
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High performance nanostructured bismuth oxide–cobaltite as a durable oxygen electrode for reversible solid oxide cells†
Na
Ai‡
ab,
Minle
Chen‡
a,
Shuai
He
c,
Kongfa
Chen
 *a,
Teng
Zhang
*a and
San Ping
Jiang
*c
*a,
Teng
Zhang
*a and
San Ping
Jiang
*c
aCollege of Materials Science and Engineering, Fuzhou University, Fuzhou, Fujian 350108, China. E-mail: kongfa.chen@fzu.edu.cn; Teng_Zhang@fzu.edu.cn; Fax: +86 591 22866537; Tel: +86 591 22866540
bTesting Center, Fuzhou University, Fuzhou, Fujian 350108, China
cFuels and Energy Technology Institute, Department of Chemical Engineering, Curtin University, Perth, WA 6102, Australia. E-mail: S.Jiang@curtin.edu.au; Fax: +61 8 9266 1138; Tel: +61 8 9266 9804
First published on 12th March 2018
Abstract
The high reactivity between bismuth oxide and cobaltite oxygen electrodes is a bottleneck in developing active and reliable bismuth oxide–cobaltite composite oxygen electrodes for solid oxide cells (SOCs). Herein, a Sr-free Sm0.95Co0.95Pd0.05O3−δ (SmCPd) oxygen electrode decorated with nanoscale Er0.4Bi1.6O3 (ESB) is synthesized and assembled on a barrier-layer-free Y2O3–ZrO2 (YSZ) electrolyte film. The cell with the ESB decorated SmCPd composite oxygen electrode exhibits a peak power density of 1.81 W cm−2 at 750 °C and 0.58 W cm−2 at 650 °C. More importantly, excellent operating stability is achieved in the fuel cell mode at 600 °C for 500 h, and in electrolysis and reversible modes at 750 °C for over 200 h. The results demonstrate the feasibility of applying bismuth oxide–cobaltite composite oxygen electrodes in developing high-performance and durable SOCs.
1. Introduction
The depletion of fossil fuels and the global climate crisis necessitate the development of innovative technologies to efficiently use renewable energy sources such as nuclear, solar and wind power. Solid oxide cells (SOCs) are such a device of choice for efficient storage and regeneration of renewable sources by alternately operating in solid oxide electrolysis cell (SOEC) and solid oxide fuel cell (SOFC) modes.1–4 Manganite and cobaltite based perovskite oxides such as La0.8Sr0.2MnO3+δ (LSM) and La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) are the most investigated oxygen electrodes for SOCs.5–9 LSM is predominantly an electronic conductor without ionic conductivity and it is favourable for use at high temperatures (800 °C and above). On the other hand, cobaltite oxides with mixed ionic/electronic conductivity (MIEC) are ideal for application at intermediate temperatures (600–800 °C). However, the surface segregation of Sr-containing cobaltite oxygen electrodes and the combination of segregated Sr with volatile species such as chromium and sulphur constitute a major degradation mechanism of SOC stacks.10–17 One straightforward means to address this critical issue is to develop Sr-free cobaltite electrodes,18 such as La(Co,Fe)xPd1−xO3−δ.19,20 Recently, we have successfully developed a high performance Sr-free Sm0.95CoO3−δ electrode doped with 5% Pd in the B-site of the perovskite structure (SmCPd) and assembled it on a barrier-layer-free Y2O3–ZrO2 (YSZ) electrolyte.21 The combination of SmCPd with an ion-conducting phase such as Gd-doped CeO2 (GDC) leads to remarkably enhanced electrocatalytic activity for the oxygen reduction reaction (ORR).21Cubic bismuth oxides such as Er and Y stabilized bismuth oxides (ESB and YSB) possess excellent surface exchange capability and the highest ionic conductivity among the electrolyte materials of SOCs.22,23 However, due to its phase instability in reducing environments, bismuth oxide is mainly applied as an interfacial layer between the electrolyte and oxygen electrode,24–26 as well as an active promoter for LSM24,27–35 and Ag electrodes.36–38 On the other hand, incorporation of bismuth oxide into cobaltite electrodes is rarely reported, possibly due to the high reactivity between bismuth oxide and cobaltite at low temperatures.24,39–41 For example, Lee et al. observed significant new phase formation after heat-treating the LSCF–ESB mixture at 850 °C,24 while Li et al. reported that LSCF started to react with YSB at 650 °C.41 On the other hand, Li et al. incorporated YSB into LSCF scaffolds by infiltration and heat-treatment at a low temperature of 600 °C, leading to a remarkably enhanced electrocatalytic activity for proton conducting SOFCs.41
Herein, we report the development of a nanostructured ESB decorated Sr-free SmCPd composite oxygen electrode, which is applied on a barrier-layer-free YSZ electrolyte via a direct assembly approach. The incorporation of the highly ion conductive ESB phase is expected to remarkably enhance the electrocatalytic activity of SmCPd, similar to the case of adding GDC.21 Our recent report has demonstrated the feasibility of preparing ESB decorated LSM at a low temperature of 600 °C.33 Furthermore, the direct assembly approach involves the in situ fabrication of oxygen electrodes under the operating conditions of SOCs (700–800 °C) by applying electrochemical polarization.42–44 Therefore, the adoption of low temperature powder synthesis and in situ electrode fabrication processes at 600–800 °C may minimize the potential chemical reaction between ESB and SmCPd, making it possible to develop a bismuth oxide–cobaltite composite oxygen electrode. The results demonstrate that the nanostructured ESB decorated SmCPd oxygen electrode is high performing and stable for running in fuel cell, electrolysis and reversible modes.
2. Experimental section
2.1 Fabrication of supported electrolyte films
Hydrogen electrode-supported YSZ films were fabricated by slurry spin coating. The hydrogen electrode supports were composed of NiO (J.T. Baker), YSZ (TZ-8Y, Tosoh) and tapioca in a mass ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:20, and the hydrogen electrode functional layer (HEFL) was composed of NiO and YSZ in a mass ratio of 50:50. The supported YSZ films were co-fired at 1450 °C in air for 5 h. The diameter of the as-sintered button cells was 14.5 mm. The thickness of the hydrogen electrode support, HEFL layer and YSZ film was 0.8 mm, 14 μm and 12 μm, respectively. The geometric surface area of the oxygen electrode was 0.23 cm2. The detailed fabrication processes are available elsewhere.45,46
:40:20, and the hydrogen electrode functional layer (HEFL) was composed of NiO and YSZ in a mass ratio of 50:50. The supported YSZ films were co-fired at 1450 °C in air for 5 h. The diameter of the as-sintered button cells was 14.5 mm. The thickness of the hydrogen electrode support, HEFL layer and YSZ film was 0.8 mm, 14 μm and 12 μm, respectively. The geometric surface area of the oxygen electrode was 0.23 cm2. The detailed fabrication processes are available elsewhere.45,46
2.2 Synthesis of powders and preparation of electrodes
Sm0.95Co0.95Pd0.05O3 (SmCPd) and Er0.4Bi1.6O3 (ESB) powders were synthesized using a modified Pechini method, with the chemicals of Sm(NO3)3·6H2O (99%, Sigma-Aldrich), Co(NO3)2·6H2O (98.0–102.0%, Alfa-Aesar), Pd(NO3)2 (10 wt% aqueous solution, Alfa Aesar), Er(NO3)3·5H2O (99.9%, Aldrich), Bi(NO3)3·5H2O (98%, Sigma-Aldrich), citric acid (CA, 99.5%, Chem Supply), ethylenediaminetetraacetic acid (EDTA, 99%, Acros Organics) and ammonia solution (28%, Ajax Finechem). The metal ions/CA/EDTA was fixed in a molar ratio of 1:1.5:1. The aqueous solution was stirred on a hot plate and the gel was heat-treated at 180 °C in an oven for 12 h. The SmCPd and ESB powders were calcined at 900 °C and 600 °C in air for 2 h, respectively.
40 wt% ESB decorated 60 wt% SmCPd (decorated ESB–SmCPd) composite powder was prepared using the same modified Pechini method mentioned above. A 0.15 M ESB aqueous precursor solution containing Er(NO3)3, Bi(NO3)3, CA, EDTA and ammonia (the same purities and suppliers as above) was prepared in a beaker. The as-synthesized SmCPd powder was placed into the beaker, magnetically stirred and heated on a hot plate to evaporate the excess water. The solution was stirred continuously until a viscous gel was obtained. The black gel was thoroughly dried at 180 °C for 12 h and the resultant ash was ground in an agate mortar, followed by calcination at 600–750 °C in air for 2 h to obtain the decorated ESB–SmCPd composite powder. The details of the decoration process have been described elsewhere.33
The ESB–SmCPd powder was combined with an ink vehicle (Fuel Cell Materials) in a mass ratio of 50:50 to form an electrode paste. The paste was coated on the YSZ electrolyte films via direct assembly by simply drying at 100 °C in air for 2 h without an additional pre-sintering process at a high temperature as in the case of conventional electrode fabrication processes.44,47 Pt paste (Gwent Electronic Materials Ltd) was painted on the electrodes as a current collector and heat-treated at 150 °C in air for 2 h.
2.3 Cell testing
The button cells were sealed on ceramic tubes using a sealant (Ceramabond 552, Aremco Products Inc.) and dried at room temperature in air for at least 12 h. Electrochemical polarization curves and impedance spectra of the cells were measured using a Gamry Potentiostat Reference 3000 and Interface 1000 in a temperature range of 600–750 °C. The hydrogen electrode was flowed with H2, H2–H2O, H2–CO2 or H2–H2O–CO2 at a rate of 25–50 ml min−1, and the oxygen electrode was open to static air. Steam was generated using a humidifier and to avoid the condensation, the stainless steel gas line was wrapped with heating tape and the temperature was maintained at 105 °C. The operating stability of the cells was investigated at a constant current density of 0.5 A cm−2 at 750 °C in the fuel cell mode, electrolysis mode and reversible mode and of 0.25 A cm−2 at 600 °C in the fuel cell mode. The impedance spectra were collected with a 10 mV AC signal applied from 100 kHz to 0.01 Hz under open circuit conditions. Three identical cells were tested to confirm the reproducibility, and the testing history and initial peak power densities are summarized in Table 1.| Cell's denotation | Cell-#1 | Cell-#2 | Cell-#3 |
|---|---|---|---|
| Testing history | Step 1. Polarization at 750 °C for 20 h in the fuel cell mode (Fig. 3a and b) | Step 1. Polarization at 750 °C for 20 h in the fuel cell mode (Fig. S6a, ESI) | Step 1. Polarization at 750 °C for 20 h in the fuel cell mode (Fig. S6b, ESI) |
| Step 2. Testing of I–V–P curves at 750–550 °C (Fig. 3c) | Step 2. Testing of I–V–P curves in varied atmospheres at 750 °C (Fig. 4) | Step 2. Polarization at 750 °C for 300 h in the fuel cell mode (Fig. 5a and c) | |
| Step 3. Polarization at 600 °C for 500 h in the fuel cell mode (Fig. 5b and d) | Step 3. Polarization at 750 °C for 80 h in the electrolysis mode and for 132 h in the reversible mode (Fig. 6) | ||
| PPD at 750 °C, polarized for 0 h/W cm−2 | 1.21 | 1.08 | 1.22 |
| PPD at 750 °C, polarized for 20 h/W cm−2 | 1.81 | 1.73 | 1.72 |
2.4 Phase, microstructure and elemental characterization
The phase of as-synthesized powders was examined using a Bruker D8 Advance X-ray diffractometer with a Cu Kα X-ray source. The morphologies of ESB and SmCPd powders, the SmCPd electrode and decorated ESB–SmCPd electrodes were characterized using field emission scanning electron microscopy (FESEM, Zeiss Neon 40EsB and Supra-55 Sapphire) combined with energy dispersive spectroscopy (EDS). The morphology and elemental distribution of the decorated ESB–SmCPd powder were measured using a high angle annular dark field scanning transmission electron microscope (HAADF-STEM, FEI Titan G2 80-200 TEM/STEM with ChemiSTEM Technology). The powders were mounted on copper grids for the SEM observations. In order to characterize the electrolyte surface after polarization, the oxygen electrodes were removed from the YSZ electrolyte surface by treatment in 32% HCl solution at room temperature for 24 h. To investigate the electrode/electrolyte interface, a lamella was milled across the tested ESB–SmCPd/YSZ interface region using an FEI Helios Nanolab G3 CX DualBeam focused ion beam (FIB)-SEM. Microstructural observation and elemental mapping analysis of the lamella were performed using the aforementioned HAADF-STEM.3. Results and discussion
3.1 Initial phase and microstructure
Fig. 1 shows the SEM micrographs of SmCPd and ESB powders, the SmCPd electrode, and the ESB–SmCPd electrode and powder. The SmCPd powder calcined at 900 °C is characterized by spherical particles in a size range of 170–600 nm (370 nm on average, Fig. 1a), while the ESB powder calcined at a lower temperature of 600 °C is irregularly shaped and in a greater size range of 20–1500 nm (630 nm on average, Fig. 1b). The greater size of the ESB powder than that of the SmCPd powder is due to the low melting point of bismuth oxide (824 °C).48 In the case of the directly assembled ESB–SmCPd electrode, the electrode is characterized by a very fine structure with ESB nanoparticles in a size range of 40–100 nm (60 nm on average, Fig. 1c), very different from the submicron structure of the directly assembled pristine SmCPd electrode (Fig. 1d and S1, ESI†). This indicates the successful decoration of SmCPd particles by the nanoscale ESB phase, which is confirmed by the HAADF micrograph and EDS maps of the ESB decorated SmCPd powder (Fig. 1e). The particle size of ESB in the decorated ESB–SmCPd electrode is much smaller as compared to that of the individual ESB powder, indicating that the presence of SmCPd has a remarkable inhibiting effect on the agglomeration and grain growth of ESB. Quantitative EDS analysis on the ESB–SmCPd electrode surface reveals that the relative cation ratios are close to those of the experimental target (Fig. S2, ESI†). | ||
| Fig. 1 SEM micrographs of the (a) SmCPd powder calcined at 900 °C, (b) ESB powder calcined at 600 °C, directly assembled (c) decorated ESB–SmCPd electrode and (d) pristine SmCPd electrode after heat-treatment at 600 °C. (e) HAADF micrograph and EDS maps of the decorated ESB–SmCPd powder. | ||
Fig. 2a and b show the XRD patterns and Rietveld refinement of as-synthesized powders. In the case of individual SmCPd and ESB powders, pure orthorhombic perovskite and cubic fluorite phases are obtained without impurity phases, respectively (Fig. 2a). For the decorated ESB–SmCPd powders calcined at 600 °C and 750 °C, in addition to the expected diffraction peaks from ESB and SmCPd, there are additional peaks related to a new phase of Bi2SmO4 (Fig. 2a). The formation of the new phase is also confirmed by Raman spectra (Fig. S3, ESI†). The occurrence of a reaction at a calcination temperature as low as 600 °C indicates that ESB is highly reactive with SmCPd. The phase identification is also confirmed by the Rietveld refinement on the decorated ESB–SmCPd powder calcined at 750 °C (Fig. 2b), which estimates the mass ratio of ESB/SmCPd/Bi2SmO4 to be 17:51:32. In such a case, the mass ratio of ESB relative to ESB + SmCPd is 25 wt%, lower than 40 wt% of the target composition. This is due to the fact that the formation of Bi2SmO4 needs more Bi from ESB than Sm from SmCPd.
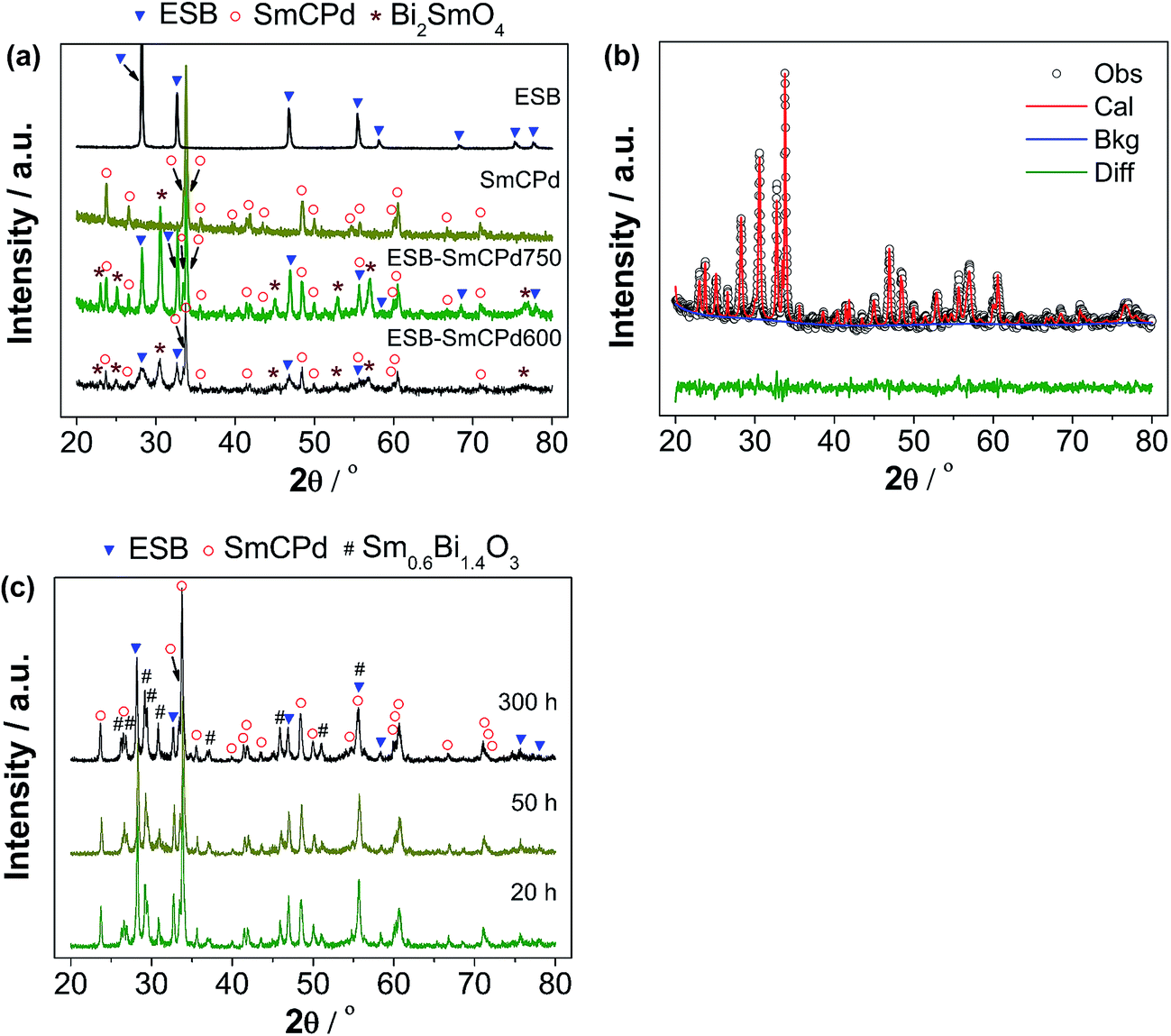 | ||
| Fig. 2 (a) XRD patterns of ESB and SmCPd powders, and the ESB decorated SmCPd composite powder calcined at 600 and 750 °C for 2 h (ESB–SmCPd600 and ESB–SmCPd750), (b) Rietveld refinement of the XRD patterns of ESB–SmCPd750, and (c) the ESB decorated SmCPd composite powder after heat-treatment at 750 °C in air for 20–300 h. | ||
The decorated ESB–SmCPd composite powder was heat-treated at 750 °C in air for 20–300 h, and the XRD patterns are shown in Fig. 2c. After the heat-treatment for 20 h and longer, Bi2SmO4 disappears and a new phase of Sm0.6Bi1.4O3 is formed, while the SmCPd and ESB phases are still observable. This indicates that Bi2SmO4 is most likely metastable, as is also confirmed by the presence of a predominant Sm0.6Bi1.4O3 phase when we tried to synthesize the Bi2SmO4 phase using the modified Pechini method (Fig. S4, ESI†). Nevertheless, Sm0.6Bi1.4O3 has been reported to possess reasonable ionic conductivity.49
3.2 Electrochemical performance and stability
Although Bi2SmO4 is formed in the as-synthesized ESB–SmCPd composite powder, the ESB–SmCPd composite electrode exhibits outstanding electrocatalytic activity. Fig. 3 shows the polarization performance of a hydrogen electrode-supported YSZ film cell with a directly assembled decorated ESB–SmCPd oxygen electrode (Cell-#1) as a function of polarization time at 0.5 A cm−2 and 750 °C. The cell produces an open circuit voltage of 1.12 V, close to the theoretical value, demonstrating that the YSZ electrolyte film is dense and crack-free with excellent gas impermeability. The initial peak power density (PPD) is 1.21 W cm−2 and rises to 1.81 W cm−2 after polarization at 750 °C for 20 h (Fig. 3a). The increase of voltage by the polarization is particularly remarkable in high current regions above 2 A cm−2 (Fig. 3a). | ||
| Fig. 3 Performance curves of the cell with the directly assembled, decorated ESB–SmCPd electrode as a function of polarization time at 0.5 A cm−2 and 750 °C. (a) Polarization curves and (b) impedance spectra; the numbers are frequency in Hz and the lines are fitting data obtained by the equivalent circuit as the inset. (c) Polarization curves as a function of temperature; prior to the test, the cell was polarized at 0.5 A cm−2 and 750 °C for 20 h. | ||
The increase of PPD is primarily related to the decrease of electrode polarization resistance (RP), as the cell ohmic resistance (RΩ) remains constant at 0.07 Ω cm2. The initial RP is 0.52 Ω cm2 and decreases to 0.43 Ω cm2 after polarization for 20 h (Fig. 3b). RΩ is mainly contributed by the ohmic resistances of the electrolyte, electrodes and contact at the interfaces of electrode/electrolyte and electrode/current collector. The insensitivity of the RΩ of the decorated ESB–SmCPd electrode to the polarization most likely indicates the instantaneous formation of the electrode/electrolyte interface of the directly assembled ESB–SmCPd electrode during the test. This is very different from the significant decrease of RΩ in the case of directly assembled pristine SmC and SmCPd–GDC composite electrodes during the polarization.21
The impedance spectra consist of two distinctly separable, depressed arcs at a high frequency of ∼1000 Hz and a low frequency of ∼0.1 Hz (Fig. 3b), indicating that there are at least two electrode processes. The electrode processes at high and low frequencies can be divided by fitting the impedance spectra using an equivalent circuit (see the inset of Fig. 3b). In the equivalent circuit, L is related to an inductance, Q and R represent the constant phase element and electrode polarization resistance, respectively, and subscripts H and L represent the high and low frequency arcs, respectively. The initial RH and RL are 0.29 and 0.23 Ω cm2, and decrease to 0.22 and 0.21 Ω cm2 after polarization at 750 °C for 20 h, respectively. Plenty of evidence including our previous studies has demonstrated that the high frequency arc is related to the reaction on the oxygen electrode, while the low frequency arc is related to the reaction on the hydrogen electrode.16,50–52 Thus the pronounced decrease of RH indicates that the increase of cell performance is mainly due to the enhanced electrocatalytic activity of the decorated ESB–SmCPd oxygen electrode during the polarization. The performance enhancement of the oxygen electrode is probably due to the conversion of Bi2SmO4 to Sm0.6Bi1.4O3 during the test at 750 °C (see the XRD data in Fig. 2c).
After polarization at 0.5 A cm−2 and 750 °C for 20 h, the cell polarization curves were measured as a function of temperature. The cell produces PPDs of 1.81, 1.12, 0.58, 0.27 and 0.12 W cm−2 at 750, 700, 650, 600 and 550 °C, respectively (Fig. 3c). The PPD of 1.81 W cm−2 at 750 °C is substantially higher than that of 0.60 W cm−2 in the case of the directly assembled pristine SmCPd electrode (Fig. S5, ESI†), indicating that the decorated ESB nanoparticles are very effective in enhancing the electrocatalytic activity of SmCPd.
Fig. 4 shows the polarization curves of another identical cell (Cell-#2) with the decorated ESB–SmCPd electrode in fuel cell and electrolysis modes with the hydrogen electrodes exposed to different atmospheres. Prior to the test, the cell was polarized at 0.5 A cm−2 and 750 °C for 20 h in the fuel cell mode (Table 1 and Fig. S6a, ESI†). In the fuel cell mode, the cell voltage decreases with increasing concentration of H2O and CO2, and there is an abrupt decrease of voltage at high currents related to concentration polarization when the concentration of H2O or CO2 exceeds 50% (Fig. 4a), due to the excess dilution of reactant (H2) for the reaction in the hydrogen electrode. In contrast, in the electrolysis mode, the cell performance is enhanced with the increase of concentration of CO2 and in particular H2O (Fig. 4b), due to the increased concentration of reactants (CO2 and H2O) for the electrolysis. The performance of co-electrolysis of 25%CO2–25%H2O–50%H2 is in between that of electrolysis of 50%CO2–50%H2 and 50%H2O–50%H2 (Fig. 4c). At an electrolysis voltage of 1.3 V, the current density is 1.24, 1.45 and 1.71 A cm−2 with 50%CO2–50%H2, 25%CO2–25%H2O–50%H2 and 50%H2O–50%H2, respectively. It is noted that the electrolysis performance is much less sensitive to the concentration of CO2 than that of H2O, most likely due to the greater diffusion coefficient of CO2 and poor adsorption capability of CO2 on the Ni surface.53–55 The electrolysis performance of the cell with the ESB–SmCPd electrode is comparable or even superior to that of the cells with other oxygen electrodes, such as mixed LSM–YSZ composites,56,57 LSM infiltrated YSZ,58 co-synthesized YSB–LSM,32 and LSCF–GDC composites.59
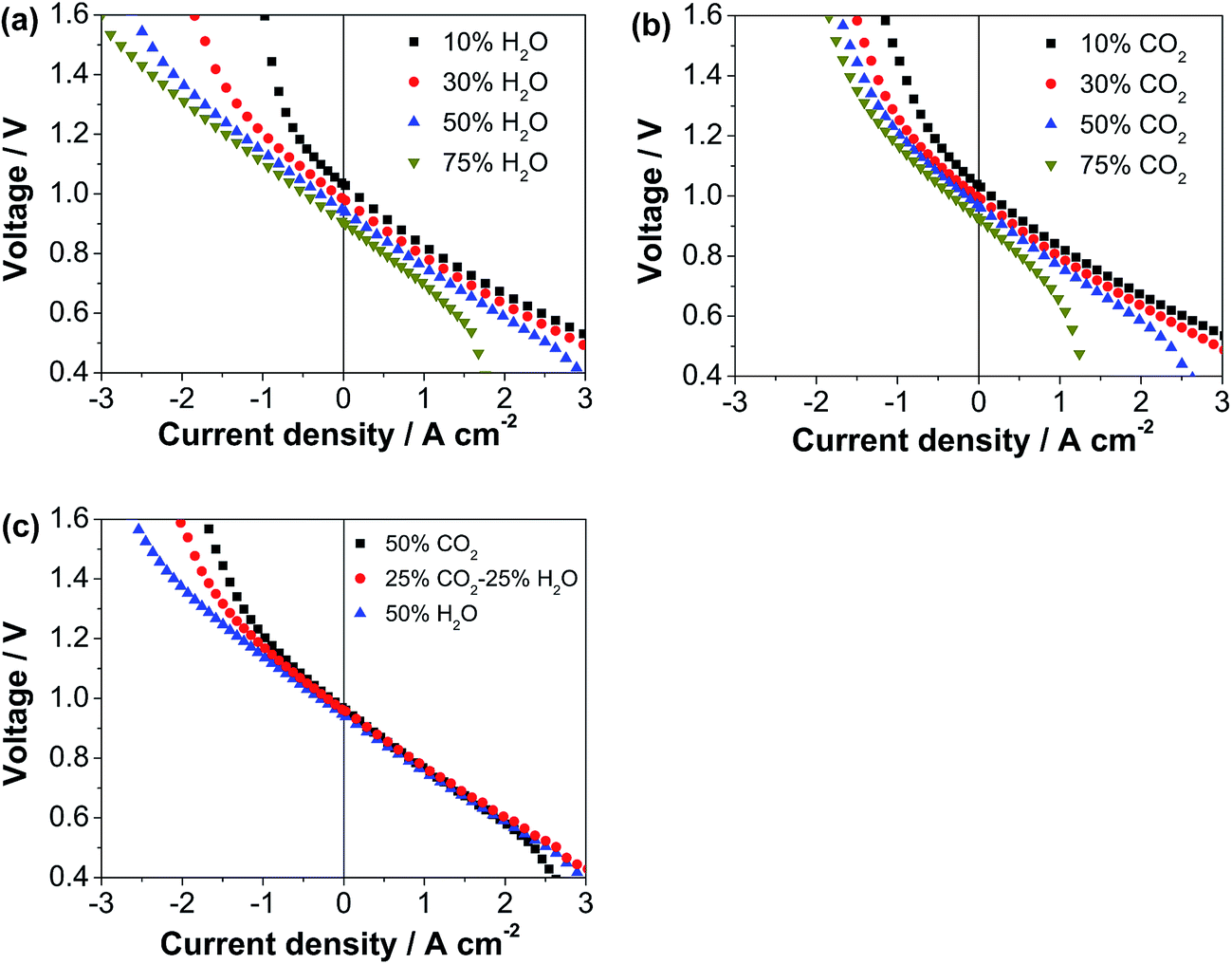 | ||
| Fig. 4 Polarization curves of the cell with the directly assembled, decorated ESB–SmCPd electrode at 750 °C: (a) as a function of steam ratio of H2O–H2 mixture gas, (b) as a function of the CO2 ratio of the CO2–H2 mixture gas, and (c) with different feedstock gases. | ||
The long-term operating stability of the cell in the fuel cell mode was measured at 0.5 A cm−2 and 750 °C (Cell-#3) as well as at 0.25 A cm−2 and 600 °C (Cell-#1), and the results are shown in Fig. 5. In the case of the stability test at 750 °C, the cell voltage increases in the first 20 h and decreases very slowly by the further polarization up to 300 h (Fig. 5a). The initial voltage is 0.94 V, and increases to 0.96 V after polarization for 20 h, but decreases to 0.93 V after polarization for 300 h. The polarization curves and PPDs of Cell-#3 during the initial polarization for 20 h can be seen in Table 1 and Fig. S6b, ESI.† The performance loss after polarization for 20 h is due to the increase of both RΩ and RP. RΩ and RP are 0.075 and 0.33 Ω cm2 after polarization for 40 h and increase to 0.109 and 0.37 Ω cm2 after polarization for 300 h, respectively (Fig. 5c). The impedance spectra are fitted using the aforementioned equivalent circuit (see Fig. 3b). The derived RH and RL are 0.18 and 0.15 Ω cm2 after polarization for 40 h, and change to 0.23 and 0.14 Ω cm2 after polarization for 300 h, respectively. Thus the increase of RP is due to the deteriorated high frequency electrode process which originates from the oxygen electrode as discussed above, indicating that the oxygen electrode is degraded during the polarization at 750 °C. On the other hand, at a lower operating temperature of 600 °C, the cell is more or less stable during the polarization at 0.25 A cm−2 for a longer period of 500 h (Fig. 5b). Consistently, the impedance spectra are unchanged by the polarization (Fig. 5d). This indicates that under the fuel cell operating conditions, the decorated ESB–SmCPd oxygen electrode is more suitable for operation at a lower temperature of 600 °C.
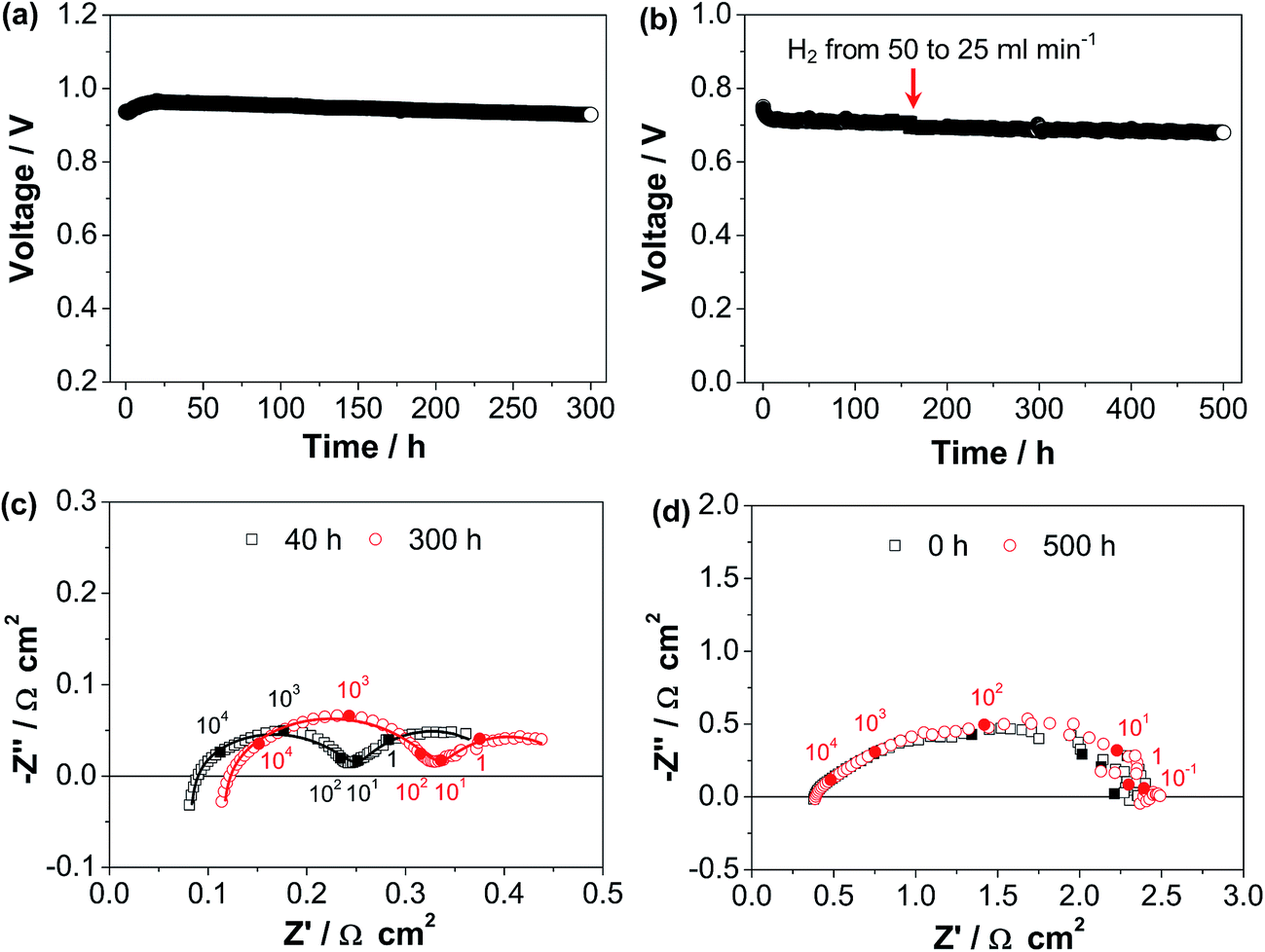 | ||
| Fig. 5 Stability curves and impedance spectra of the cells with the directly assembled, decorated ESB–SmCPd electrode in the fuel cell mode: (a and c) at 0.5 A cm−2 and 750 °C and (b and d) at 0.25 A cm−2 and 600 °C. In (c and d), the numbers are frequency in Hz. During the tests, the hydrogen electrodes were flowed with hydrogen. | ||
The operating stability of the cell with the decorated ESB–SmCPd electrode (Cell-#2) was also investigated in electrolysis and reversible modes at a current density of 0.5 A cm−2 at 750 °C, and the results are shown in Fig. 6. The cell exhibits good stability during the electrolysis operation for 80 h (Fig. 6a), much better than the significant performance decay in the case of LSM-based oxygen electrodes in the electrolysis mode.60–65 In addition, the reversible operation, i.e., 4 h in the fuel cell mode and 4 h in the electrolysis mode in each cycle is more or less stable over 16 cycles for 132 h (Fig. 6b). The high performance and the excellent operating stability demonstrate that the decorated ESB–SmCPd is a potential active oxygen electrode for durable SOCs.
 | ||
| Fig. 6 Stability curves of the cell with the directly assembled, decorated ESB–SmCPd electrode. (a) In the electrolysis mode and (b) in the fuel cell/electrolysis reversible operation mode. During the tests, the hydrogen electrodes were flowed with a 50%CO2–50%H2 mixture. | ||
3.3 Microstructure and cation interdiffusion at the interface
Fig. 7 shows the electrolyte surface in contact with the decorated ESB–SmCPd oxygen electrodes after the stability tests at 750 and 600 °C in the fuel cell mode. In this case, the oxygen electrodes were removed by acid treatment. After polarization at 0.5 A cm−2 and 750 °C for 300 h (Cell-#3, see Fig. 5a and c), the electrolyte surface is covered by a rough deposition layer and the grain boundaries of the YSZ electrolyte are invisible (Fig. 7a and b). On the other hand, after polarization at 0.25 A cm−2 and 600 °C for 500 h (Cell-#1, see Fig. 5b and d), there is no such deposition layer, as is evident by the relatively clean surface and clear grain boundaries (Fig. 7c and d). The electrode was polarized at 750 °C for 20 h prior to the stability test at 600 °C, and therefore, the clean electrolyte surface indicates that there is no severe interfacial reaction between the oxygen electrode and the YSZ electrolyte during such a short period of test at 750 °C. There are also irregularly shaped contact marks on the YSZ electrolyte surface (indicated by the arrows, Fig. 7d), an indication of the in situ formation of the electrode/electrolyte interface under the influence of electrochemical polarization. The absence of the deposition layer and the presence of contact marks on the electrolyte surface are consistent with the good operating stability at 600 °C in the fuel cell mode (see Fig. 5b and d). | ||
| Fig. 7 SEM micrographs of the acid cleaned YSZ electrolyte surface of the cells with directly assembled and decorated ESB–SmCPd electrodes after polarization in the fuel cell mode: (a and b) at 0.5 A cm−2 and 750 °C for 300 h and (c and d) at 0.25 A cm−2 and 600 °C for 500 h. | ||
To gain compositional information on the deposition layer on the YSZ electrolyte surface of Cell-#3 after polarization at 0.5 A cm−2 and 750 °C for 300 h (see Fig. 7a), FIB-SEM was used to prepare a lamella sample across the electrolyte surface and the STEM-EDS results are shown in Fig. 8. The electrolyte surface is covered by a dense deposition layer with a loose cluster located on the layer (see the HADDF image, Fig. 8a). The cluster is identified as Pd by EDS analysis (spectrum of spot 1, Fig. 8b). Previous XPS analysis has shown that for as-prepared SmCPd, Pd exists in the lattice of the SmCPd perovskite structure.21 Thus the observation of the Pd phase is because of the occurrence of Pd exsolution out of the SmCPd lattice by the electrochemical polarization. Pd exsolution was also observed in the directly assembled SmCPd–GDC electrode after the cathodic polarization.21 The great size of the Pd phase is most likely due to the high tendency of agglomeration and grain growth of Pd at the high operating temperature of SOCs.66,67 The deposition layer has a nominal composition of Sm0.53Er0.51Y0.06Bi0.44Zr0.46O3+δ (spectrum of spot 2, Fig. 8b), which may belong to a (Bi,Zr)2O3+δ solid solution co-doped with Sm and Er. There is a lack of the conductivity properties of (Bi,Zr)2O3+δ in the literature, but its formation probably leads to the increase of cell ohmic resistance after polarization at 750 °C for 300 h (Fig. 5c).
 | ||
| Fig. 8 (a) HADDF micrograph and EDS maps at the electrode/electrolyte interface of the cell with the directly assembled, decorated ESB–SmCPd electrode after polarization at 0.5 A cm−2 and 750 °C for 300 h in the fuel cell mode, and (b) EDS spectra of the selected spots marked in (a). | ||
We have successfully synthesized a nanostructured ESB decorated SmCPd composite electrode, which is directly assembled on a barrier-layer-free YSZ electrolyte film. Although the powder synthesis and cell operation at low temperatures (600–750 °C) cannot eliminate the chemical reaction between ESB and SmCPd, forming metastable Bi2SmO4 and later thermodynamically stable Sm0.6Bi1.4O3, the ESB–SmCPd electrode exhibits exceptionally higher electrocatalytic activity in comparison with the pristine SmCPd. This implies that the chemical reaction between ESB and SmCPd is not that harmful as originally expected. More importantly, the partial consumption of ESB and formation of a new Sm0.6Bi1.4O3 phase in the composite electrode are beneficial to the stable operation in particular at 600 °C for 500 h (Fig. 5b). In the case of ESB, it tends to undergo drastic conductivity degradation at 600 °C and below, due to the transformation in the arrangement of oxygen vacancies from the disordered state to the ordered state68 or due to the transformation from the cubic phase to the rhombohedral phase.69 On the other hand, the consequence of losing the highly ion conductive ESB phase is that at a reduced temperature of 650 °C and below, the cell performance with ESB decorated SmCPd is lower than that with ESB decorated LSM33 (PPDs at 650 °C: 0.58 vs. 0.71 W cm−2, see Fig. S7, ESI†). This is most likely due to the higher activation energy for the electrocatalytic reaction on decorated ESB–SmCPd than on decorated ESB–LSM (Fig. S8 and S9, ESI†). In the decorated ESB–LSM electrode, ESB is retained due to its chemical compatibility with LSM.33
4. Conclusions
Nanostructured ESB decorated SmCPd is investigated as a potential oxygen electrode for SOCs and directly assembled on a barrier-layer-free YSZ electrolyte film. A metastable Bi2SmO4 phase is formed in the decorated ESB–SmCPd and is transformed to Sm0.6Bi1.4O3 after dwelling at 750 °C. The presence of Sm0.6Bi1.4O3 in the decorated ESB–SmCPd oxygen electrode has no detrimental effect on the electrode activity, as the cells with the directly assembled ESB–SmCPd oxygen electrodes exhibit exceptionally high electrocatalytic activity and operating stability for power generation and high temperature electrolysis, with excellent stability at 600 °C for 500 h in the fuel cell mode, and in electrolysis and reversible modes at 750 °C for over 200 h. The present study demonstrates the feasibility of developing a Sr-free cobaltite oxygen electrode combined with bismuth oxide for active and stable SOCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the Fuzhou University Qishan Scholarship Program (XRC-17010), the National Natural Science Foundation of China (51672045 and U1732155), the Natural Science Foundation of Fujian Province (2018J01678), the Testing Fund of Fuzhou University (2018T002), and the Australian Research Council under the Discovery Project Scheme (DP150102044, DP150102025, DP180100568 and DP180100731). The authors thank Zhenhuan Zheng for XRD analysis, and acknowledge the facilities, scientific and technical assistance of the Curtin University Microscopy & Microanalysis Facility and UWA Centre for Microscopy, Characterisation and Analysis, both of which are funded by the University, State and Commonwealth Governments.References
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 15014 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- B. Yu, W. Q. Zhang, J. M. Xu and J. Chen, Int. J. Hydrogen Energy, 2008, 33, 6873–6877 CrossRef CAS.
- M. Ni, M. K. H. Leung and D. Y. C. Leung, Int. J. Hydrogen Energy, 2008, 33, 2337–2354 CrossRef CAS.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- C. W. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2010, 14, 1125–1144 CrossRef CAS.
- S. P. Jiang, J. Mater. Sci., 2008, 43, 6799–6833 CrossRef CAS.
- H. Ding, A. V. Virkar, M. Liu and F. Liu, Phys. Chem. Chem. Phys., 2013, 15, 489–496 RSC.
- G. Vovk, X. Chen and C. A. Mims, J. Phys. Chem. B, 2004, 109, 2445–2454 CrossRef PubMed.
- J. Kuyyalil, D. Newby, J. Laverock, Y. Yu, D. Cetin, S. Basu, K. Ludwig and K. Smith, Surf. Sci., 2015, 642, 33–38 CrossRef CAS.
- Y. Liu, K. Chen, L. Zhao, B. Chi, J. Pu, S. P. Jiang and L. Jian, Int. J. Hydrogen Energy, 2014, 39, 15868–15876 CrossRef CAS.
- N. Ni, S. J. Cooper, R. Williams, N. Kemen, D. W. McComb and S. J. Skinner, ACS Appl. Mater. Interfaces, 2016, 8, 17360–17370 CAS.
- N. Ni and S. Skinner, Solid State Ionics, 2016, 288, 28–31 CrossRef CAS.
- K. Chen, N. Li, N. Ai, Y. Cheng, W. D. A. Rickard and S. P. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 31729–31737 CAS.
- Y.-L. Huang, A. M. Hussain, C. Pellegrinelli, C. Xiong and E. D. Wachsman, ACS Appl. Mater. Interfaces, 2017, 9, 16660–16668 CAS.
- Y. Chen, W. Zhou, D. Ding, M. Liu, F. Ciucci, M. Tade and Z. Shao, Adv. Energy Mater., 2015, 5, 1500537 CrossRef.
- N. Li, N. Ai, S. He, Y. Cheng, W. D. A. Rickard, K. Chen, T. Zhang and S. P. Jiang, Solid State Ionics, 2018, 316, 38–46 CrossRef CAS.
- J. Shen, Y. Chen, G. Yang, W. Zhou, M. O. Tadé and Z. Shao, J. Power Sources, 2016, 306, 92–99 CrossRef CAS.
- N. Ai, N. Li, W. D. Rickard, Y. Cheng, K. Chen and S. P. Jiang, ChemSusChem, 2017, 10, 993–1003 CrossRef CAS PubMed.
- B. Steele, J. Kilner, P. Dennis, A. McHale, M. Van Hemert and A. Burggraaf, Solid State Ionics, 1986, 18, 1038–1044 CrossRef.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- K. T. Lee, D. W. Jung, H. S. Yoon, A. A. Lidie, M. A. Camaratta and E. D. Wachsman, J. Power Sources, 2012, 220, 324–330 CrossRef CAS.
- J. Hou, L. Bi, J. Qian, Z. Zhu, J. Zhang and W. Liu, J. Mater. Chem. A, 2015, 3, 10219–10224 CAS.
- D. W. Joh, J. H. Park, D. Kim, E. D. Wachsman and K. T. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 8443–8449 CAS.
- J. Li, S. Wang, X. Sun, R. Liu, X. Ye and Z. Wen, J. Power Sources, 2008, 185, 649–655 CrossRef CAS.
- Z. Y. Jiang, L. Zhang, L. L. Cai and C. R. Xia, Electrochim. Acta, 2009, 54, 3059–3065 CrossRef CAS.
- L. Wu, Z. Jiang, S. Wang and C. Xia, Int. J. Hydrogen Energy, 2013, 38, 2398–2406 CrossRef CAS.
- K. T. Lee, A. A. Lidie, H. S. Yoon and E. D. Wachsman, Angew. Chem., Int. Ed., 2014, 49, 13463–13467 CrossRef PubMed.
- J. Hou, L. Bi, J. Qian, Z. Gong, Z. Zhu and W. Liu, J. Power Sources, 2016, 301, 306–311 CrossRef CAS.
- J. Yan, Z. Zhao, L. Shang, D. Ou and M. Cheng, J. Power Sources, 2016, 319, 124–130 CrossRef CAS.
- N. Ai, N. Li, S. He, Y. Cheng, M. Saunders, K. Chen, T. Zhang and S. P. Jiang, J. Mater. Chem. A, 2017, 5, 12149–12157 CAS.
- N. Ai, J.-P. Veder, Y. Cheng, M. Chen, K. Chen, T. Zhang and S. P. Jiang, J. Electrochem. Soc., 2017, 164, F1471–F1477 CrossRef CAS.
- A. S. Painter, Y.-L. Huang and E. D. Wachsman, J. Power Sources, 2017, 360, 391–398 CrossRef CAS.
- Z. Wu and M. Liu, J. Am. Ceram. Soc., 1998, 81, 1215–1220 CrossRef CAS.
- Y. Zhou, Z. Zhang, C. Yuan, J. Li, C. Xia, Z. Zhan and S. Wang, Int. J. Hydrogen Energy, 2013, 38, 16579–16583 CrossRef CAS.
- C. R. Xia and M. L. Liu, Adv. Mater., 2002, 14, 521–523 CrossRef CAS.
- V. Kharton, E. Naumovich and V. Samokhval, Solid State Ionics, 1997, 99, 269–280 CrossRef CAS.
- V. V. Kharton, E. N. Naumovich, A. A. Yaremchenko and F. M. B. Marques, J. Solid State Electrochem., 2001, 5, 160–187 CrossRef CAS.
- G. Li, B. He, Y. Ling, J. Xu and L. Zhao, Int. J. Hydrogen Energy, 2015, 40, 13576–13582 CrossRef CAS.
- K. Chen, N. Li, N. Ai, M. Li, Y. Cheng, W. D. A. Rickard, J. Li and S. P. Jiang, J. Mater. Chem. A, 2016, 4, 17678–17685 CAS.
- M. Li, K. Chen, B. Hua, J.-l. Luo, W. D. A. Rickard, J. Li, J. T. S. Irvine and S. P. Jiang, J. Mater. Chem. A, 2016, 4, 19019–19025 CAS.
- N. Li, N. Ai, K. Chen, Y. Cheng, S. He, M. Saunders, A. Dodd, A. Suvorova and S. P. Jiang, RSC Adv., 2016, 6, 99211–99219 RSC.
- K. Chen, Z. Lu, N. Ai, X. Huang, Y. Zhang, X. Ge, X. Xin, X. Chen and W. Su, Solid State Ionics, 2007, 177, 3455–3460 CrossRef CAS.
- K. Chen, X. Chen, Z. Lu, N. Ai, X. Huang and W. Su, Electrochim. Acta, 2008, 53, 7825–7830 CrossRef CAS.
- S. P. Jiang, J. Electrochem. Soc., 2015, 162, F1119–F1128 CrossRef CAS.
- N. M. Sammes, G. A. Tompsett, H. Nafe and F. Aldinger, J. Eur. Ceram. Soc., 1999, 19, 1801–1826 CrossRef CAS.
- M. Drache, S. Obbade, J. Wignacourt and P. Conflant, J. Solid State Chem., 1999, 142, 349–359 CrossRef CAS.
- N. Ai, K. Chen, S. P. Jiang, Z. Lü and W. Su, Int. J. Hydrogen Energy, 2011, 36, 7661–7669 CrossRef CAS.
- K. Chen, L. Zhang, N. Ai, S. Zhang, Y. Song, Y. Song, Q. Yi, C.-Z. Li and S. P. Jiang, Energy Fuels, 2016, 30, 1849–1857 CrossRef CAS.
- M. Riegraf, V. Yurkiv, R. Costa, G. Schiller and K. A. Friedrich, ChemSusChem, 2017, 10, 587–599 CrossRef CAS PubMed.
- Y. Li, P. Li, B. Hu and C. Xia, J. Mater. Chem. A, 2016, 4, 9236–9243 CAS.
- Z. L. Zhan and L. Zhao, J. Power Sources, 2010, 195, 7250–7254 CrossRef CAS.
- Y.-F. Sun, Y.-Q. Zhang, J. Chen, J.-H. Li, Y.-T. Zhu, Y.-M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J.-L. Luo, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS PubMed.
- T. Liu, Y. Wang, Y. Zhang, S. Fang, L. Lei, C. Ren and F. Chen, Electrochem. Commun., 2015, 61, 106–109 CrossRef CAS.
- C. Graves, S. D. Ebbesen and M. Mogensen, Solid State Ionics, 2011, 192, 398–403 CrossRef CAS.
- C. H. Yang, C. Jin, A. Coffin and F. L. Chen, Int. J. Hydrogen Energy, 2010, 35, 5187–5193 CrossRef CAS.
- M. J. López-Robledo, M. A. Laguna-Bercero, A. Larrea and V. M. Orera, J. Power Sources, 2018, 378, 184–189 CrossRef.
- J. R. Mawdsley, J. D. Carter, A. J. Kropf, B. Yildiz and V. A. Maroni, Int. J. Hydrogen Energy, 2009, 34, 4198–4207 CrossRef CAS.
- K. Chen, N. Ai and S. P. Jiang, Int. J. Hydrogen Energy, 2012, 37, 10517–10525 CrossRef CAS.
- Y. Zhang, K. Chen, C. Xia, S. P. Jiang and M. Ni, Int. J. Hydrogen Energy, 2012, 37, 13914–13920 CrossRef CAS.
- Y. Zheng, Q. Li, T. Chen, W. Wu, C. Xu and W. G. Wang, Int. J. Hydrogen Energy, 2015, 40, 2460–2472 CrossRef CAS.
- G. A. Hughes, K. Yakal-Kremski and S. A. Barnett, Phys. Chem. Chem. Phys., 2013, 15, 17257–17262 RSC.
- C. Graves, S. D. Ebbesen, S. H. Jensen, S. B. Simonsen and M. B. Mogensen, Nat. Mater., 2015, 14, 239–244 CrossRef CAS PubMed.
- N. Ai, K. Chen, S. Liu and S. P. Jiang, Int. J. Hydrogen Energy, 2013, 38, 16569–16578 CrossRef CAS.
- F. Liang, Z. Wang, J. Sunarso, J. Mao and W. Zhou, Int. J. Hydrogen Energy, 2018, 43, 53–63 CrossRef CAS.
- N. Jiang, R. M. Buchanan, D. A. Stevenson, W. D. Nix, J.-Z. Li and J.-L. Yang, Mater. Lett., 1995, 22, 215–219 CrossRef CAS.
- K. Huang, M. Feng and J. B. Goodenough, Solid State Ionics, 1996, 89, 17–24 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta00370j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |