
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of polymer molecular weight on the performance of PTB7-Th:O-IDTBR non-fullerene organic solar cells†
Sebastian F.
Hoefler
 a,
Thomas
Rath
*a,
Nadiia
Pastukhova
b,
Egon
Pavlica
b,
Dorothea
Scheunemann
c,
Sebastian
Wilken
c,
Birgit
Kunert
d,
Roland
Resel
d,
Mathias
Hobisch
ae,
Steven
Xiao
f,
Gvido
Bratina
b and
Gregor
Trimmel
a
a,
Thomas
Rath
*a,
Nadiia
Pastukhova
b,
Egon
Pavlica
b,
Dorothea
Scheunemann
c,
Sebastian
Wilken
c,
Birgit
Kunert
d,
Roland
Resel
d,
Mathias
Hobisch
ae,
Steven
Xiao
f,
Gvido
Bratina
b and
Gregor
Trimmel
a
aInstitute for Chemistry and Technology of Materials (ICTM), NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at
bLaboratory of Organic Matter Physics, University of Nova Gorica, Vipavska 11c, 5270 Ajdovščina, Slovenia
cDepartment of Physics, Energy and Semiconductor Research Laboratory, Carl von Ossietzky University of Oldenburg, Carl-von-Ossietzky-Straße 9-11, 26129 Oldenburg, Germany
dInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
eInstitute of Paper, Pulp and Fibre Technology, Graz University of Technology, Inffeldgasse 23, 8010 Graz, Austria
f1-Material Inc., 2290 Chemin St-François, Dorval, Quebec H9P 1K2, Canada
First published on 17th April 2018
Abstract
Recent advances in the development of non-fullerene acceptors have increased the power conversion efficiency of organic solar cells to approximately 13%. Fullerene-derivatives and non-fullerene acceptors possess distinctively different structural, optical and electronic properties, which also change the requirements on the polymer donor in non-fullerene organic solar cells. Therefore, in this study, the effect of the molecular weight of the conjugated polymer on the photovoltaic performance, charge carrier mobility, crystallization properties, film morphology, and non-geminate recombination dynamics is systematically investigated in polymer:small molecule organic solar cells using the low bandgap polymer PTB7-Th as the donor and the non-fullerene indacenodithiophene-based small molecule O-IDTBR as the acceptor. Among the examined polymer samples (50–300 kDa), high molecular weights of PTB7-Th (with an optimum molecular weight of 200 kDa) are advantageous to achieve high efficiencies up to 10%, which can be correlated with an increased crystallinity, an improved field-effect hole mobility (1.05 × 10−2 cm2 V−1 s−1), lower charge carrier trapping and a reduced activation energy of charge transport (98 meV). Bias-assisted charge extraction and transient photovoltage measurements reveal higher carrier concentrations (1016 cm−3) and long lifetimes (4.5 μs) as well as lower non-geminate recombination rate constants in the corresponding devices, supporting the high photocurrents (ca. 15.2 mA cm−2) and fill factors (>60%).
Introduction
Non-fullerene acceptors (NFAs) have attracted considerable attention as an alternative to commonly used fullerene-based acceptor materials over the last few years. In particular, solution-processable small molecule electron acceptors have contributed significantly to the recent progress in the emerging field of organic photovoltaics with a power conversion efficiency (PCE) exceeding 13%.1 These remarkable advances with efficiencies already exceeding those of fullerene-based organic solar cells can be related to the various beneficial aspects of non-fullerene electron acceptors such as excellent charge transport, tunable optoelectronic properties, and variable energy levels, which originate from the rational molecular design by means of chemical modification and functionalization.1–5 Beside molecular design considerations regarding both donor and acceptor species, film morphology and interfacial layer engineering have been in the focus more recently.6–8Bulk-heterojunction blend systems with complementary absorption properties of donor and acceptor materials, perfectly aligned energy levels, high charge carrier mobilities and favorable phase separation are essential for highly efficient photovoltaic devices. Because of the differences of polymer donors and small molecule acceptors in terms of solubility, aggregation behavior, and other physicochemical properties, considerable attention needs to be drawn to the structure–property relationship of bulk-heterojunction blend systems in order to gain fundamental understanding of the molecular interactions and to improve the photovoltaic performance.
Fullerene-derivatives and non-fullerene acceptors have significantly different structural, optical and also electronic properties. For example, the fullerene moiety of PCBM has an isotropic conjugated spherical structure and shows a different behavior in terms of charge carrier mobility and phase separation/agglomeration compared to organic non-fullerene acceptors, which exhibit anisotropic conjugated structures. This also implies differences in π–π interactions between the conjugated polymer and the acceptor.9,10
Li et al. already investigated differences in polymer:fullerene and polymer:non-fullerene solar cells and found that the high mobility of the conjugated polymer is considerably more important in polymer:fullerene solar cells as the electron mobility in the fullerene phase is typically higher than the hole mobility in the polymer phase.11 On the other hand, due to the intrinsically lower mobilities of NFAs compared to those of fullerenes, the mobility of the conjugated polymer might not be that critical in polymer:non-fullerene solar cells.
Notable efforts have been made to elucidate the role of the polymer molecular weight in combination with fullerene derivatives (e.g., PC61BM12–14 and PC71BM15–17) or polymeric acceptor materials (e.g. P(NDI2OD-T2)18,19). It was found that the molecular weight of the conjugated polymer is an essential parameter affecting various crucial aspects such as charge carrier mobility, molecular ordering and packing, optoelectronic and physicochemical properties, film morphology, and photovoltaic performance.12–19 For example, conjugated polymers with high molecular weights typically exhibit enhanced absorption properties, improved charge carrier mobility values, and a favorable film morphology, which contributes positively to the solar cell performance.17–21 However, the solubility in common organic solvents is typically lower arising from molecular aggregation and gelation, which causes issues in terms of processability.13–15,22 In combination with fullerene derivatives, the molecular weight dependence was first investigated by Schilinsky et al. using P3HT as the polymeric donor species.12 Higher molecular weights of P3HT (>10 kDa) were demonstrated to be beneficial for obtaining high PCE values due to improved optical absorption properties, enhanced intermolecular ordering, and higher charge carrier mobility.12 By increasing the molecular weight further, a decrease in the device performance was observed mainly originating from changes in the blend film morphology and reduced charge carrier mobility values.23 An optimum regime was also reported for other conjugated polymers in fullerene-based organic solar cells.13,14 This highlights the importance of molecular weight optimization of conjugated polymers in order to achieve high-performance organic solar cells.
Despite the recent progress and enormous potential of non-fullerene organic solar cells (NF-OSCs), the molecular weight dependence of the conjugated polymer in blend systems with non-fullerene acceptors has not been studied so far, even though it cannot be assumed that the influence of the molecular weight of the conjugated polymer is similar in fullerene-based and non-fullerene organic solar cells.
Therefore, we thoroughly investigated bulk-heterojunction blends based on the polymeric donor PTB7-Th and the small molecule non-fullerene acceptor O-IDTBR, which can act as a valuable model system to examine the molecular weight dependence and provide guidance for various other material combinations. PTB7-Th (poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2-6-diyl]), also known as PCE-10, PBDTTT-EFT or PBDTT-FTTE, is a high-performance donor–acceptor copolymer with a two-dimensional conjugated structure consisting of benzo[1,2-b;4,5-b′]dithiophene (BDT) units with 5-(2-ethylhexyl)thiophen-2-yl side chains and fluorinated thieno[3,4-b]thiophene (TT) units modified with a branched 2-ethylhexyl carboxylate moiety, which are used as donor and acceptor building blocks, respectively (Fig. 1A). The polymer exhibits a low optical bandgap (ca. 1.58 eV),8,24–28 a comparably high charge mobility (ca. 10−3 cm2 V−1 s−1),27–31 good solubility in common organic solvents (e.g., chloroform, chlorobenzene, and ortho-dichlorobenzene),26,27 a moderate thermal stability (ca. 350 °C),25–27 and an excellent photovoltaic performance in fullerene-based (e.g. PTB7-Th:PC71BM)32–34 as well as fullerene-free (e.g., PTB7-Th:ITCT,35 PTB7-Th:COi8DFIC,36 and PTB7-Th:IDTBR:IDFBR37) organic solar cells with PCE values up to 12.2%.36 The relatively low-lying highest occupied molecular orbital (HOMO) energy level of PTB7-Th (−5.24 eV)25 is favorable for realizing high VOC values in comparison to other low bandgap polymers.26 The indacenodithiophene (IDT)-based small molecule O-IDTBR ((5Z,5′Z)-5,5′-(((4,4,9,9-tetraoctyl-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene-2,7-diyl)bis(benzo[c][1,2,5]thiadiazole-7,4-diyl))bis(methanylylidene))bis(3-ethyl-2-thioxothiazolidin-4-one)), recently introduced by the McCulloch group,38 was examined as a non-fullerene electron acceptor. O-IDTBR exhibits an acceptor–donor–acceptor (A–D–A) structure consisting of an electron-donating ladder-type fused five-ring indaceno[1,2-b:5,6-b′]dithiophene core with an aliphatic n-octyl side chain, flanked by two benzothiadiazole moieties and two electron-withdrawing rhodanine terminal groups (Fig. 1A). The small molecule acceptor has been in the focus as a high-performance n-type semiconductor in organic photovoltaics due to favorable absorption and charge transport properties,39 reduced charge recombination losses,37,40 a low voltage loss,39,40 a promising stability under ambient conditions,37,38 and an excellent photovoltaic performance exceeding 11%.37,39
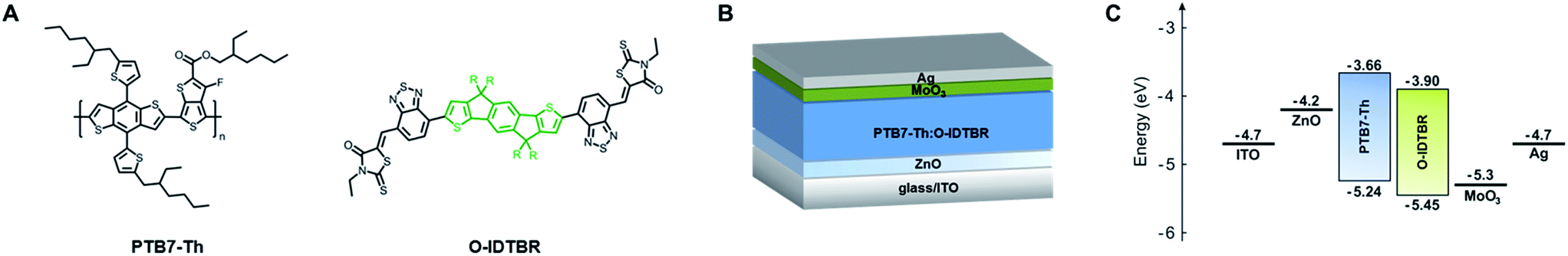 | ||
| Fig. 1 (A) Chemical structures of PTB7-Th and O-IDTBR (R = n-octyl). The fused five-ring indaceno[1,2-b:5,6-b′]dithiophene core is highlighted in green. (B) Schematic representation of the examined bulk-heterojunction-type PTB7-Th:O-IDTBR solar cell in inverted architecture and (C) the corresponding energy level diagram related to the vacuum level. The energy levels of PTB7-Th and O-IDTBR were adopted from the literature.25,37 | ||
In the course of this study, we systematically investigated the influence of the molecular weight of PTB7-Th on the photovoltaic performance of polymer:small molecule NF-OSCs using O-IDTBR as the electron acceptor. For this purpose, a series of PTB7-Th polymers with different molecular weights were examined including low (∼50 kDa), medium (∼100 kDa), high (∼200 kDa), and very high (∼300 kDa) molar mass fractions. The high molecular weights of PTB7-Th (with an optimum molecular weight of 200 kDa) were found to be advantageous to achieve high PCE values of almost 10%, while polymers with lower and higher molecular weights exhibited significantly lower PCE values. In order to elucidate the origin of the substantial differences in the solar cell performance, the molecular weight dependence of the charge carrier mobility, crystallization behavior and molecular packing, film morphology, and non-geminate recombination dynamics were examined in detail.
Results and discussion
Photovoltaic performance
Bulk-heterojunction solar cells were fabricated in an inverted architecture consisting of glass/ITO (indium tin oxide, ca. 140 nm)/ZnO (30–40 nm)/PTB7-Th:O-IDTBR (80–90 nm)/MoO3 (10 nm)/Ag (100 nm) using different molar mass fractions of PTB7-Th (in the range between 50 kDa and 300 kDa) and a donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor ratio of 1:1.5 by weight. A schematic representation of the device architecture and the corresponding energy level diagram are shown in Fig. 1B and C, respectively. According to previous studies, the higher amount of O-IDTBR within the active layer is suggested to compensate its lower electron mobility (μe = 6.3 × 10−4 cm2 V−1 s−1)39 and is therefore beneficial to obtain a more balanced charge carrier mobility, to reduce space charge accumulation, and to improve the solar cell performance.41
:acceptor ratio of 1:1.5 by weight. A schematic representation of the device architecture and the corresponding energy level diagram are shown in Fig. 1B and C, respectively. According to previous studies, the higher amount of O-IDTBR within the active layer is suggested to compensate its lower electron mobility (μe = 6.3 × 10−4 cm2 V−1 s−1)39 and is therefore beneficial to obtain a more balanced charge carrier mobility, to reduce space charge accumulation, and to improve the solar cell performance.41
The current density–voltage (J–V) curves of PTB7-Th:O-IDTBR solar cells under illumination and dark conditions are shown in Fig. 2A, while the characteristic photovoltaic performance parameters averaged over ten devices are summarized in Table 1. The average efficiency significantly improves with increasing the molecular weight of PTB7-Th from 8.44 ± 0.21% (50 kDa) to 9.57 ± 0.25% (200 kDa) due to a higher photocurrent generation, followed by a decline to 7.73 ± 0.18% for the very high molecular weight polymer (300 kDa) arising from substantially lower fill factor (FF) values. The best performance was obtained using the high molecular weight polymer (200 kDa) with a VOC of 1.00 V, a JSC of 15.44 mA cm−2, a FF of 64.6%, and a PCE of 9.94%, which are among the highest values for fullerene-based and fullerene-free OSCs in the same device geometry using the low bandgap polymer PTB7-Th as the donor.32,37 This remarkable performance is mainly attributable to the reasonably high JSC and FF values together with a low series resistance (RS, 6.0 Ω cm2) and a comparably high shunt resistance (RSH, 0.8 kΩ cm2). By comparing the individual photovoltaic performance parameters, it is found that the VOC slightly decreases from 1.01 V (50 kDa) to 0.99 V (300 kDa), while the JSC value significantly increases up to about 15.2 mA cm−2 for higher molecular weight polymers due to enhanced charge mobility values typically found for polymers with higher molar mass fractions (Fig. 2C, D and S1†).20,21 High FF values above 60% were found for polymer samples up to 200 kDa, while a FF of only 52% was obtained with the very high molecular weight polymer (300 kDa).
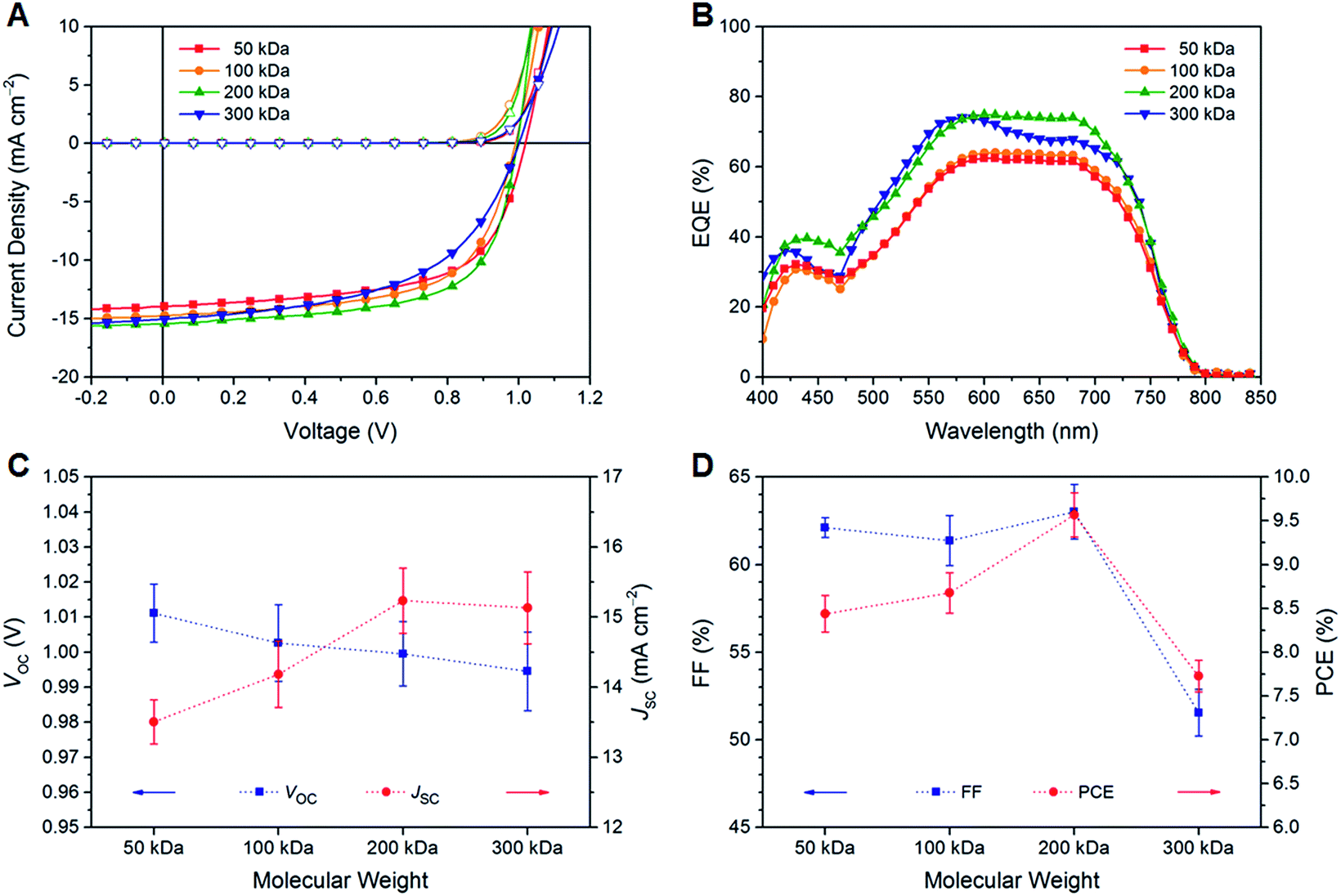 | ||
| Fig. 2 (A) J–V curves of PTB7-Th:O-IDTBR solar cells under illumination (full symbols, 1000 W m−2, AM 1.5 G) and dark conditions (empty symbols) and (B) EQE spectra of the corresponding photovoltaic devices with different molecular weights of PTB7-Th. (C and D) Effect of the molecular weight of PTB7-Th on the characteristic photovoltaic performance parameters (VOC, JSC, FF, and PCE) averaged over ten devices. Dotted lines are drawn to aid the eye. | ||
| Molecular weight | PDIa | V OC [V] | J SC [mA cm−2] | FF [%] | PCE [%] | R S [Ω cm2] | R SH [kΩ cm2] |
|---|---|---|---|---|---|---|---|
| a Polydispersity index. | |||||||
| 50 kDa | ca. 2 | 1.01 ± 0.01 | 13.5 ± 0.3 | 62.1 ± 0.6 | 8.44 ± 0.21 (max. 8.84) | 7.8 ± 0.7 | 0.53 ± 0.21 |
| 100 kDa | ca. 2 | 1.00 ± 0.01 | 14.2 ± 0.5 | 61.4 ± 1.4 | 8.68 ± 0.23 (max. 9.08) | 9.5 ± 1.7 | 0.40 ± 0.18 |
| 200 kDa | ca. 3 | 1.00 ± 0.01 | 15.2 ± 0.5 | 63.0 ± 1.6 | 9.57 ± 0.25 (max. 9.94) | 6.0 ± 0.7 | 0.81 ± 0.34 |
| 300 kDa | ca. 4 | 0.99 ± 0.01 | 15.1 ± 0.5 | 51.6 ± 1.3 | 7.73 ± 0.18 (max. 8.09) | 12.6 ± 1.1 | 0.42 ± 0.09 |
A similar trend in the photovoltaic performance has already been reported for fullerene-based bulk-heterojunction solar cells in the same architecture using PC71BM as the electron acceptor.8 Liao et al. found that the efficiency was improved with increasing the molecular weight of PTB7-Th from 7.02% (78 kDa) to 7.64% (124 kDa), followed by a decline for higher molar mass fractions to 5.35% (375 kDa).8 The considerably enhanced performance of the PTB7-Th:O-IDTBR solar cells compared to PTB7-Th:PC71BM-based devices is attributable to a substantially higher VOC of about 1.00 V compared to 0.79 V,8 which originates from the high-lying lowest unoccupied molecular orbital (LUMO) energy level of O-IDTBR (−3.9 eV),37,39,40,42 concomitant with a very small voltage loss of only ca. 0.59 V.42 An optimum range of the molecular weight was also found for other donor polymers (e.g., PTzBT-14HD,13 PBnDT-FTAZ,14 and P3HT23,43) in fullerene-based bulk-heterojunction solar cells. For very high or very low molecular weight polymers, however, solubility or processability issues, high polydispersity indices, differences in film morphology evolution or the degree of crystallinity were reported to have a detrimental effect on the device performance.13,14,43
The external quantum efficiency (EQE) spectra indicate a broad and strong photoresponse in the spectral region from 400–800 nm with a plateau in the range of 550–700 nm along with a shoulder at approximately 480–500 nm. A second maximum is found in the lower wavelength region at about 430 nm (Fig. 2B). The EQE spectra are in good agreement with the optical absorption properties of PTB7-Th (Table S1†). The optical absorption coefficient α of thin polymer films at the absorption maximum (ca. 700 nm) is in the range of 105 cm−1, and the optical bandgap (Eoptg) determined from the absorption onset is about 1.59 eV (Fig. S3 and S4†). Both values are in line with the data reported in the literature; however, no notable trend with regard to the polymer molecular weight was found.8,24–28 The intensities of the EQE spectra are consistent with the experimental JSC values. The higher EQE values of the solar cells based on the higher molecular weight polymers correlate well with the higher JSC values in these samples. However, it should be noted that the EQE spectrum of the solar cells implementing the 300 kDa polymer sample exhibits a different shape with a more pronounced maximum at a wavelength of about 580 nm.
Charge carrier mobility
To understand the origin of the substantial differences in the photovoltaic performance depending on the different molecular weights, in particular the decrease in efficiency for the 300 kDa polymer, the charge carrier mobility of PTB7-Th was examined using organic field-effect transistor (OFET) devices in bottom-contact bottom-gate configuration. Fig. 3A shows typical transconductance characteristics, while the extracted field-effect hole mobility (μFETh) values can be found in Table 2 and Fig. S5.† The hole mobility gradually improves with increasing the molecular weight of PTB7-Th from 6.2 × 10−3 cm2 V−1 s−1 (50 kDa) to 1.2 × 10−2 cm2 V−1 s−1 (300 kDa), which perfectly coincides with the expectation that hole mobility values typically increase for conjugated polymers with higher molecular weights.20 This behavior is described in several studies to typically originate from a higher effective conjugation length, an extended interconnectivity between relatively ordered polymer aggregates and a lower amount of terminal groups in high molecular weight polymers.20,21,44,45 The highest field-effect mobility values within this study for the polymer with the highest molar mass fraction (300 kDa) are about one order of magnitude higher than hole mobilities derived from the space-charge-limited current (SCLC) method in the literature (ca. 10−3 cm2 V−1 s−1).27–31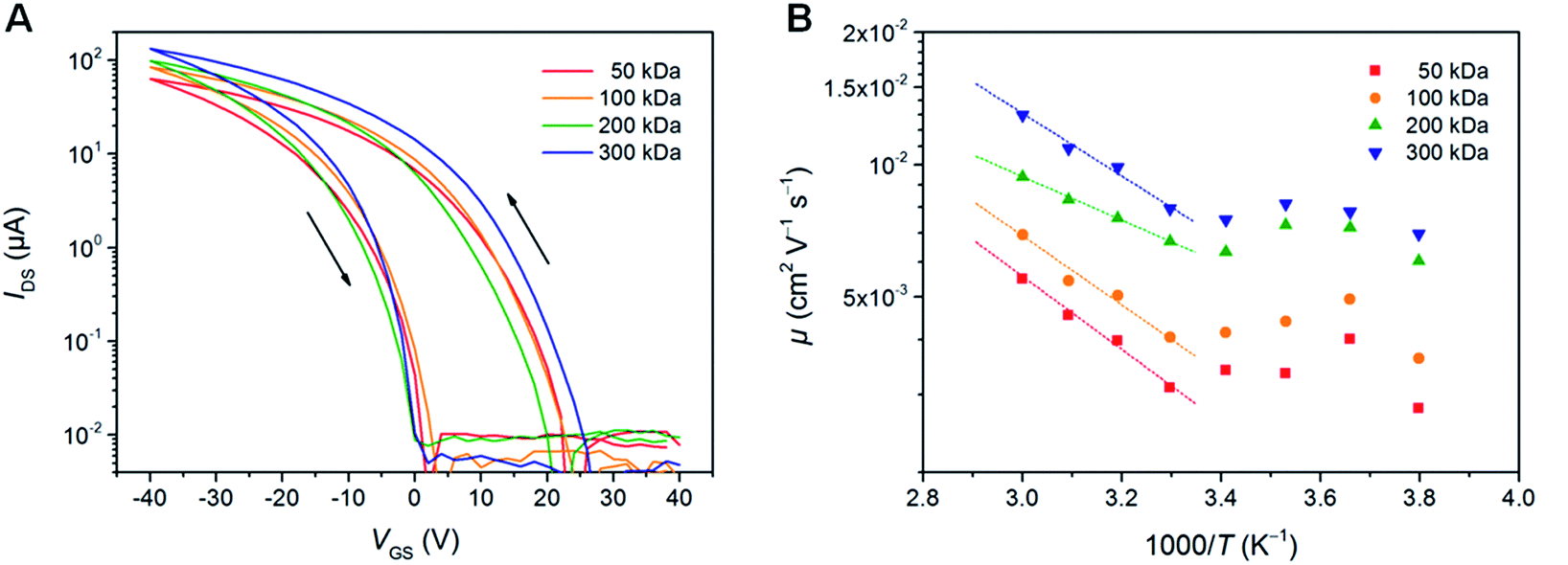 | ||
| Fig. 3 Charge transport characterization of PTB7-Th thin films with different molecular weights in 20 μm OFETs. (A) Typical transconductance characteristics (IDS–VGS) measured at a drain voltage of −40 V. (B) Field-effect mobility as a function of temperature of PTB7-Th thin films with different molecular weights in the saturation regime measured at a drain voltage of −40 V. Dotted lines represent the Arrhenius-type dependence log(μ) ∝ Ea/kBT. | ||
| Molecular weight | μ FETh [cm2 V−1 s−1] | On/off ratio | V th [V] | E a [meV] |
|---|---|---|---|---|
| a Field-effect hole mobility. b Threshold voltage. c Arrhenius-type activation energy. | ||||
| 50 kDa | 0.62 ± 0.01 × 10−2 | 1.9 ± 0.3 × 103 | 17.1 ± 0.5 | 165 ± 6 |
| 100 kDa | 0.78 ± 0.03 × 10−2 | 3.8 ± 1.0 × 103 | 17.3 ± 0.2 | 156 ± 12 |
| 200 kDa | 1.05 ± 0.03 × 10−2 | 3.1 ± 0.4 × 103 | 12.9 ± 0.5 | 98 ± 2 |
| 300 kDa | 1.16 ± 0.01 × 10−2 | 5.0 ± 1.3 × 103 | 17.7 ± 0.4 | 142 ± 7 |
Comparing the hole mobility values with the obtained solar cell parameters, the improved hole mobility for the higher molecular weight polymer samples can be correlated to the enhanced JSC values. However, the improved charge mobility cannot explain the decline of FF and PCE values of the very high molecular weight polymer sample (300 kDa). Rather than the charge carrier mobility, the low FF and PCE values are suggested to be resulting from the increased activation energy (Ea) in the 300 kDa polymer (142 ± 7 meV), which was determined from temperature-dependent field-effect mobility measurements (Fig. 3B). In case of the 200 kDa sample, we assume that the improved performance can also be correlated with the Ea and the threshold voltage (Vth). In OFETs, the injected charges first fill the traps, which are in the tail of the energy distribution of states (EDS). The states in the tail of the EDS are protruding into the energy gap of the polymer and are therefore less likely to act as charge transporting states in hopping. Thus, the charge transport process occurs only after the injected charge starts to occupy energetically more favorable electronic states in terms of energetic overlap of transporting states. Hence, when the measured Ea is low, the EDS tails comprise less states, which could be considered as deep-traps. In this case, the Vth, which reflects the density of immobile charges in the active channel, is also reduced. As can be seen in Table 2, the best-performing polymer sample (200 kDa) exhibits a 26% lower Vth and 30% lower Ea compared to the other polymers. Based on these data, we can speculate that this correlation could potentially result from the reduced trapping in PTB7-Th, which in turn reduces charge accumulation and recombination losses in the solar cells.
Grazing incidence X-ray diffraction
Grazing incidence X-ray diffraction (GIXRD) measurements of polymer thin films were performed in order to determine possible differences in the aggregation properties, crystallization behavior, and molecular packing. Fig. 4A shows the X-ray diffractograms of PTB7-Th thin films with different molecular weights on silicon substrates spin-coated from ortho-dichlorobenzene solutions with layer thicknesses of 70 ± 15 nm. The major diffraction peak with a maximum at ca. 22.2° 2θ, which corresponds to a d-spacing of about 4.00 Å, can be attributed to intermolecular van der Waals interactions (C–H⋯H–C) together with contributions from π–π stacking of the polymer backbone.6,7,46 According to GIXRD and grazing incidence wide-angle X-ray scattering (GIWAXS) measurements reported in the literature, the diffraction peak can be correlated with the (010) reflection in the out-of-plane direction and is due to the preferential alignment of the polymer in the face-on orientation.6,7,46 Comparing the PTB7-Th thin films with different molecular weights, only negligible differences with regard to the d-spacing distance can be observed (Table S3†), which are in line with the values reported in the literature (d = 3.94–4.13 Å).7,25,46 However, it should be noted that the high molecular weight polymer sample (200 kDa) exhibits a slightly less compact packing (4.03 Å) compared to the other polymers (ca. 3.96–4.02 Å). In addition, the intensity of the diffraction peak at 22.2° 2θ increases for higher molecular weight polymers indicating an overall higher degree of crystallinity, which is originating from a more pronounced correlation between the polymer backbones. The highest crystallinity was found for the 200 kDa polymer sample, which supports the improved charge transport properties and efficiency of the corresponding solar cells. The slightly decreased crystallinity of the 300 kDa polymer sample can be possibly explained by a more pronounced chain entanglement, which hinders the π–π stacking interactions of the polymer chain.19,23,44,47 Since a higher degree of chain entanglement promotes trapping during intra- and inter-molecular charge transport processes and thus induces recombination losses, a negative effect on the photovoltaic performance, in particular the FF values, can be expected.19,23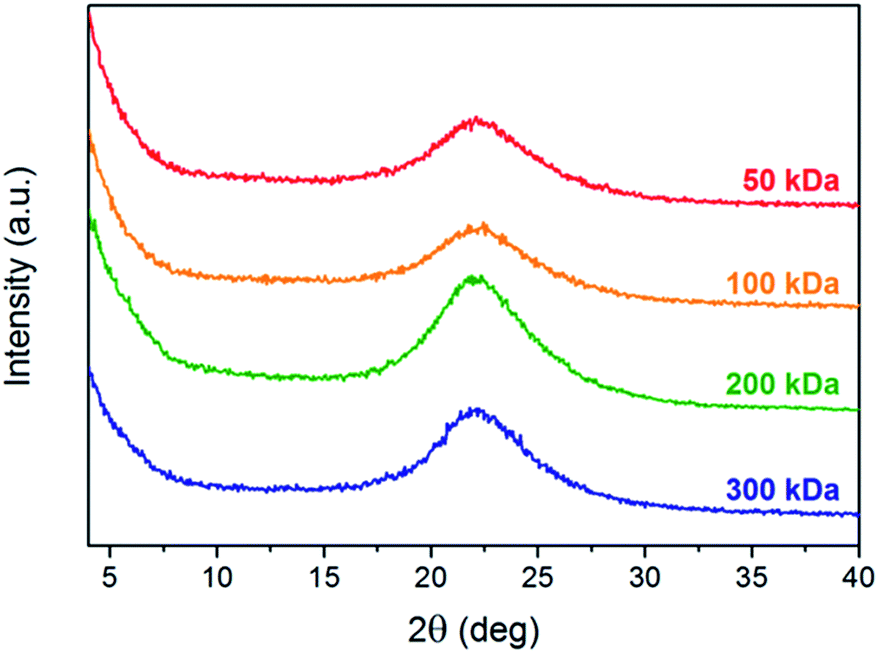 | ||
| Fig. 4 Grazing incidence X-ray diffraction patterns of PTB7-Th thin films with different molecular weights (Cu Kα radiation, λ = 1.5406 Å) deposited on silicon substrates by spin coating from ortho-dichlorobenzene solutions (normalized to layer thickness). | ||
Morphological characterization
Since the morphology of the active layer, i.e., the nanometer-scale phase separation of donor and acceptor domains, is a crucial parameter affecting the photovoltaic performance of bulk-heterojunction solar cells, the impact of the polymer molecular weight on the film morphology and phase separation properties was investigated. For this purpose, the surface morphology of PTB7-Th:O-IDTBR blend thin films was examined via atomic force microscopy (AFM) measured in tapping mode. The topography images of the blend thin films reveal a very smooth surface with a root-mean-square roughness (Rq) in the range of 2.37–3.76 nm (Fig. 5A–D). The roughness decreases for blend thin films with higher molecular weight polymers, also indicating reduced domain sizes in comparison to their lower molecular weight analogs. The more pronounced phase separation in the lower molecular weight polymer blends might originate from a faster diffusion of donor and acceptor molecules inducing the formation of distinct phase-separated domains.18 | ||
| Fig. 5 AFM topography images (A–D) and phase images (E–H) of PTB7-Th:O-IDTBR blend thin films with a molecular weight of 50 kDa (A and E), 100 kDa (B and F), 200 kDa (C and G), and 300 kDa (D and H) of PTB7-Th (image size: 2 × 2 μm). | ||
A very low phase contrast is observed in the phase images (Fig. 5E–H) as expected due to the rather similar chemical composition of the donor and acceptor in the blend, which makes it difficult to draw additional conclusions from these data. Nevertheless, the topography images reveal that the excellent photovoltaic performance of the solar cells prepared with the high molecular weight polymer (200 kDa) might not only be due to the enhanced charge carrier mobility and higher crystallinity but could also be influenced by the favorable film morphology of the active layer with a less pronounced phase separation and reduced domain sizes. Typically, a finely distributed, bicontinuous interpenetrating network of donor and acceptor domains with an enhanced interfacial area is beneficial for exciton dissociation and charge carrier transport and thus can lead to higher charge generation yields as well as an improved solar cell performance. However, bulk-heterojunction systems with smaller domain sizes or disordered percolation pathways are also more prone towards recombination losses due to the formation of isolated domains without thoroughly interconnected pathways acting as recombination sites, which can reduce the FF values significantly.19 Since no substantial differences are observable in the AFM images of the blend thin films of the higher molecular weight polymers, we assume that the reduced FF value and lower efficiency of the solar cells based on the very high molecular weight polymer (300 kDa) originate from a higher degree of recombination or differences in the charge recombination dynamics, which could arise from the reduced crystallinity as discussed before.
Non-geminate recombination dynamics
To study the effect of the molecular weight of PTB7-Th on the non-geminate recombination behavior, bias-assisted charge extraction (BACE) and transient photovoltage (TPV) measurements were performed. Fig. 6A shows BACE photocurrent transients that were recorded after switching off the illumination and simultaneously changing the external bias from open circuit to an extraction voltage of −5 V. Integrating the transient current with respect to time yields the carrier concentration n, which was determined as a function of VOC by varying the light intensity (Fig. 6B). For the PTB7-Th:O-IDTBR solar cells, the carrier concentration at open circuit increases significantly from low (50 kDa) to high molecular weight polymers (200 kDa) but then decreases again for the very high molecular weight polymer (300 kDa). This trend is consistent with the steady-state photovoltaic performance. In particular, the maximum carrier concentration of the solar cells based on the 200 kDa polymer corresponds well with the highest photocurrent and fill factor under one sun.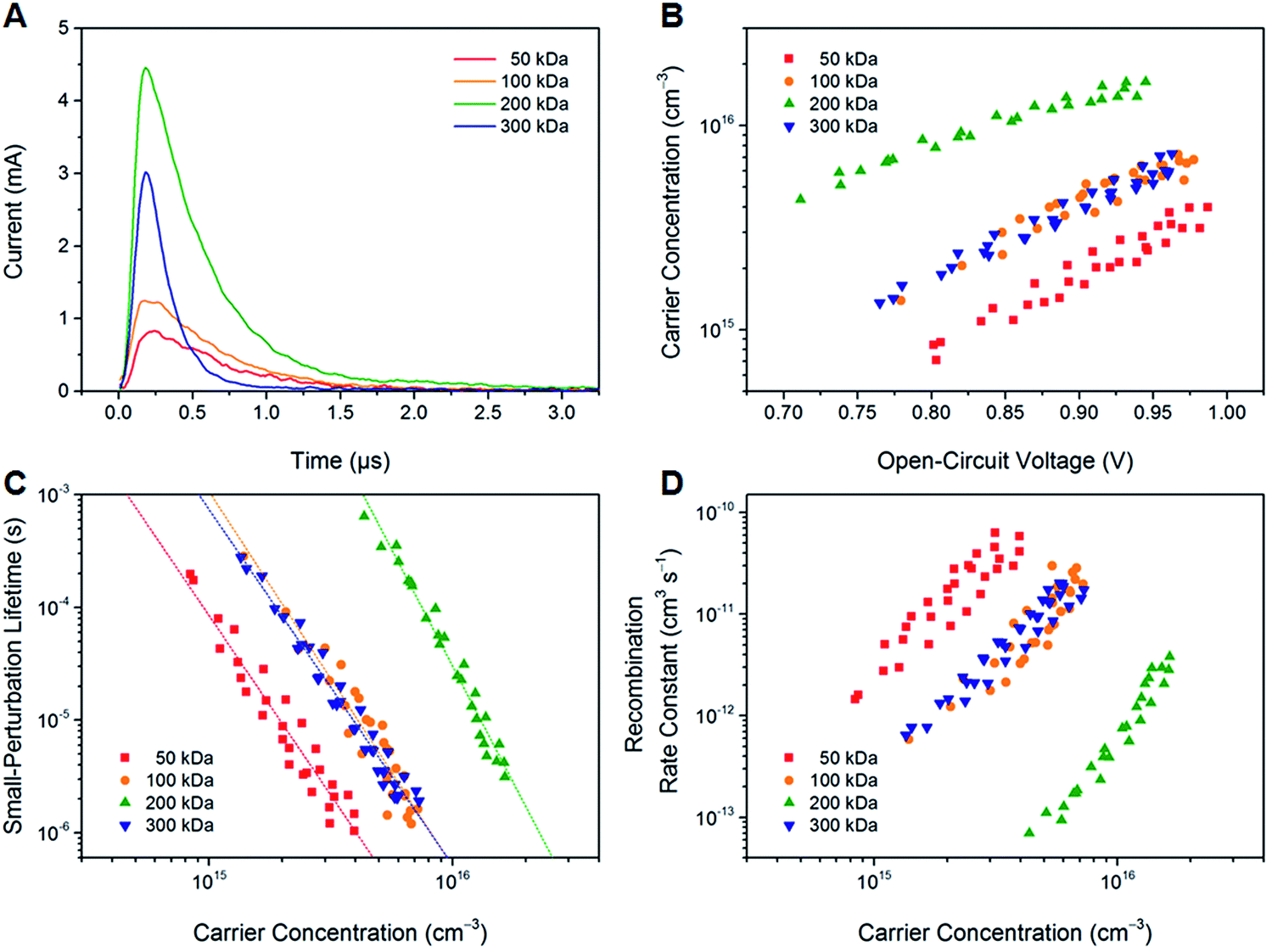 | ||
| Fig. 6 (A) BACE photocurrent transients measured at a light intensity close to one sun and an extraction voltage of −5 V. (B) Extracted charge carrier concentration n as a function of VOC. (C) Small-perturbation lifetime τΔn determined from TPV measurements versus carrier concentration n. Dotted lines indicate fits to a power law. (D) Non-geminate recombination rate constant krec as a function of the carrier concentration n. The data shown in panels (B–D) correspond to four individual devices for each molecular weight of PTB7-Th. | ||
In addition, the carrier lifetime was determined using TPV measurements. For this purpose, the illumination present during the BACE experiment was superimposed with a light pulse to add an extra amount of carriers Δn to the carrier concentration n. Light intensities were chosen such that Δn ≪ n. It was found that the decay dynamics after switching off the pulse were mono-exponential with the small-perturbation lifetime τΔn for all samples under investigation. Fig. 6C shows the dependence of τΔn on the total carrier concentration. At fixed n, the longest small-perturbation lifetimes were obtained for the devices implementing the 200 kDa polymer. To evaluate the overall carrier lifetime τ, the relationship τ = (λ + 1)τΔn was used,48 where λ is an empirical parameter representing the slope of the data in Fig. 6C. We find that λ varies between 3.2 and 4.1 for the samples investigated. The corresponding carrier lifetimes under one sun are 3.0 μs (50 kDa), 2.8 μs (100 kDa), 4.5 μs (200 kDa) and 3.2 μs (300 kDa). Again, the lifetime is maximal for the 200 kDa polymer sample. It should be noted that the parameter λ is also characteristic of the apparent recombination mechanism, since R(n) ∝ nλ+1, where R is the non-geminate recombination rate of photogenerated charge carriers. Hence, it can be concluded that the non-geminate recombination order in the PTB7-Th:O-IDTBR devices is larger than 2, which is in agreement with previous reports on similar systems.46,49 Such a behavior is often attributed to charge carrier trapping in exponential tail states.50 This explanation would be consistent with the finding that charge transport requires thermal activation (Fig. 3B). However, assuming the relationship λ = Ea/kBT,50 there is a quantitative disagreement with the energy values reported in Table 2. The difference might be related to the fact that the mobility was measured for pure PTB7-Th films, while BACE and TPV studies were performed on PTB7-Th:O-IDTBR blends. Hence, it appears likely that the presence of the O-IDTBR acceptor alters the density of states distribution of the polymer.
From the carrier lifetimes and concentrations, we calculated the non-geminate recombination rate constant krec according to krec = 1/nτ(n),51 as shown in Fig. 6D. The devices with the high molecular weight polymer (200 kDa) exhibit much lower recombination rate constants than the other samples. Nevertheless, in all cases, krec is reduced compared to the Langevin rate constant, kL = q/ε(μe + μh), where q is the elementary charge and ε is the dielectric constant. Depending on the exact value of the hole mobility, kL is in the order of 10−9 cm3 s−1. The reduction is still valid if we consider only the minimum mobility (here: the electron mobility), according to Koster et al.,52 which would result in a recombination rate constant of 3 × 10−10 cm3 s−1. Hence, it can be concluded from BACE and TPV measurements that the molecular weight has only a little effect on the recombination mechanism by itself (as indicated by the similar values of λ) but a large influence on the recombination strength. The best-performing polymer samples (200 kDa) were found to exhibit the lowest recombination rate constants resulting in the highest carrier concentration and carrier lifetime under normal operating conditions.
Conclusions
This study demonstrated a remarkable influence of the molecular weight of PTB7-Th on the photovoltaic performance of PTB7-Th:O-IDTBR non-fullerene organic solar cells. It was found that the performance change of polymer:non-fullerene solar cells is not primarily governed by the charge carrier mobility or the crystallinity of the conjugated polymer. Instead, parameters like activation energy for carrier transport in the conjugated polymer, carrier concentration and recombination rate were identified to strongly influence the performance.Even though the highest charge carrier mobilities were found for the 300 kDa polymer, the 200 kDa polymer showed the highest efficiency in the solar cells. The reasons for this behavior are (i) the 200 kDa polymer exhibited by far the lowest activation energy, and (ii) the devices based on the 200 kDa polymer had clearly the lowest recombination rate constants as well as the highest carrier concentrations and carrier lifetimes. This suggests that charge carrier trapping also plays a major role in polymer:non-fullerene acceptor organic solar cells.
The presented results highlight the importance of the molecular weight of polymers as a crucial factor for high-performance NF-OSCs. Thus, besides the chemical design of the polymer, the optimum molecular weight fraction has to be found as well in order to promote further advances in the field of polymer-based photovoltaics.
Experimental section
Chemicals and materials
PTB7-Th (low, medium, high and very high molecular weight, 50–300 kDa) and O-IDTBR were provided by 1-Material Inc. ITO-coated glass substrates (15 × 15 mm, 15 Ω sq−1) were purchased from Lumtec; zinc acetate dihydrate (Zn(CH3COO)2·2H2O, ≥99.5%) was obtained from Fluka; 2-propanol (≥99.8%) was procured from Roth; 2-methoxyethanol (99.8%, anhydrous), ethanolamine (≥98%), ortho-dichlorobenzene (99%), chloroform (≥99.9%), molybdenum(VI) oxide (99.98%, trace metals basis) were purchased from Sigma-Aldrich; silver (99.99%) was obtained from Kurt J. Lesker Company Ltd. All chemicals were used as received without further purification.Fabrication of non-fullerene organic solar cells
Pre-patterned ITO-coated glass substrates were cleaned by sonication in 2-propanol (60 min, 40–50 °C) and oxygen plasma treatment (FEMTO, Diener Electronic, 3 min). ZnO thin films acting as the electron transport layer were derived from a sol–gel reaction of a zinc oxide precursor solution.53 The zinc oxide precursor was prepared by dissolving zinc acetate dihydrate (0.5 g, 2.3 mmol) in 2-methoxyethanol (5 mL) using ethanolamine (150 μL, 2.5 mmol) as the stabilizer.53 The solution was vigorously stirred overnight under ambient conditions for the hydrolysis reaction, followed by filtration through a 0.45 μm PTFE syringe filter before spin coating (4000 rpm, 30 s). The ZnO films were annealed under ambient conditions (150 °C, 15 min) to achieve layer thicknesses in the range of 30–40 nm. PTB7-Th was dissolved in ortho-dichlorobenzene at 70 °C, blended with O-IDTBR in a 1:1.5 (w/w) donor:acceptor ratio (20.0–37.5 mg mL−1 total concentration), and spin-coated to obtain active layer thicknesses in the range of 80–90 nm. A molybdenum(VI) oxide anode interfacial layer (10 nm, deposition rate: ca. 0.1–0.2 Å s−1) and a silver anode (100 nm, 0.1–1.0 Å s−1) were deposited by thermal evaporation under reduced pressure (ca. 10−5 mbar) through a shadow mask, defining the active area (9 mm2).
Solar cell characterization
J–V curves were recorded under illuminated and dark conditions in an inert atmosphere using a Keithley 2400 source meter and a Dedolight DLH400D xenon lamp (1000 W m−2, AM 1.5 G), which was calibrated with a standard reference silicon solar cell (Fraunhofer ISE). A metal mask (2.9 × 2.9 mm) was used to define the illuminated area to be 8.41 mm2. The solar cells were illuminated continuously for 5–10 min (1000 W m−2, AM 1.5 G) prior to the measurement of the J–V curves (Fig. S2†). Photovoltaic characteristic parameters such as VOC, JSC, FF, and PCE were determined from the J–V curves under illumination and were averaged over ten devices. Series (RS) and shunt (RSH) resistance values were extracted from the J–V curves under illumination. EQE measurements were performed under ambient conditions using a MuLTImode 4-AT monochromator (Amko) equipped with a 75 W xenon lamp (LPS 210-U, Amko), a lock-in amplifier (Stanford Research Systems, Model SR830), and a Keithley 2400 source meter. The monochromatic light was chopped at a frequency of 30 Hz, and constant background illumination was provided by white light-emitting diodes (LEDs). The EQE spectra were measured in the wavelength range of 350–1000 nm (increment: 10 nm). The measurement set-up was spectrally calibrated with a reference photodiode (Newport Corporation, 818-UV/DB).Thin film characterization
Absorption spectra were recorded on a UV-1800 UV-vis spectrophotometer (Shimadzu) in the range of 350–1000 nm. Absorption coefficients were determined from thin films deposited by spin coating from ortho-dichlorobenzene solutions. Surface profilometry measurements were performed on a DektakXT stylus surface profiling system (Bruker) equipped with a 12.5 μm-radius stylus tip in order to determine the layer thickness of the thin film samples. Line scans were recorded over a length of 1000 μm, with a stylus force of 3 mg, and a resolution of 0.33 μm pt−1. Layer thickness values were derived from two-dimensional surface profiles using Vision 64 software (Bruker).Charge carrier mobility
Organic field-effect transistors with a bottom-contact bottom-gate configuration (IPMS Fraunhofer Institute) were used consisting of n++-Si substrates with 230 nm of thermally grown SiO2 as the gate dielectric (15 nF capacitance Ci) and pre-patterned pairs of gold electrodes with an interdigitated geometry as the source and drain. The channel length and width were 20 μm and 10 mm, respectively. PTB7-Th thin films were spin-coated from ortho-dichlorobenzene to achieve layer thicknesses of 60 nm. For each polymer sample, four different OFETs were characterized. All solutions, samples and devices were prepared and measured in a N2-filled glovebox with H2O and O2 levels below 10 ppm to avoid oxidative doping of the materials and ensure reproducibility of the experiments. The device characteristics were measured by contacting the source, drain and gate electrodes and applying different voltages to obtain transconductance (IDS–VGS) graphs. All measurements were performed using Keithley devices (Keithley 2400 source meter and Keithley 6487 picoammeter). Each IDS–VGS graph (transfer curve) was constructed by sweeping the gate voltages from +40 V to −40 V and back to +40 V, with one measurement every 2 V. The drain voltage was either −5 V (linear regime) or −40 V (saturation regime). Transfer curves were plotted and fitted to extract the value of the mobility (μ) and the threshold voltage (Vth). The transfer curves IDS–VGS were used to extract the value of the ION/IOFF ratio by dividing the value of the observed current in the saturation regime when the transistor is on (VGS = −20 V) by the value of the current when the transistor is off (VGS = +20 V). Experimental data were analyzed using standard field-effect transistor equations. For temperature-dependent field-effect mobility measurements, the device characteristics were measured by sweeping the temperature from −20 °C to 60 °C and back from 50 °C to −10 °C using a temperature step of 20 °C with the device temperature being controlled using a Peltier element. Details of the calculation of the Arrhenius-type activation energy (Ea) are provided in the ESI.†Grazing incidence X-ray diffraction (GIXRD)
XRD measurements of PTB7-Th thin films deposited on silicon substrates were performed on a PANalytical Empyrean diffractometer (Cu Kα radiation, λ = 1.5406 Å) in grazing incidence configuration in coplanar geometry using a fixed incidence angle of 0.29° well above the critical angle of the polymer measured at 0.171°. The scattered intensity was measured with a PIXcel3D 1 × 1 detector in scanning line mode.Atomic force microscopy (AFM)
Surface morphology and roughness measurements of the PTB7-Th:O-IDTBR films on glass/ITO/ZnO substrates were performed on a Veeco Multimode Quadrax MM atomic force microscope (Bruker) in tapping mode using non-coated Si-cantilevers (NCH-VS1-W, NanoWorld AG) with a resonance frequency of 291 kHz and a force constant of 42 N m−1. All measurements were acquired at room temperature under ambient conditions. Root-mean-square roughness calculations and image processing were done using Nanoscope software (V7.30r1sr3, Veeco).Bias-assisted charge extraction (BACE) and transient photovoltage (TPV)
Carrier concentrations were estimated using the BACE technique, which is described elsewhere.54 In brief, the samples were illuminated with a Seoul P4 LED (wavelength: 625 nm) until reaching the steady state. During illumination, the samples were held at a fixed bias, which corresponds exactly to the VOC at the given light intensity. The bias was applied using a pulse generator (Agilent 81150A). When the LED was switched off (fall time <200 ns), the applied voltage was simultaneously changed to the reverse direction to accelerate the charge carrier extraction. The current transient resulting from the extraction process was recorded with a digital storage oscilloscope (Tektronix DPO7104) using an input impedance of 50 Ω. To account for the parasitic capacitance of the samples, the experiment was repeated without illumination using the same voltage step and zero pre-bias. The original signal was then corrected with the signal from the dark measurements. The charge carrier concentration n was calculated via , where j(t) is the corrected current density, q is the elementary charge, A is the device area, and d is the thickness of the active layer. For TPV measurements, the constant illumination by the LED was superimposed with a light pulse of low intensity. The decay of the open-circuit voltage after switching off the pulse was recorded with the oscilloscope (input impedance: 1 MΩ). The experiment was repeated for different values of VOC, corresponding to different intensities of the continuous illumination. Voltage transients were fitted to ΔVOC ∝ exp(−t/τΔn) to yield the small-perturbation lifetime.
, where j(t) is the corrected current density, q is the elementary charge, A is the device area, and d is the thickness of the active layer. For TPV measurements, the constant illumination by the LED was superimposed with a light pulse of low intensity. The decay of the open-circuit voltage after switching off the pulse was recorded with the oscilloscope (input impedance: 1 MΩ). The experiment was repeated for different values of VOC, corresponding to different intensities of the continuous illumination. Voltage transients were fitted to ΔVOC ∝ exp(−t/τΔn) to yield the small-perturbation lifetime.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was carried out within the project “SolaBat – Solar cell meets battery – Realization of a hybrid energy system” funded by the Austrian “Climate and Energy Fund” within the program Energy Emission Austria (FFG No. 853 627). This work was funded in part by the Slovenian Research Agency through the research program P1-0055. N. P. acknowledges the Slovene Human Resources Development and Scholarship Fund. The research was also supported by the project RETINA, which is being implemented and co-financed by the European Union – European Regional Development Fund in the frame of the Cooperation Programme Interreg V-A Slovenia-Austria in the programme period 2014–2020. D. S. and S. W. thank Jürgen Parisi for constant support.Notes and references
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803 CrossRef CAS PubMed.
- N. Liang, W. Jiang, J. Hou and Z. Wang, Mater. Chem. Front., 2017, 1, 1291 RSC.
- W. Chen and Q. Zhang, J. Mater. Chem. C, 2017, 5, 1275 RSC.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X.-Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- W. Huang, E. Gann, L. Thomsen, C. Dong, Y.-B. Cheng and C. R. McNeill, Adv. Energy Mater., 2015, 5, 1401259 CrossRef.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119 CrossRef CAS PubMed.
- C. Zhan, X. Zhang and J. Yao, RSC Adv., 2015, 5, 93002 RSC.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- P. Schilinsky, U. Asawapirom, U. Scherf, M. Biele and C. J. Brabec, Chem. Mater., 2005, 17, 2175 CrossRef CAS.
- I. Osaka, M. Saito, H. Mori, T. Koganezawa and K. Takimiya, Adv. Mater., 2012, 24, 425 CrossRef CAS PubMed.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345 CrossRef CAS.
- Z. Li, D. Yang, X. Zhao, T. Zhang, J. Zhang and X. Yang, Adv. Funct. Mater., 2018, 1705257 CrossRef.
- C. Liu, K. Wang, X. Hu, Y. Yang, C.-H. Hsu, W. Zhang, S. Xiao, X. Gong and Y. Cao, ACS Appl. Mater. Interfaces, 2013, 5, 12163 CAS.
- H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359 CrossRef CAS PubMed.
- N. Zhou, A. S. Dudnik, T. I. N. G. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. Olvera De La Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240 CrossRef CAS PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038 CrossRef CAS PubMed.
- M. Koppe, C. J. Brabec, S. Heiml, A. Schausberger, W. Duffy, M. Heeney and I. McCulloch, Macromolecules, 2009, 42, 4661 CrossRef CAS.
- A. M. Ballantyne, L. Chen, J. Dane, T. Hammant, F. M. Braun, M. Heeney, W. Duffy, I. McCulloch, D. D. C. Bradley and J. Nelson, Adv. Funct. Mater., 2008, 18, 2373 CrossRef CAS.
- F. Bencheikh, D. Duché, C. M. Ruiz, J.-J. Simon and L. Escoubas, J. Phys. Chem. C, 2015, 119, 24643 CAS.
- S. Zhang, L. Ye, W. Zhao, D. Liu, H. Yao and J. Hou, Macromolecules, 2014, 47, 4653 CrossRef CAS.
- D. Wang, F. Zhang, L. Li, J. Yu, J. Wang, Q. An and W. Tang, RSC Adv., 2014, 4, 48724 RSC.
- C. Cui, W.-Y. Wong and Y. Li, Energy Environ. Sci., 2014, 7, 2276 CAS.
- L. Ye, W. Jiang, W. Zhao, S. Zhang, Y. Cui, Z. Wang and J. Hou, Org. Electron., 2015, 17, 295 CrossRef CAS.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Energy Environ. Sci., 2014, 7, 2939 CAS.
- K. D. Deshmukh, S. K. K. Prasad, N. Chandrasekaran, A. C. Y. Liu, E. Gann, L. Thomsen, D. Kabra, J. M. Hodgkiss and C. R. McNeill, Chem. Mater., 2017, 29, 804 CrossRef CAS.
- Q. Cao, W. Xiong, H. Chen, G. Cai, G. Wang, L. Zheng and Y. Sun, J. Mater. Chem. A, 2017, 5, 7451 CAS.
- J. Huang, J. H. Carpenter, C.-Z. Li, J.-S. Yu, H. Ade and A. K.-Y. Jen, Adv. Mater., 2016, 28, 967 CrossRef CAS PubMed.
- Q. Wan, X. Guo, Z. Wang, W. Li, B. Guo, W. Ma, M. Zhang and Y. Li, Adv. Funct. Mater., 2016, 26, 6635 CrossRef CAS.
- Q. Liu, P. Romero-Gomez, P. Mantilla-Perez, S. Colodrero, J. Toudert and J. Martorell, Adv. Energy Mater., 2017, 7, 1700356 CrossRef.
- J. Sun, Z. Zhang, X. Yin, J. Zhou, L. Yang, R. Geng, F. Zhang, R. Zhu, J. Yu and W. Tang, J. Mater. Chem. A, 2018, 6, 2549–2554 CAS.
- Z. Xiao, X. Jia, D. Li, S. Wang, X. Geng, F. Liu, J. Chen, S. Yang, T. P. Russell and L. Ding, Sci. Bull., 2017, 62, 1494 CrossRef.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2016, 16, 363 CrossRef PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298 CrossRef CAS PubMed.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth, N. Gasparini, P. Kaienburg, H. Yan, A. Amassian, C. J. Brabec, J. Durrant and I. McCulloch, Energy Environ. Sci., 2016, 9, 3783 CAS.
- J. A. Bartelt, D. Lam, T. M. Burke, S. M. Sweetnam and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500577 CrossRef.
- S. Badgujar, C. E. Song, S. Oh, W. S. Shin, S.-J. Moon, J.-C. Lee, I. H. Jung and S. K. Lee, J. Mater. Chem. A, 2016, 4, 16335 CAS.
- W. Ma, J. Y. Kim, K. Lee and A. J. Heeger, Macromol. Rapid Commun., 2007, 28, 1776 CrossRef CAS.
- Y. Lei, J. Sun, J. Yuan, J. Gu, G. Ding and W. Ma, J. Mater. Sci. Technol., 2017, 33, 411 Search PubMed.
- J. Kuwabara, T. Yasuda, N. Takase and T. Kanbara, ACS Appl. Mater. Interfaces, 2016, 8, 1752 CAS.
- R. Singh, J. Lee, M. Kim, P. E. Keivanidis and K. Cho, J. Mater. Chem. A, 2017, 5, 210 CAS.
- J.-F. Chang, J. Clark, N. Zhao, H. Sirringhaus, D. W. Breiby, J. W. Andreasen, M. M. Nielsen, M. Giles, M. Heeney and I. McCulloch, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 115318 CrossRef.
- A. Maurano, C. G. Shuttle, R. Hamilton, A. M. Ballantyne, J. Nelson, W. Zhang, M. Heeney and J. R. Durrant, J. Phys. Chem. C, 2011, 115, 5947 CAS.
- A. Foertig, J. Kniepert, M. Gluecker, T. Brenner, V. Dyakonov, D. Neher and C. Deibel, Adv. Funct. Mater., 2014, 24, 1306 CrossRef CAS.
- T. Kirchartz, B. E. Pieters, J. Kirkpatrick, U. Rau and J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115209 CrossRef.
- C. G. Shuttle, B. O'Regan, A. M. Ballantyne, J. Nelson, D. D. C. Bradley and J. R. Durrant, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 113201 CrossRef.
- L. J. A. Koster, V. D. Mihailetchi and P. W. M. Blom, Appl. Phys. Lett., 2006, 88, 052104 CrossRef.
- Y. Sun, J. H. Seo, C. J. Takacs, J. Seifter and A. J. Heeger, Adv. Mater., 2011, 23, 1679 CrossRef CAS PubMed.
- J. Kniepert, I. Lange, N. J. van der Kaap, L. J. A. Koster and D. Neher, Adv. Energy Mater., 2014, 4, 1301401 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional photovoltaic performance parameters, J–V curves, UV-vis absorption spectra, optical properties, charge carrier mobility and GIXRD data. See DOI: 10.1039/c8ta02467g |
| This journal is © The Royal Society of Chemistry 2018 |