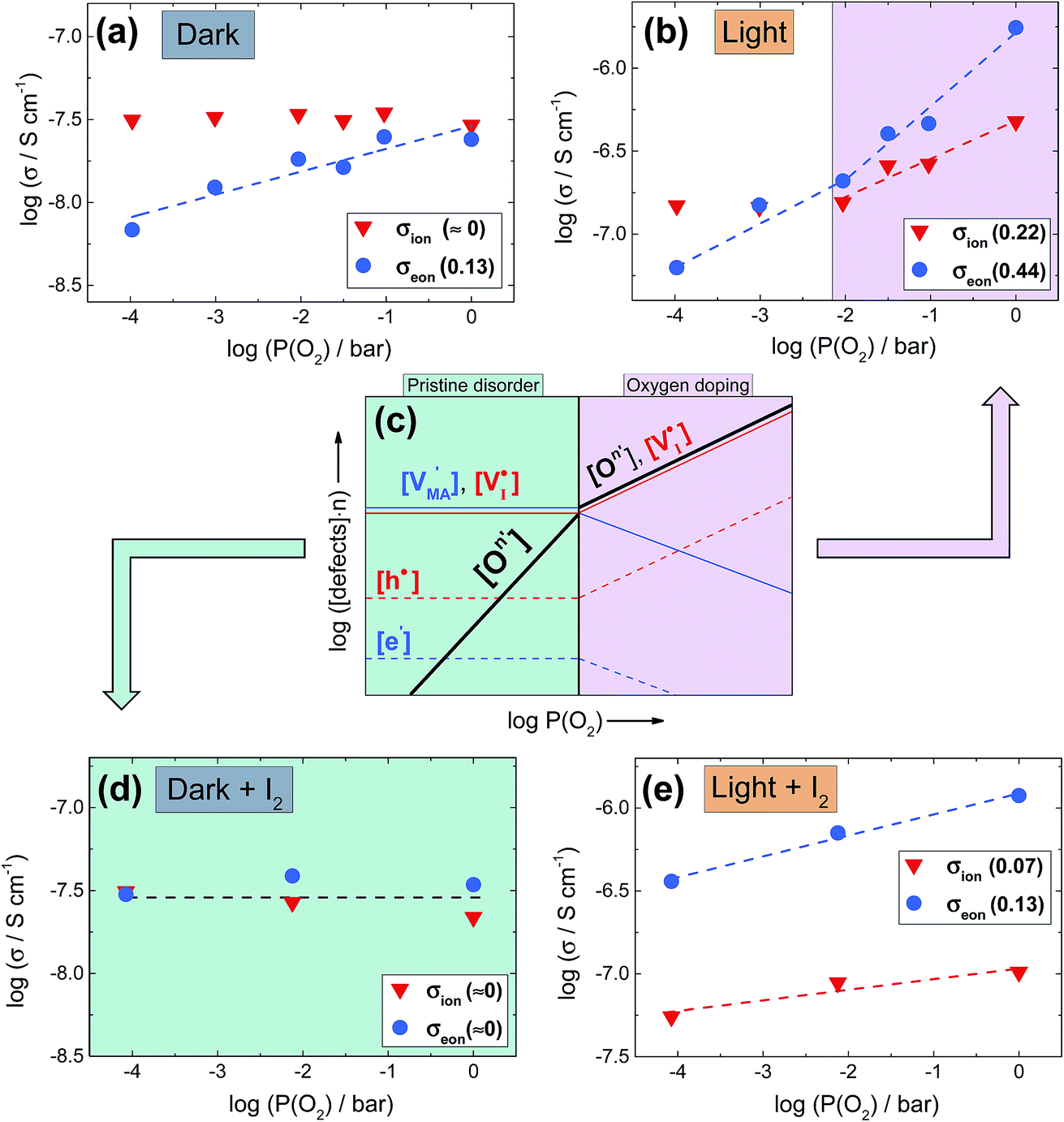 | ||
Fig. 2 (a and b) Ionic and electronic contribution to the conductivity of MAPbI3 thin films as a function of oxygen partial pressure, extracted from d.c. galvanostatic polarization experiments performed at 333 K, in the dark and under 0.5 mW cm−2 illumination. Slopes are given in the legends. The behavior under higher light intensity is reported in Fig. S5.† (c) Kröger–Vink diagram representing defect concentrations as a function of oxygen partial pressure. The incorporation of O2 is noted as a general atomic O-defect of effective negative n-charge (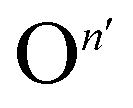 ). The relative slopes obtained when this defect overcomes the intrinsic equilibrium and achieves doping (violet region) are dependent on the charge of the defect, and are given in ESI Fig. S2.† (d and e) Conductivity measurements as a function of P(O2) (analogous as panel (a) and (b)), this time carried out under constant P(I2) (1.4 × 10−6 bar), in the dark and under 0.5 mw cm−2 illumination. Note that, in the dark, conductivity is not affected by oxygen pressure variations, while under illumination only a weak dependence remains. ). The relative slopes obtained when this defect overcomes the intrinsic equilibrium and achieves doping (violet region) are dependent on the charge of the defect, and are given in ESI Fig. S2.† (d and e) Conductivity measurements as a function of P(O2) (analogous as panel (a) and (b)), this time carried out under constant P(I2) (1.4 × 10−6 bar), in the dark and under 0.5 mw cm−2 illumination. Note that, in the dark, conductivity is not affected by oxygen pressure variations, while under illumination only a weak dependence remains. | ||
Degradation thermodynamics
We now discuss the phase stability of halide perovskites with respect to oxygen. Thermodynamically, here we refer to oxygen partial pressures that lead to an oxygen content exceeding the limit of oxygen solubility. Using recent experimental values4,32,33 in conjunction with tabulated standard data (ESI Section 2†) we can give standard reaction Gibbs energies for a variety of possible decomposition reactions (ESI Table S3†). Solubility effects, though highly relevant for transport issues, are negligible as far as their impact on phase energetics is concerned. As pointed out in ref. 34, reactions that involve C–N bond breaking (and conversion of MAPbI3 into CH3I and NH3) are kinetically hindered and not relevant for room temperature, while the expected decomposition reactions gives PbI2, HI and CH3NH2 (Table S3, eqn (S2)†). The related ΔrG0 value is distinctly positive (>120 kJ mol−1), so that in spite of the fact that the educts can be removed under a dynamic gas flow, this reaction can only occur to a very minor extent. A closer look into the thermodynamics shows that HI is marginally unstable with respect to decomposition, slightly facilitating the MAPbI3 decomposition reaction into I2 and H2 (Table S3, eqn (S3)†). Much more energetically favourable, however, is the decomposition if O2 is involved (ΔrG0 = 5.3 kJ mol−1), owing to the enormous energy gained when instead of HI (or H2), water is formed: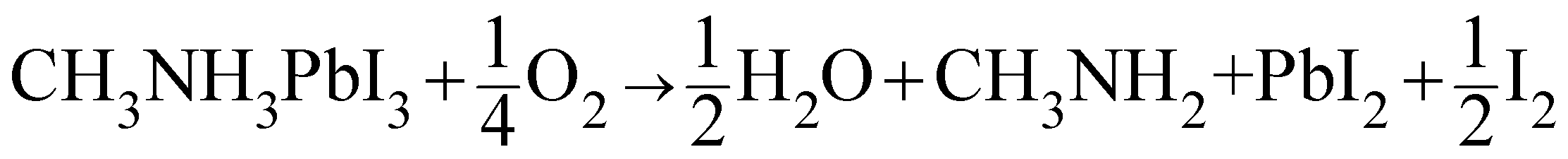 | (1) |
We note that the same reaction was proposed for superoxide-induced degradation.10,17 Indeed, the kinetic mechanism behind phase decomposition (but also behind oxygen incorporation) is expected to involve reactive intermediates such as superoxide species (as it is also the case for perovskites at high temperatures),35 while leaving the thermodynamic picture unchanged. It is also conceivable to further oxidize the products of eqn (1), in particular PbI2 to PbO, resulting in an additional modest free energy gain (Table S3, eqn (S5)†). The fact that only PbI2 is found as degradation product (as reported later) shows that such oxidation is, as expected, kinetically difficult. Moreover, other reaction channels can be opened by the presence of water6,7 formed in situ, and also by illumination.8,9,27 Focusing on eqn (1), the aforementioned only slightly positive value of Gibbs energy for the decomposition reaction involving O2 is easily turned into a negative balance if the produced gases are far from 1 bar of partial pressure (note that a reduction of the partial pressure by only one order of magnitude is already equivalent to a variation of −5.7 kJ mol−1 per gas molecule). Hence a slightly positive standard reaction Gibbs energy – given the dynamic gas atmosphere – demands a nearly complete exclusion of oxygen for the reaction in eqn (1) not to be of relevance; without such exclusion, O2 is thermodynamically expected to degrade MAPbI3 to PbI2 and release I2. Given this clear issue, it is worth considering alternative MOIHP compositions that can avoid a similar degradation. In this respect, we note that other halides, such as MAPbBr3 and MAPbCl3 are thermodynamically expected to be much more stable with respect to O2 (ESI Table S4†). Additionally, even though no thermodynamic data for formamidinium – (FA) containing perovskites have been reported, several works observed an increased stability of pure FAPbI3 and or FA, MA mixtures with respect to pure MAPbI3.36,37 We will experimentally address these points below.
Degradation kinetics
Now we are armed to discuss the kinetics of degradation of MAPbI3 under O2 exposure. Degradation kinetics is experimentally followed by monitoring in situ the UV-Vis absorption of a MAPbI3 film kept under constant gas flow, with conditions purposefully chosen to induce a degradation of the films in a reasonable amount of time. As shown in Fig. 3, no degradation is observable even after more than 150 hours when exposing films to O2 in dark conditions (at 333 K). When exposing the samples simultaneously to O2 and light, however, full degradation to PbI2 is achieved in less than 40 hours. As a reference, we exposed films to Argon under identical illumination conditions (Fig. S8a†), and no degradation was observed. These results are not unexpected, since the rather stable behavior in the dark – even under conditions where the whole phase should be converted into the products of eqn (1) – can be consistently attributed to the kinetic hindrances discussed above. Nonetheless, in the dark one may still expect this reaction to be of relevance for the surface layer, for which a transfer step into the bulk is not necessary. This straightforwardly explains the oxygen-induced “iodide-to-iodine conversion” observed when analyzing the transport properties (Fig. 2a). The aforementioned kinetic hindrances are, however, removed by illumination, allowing the sample to undergo a quick degradation. Interestingly, when following the degradation kinetics under illumination as a function of time (Fig. 3c), one clearly observes an initial relatively constant degradation rate, followed by a sudden accelerated drop, suggesting the opening of a parallel degradation path, such as one involving the water that is generated in situ by oxygen exposure. As expected from eqn (1), O2-induced degradation can be readily slowed down by exposing the film simultaneously to iodine partial pressure (Fig. 3c). Unsurprisingly, exposure to similar iodine partial pressures in an otherwise inert atmosphere (Ar) does not lead to degradation (Fig. S8b†). For very high I2 partial pressures, (≥1 bar), degradation of MOIHPs was observed,38 which would imply another degradation path under such harsh conditions. The effect of simultaneous iodine exposure on the oxygen interaction with MAPbI3 is visible also in photoluminescence (PL) spectra, where the enhancement of the PL response observed under pure O2 (ref. 14–16) is weakened by I2 (ESI Section 14, Fig S13 and S14†).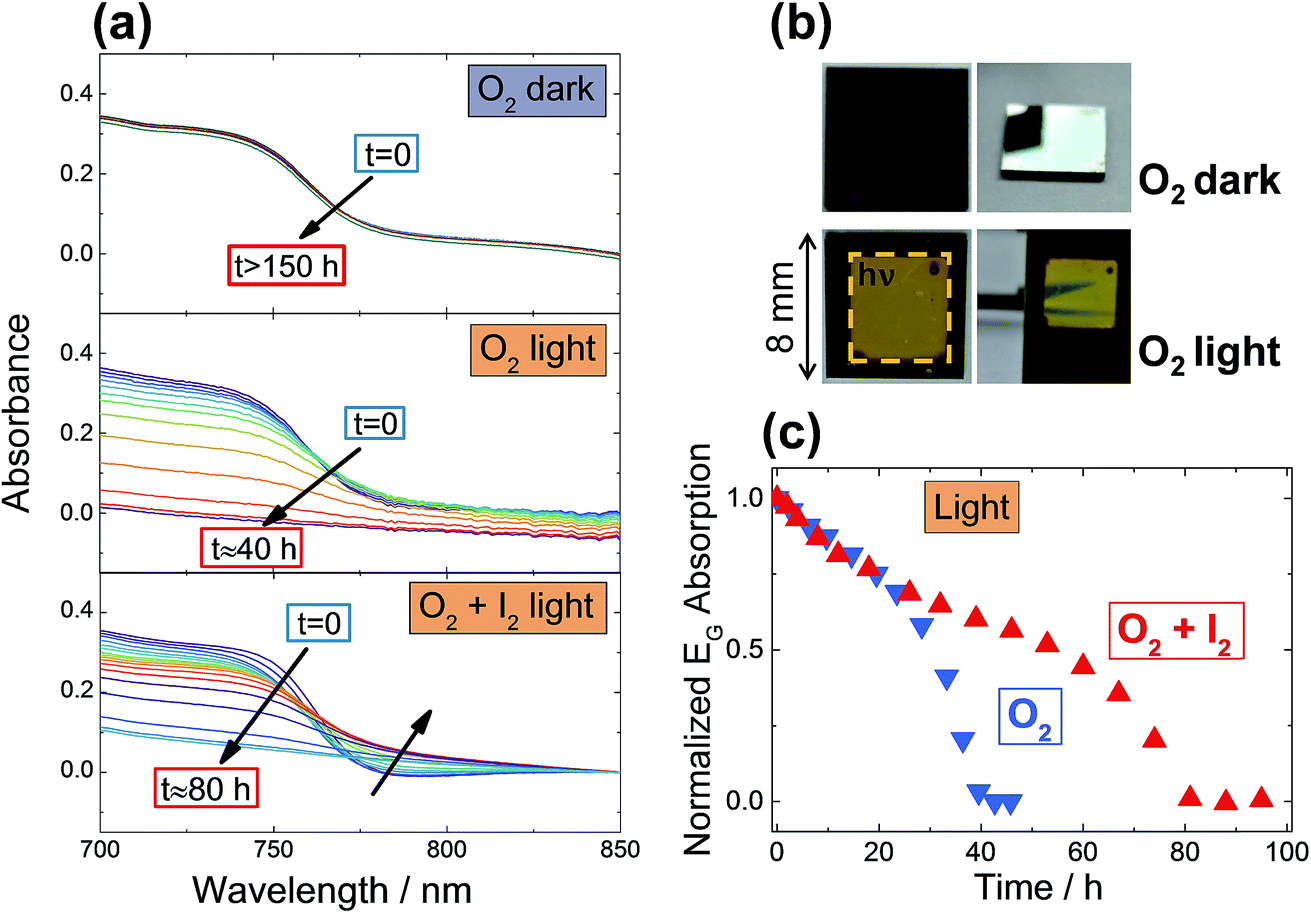 | ||
| Fig. 3 (a) Stability of MAPbI3 films under O2 exposure, in dark and under illumination (4.6 mW cm−2), monitored by UV-Vis spectroscopy. The samples are kept at 333 K under constant gas flow. After more than 150 hours, no changes are visible for samples kept in the dark between measurements, while 40 hours are sufficient to completely degrade samples exposed to light. As a comparison, a film kept under the same conditions (O2 and light) but also under constant iodine partial pressure (P(I2) = 1.4 × 10−5 bar), shows doubled stability, with full degradation achieved after 80 hours. Note the formation of a tail in the absorption, indicating amorphization of MAPbI3. (b) Photos of the sample after the O2 exposure under dark and light. Note the clear difference between illuminated and non-illuminated area. The sample kept in dark is identical to a pristine sample, with good transparency and reflectivity. (c) shows the degradation rate (here taken as the bandgap absorption of MAPbI3, see details in ESI Section 1†) as a function of time under light and O2, with and without simultaneous iodine exposure. These data are extracted from the spectra of panel (a). | ||
An analogous degradation study has been performed using in situ XRD measurements (Fig. 4a and b). The results obtained are in full agreement with the UV-Vis study, and here once again we observe an enhanced stability under simultaneous iodine and oxygen exposure. Interestingly, while under O2 exposure the film degrades yielding crystalline PbI2, under simultaneous exposure to O2 and I2 no PbI2 peaks are visible after the treatment (Fig. 4a, S9c†). The color change from black (MAPbI3) to yellow (PbI2) occurs as expected, but the absence of diffraction peaks speaks for the formation of an amorphous phase. Notably, UV-Vis spectra under O2 and I2 (Fig. 3a) show the formation of an absorption tail at longer wavelengths, in line with a loss of crystallinity of the perovskite phase. A similar behavior – though much less pronounced – is present when exposing for long times MAPbI3 to I2 in inert atmosphere (under illumination, see Fig. S8b†). We recognize that this finding needs additional experimental studies, hence we will not discuss it further here.
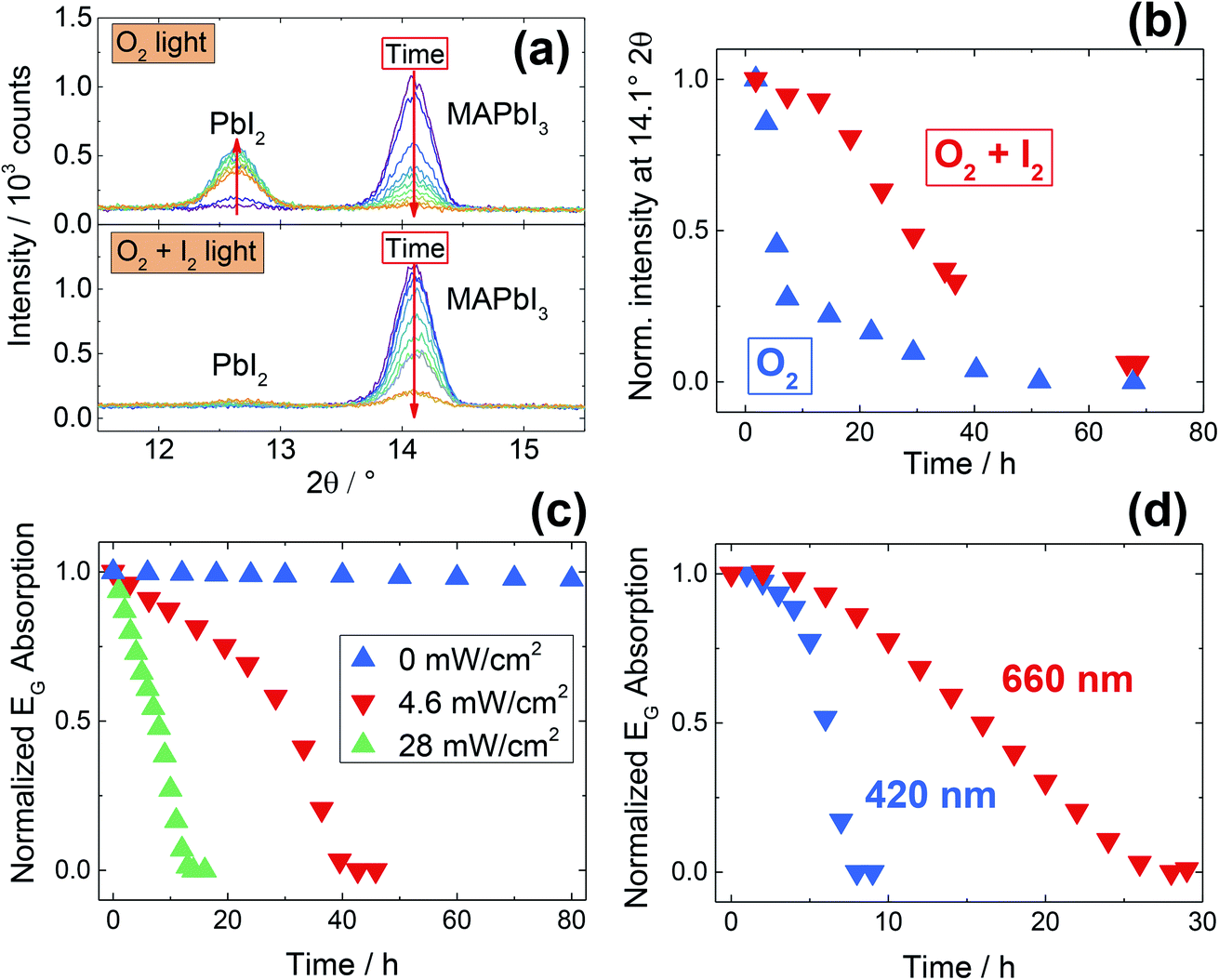 | ||
| Fig. 4 (a) Stability of MAPbI3 films under O2 exposure under illumination (43 mW cm−2), with and without simultaneous iodine exposure (P(I2) = 4.5 × 10−6 bar), measured by in situ XRD at room temperature. Exposure to O2 in the dark gives no significant degradation (Fig. S9a†), while under illumination a severe degradation takes place, with full disappearance of all MAPbI3 reflections after 50 hours (see also Fig. S9b†). After degradation under oxygen and iodine, no PbI2 peaks are visible, indicating the formation of an amorphous phase. (b) Normalized intensity of the strongest MAPbI3 reflection (at 14.1° 2θ), as a function of degradation time, extracted from the diffractograms of panel (a). The film kept under simultaneous iodine exposure shows enhanced stability. We recognize that, due to preferred orientations of the MAPbI3 crystals, the analysis of the intensity of a single peak can only be semi-quantitative. (c) Degradation kinetics of pure MAPbI3 films under O2 measured with UV-Vis spectroscopy under different light intensities. (d) Wavelength dependence of the degradation kinetics, monitored using UV-Vis spectroscopy, under 4 mW cm−2 illumination supplied by different LED lamps. | ||
With the intent of obtaining a deeper understanding of the O2 degradation processes under conditions that are relevant for MOIHPs application, we investigated the influence of the spectral content and of the illumination intensity. As previously shown (Fig. 3), degradation quickly takes place even at low light intensity (4.6 mW cm−2) when coupled with a slight heating (333 K). A stronger light intensity (43 mW cm−2) can induce a quicker degradation even at room temperature (Fig. 4a and b). In both cases, no UV-light is present, clearly indicating that this is not necessary to induce degradation of MOIHPs under O2; additionally, the illumination intensity used is significantly below 1 Sun (100 mW cm−2). We thus probe the intensity dependence of the degradation process (Fig. 4c); as expected, higher intensities accelerate the degradation process. Interestingly, as shown in Fig. 4d, we also observe a clear wavelength dependence for the degradation. Under identical light intensities, violet (420 nm) light is much more effective than red (660 nm) in inducing degradation. As local heating of the MOIHP surface is not a probable reason for this phenomenon (the samples are at 333 K), we postulate that it is the result of a favored electron transfer to the adsorbed O2 molecules due to high-energy electrons. We can thus conclude that, under real operative conditions (1 Sun, including high-energy UV-light) the degradation processes will be much more severe than what we observe here, and this makes the O2-induced degradation of MOIHP most relevant for real photovoltaic devices. In real devices, additional degradation pathways may occur, but note that the increased complexity of a solar cell cannot suppress the inherent instability of the photo-absorber if oxygen access is possible.
Since for MAPbI3 the degradation processes are clearly very severe, as a last analysis we investigate the stability of a mixed-halide (I, Br) and of a mixed-cation (MA, FA) MOIHP under O2. Starting with the mixed-halide perovskite, we chose a phase-stable formulation, i.e. MAPb(Br0.2I0.8)3, in order to avoid halide de-mixing under illumination.39 Interestingly, the sample does not show any enhanced stability with respect to the pure iodide (Fig. 5a). XRD analysis of the film after degradation, reported in Fig. 5b, shows that the expected removal of I2 (due to O2 incorporation in the perovskite) takes place in the mixed-halides as well, resulting in the formation of PbI2 and MAPbBr3, while the pristine sample displayed only the crystallographic phase of tetragonal MAPbI3. This clearly indicates that mixed-halide perovskites still suffer from severe O2-induced degradation, notwithstanding a free energy gain obtained from mixing entropy (which is expected however to be rather small). As previously predicted by thermodynamics considerations, the newly formed MAPbBr3 phase easily survives the O2 exposure, since no PbBr2 reflections are present after the treatment. As far as the mixed-cation perovskites are concerned, we choose a formulation for which the perovskite phase is structurally stable at room temperature, namely (MA0.8FA0.2)PbI3 (note that pure FAPbI3 crystallizes in a non-perovskite phase).40 Notably, in this case the degradation rate is significantly lowered – even though not completely suppressed – by mixing FA with MA (Fig. 5c and d, also Fig. S10†). As it is not the minor mixing free energy that can be responsible for the enhanced stability (cf. results on mixed halide formulations), we can attribute this finding to the following reasons: (i) thermodynamically, the stability is increased as deprotonation of the FA cation is less favored (formamidine is a stronger base than methylamine),41 and so will be the final oxidation driving force. (ii) Kinetically, we found a diminished ion conductivity by FA admixing36 that we directly observe in electrical measurements (Fig. S11†). This leads to a lowering of the rate of the transport step of the degradation. For all these reasons, we expect that higher FA contents, such as the one commonly used in state-of-the-art perovskite solar cells (FA > 80%), could potentially yield more stable, but possibly not phase-pure compounds.42 The analysis given in the ESI (Fig. S15†), shows that higher FA contents indeed improve the oxygen stability. Future work is needed to decide if this is due to the chemistry or to the possible two-phase nature of the material. Lastly, we note that in the case of (MA0.8FA0.2)PbI3, no peak belonging to PbI2 are observed after degradation, indicating formation of an amorphous phase (ESI Section 11†) or pure FAPbI3. Moreover, UV-Vis spectra (Fig. 5c) show the formation of an absorption tail at longer wavelengths, indicating a loss of crystallinity of the perovskite phase.
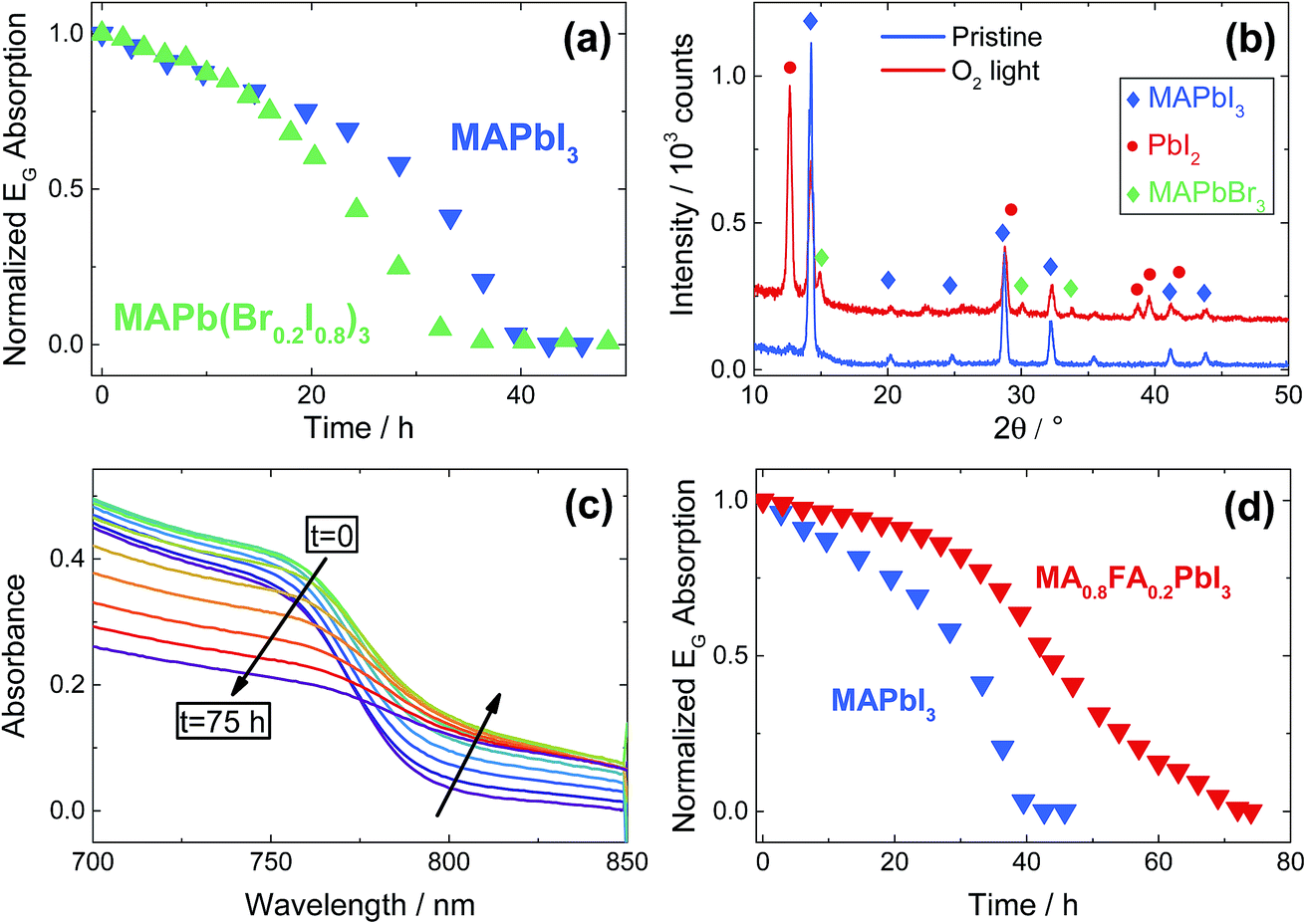 | ||
| Fig. 5 (a) Comparison between the stability of a pure MAPbI3 film with a sample containing 20% Br, i.e. MAPb(Br0.2I0.8)3. Both films were exposed to O2 under illumination (4.6 mW cm−2) at 333 K and measured by UV-Vis. (b) XRD pattern of the bromine-containing film before and after O2-induced degradation. Note the appearance of peaks pertaining to the pure MAPbBr3 phase, that were absent in the pristine film. The presence of weak MAPbI3 peaks after degradation is because part of the film is shaded from the light by the sample holder, thus avoiding degradation (see Fig. 3b). (c) UV-Vis spectra of (MA0.8FA0.2)PbI3 exposed to O2 under illumination (4.6 mW cm−2) at 333 K. Note the increase of the absorbance at high wavelengths. (d) Comparison between the degradation of pure MAPbI3 and of (MA0.8FA0.2)PbI3 under O2 exposure, monitored with UV-Vis under illumination (4.6 mW cm−2) at 333 K. | ||
As a conclusion, we end up with the following picture: MAPbI3 shows a certain oxygen solubility, which would be the stable state only for extremely low oxygen partial pressures. Interestingly, this dissolution is kinetically feasible only under light because otherwise the surface reaction steps are too sluggish (while the bulk diffusion is still acceptable). Oxygen, once introduced, acts as an acceptor dopant owing to O2− replacing I− or being accommodated interstitially. Under realistic conditions, MAPbI3 is thermodynamically expected to degrade forming water and iodine. Consequently, the degradation can be significantly slowed down by simultaneous mild iodine treatment. In agreement with the above, this reaction proceeds quickly under illumination, while in the dark the surface reaction is sluggish and keeps the material metastable. The kinetic mechanism for both the oxygen incorporation and phase decomposition is expected to involve reactive intermediate such as superoxide species, as well studied for perovskites at high temperatures35 and also in agreement with ref. 10 and 17. Forming mixed anionic compositions with other halides does not substantially improve stability since here the MAPbI3 portion is selectively and quickly degraded. More encouraging is the cation mixing, where we observed a significant reduction of the degradation rate. Nevertheless, the major tendency still remains and we conclude that under illumination, oxygen exposure is to be avoided to prevent degradation of MAPbI3 or its mixtures. Naturally, this process must also have a major impact on the performance stability of the related photovoltaic devices, notwithstanding additional effects on the other solar cell components. Hence, full encapsulation or a completely oxygen-free environment is required for a stable performance of perovskite solar cells.
Methods
Experimental methods are given in the ESI Section 1.†Author contributions
A. S. synthetized the materials and carried out the stability characterizations. T. A. performed the ToF-SIMS measurements. A. S. and G. Y. K. performed the electrical experiments. U. S. and R. M. supervised and discussed the chemical diffusion experiments. J. M. and M. G. supervised and discussed the stability characterization and the electrical measurements. A. S., R. M., G. Y. K. and J. M. wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are very grateful to Dr Alberto Jiménez-Solano for performing the photoluminescence measurements. Dr Helga Hoier is thanked for carrying out the XRD measurements, and Dr Giuliano Gregori and Dr Tae-Youl Yang are acknowledged for their help in the initial characterization of the samples. Achim Güth is thanked for his help in the electrode deposition. This work was performed within the framework of the Max Planck-EPFL Center for Molecular Nanoscience and Technology. Open Access funding by the Max Planck Society.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- H. S. Kim, et al., Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9{\%}, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- B. Conings, et al., Intrinsic Thermal Instability of Methylammonium Lead Trihalide Perovskite, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- B. Brunetti, C. Cavallo, A. Ciccioli, G. Gigli and A. Latini, On the Thermal and Thermodynamic (In)Stability of Methylammonium Lead Halide Perovskites, Sci. Rep., 2016, 6, 31896 CrossRef PubMed.
- G. Divitini, et al., In situ observation of heat-induced degradation of perovskite solar cells, Nat. Energy, 2016, 1, 15012 CrossRef.
- G. Niu, et al., Study on the stability of CH3NH3PbI3 films and the effect of post-modification by aluminum oxide in all-solid-state hybrid solar cells, J. Mater. Chem. A, 2014, 2, 705–710 Search PubMed.
- A. M. A. Leguy, et al., Reversible Hydration of CH3NH3PbI3$ in Films, Single Crystals, and Solar Cells, Chem. Mater., 2015, 27, 3397–3407 CrossRef.
- P. Pistor, A. Ruiz, A. Cabot and V. Izquierdo-Roca, Advanced Raman Spectroscopy of Methylammonium Lead Iodide: Development of a Non-destructive Characterisation Methodology, Sci. Rep., 2016, 6, 35973 CrossRef PubMed.
- Y. Li, et al., Light Induced Degradation of CH3NH3PbI3$ Thin Film, J. Phys. Chem. C, 2017, 121, 3904–3910 Search PubMed.
- N. Aristidou, et al., The Role of Oxygen in the Degradation of Methylammonium Lead Trihalide Perovskite Photoactive Layers, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef PubMed.
- A. J. Pearson, et al., Oxygen Degradation in Mesoporous Al2O3/CH3NH3PbI3−xClx Perovskite Solar Cells: Kinetics and Mechanisms, Adv. Energy Mater., 2016, 6, 1600014 CrossRef.
- X. Tang, et al., Photoinduced degradation of methylammonium lead triiodide perovskite semiconductors, J. Mater. Chem. A, 2016, 4, 15896–15903 Search PubMed.
- Q. Sun, et al., Role of Microstructure in Oxygen Induced Photodegradation of Methylammonium Lead Triiodide Perovskite Films, Adv. Energy Mater., 2017, 7, 1700977 CrossRef.
- Y. Tian, et al., Mechanistic insights into perovskite photoluminescence enhancement: light curing with oxygen can boost yield thousandfold, Phys. Chem. Chem. Phys., 2015, 17, 24978–24987 RSC.
- J. F. Galisteo-López, M. Anaya, M. E. Calvo and H. Míguez, Environmental Effects on the Photophysics of Organic–Inorganic Halide Perovskites, J. Phys. Chem. Lett., 2015, 2200–2205 CrossRef PubMed.
- R. Brenes, et al., Metal Halide Perovskite Polycrystalline Films Exhibiting Properties of Single Crystals, Joule, 2017, 1, 155–167 CrossRef.
- N. Aristidou, et al., Fast oxygen diffusion and iodide defects mediate oxygen-induced degradation of perovskite solar cells, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- S. Pont, et al., Tuning CH3NH3Pb(I1−xBrx)3 perovskite oxygen stability in thin films and solar cells, J. Mater. Chem. A, 2017, 5, 9553–9560 Search PubMed.
- F. T. F. O'Mahony, et al., Improved environmental stability of organic lead trihalide perovskite-based photoactive-layers in the presence of mesoporous TiO2, J. Mater. Chem. A, 2015, 3, 7219–7223 Search PubMed.
- D. Bryant, et al., Light and oxygen induced degradation limits the operational stability of methylammonium lead triiodide perovskite solar cells, Energy Environ. Sci., 2016, 9, 1655–1660 Search PubMed.
- M. Manceau, et al., Effects of long-term UVvisible light irradiation in the absence of oxygen on P3HT and P3HT:PCBM blend, Sol. Energy Mater. Sol. Cells, 2010, 94, 1572–1577 CrossRef.
- M. Hermenau, et al., Water and oxygen induced degradation of small molecule organic solar cells, Sol. Energy Mater. Sol. Cells, 2011, 95, 1268–1277 CrossRef.
- F. C. Krebs and K. Norrman, Analysis of the failure mechanism for a stable organic photovoltaic during 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h of testing, Prog. Photovoltaics, 2007, 15, 697–712 Search PubMed.
000 h of testing, Prog. Photovoltaics, 2007, 15, 697–712 Search PubMed. - M. O. Reese, et al., Photoinduced degradation of polymer and polymer-fullerene active layers: experiment and theory, Adv. Funct. Mater., 2010, 20, 3476–3483 CrossRef.
- A. Seemann, H. J. Egelhaaf, C. J. Brabec and J. A. Hauch, Influence of oxygen on semi-transparent organic solar cells with gas permeable electrodes, Org. Electron., 2009, 10, 1424–1428 CrossRef.
- A. Zohar, et al., Impedance Spectroscopic Indication for Solid State Electrochemical Reaction in CH3NH3PbI3 Films, J. Phys. Chem. Lett., 2016, 7, 191–197 CrossRef PubMed.
- G. Y. Kim, et al., Large tunable photoeffect on ion conduction in halide perovskites and implications for photodecomposition, Nat. Mater., 2018, 17, 445 CrossRef PubMed.
- R. Merkle, R. A. De Souza and J. Maier, Optically Tuning the Rate of Stoichiometry Changes: Surface-Controlled Oxygen Incorporation into Oxides under UV Irradiation, Angew. Chem., Int. Ed., 2001, 40, 2126–2129 CrossRef PubMed.
- Y. Shao, et al., Grain boundary dominated ion migration in polycrystalline organic–inorganic halide perovskite films, Energy Environ. Sci., 2016, 9, 1752–1759 Search PubMed.
- J. S. Yun, et al., Critical Role of Grain Boundaries for Ion Migration in Formamidinium and Methylammonium Lead Halide Perovskite Solar Cells, Adv. Energy Mater., 2016, 6, 1600330 CrossRef.
- A. Senocrate, et al., The Nature of Ion Conduction in Methylammonium Lead Iodide: A Multimethod Approach, Angew. Chem., Int. Ed., 2017, 56, 7755–7759 CrossRef PubMed.
- G. P. Nagabhushana, R. Shivaramaiah and A. Navrotsky, Direct calorimetric verification of thermodynamic instability of lead halide hybrid perovskites, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7717–7721 CrossRef PubMed.
- I. L. Ivanov, et al., Thermodynamics of formation of hybrid perovskite-type methylammonium lead halides, J. Chem. Thermodyn., 2018, 116, 253–258 CrossRef.
- A. Latini, G. Gigli and A. Ciccioli, A study on the nature of the thermal decomposition of methylammonium lead iodide perovskite, CH3NH3PbI3: an attempt to rationalise contradictory experimental results, Sustainable Energy Fuels, 2017, 1, 1351–1357 Search PubMed.
- R. Merkle and J. Maier, How is oxygen incorporated into oxides? A comprehensive kinetic study of a simple solid-state reaction with SrTiO3 as a model material, Angew. Chem., Int. Ed., 2008, 47, 3874–3894 CrossRef PubMed.
- T. Zhang, et al., Profiling the organic cation-dependent degradation of organolead halide perovskite solar cells, J. Mater. Chem. A, 2017, 5, 1103–1111 Search PubMed.
- G. Murugadoss, et al., Light stability tests of methylammonium and formamidinium Pb-halide perovskites for solar cell applications, Jpn. J. Appl. Phys., 2015, 54, 08KF08 CrossRef.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour, Nat. Energy, 2016, 2, 16195 CrossRef.
- E. T. Hoke, et al., Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics, Chem. Sci., 2015, 6, 613–617 RSC.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties, Inorg. Chem., 2013, 52, 9019–9038 CrossRef PubMed.
- pKb value for Methylamine and Formamidine. Chemical Abstract Service – SciFinder – Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994-2018 ACD/Labs).
- D. J. Kubicki, et al., Cation Dynamics in Mixed-Cation (MA)x(FA)1−xPbI3 Hybrid Perovskites from Solid-State NMR, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta04537b |
| This journal is © The Royal Society of Chemistry 2018 |