
Enhancing the real-time detection of phase changes in lithium–graphite intercalated compounds through derivative operando (dOp) NMR cyclic voltammetry†
Jose L.
Lorie Lopez
 ,
Philip J.
Grandinetti
* and
Anne C.
Co
*
,
Philip J.
Grandinetti
* and
Anne C.
Co
*
Department of Chemistry and Biochemistry, Ohio State University, USA. E-mail: grandinetti.1@osu.edu; co.5@osu.edu
First published on 27th November 2017
Abstract
Robust and reliable diagnostic tools that can be employed under operating conditions are crucial to understanding performance and failure mechanisms in battery processes. The operando spectrum of a battery often consists of a strongly overlapping mixture of time dependent and independent resonances due to the compositional complexity. Here we report a new method called derivative operando (dOp) that improves the resolution of operando nuclear magnetic resonance (NMR) spectra by removing time independent signals and further distinguishes between time dependent signals associated with the formation and removal of species. This approach not only provides better resolution but also more clearly reveals correlations between resonances and the chemical transformations occurring at a specific potential. With the dOp-NMR method we detect the formation of lithium graphite intercalation compounds (GICs), including the signatures of LiC72 and its precursors, which have been previously undetected. We also observe a clear correlation of the dOp 7Li NMR spectra of lithium metal dendrites on the counter electrode with the chemistry of the working electrode.
1 Introduction
The growing demand for batteries with higher energy and power densities has stimulated considerable efforts towards the development of new materials.1–3 Concurrently, advances in robust, non-destructive in situ and operando characterization techniques4 are becoming critical as we attempt to improve our understanding of how structural transformations, reaction dynamics, and transient non-equilibrium processes at various length and time scales determine battery performance and failure mechanisms. A number of complementary operando techniques have been developed to probe specific chemical processes, each with its respective advantages and limitations. For instance, operando X-ray scattering5–7 has been invaluable for identifying and detecting transformations among various crystalline phases during cycling. Another example is operando scanning probe microscopy,8 which monitors the expansion and contraction of electrode materials. Neutron depth profiling9–13 has been used to quantify lithium accumulation/depletion, lithium rearrangement, and lithium transport during lithiation and delithiation processes in lithium ion batteries. Nuclear magnetic resonance (NMR) spectroscopy and imaging (MRI), in particular, have become important new tools in operando studies of battery materials using NMR active nuclei to provide quantitative identification of structural species in crystalline and non-crystalline solids as well as in fluid phases, including anode materials,14–16 metallic dendrites,17 electrolytes,18 and electrolyte decomposition products.19In spite of these successes, the interpretation and assignment of an operando signal from a battery is challenging due to overlapping signals arising from the electrolyte, cathode, anode, and solid–electrolyte interface (SEI) phases. In the case of NMR the higher spectral resolution of magic-angle spinning (MAS) has remained out of reach due to a number of technical hurdles, notably the magnetic braking due to eddy currents arising in metallic current collectors as well as in metallic electrode materials during sample rotation.
Reported herein is an NMR method we have developed that (1) improves the resolution of overlapping resonances – aiding in spectral assignments – and more importantly, (2) differentiates the signals due to the formation and removal of species by eliminating all signals that are constant during the battery processes through signal processing. This approach, which we have coined “derivative operando” (dOp) spectroscopy, is not limited to processing NMR spectra but can be generally applied to other operando methods to remove sizable signals that remain invariant during the operando process. The dOp spectrum not only provides better resolution but also offers a more direct correlation of the species formed and removed with the electrochemical current measured at each potential/time.
2 Methods
2.1 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio of ethyl carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte (Purolyte A5 Series, Novolyte Technologies). The carbon and Li electrodes were separated by a Celgard® 2400 separator soaked in the electrolyte prior to assembly. The lithium electrode was pressed onto a copper mesh current collector. The cell configuration was similar to that shown by Letellier et al.,21 but two working electrodes were used to increase the NMR signal as illustrated in Fig. 1A. The stack was vacuum sealed in a polyethylene plastic bag inside the glove box. The electrochemical cell was 8 mm wide and 10 mm long and contained a total of 18.1 mg of MCMB.
:1 volume ratio of ethyl carbonate (EC) and dimethyl carbonate (DMC) was used as the electrolyte (Purolyte A5 Series, Novolyte Technologies). The carbon and Li electrodes were separated by a Celgard® 2400 separator soaked in the electrolyte prior to assembly. The lithium electrode was pressed onto a copper mesh current collector. The cell configuration was similar to that shown by Letellier et al.,21 but two working electrodes were used to increase the NMR signal as illustrated in Fig. 1A. The stack was vacuum sealed in a polyethylene plastic bag inside the glove box. The electrochemical cell was 8 mm wide and 10 mm long and contained a total of 18.1 mg of MCMB.
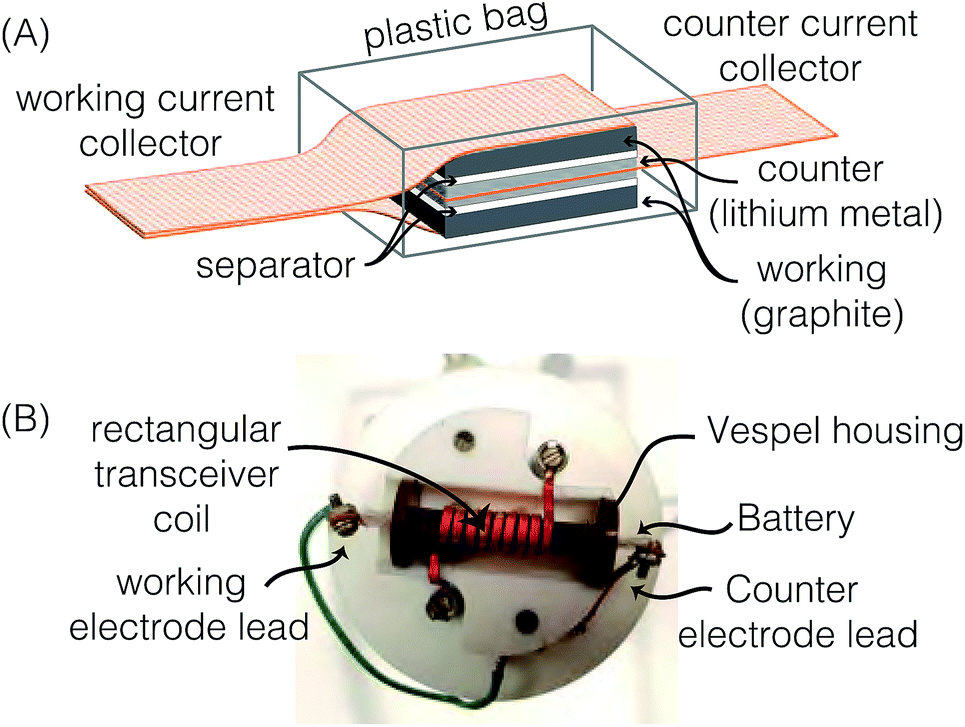 | ||
| Fig. 1 (A) Schematic drawing of a bag cell configuration with two working electrodes. (B) Labeled photo of the modified probe head with a rectangular coil and Vespel housing. | ||
The data presented here were measured using the same carbon electrode. We have done this to ensure consistency in our comparison of NMR and electrochemical data. We have repeated all experiments reported here with several identical carbon electrode systems to confirm that all the trends reported are statistically significant and reproducible.
2.2 NMR measurements
NMR measurements were performed on a hybrid Tecmag Apollo-Chemagnetics CMX II 9.4 T (155.48 MHz for 7Li) NMR spectrometer. All frequency shifts are given in terms of the dimensionless quantity,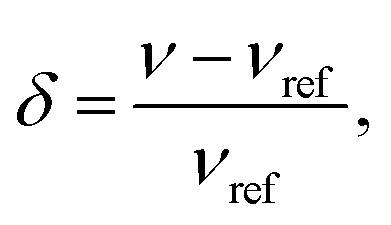 | (1) |
| Li site | Stage | Potential/V (vs. Li/Li+) | δ Li/ppm | C q/kHz | T 1/s | Fraction of transverse magnetization | Predicted integrated intensity |
|---|---|---|---|---|---|---|---|
| Li metal | — | — | 274 | — | 0.140 | 0.369 | — |
| Precursors | >8 | >0.47 | ∼−82 to −2.5 | — | — | — | — |
| LiC72 | 8 | 0.47 | −2.5 to −1.0 | — | — | — | — |
| LiC36 | 4 | 0.189 | ∼2 | 36 | 3.15 | 0.096 | 0.2889 |
| LiC27 | 3 | 0.137 | 6.3 | 36 | 3.87 | 0.085 | 0.1702 |
| LiC18 | 2L | 0.116 | 12.5 | 36 | — | — | — |
| LiC12 | 2 | 0.086 | 48.3 | 34 | 2.14 | 0.118 | 0.7074 |
| LiC6 | 1 | 0.047 | 45.2 | 44 | 1.80 | 0.128 | 2.2968 |
The 7Li nucleus has spin S = 3/2 and has three NMR active transitions: the central transition 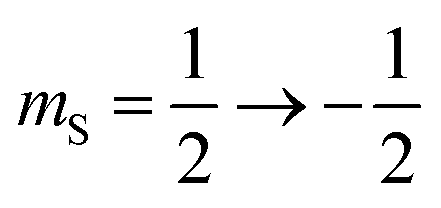 and two satellite transitions,
and two satellite transitions, 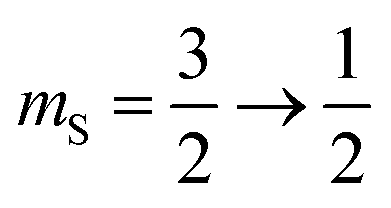 and
and 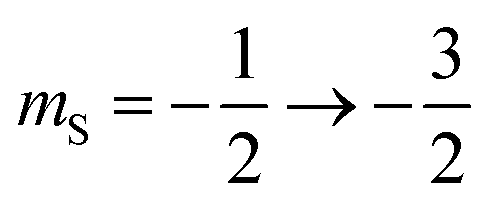 . In addition to having a nuclear magnetic dipole moment, the 7Li nucleus also has an electric quadrupole moment. The coupling between this quadrupole moment and local electric field gradients can lead to an anisotropic broadening of the satellite transition resonances as illustrated in Fig. 2A. These line shapes can appear somewhat distorted with intensity loss from the broad component of the anisotropic line shape due to receiver dead time at the beginning of signal acquisition. In contrast, the central transition experiences no anisotropic quadrupolar broadening to first order.23
. In addition to having a nuclear magnetic dipole moment, the 7Li nucleus also has an electric quadrupole moment. The coupling between this quadrupole moment and local electric field gradients can lead to an anisotropic broadening of the satellite transition resonances as illustrated in Fig. 2A. These line shapes can appear somewhat distorted with intensity loss from the broad component of the anisotropic line shape due to receiver dead time at the beginning of signal acquisition. In contrast, the central transition experiences no anisotropic quadrupolar broadening to first order.23
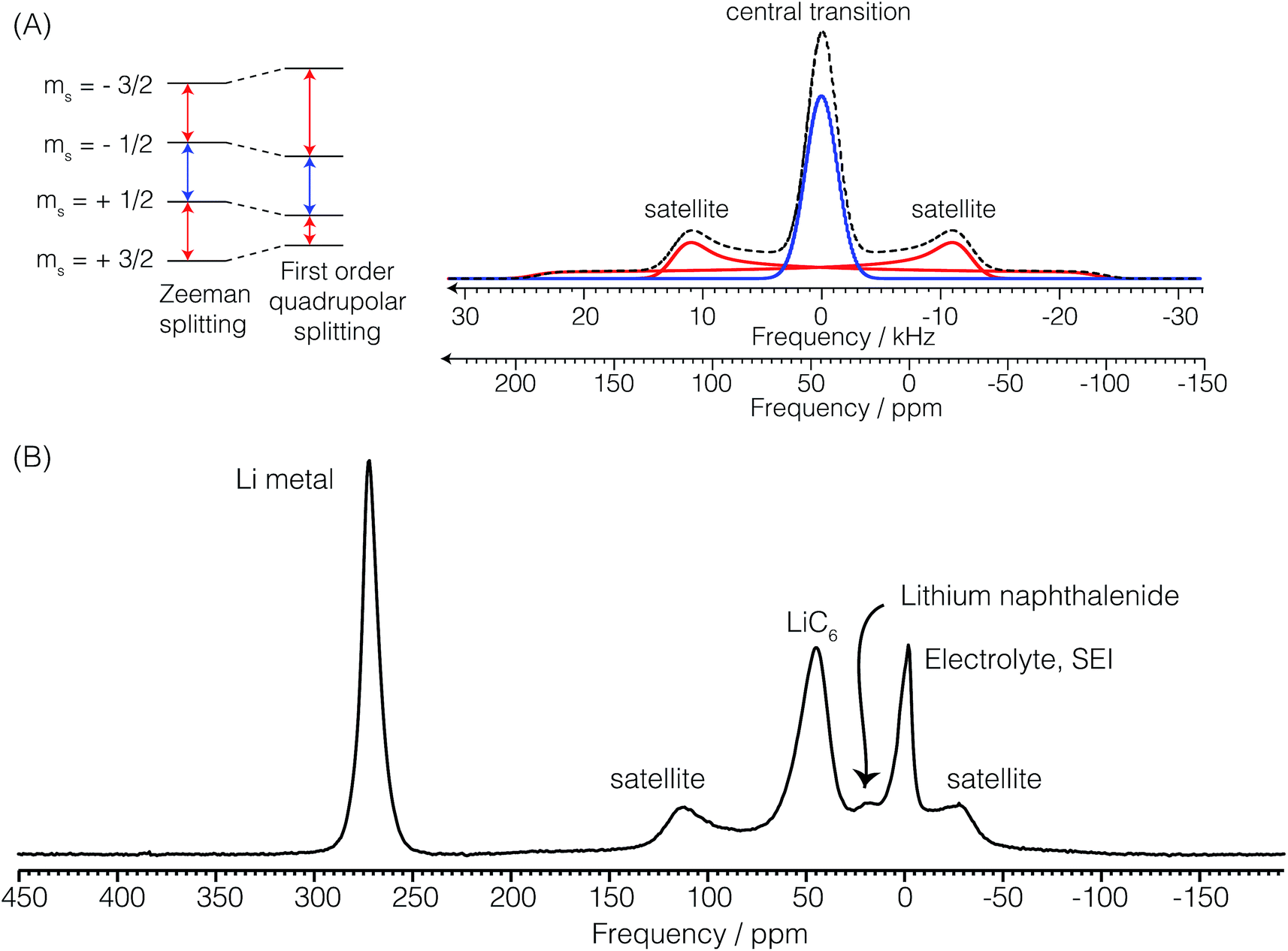 | ||
| Fig. 2 (A) Component and overall powder pattern NMR line shapes for the two satellite transitions and the central transition of the 7Li nucleus with a quadrupolar coupling constant of Cq = 44 kHz and a quadrupolar asymmetry parameter of zero. (B) In situ7Li NMR spectrum of the cell during the formation of LiC6 showing the resonant frequencies of the lithium metal, electrolyte and SEI, lithium naphthalenide, and the quadrupolar powder pattern of LiC6. | ||
Radio frequency power levels were reduced to 25 watts to eliminate arcing in the coil. This corresponds to a rf field strength of ω1/(2π) = 14.4 kHz. At this power, nutation experiments show that the lithium metal resonance nutates at twice the rate of the lithium sites in the electrolyte and the graphite intercalated compounds (GICs). An increase by a factor of (S + 1/2) in the nutation frequency (2 for 7Li) indicates that the rf pulse excites only the central transition of the Li metal resonance due to its larger quadrupole splitting ωq ≫ ω1. This observation of faster nutation of the lithium metal resonance conflicts with the conclusion of Chevallier et al.16 who noted that the lithium nuclei in the metal experience no electric field gradient. At this power a 2.5 μs pulse length corresponds to a 13° rotation of all lithium transitions for sites in the electrolyte and GICs and a 26° rotation of the central transition for sites in the lithium metal.
Fig. 2B shows a typical in situ7Li NMR spectrum of the Li ion cell with a lithium counter electrode and graphite as the working electrode at a potential where LiC6 is the dominant form of lithiated graphite. The 7Li NMR resonance at δ = 274 ppm is assigned to the central transition of 7Li in the metallic lithium counter electrode. The 7Li satellite transitions in the metal are too broad for detection. The strongly positive shift from 0 ppm arises from an increased magnetic field at the nucleus due to an interaction with the conduction electrons called the Knight shift.24 The lithium metal sites have a spin–lattice relaxation time of T1 = 140 ms. The electrolyte, LiPF6, and SEI appear around −2.5 ppm. As noted earlier these resonances nutate at a rate of ω1. The additional lack of any observable anisotropic broadening indicates that these 7Li sites experience little to no quadrupolar coupling to local electric field gradients as would be expected for a Li+ cation in an isotropic environment. The 7Li spin–lattice relaxation time of LiPF6 is 675 ms. Most notably, seen in this spectrum are central and satellite transitions for 7Li of LiC6. The central transition peaks at approximately 45 ppm. The quadrupolar coupling—responsible for the anisotropic broadening of the satellite transitions—is characterized by a constant of Cq = 44 kHz and quadrupole asymmetry parameter of approximately ηq = 0. The 7Li spin–lattice relaxation time of LiC6 is 1.8 s. Finally, we note that the lower intensity resonance at approximately 18 ppm is assigned to lithium naphthalenide which is produced during the formation of LiC12.
A CHI760D (CH Instruments, Inc., Austin, TX) galvanostat/potentiostat was used to perform the electrochemical measurements. All voltages were measured and reported against Li/Li+. The initial open circuit potential (OCP) of the cell was approximately 3.1 V vs. Li/Li+. The OCP was measured over a period of 15 minutes to ensure steady state potential before the beginning of the operando measurements. The NMR signal was simultaneously acquired with three cyclic voltammograms (CV) between 700 and 5 mV at a potential sweep of 2 μV s−1.
For operando NMR/cyclic voltammetry experiments the acquisition time of a single free induction decay was 10.24 ms. With a 13° pulse and a T1 of 1.8 s for the LiC6 we set the recycle delay time to 49.4 ms to optimize sensitivity according to the Ernst angle equation26 and averaged 15084 scans for each spectrum. Because of the differential 7Li relaxation times of the GICs we calculate and present in Table 1 the fraction of transverse magnetization after the steady state has been reached for the excitation pulse length and recycle delay used in the operando NMR measurements. A fraction of 1 corresponds to the excitation of equilibrium magnetization. There is a systematic decrease in 7Li T1 with increasing lithiation of the GICs leading to a slight but not insignificant loss in GIC resonance intensity (quantitative areas) with decreasing lithiation.
With these parameters each spectrum was acquired in Δtop = 15 min giving an electrochemical resolution of 1.8 mV per NMR spectra. A total of Nop = 2317 spectra were acquired for a total measurement (operando) time, top, of 580 h (approx. 24 days).
The derivative of the operando signal as a function of operando time, top, and NMR transition frequency, ω, is calculated via the Fourier derivative theorem which states
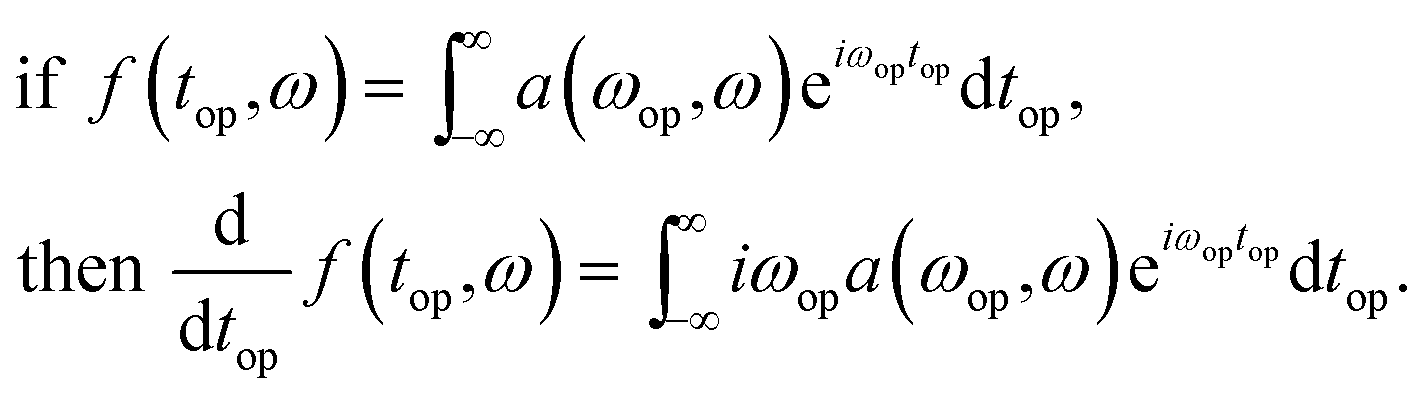 | (2) |
In other words, the derivative of the signal, f(top,ω), is a convolution of the signal with the inverse Fourier transform of the function iωop. This function, however, will amplify high frequency noise so a Hamming function is applied to cut off the undesired higher frequencies. In practice this is implemented in four steps: (1) application of a discrete Nop point Fourier transform of the signal with respect to the battery operando time, top; (2) multiplication of the signal by iωop; (3) multiplication of the signal by a Hamming function given by
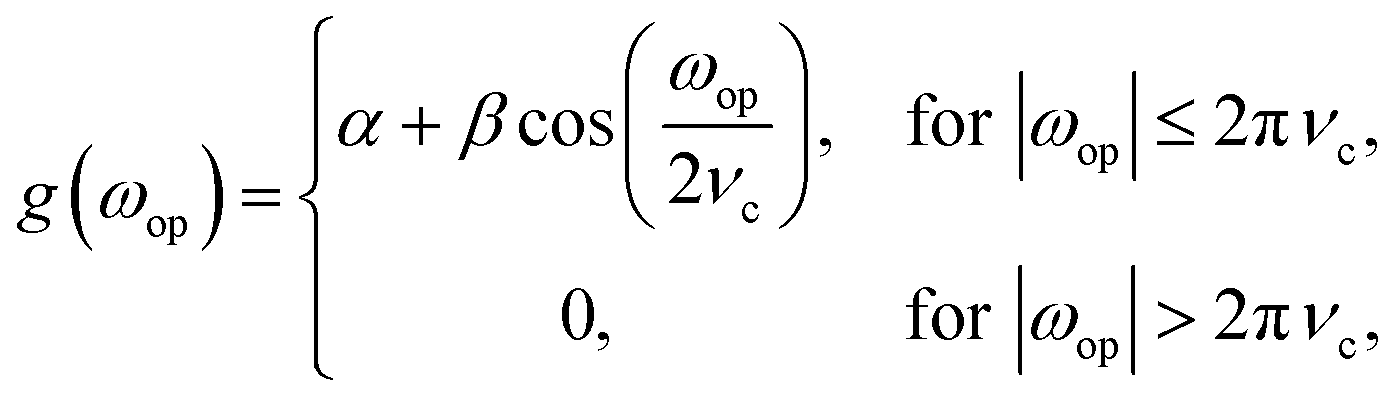 | (3) |
3 Results and discussion
3.1 Cyclic voltammetry
Fig. 3A shows the cyclic voltammogram of the carbon electrode measured at a sweep rate of 2 μV s−1 from 700 to 5 mV vs. Li/Li+. The direction of the scan is indicated in the figure. Negative currents indicate lithiation or reduction and positive currents indicate delithiation or oxidation processes. The peak assignments were based on in situ XRD measurements.5,7 The first cycle (blue dashed line) shows broader and shifted peaks as the freshly prepared battery is negatively polarized. A solid electrolyte interface (SEI) slowly forms on the surface of both electrodes, changing their surface chemistry, and therefore, resulting in a broader distribution of potentials that shift with respect to each other. As the electrode gets passivated, the potential of the lithium anode reaches a steady state, and the observed peak potentials for the subsequent cycles on lithium do not change substantially, reflecting the electrochemical reactions occurring on MCMB. The second (red dotted line) and third (green solid line) cycles show reproducible peak potentials. The electrochemical reactions have been previously described.5,7,28 In order to aid in the discussion in Section 3.3, the stoichiometric coefficients of the reactants and products in the electrochemical equations given below have been normalized to reflect the predicted relative intensities in the dOp NMR spectrum for a fixed amount of graphite. The electrochemical half-reaction for the peak labelled A corresponds to the transition to stage 4 (LiC36) from stage 8 (LiC72), and is described by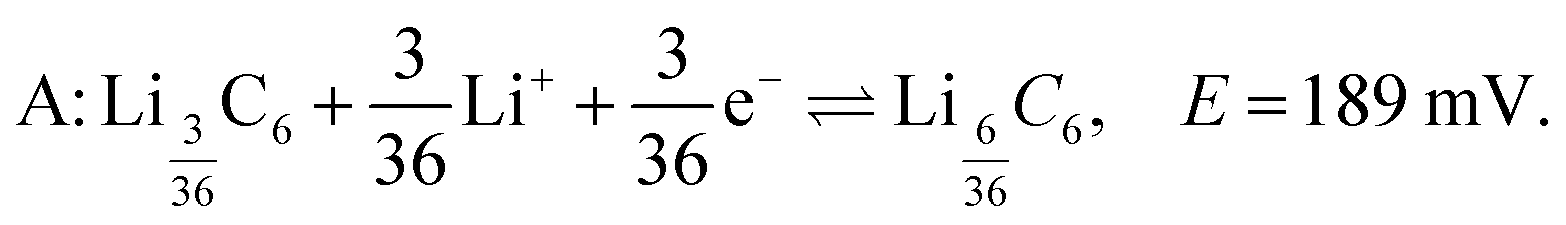 | (4) |
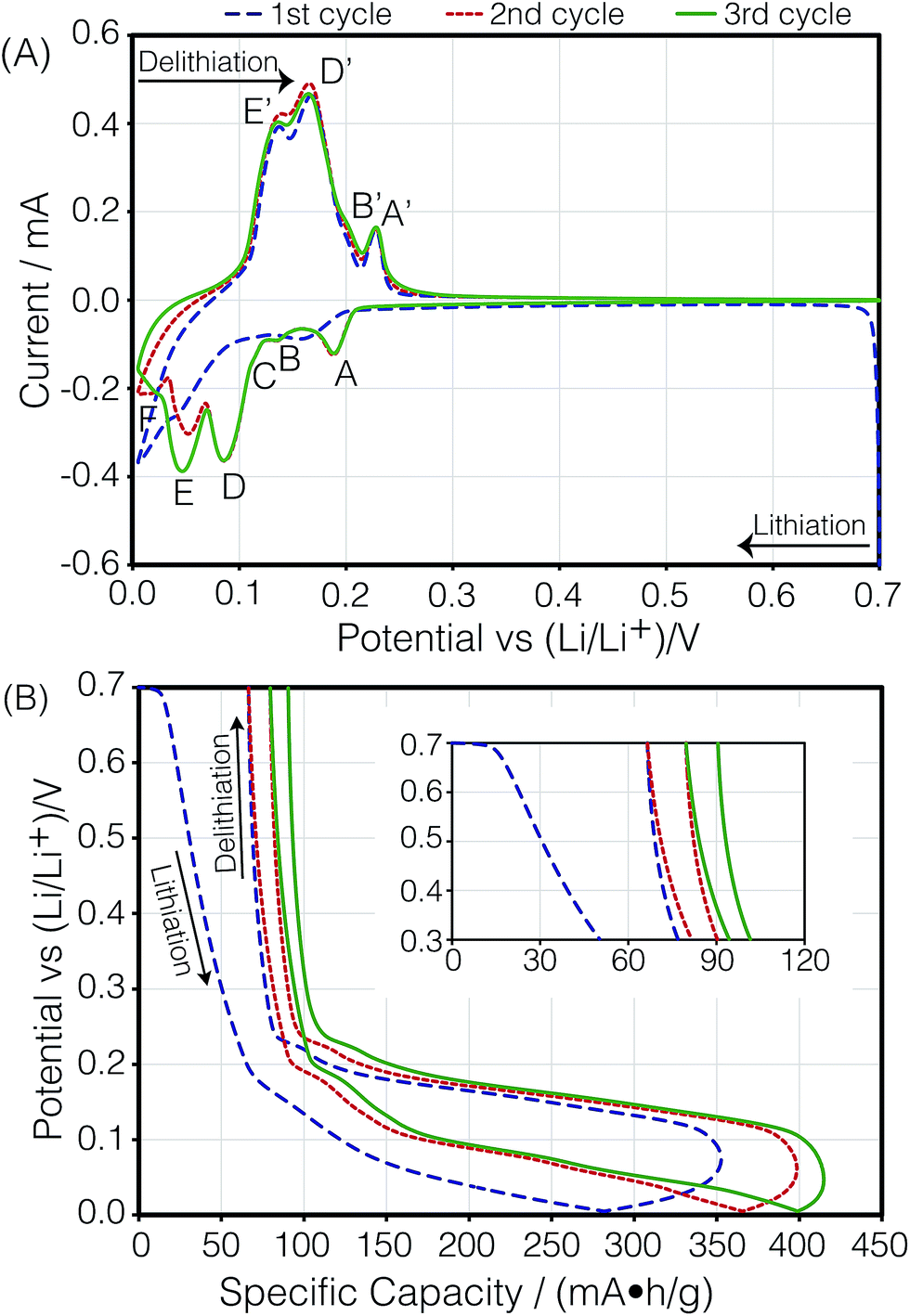 | ||
| Fig. 3 (A) Cyclic voltammogram of the carbon/Li electrochemical cell depicting lithiation as a function of potential at 2 μV s−1. (B) Specific capacity during the cycling of the cell as a function of cell potential. The peaks labelled A, B, C, D, E, and F are described in the text. | ||
Similarly, the peak labelled B corresponds to the formation of stage 3 (LiC27), and is described by
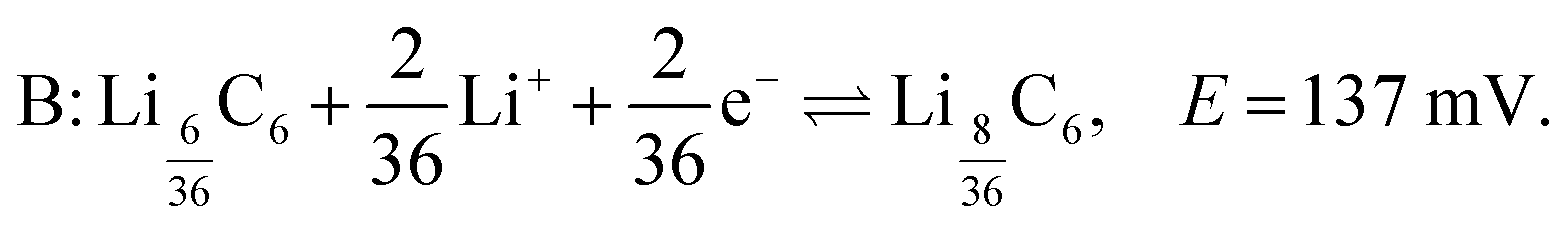 | (5) |
The shoulder labelled C at 116 mV suggests the transition to stage 2L (LiC18), and is described by
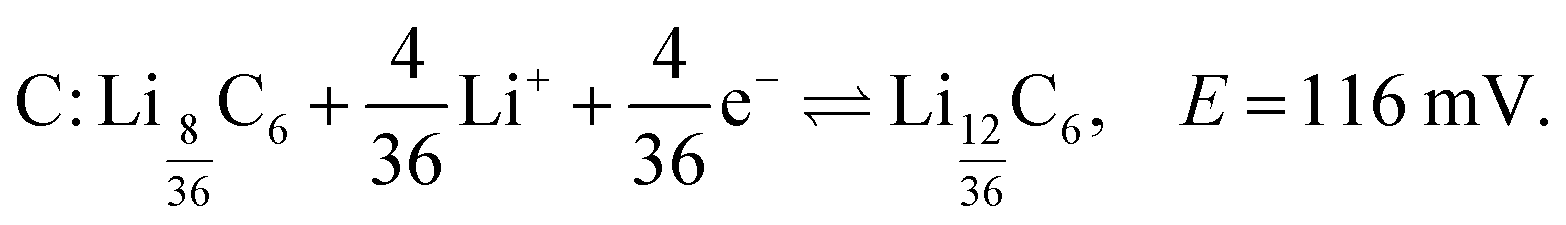 | (6) |
The peak labelled D corresponds to the formation of stage 2 (LiC12), and is described by
 | (7) |
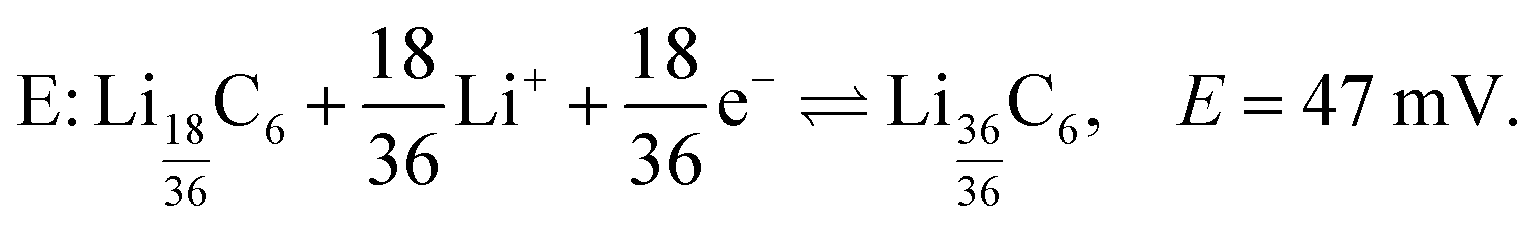 | (8) |
The transition to form stage 8 (LiC72), also known as dilute stage 1, has no evident peak in the CV. Peak F at near 15 mV will be discussed later in the text.
Fig. 3B shows the cell's specific capacity as a function of cell potential. The plot was obtained by integrating the currents in the CVs as a function of potential and normalizing them to the sweep rate. Even though the first cycle achieved a specific capacity of 353 mA h g−1 during lithiation, which is higher than the reported value from the manufacturer of 345 mA h g−1, the delithiation capacity for the cycle was 286.5 mA h g−1, giving an irreversible capacity of 66.5 mA h g−1. Specific capacities of 332 and 335 mA h g−1 were attained for the second and third lithiation of the cell, with smaller irreversible capacities of 13.2 and 10.8 mA h g−1, respectively. This trend was expected as it has been previously shown that the SEI is primarily formed during the first cycle of the cell, but small amounts continue to form during the subsequent cycles.29,30
3.2 Conventional NMR spectrum
Before discussing the dOp spectrum we first review the spectral features of the conventional operando 7Li NMR spectrum as a function of operando time or cell potential. The operando 7Li NMR spectrum for the second cycle is shown in Fig. 4. The operando 7Li NMR spectrum for all three cycles is given in the ESI.† The corresponding potential at which lithiation and delithiation occur is shown on the left vertical axis of Fig. 4 along with the corresponding cyclic voltammogram. The sharp 7Li NMR resonances due to the electrolyte and SEI resonances near δ = 0 ppm remain relatively constant at all operando times.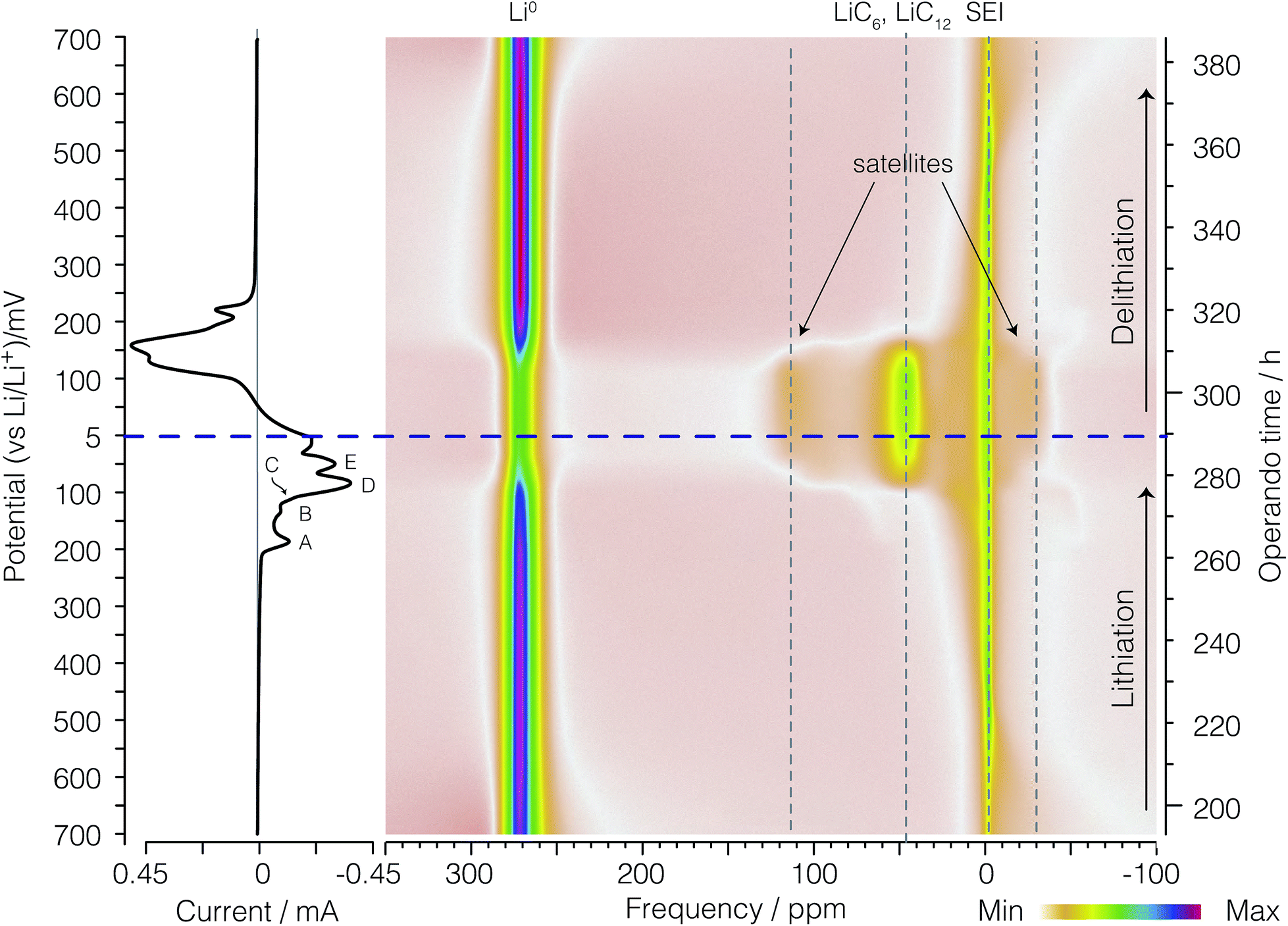 | ||
| Fig. 4 A color intensity plot of the operando 7Li NMR spectra of the second cycle of the electrochemical cell. Light colors represent low peak intensities and darker colors represent higher peak intensities. The horizontal axis shows the NMR frequency shift in ppm with reference to 1 M LiCl. The vertical axis shows the operando time for the second cycle of the cell. The horizontal blue (dashed) line represents the time at which the current polarity is switched. On the left is the corresponding cyclic voltammogram. Peaks labelled A, B, C, D, and E represent the formation of LiC36, LiC27, LiC18, LiC12, and LiC6 respectively as shown in eqn (4), (5), (6), (7), and (8), respectively. | ||
The signal intensity at 274 ppm from lithium metal arises from sites inside a skin depth of 12.3 μm even though the lithium electrode thickness is 0.3 mm. As shown earlier16,21 with the growth of lithium dendrites during the delithiation process on each cycle there is a dramatic increase in signal intensity as the lithium dendrites can be fully penetrated by the radio frequency of the NMR. Due to the fact that no dendrites are present during lithiation on the first cycle, no changes in the metallic lithium signal intensity are observed (see Fig. S1 of ESI†). The noticeable drop in lithium metal signal intensity at top = 280 h (Eop = 74 mV) corresponds to the complete removal of deposited lithium dendrites on the counter electrode during lithiation of the graphite. Conversely, the reappearance of lithium dendrite resonance during the delithiation of carbon in the 2nd cycle occurs at approximately top = 310 h (Eop = 152 mV).
At the beginning of the second cycle—the bottom of Fig. 4—between top = 193 h (Eop = 700 mV) and top = 225 h (Eop = 470 mV), we observe a broad featureless resonance of low intensity spanning a range of negative (diamagnetic) shifts from δ = −70 ppm to 0 ppm that progressively narrows while shifting towards 0 ppm during lithiation. On delithiation this peak reappears at top = 365 h (Eop = 549 mV), broadening and moving to more negative (diamagnetic) shifts until the end of the cycle at top = 386 h (Eop = 700 mV). This broad resonance does not appear as mentioned in previous work. The assignment of this resonance is discussed in Section 3.3.
Between top = 222 h (Eop = 490 mV) and top = 234 h (Eop = 400 mV) a boost in the signal intensity around −2.5 ppm to −1 ppm is observed in a region also associated with the electrolyte and SEI resonances and appears with maximum intensity around top = 226 h (Eop = 460 mV). On delithiation this peak appears at top = 341 h (Eop = 376 mV) and lasts until top = 361 h (Eop = 520 mV). This resonance was tentatively assigned to the gas-like stage, LiC72, by Chevallier et al.16 In contrast to this previous work,16 we observe no discernible quadrupolar satellite transition singularities in the line shape of this phase, although the sensitivity of our NMR spectrum may not be sufficient to observe these low lying intensities.
After the formation of LiC72, between top = 226 h (Eop = 460 mV) and top = 263 h (Eop = 196 mV), the intercalated lithium resonances continue to move to more positive shifts while approximately doubling in line width from a fwhm of 10 ppm to 20 ppm. On delithiation these resonances appear at top = 321 h (Eop = 232 mV) and last until top = 345 h (Eop = 405 mV). As with the LiC72 we observe no discernible quadrupolar satellite transition singularities in the line shape of these resonances.
The 7Li NMR resonance of the first dilute stage of lithiated graphite, LiC36, begins appearing at top = 263 h (Eop = 196 mV) near δ = 1 ppm with quadrupolar satellite transition singularities flanking at ±9 kHz (±58 ppm). The appearance of this resonance coincides with the appearance of peak A in the CV on the left of the spectrum. This intercalated lithium resonance continues to move to more positive shifts from 1 to about 6 ppm while maintaining the same quadrupolar splitting until the resonance becomes obscured by its disappearance and the growth of the overlapping resonance from LiC27 at top = 271 h (Eop = 137 mV) and 6 ppm with quadrupolar satellite transition singularities flanking at ±9 kHz (±58 ppm).
The dense stages 1 and 2 appear with the strongest intensity at δ = −50 ppm to 120 ppm between Eop = 74 mV (top = 280 h) and top = 310 h (Eop = 152 mV). The resonances for the two 7Li satellite transitions of stages 4 through 1 have characteristic quadrupolar powder pattern line shapes associated with ηq ≈ 0. The two perpendicular powder pattern singularities are observed at frequencies that flank the central transition at ±8 kHz to ±11 kHz depending on the site.
3.3 dOp NMR spectrum
Fig. 5 and 7 show the dOp 7Li NMR spectrum split into two parts for the late and early stages of lithiation, respectively. The dOp NMR spectrum is obtained by taking the first derivative of the conventional two-dimensional NMR spectrum with respect to top as described in Section 2.2.2. Positive derivative intensities are shown in green and represent the formation of lithium species (henceforth called formation peaks) whereas negative derivative intensities are shown in red and represent the removal of lithium species (henceforth called removal peaks). As noted earlier the signal at 274 ppm arises from the lithium metal. In stark contrast to the conventional NMR spectrum in Fig. 4 we clearly see in the dOp NMR spectrum of Fig. 5 the high rate of removal of lithium metal during lithiation as the intense red resonance region from top = 250 h (Eop = 300 mV) to top = 290 h (Eop = 5 mV) and the strong rate of formation of lithium metal during delithiation as the intense green resonance region from top = 290 h (Eop = 5 mV) to top = 326 h (Eop = 268 mV). The intensity of the lithium signal (rate of dendrite removal) behaves in a manner completely analogous to a CV with the lithium metal NMR derivative intensity correlated with electrochemical current as shown in Fig. 6A (vide infra). Similarly, the 7Li dOp NMR resonances assigned to the formation and removal of the intercalated graphite stages are clearly visible as the green and red resonances in the region from −50 ppm to 150 ppm. As expected, the formation peaks of the GIC are correlated with the removal peaks of the lithium metal and vice versa. Compared to the conventional NMR spectrum, however, the dOp spectrum reveals considerably more details about the chemistry of the lithiation and delithiation processes as we describe in further detail below.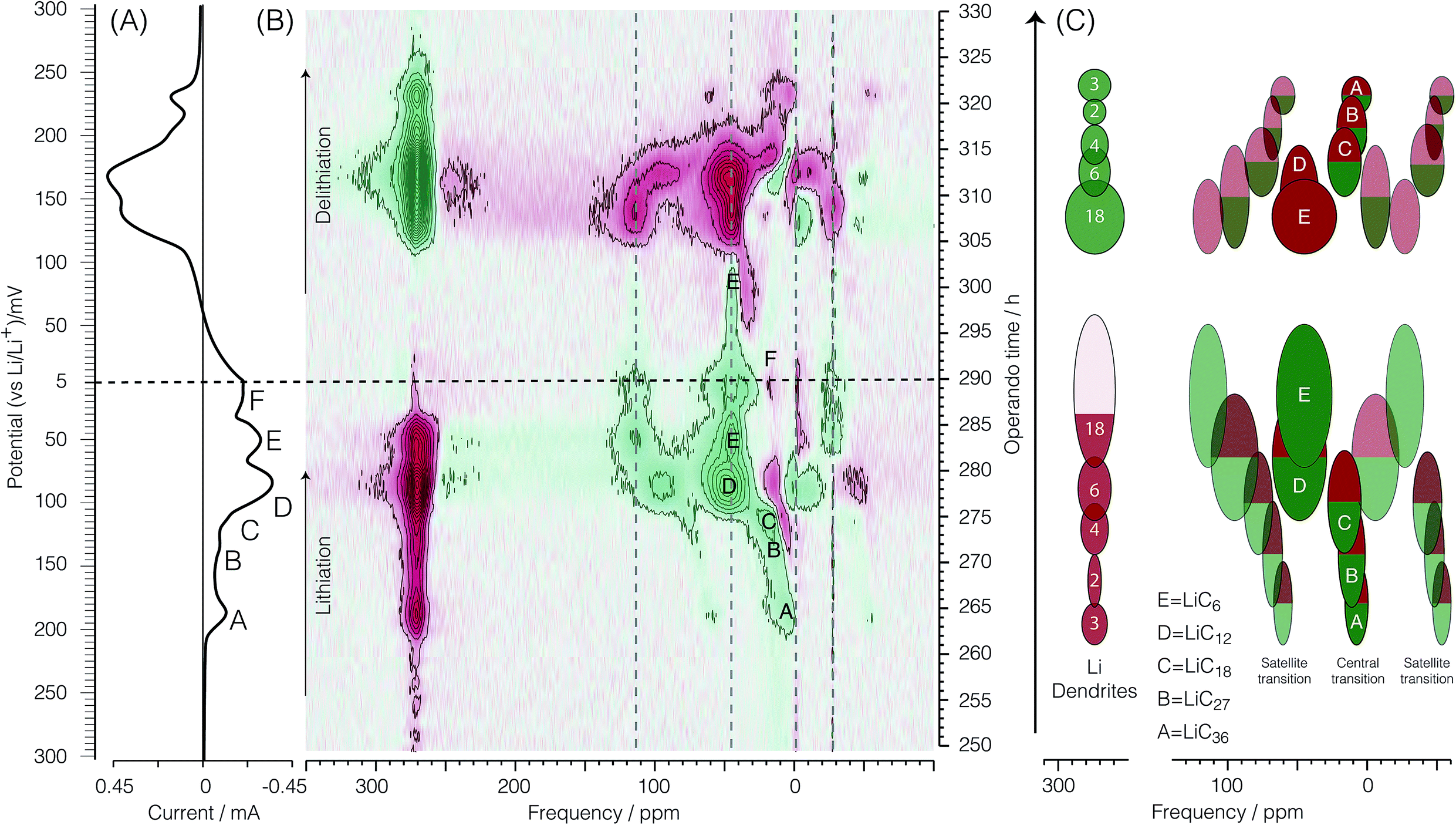 | ||
| Fig. 5 (A) Cyclic voltammogram of the second cycle of the cell. (B) Corresponding bicolor dOp NMR spectrum as a function of time in the potential range between 300 and 5 mV. (C) Schematic representation of the dOp spectrum of the lithiation and delithiation of a carbon electrode. The colored areas have been normalized to reflect the relative intensities in the NMR spectrum for a fixed amount of graphite and to match the stoichiometric coefficients of the reactants and products in the electrochemical equations. | ||
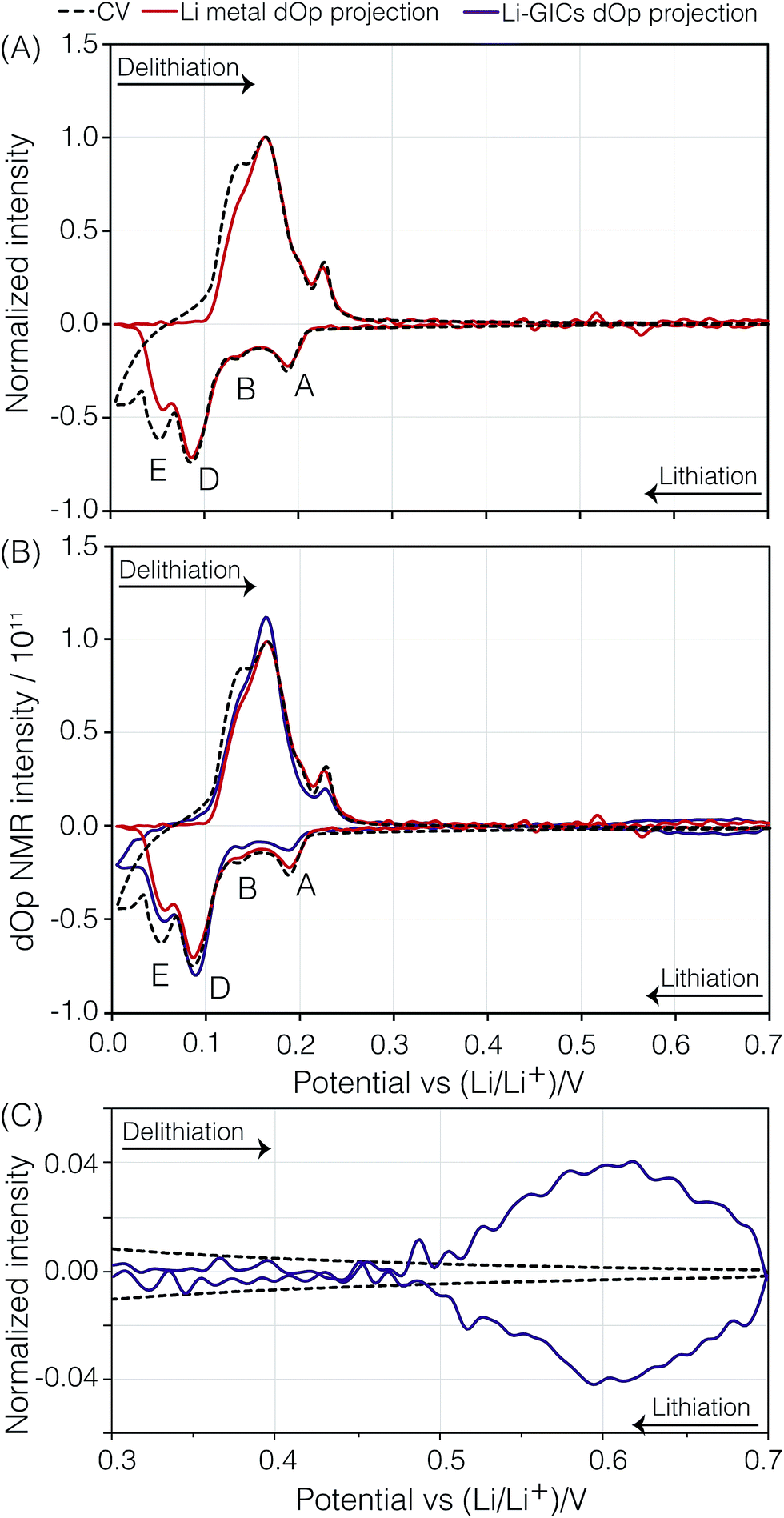 | ||
| Fig. 6 (A) Electrochemical current CV (black dashed line) and the “dOp-NMR cyclic voltammogram” (red line) taken from the projection of the 7Li dOp-NMR signal across the lithium metal for the second cycle of the carbon/Li electrochemical cell. (B) Comparison of the dOp-NMR CV taken from the projection of the 7Li dOp-NMR signal across the lithium metal (red dashed line) and the projection of the Li–GICs (purple line) to the electrochemical CV. (C) Comparison of the dOp NMR CV and the electrochemical CV in the region between 700 and 300 mV depicting the formation of LiC72 and precursors. The peaks labelled A, B, D, and E were previously described in the text. | ||
Presuming that the concentration of each lithium species in the cell is proportional to the NMR signal intensity in the conventional NMR spectrum, then the change in the NMR signal as a function of time is correlated with the electrochemical current measured at each operando time/potential. If we take the projection of the dOp spectra in the frequency range between 200 ppm and 350 ppm and plot it in a cyclic manner we obtain the “dOp-NMR Cyclic Voltammogram” shown in Fig. 6A. The reaction rates along the vertical axis of both the dOp-NMR CV and electrochemical current CV were normalized for better comparison. During lithiation of carbon, we see excellent agreement between the CV and the dOp-NMR CV of the lithium metal until about 50 mV where dendrites start to deplete and the dOp NMR signal dies. Similarly at the beginning of delithiation, mossy like structures grow on the surface of the lithium metal electrode that do not get fully penetrated by the NMR radio frequency like dendrites, and result in a weaker signal in the dOp NMR spectrum than that observed in the CV. Overall, the dOp NMR spectrum shows a strong correlation with the electrochemical currents implying that for every electron transferred from the system, a lithium ion is removed from the counter electrode. The numbers in circles in the metal signal in the schematic on the right of Fig. 5 correspond to the numerator of the lithium ion fraction in the corresponding half-reaction. These numbers roughly correspond to the area of the dOp lithium metal resonance in the region correlated with each half-reaction.
LiC36. Fig. 5 clearly shows a formation peak labelled A around δ = 4 ppm at top = 264 h (Eop = 189 mV). This peak correlates with the cathodic peak labelled A in the electrochemical voltammogram and is assigned to the formation of LiC36 which is described by eqn (4). Formation peak A is also more clearly seen in the dOp NMR spectrum to be flanked on both sides by two peaks appearing at δ ≈ 4 ± 60 ppm due to the satellite transitions of 7Li, a spin I = 3/2 nucleus. Associated with this formation peak is a corresponding removal peak (red) at δ = 274 ppm from lithium dendrite removal. Removal of the previous phase LiC72 is also observed by a faint removal peak at 0 ppm. In the schematic diagram on the right of Fig. 5 we indicate the LiC36 (green) formation peak with a green central transition line and its two satellites along with a corresponding lithium dendrite (red) removal peak. In the schematic on the right of Fig. 5 the satellite areas are made transparent to match their lower peak intensity compared to the central transition line. This process of formation of LiC36 will be further discussed in Section 3.3.5.
LiC27. The formation peak labelled B at δ ≈ 10 ppm and top = 270–272 h (Eop = 150–137 mV) in the dOp NMR spectrum in Fig. 5 correlates with the formation of the cathodic peak labelled B in the electrochemical voltammogram. This peak is assigned to the formation of LiC27 associated with the half-reaction of eqn (5). This formation peak is also flanked on both sides by two formation peaks appearing at δ ≈ 10 ± 60 ppm due to the satellite transitions. Homologous removal peaks for LiC36 at 10 ppm and lithium dendrites at δ = 274 ppm are also observed. Note that the corresponding removal peak of lithium dendrite drops in intensity compared to the previous reaction, which is consistent with the reduction of the stoichiometric coefficient of Li+ by 1/3 from the half reaction A to B.
LiC18. The strong formation peak labelled C at 15 ppm and top = 274 h (Eop = 117 mV) in the dOp NMR spectrum correlates with the formation of the cathodic peak labelled C in the electrochemical voltammogram. As previously mentioned, the electrochemical voltammogram shows LiC18 as a shoulder (peak C) during the formation of LiC12. Therefore, this dOp peak is assigned to the formation of LiC18 as part of the half-reaction of eqn (6). Associated with this half-reaction is the removal peak of LiC27 at δ ≈ 10 ppm for its central transition. Again, the power of the dOp approach is apparent considering that the transition from LiC36 to LiC18 is not discernible in the conventional NMR spectrum in Section 3.2. The higher intensity of the removal peak for LiC27 in the dOp spectrum is due to the higher lithium concentration of the phase compared to the former phases. LiC18 also shows two satellite transition peaks appearing at δ ≈ 15 ± 60 ppm. During its formation the lithium dendrite removal peak does not show a well-defined area due to the low electrochemical resolution between this phase and the next (LiC12).
LiC12. Fig. 5 also shows the dOp NMR spectrum of the dense stages of lithiation LiC12 and LiC6. These phases are seen in the conventional NMR spectrum (Section 3.2) by a broad, strong signal at δ ≈ 45 ppm with two perpendicular powder pattern singularities at frequencies that flank the central transition at ±8 to ±11 kHz depending on the site. The dOp spectrum, in contrast, shows well resolved areas for both phases. Accumulation of LiC12 is visible as a strong formation peak at top = 275 h (Eop = 112 mV) at δ ≈ 48.3 ± 48 ppm, and as seen in eqn (7), results from further lithiation of graphite as lithium metal and LiC18 are removed. The LiC18 removal peaks for the central and one of the satellite transitions occur at 15 ppm and −50 ppm, respectively. The dOp intensity of lithium dendrites decreased (intense red) drastically due to the faster rates of removal of lithium to form LiC12. This transition is depicted in the CV by a broad reduction peak at Eop = 85 mV labelled D. Due to the higher NMR shift and similar quadrupolar frequencies of this phase compared to those of the dilute stages, it is easier to discern the formation of the LiC12 central transition and both satellites as the previous LiC18 phase (peak C) is removed. This behavior is illustrated in the schematic diagram on the right of Fig. 5, where the satellite at δ ≈ 0 ppm from LiC12 (peak D) grows at an NMR shift that is between the central and right satellite removal peaks of C.
LiC6. As lithiation continues LiC6 is formed according to eqn (8) and peak E appears at δ ≈ 42 ppm and top = 281 h (Eop = 66 mV) in the dOp spectrum. Due to the close proximity of the central transitions of LiC6 and LiC12 there is strong overlap of their corresponding formation and removal peaks, respectively, making the central transition of the LiC6 in the dOp spectrum appear narrower and lower in intensity than expected based on the stoichiometry. The strong growth of LiC6 is, however, observed in the dOp spectrum at both its flanking satellite transition formation peaks at 110 ppm and −26 ppm while a red peak at 0 ppm confirms the removal of the right satellite LiC12. The formation of LiC6 is observed until top = 296 h (Eop = 51 mV). In this potential range thermodynamics still favors the Li reduction even though the cell is being swept in the opposite (oxidation) direction as the potential is not sufficiently positive to oxidize (or delithiate) Li from the carbon structure due to the irreversible nature of the chemical reaction. In other words, at potentials where Li insertion is favorable, and if the underlying carbon is not fully lithiated to 100%, Li will continue to move into the carbon structure and form LiC6 at these potentials regardless of the direction of the potential sweep. Since the lithium electrode has run out of dendrites at this stage an expected lithium metal removal peak at δ = 274 ppm correlated with LiC6 formation is not observed.
Lithium naphthalenide. At the end of lithiation a cathodic peak at top = 288.5 h (Eop = 15 mV), labelled F, is noted in the CV. It has been previously reported that at these negative potentials Li–Li clusters contained in the structure LiC2 can be formed as well as the formation of lithium naphthalenide by the bonding of lithium atoms to the edging carbons of the graphene sheets in MCMB.31,32 The formation of Li–Li clusters is characterized by an NMR shift close to that of metallic lithium due to the strong metallic character of the Li–Li bond. Our experiments do not reveal such an NMR shift and therefore, we can infer that the cathodic peak in the CV may be related to the formation of the ionic complex lithium naphthalenide. This compound has no discernible quadrupolar satellite transition singularities in the line shape and has been previously explained by Chevallier et al.16 to form as the formation of LiC12 starts. Although there is an NMR signature for lithium naphthalenide present in the conventional NMR spectrum, there is no clear evidence of such a signature in the dOp spectrum due to the overlapping NMR shift for the delithiation of LiC18. The formation of this compound can be inferred by the smaller removal peak width of the LiC12 satellite at 0 ppm, contrary to what has been illustrated in the schematic of Fig. 5.
Further delithiation results in a formation peak at approximately 15 ppm at top = 310 h (Eop = 152 mV) for the evolution of the LiC18 central transition as well as at 274 ppm for the deposition of more dendritic lithium. It is important to note that on delithiation, the peaks for LiC27 and LiC18 cannot be distinguished in the dOp spectrum due to the fast rates of delithiation of the carbon electrode. However, our potential window experiments have shown that the oxidation peak that appears as a shoulder during the delithiation of LiC12 in the CV at top = 317 h (Eop = 205 mV) corresponds to the delithiation of LiC27 and the oxidation peak for the delithiation of LiC18 is buried underneath the removal peak of LiC12.
The removal peak around δ = 4 ppm for LiC36 during delithiation appears at top = 321 h (Eop = 232 mV), while formation peaks are observed at 274 and 0 ppm representative of dendrite and LiC72 formation, respectively. This transition is correlated with the oxidation peak seen in the electrochemical voltammogram at 230 mV.
Precursors to LiC72. Fig. 7 shows the dOp spectrum of the lithiation processes occurring at potentials between 700 and ∼200 mV—where the formation of LiC36 occurs—and the delithiation processes occurring between ∼200 mV and the end of the cycle at 700 mV.
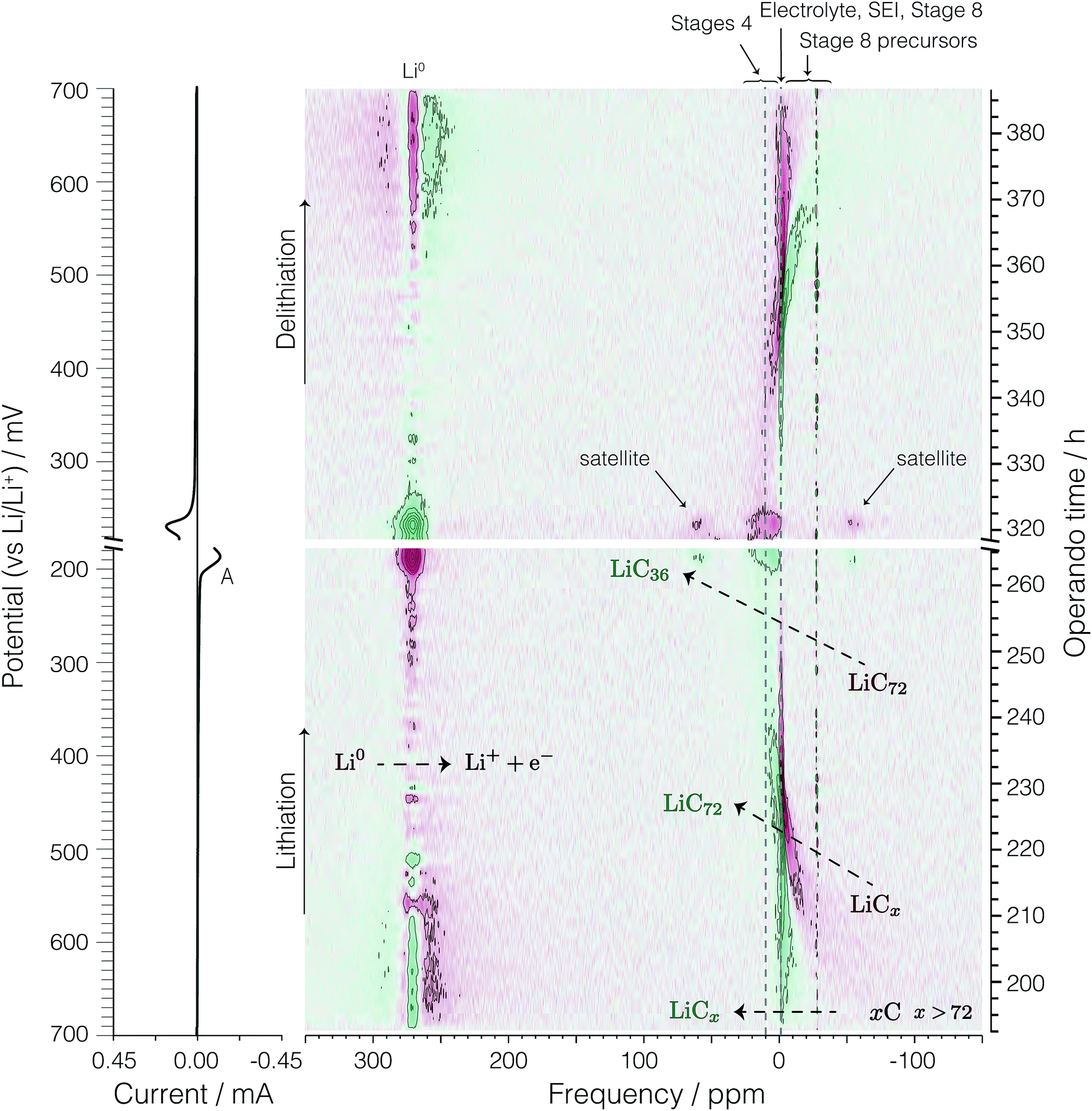 | ||
| Fig. 7 On the left is the cyclic voltammogram of the second cycle of the cell and on the right is the corresponding bicolor dOp NMR spectrum as a function of time in the potential range between 700 and 200 mV during lithiation and delithiation. | ||
At the beginning of the cycle at top = 193 h (Eop = 700 mV) and up to about top = 230 h (Eop = 430 mV) the conventional NMR spectrum in Fig. 4 shows a broad area of negative shifts that continuously narrows while shifting more positively. The dOp NMR spectrum in Fig. 7, in contrast, shows the appearance of a sharp formation peak with shifts between 0 and −15 ppm while broad, low intensity red peaks with negative (diamagnetic) shifts disappear. These negatively shifted peaks are observed to progressively narrow while shifting towards 0 ppm during lithiation, indicating the formation of GICs in stages higher than 8, which we call “precursors” for simplicity.
| xC + Li+ + e− ⇌ LiCx, x > 72. | (9) |
The broad NMR line shape of these precursors indicates a distribution of NMR signals arising from the different lithium environments that conform to the precursors.
Theoretical and computational calculations33–35 have shown that the band structure of graphite changes from a narrow band semiconductor to a conductor when lithium ions intercalate into the graphene sheets due to the injection of electrons from the external circuit that are delocalized within the graphene layers. The magnetization of free conduction electrons on these layers results in an enhancement of the magnetic field experienced by the lithium nuclei, explaining the 7Li NMR Knight shifts observed for the GICs with stages lower than 4.36 The diamagnetic shifts observed for the Li–GIC precursors suggest that at this stage during lithiation not enough electrons have been injected into the graphene layers to make them conducting and therefore, no Knight shifts are observed in the 7Li NMR of the Li–GIC precursors.
LiC72. Between top = 215 h (Eop = 542 mV) and top = 234 h (Eop = 400 mV) the intensity of a formation peak at around −2.5 to −1 ppm, tentatively assigned to LiC72 by Chevallier et al.,16 intensifies while the broad diamagnetic removal peaks narrow and shift to more positive values. This formation peak is observed to reach maximum intensity at top = 225 h (Eop = 470 mV). No discernible quadrupolar satellite transition singularities are observed in the line shape of this formation peak. Assuming that the total lithium transferred to the system up to this potential (Eop = 470 mV) is used to form Li–GICs then the total charge and moles of lithium calculated result in a Li–C stoichiometry of LiC69, which is close to that of stage 8 LiC72. Thus, we confirm the assignment of this unknown formation peak to LiC72.
Pre-dilute stages. After the formation of LiC72 at top = 225 h (Eop = 470 mV) its corresponding formation peak continues to move towards more positive shifts as it broadens and disappears from top = 237 h (Eop = 383 mV) to top = 260 h (Eop = 218 mV). Similarly, the loss of LiC72 is seen by the red removal peak at around 0 ppm from top = 225 h (Eop = 470 mV) to top = 260 h (Eop = 218 mV) where the formation peak of LiC36 becomes visible. The broadening in the line shape might be due to a continuous lithium packing into the graphene layers, resulting in broad, low intensity NMR peaks that continuously shift from 1 ppm to 6.3 ppm from the distribution of phases occurring during this transition. No discernible quadrupolar satellite transition singularities are observed in the line shape of these phases. On the basis of these results we write the electrochemical half-reactions during lithiation of the GICs for the precursors as
| xC + Li+ + e− ⇌ LiCx, x > 72, E > 470 mV, | (10) |
| LiCx + nLi+ + ne− ⇌ LiC72, E = 470 mV, | (11) |
| LiC72 + Li+ + e− ⇌ LiCy, 72 > y > 36, 470 mV > E > 189 mV, |
| LiCy + Li+ + e− ⇌ LiC36, E = 189 mV. | (12) |
Although the dOp NMR CV of the GICs shows similar behavior for both the electrochemical current CV and the lithium metal dOp NMR CV, there are minor discrepancies among the relative intensities of the GIC CV peaks A, B, C, D, and E. The lower relative intensity of the GIC CV peaks A and B compared to the corresponding electrochemical current CV and the lithium metal dOp NMR CV peaks arises from the longer GIC relaxation times of A and B, and the subsequent lower fraction of transverse magnetization excited, as noted in Table 1. On the other hand, the GIC CV peak for D, with its shorter relaxation time, is reasonably close to the corresponding electrochemical current CV and the lithium metal dOp NMR CV peaks for D. Based on stoichiometry one would expect a lower intensity for D in all CVs. The apparent boost in D intensity is attributed to broader CV peaks of C and E which extend under the CV peak of D. This is more clearly visible in the 2D dOp NMR spectrum in Fig. 5 which shows that LiC12 (peak D) begins forming at top = 275 h (Eop = 112 mV), where the previous phase LiC18 (peak C) is still forming. Additionally, the formation of LiC6 (peak E) is seen to begin at top = 281 h (Eop = 66 mV) while LiC12 is still being formed. Both of these overlapping formations result in a higher CV intensity for D due to possible inhomogeneity of the cell voltage within the electrode.
A direct comparison of the relative intensities or areas of the dOp NMR CV of the lithium metal to the GICs in Fig. 6B is complicated by a number of factors. The first is that the central transition equilibrium magnetization of the lithium metal is a factor of S + 1/2 = 2 times smaller than the full nuclear magnetization of the lithium in the GIC. The second is that the excitation pulse length used in these experiments corresponds to a 26° rotation of the central transition magnetization of the lithium metal and a 13° rotation of the full nuclear magnetization of the lithium in the GIC—giving approximately a factor of two better excitation efficiency for the lithium metal. Finally, the significantly shorter T1 of lithium metal leads to it having an ∼3 times greater fraction of steady-state transverse magnetization than the GICs. The first two factors effectively cancel each other and the last factor, predicting the lithium metal signal area to be ∼3 times greater than the GIC signal areas, is not what is observed. The most likely explanation is that two thirds of the expected lithium metal dOp signal is not observed due to skin depth effects.
Finally, we note that unrelated but another potential source of intensity error in the NMR can arise from drifts in the receiver phase during the operando experiment. Possible remedies for this source of error could include (1) auto tuning the probe during the acquisition,37 (2) employing post-acquisition auto-phasing the spectra, or (3) in the case of spin I ≤ 3/2, like 7Li, the use of a total echo experiment38 to eliminate dead time issues.
4 Conclusions
Although operando 7Li NMR is a powerful technique for probing the changes that occur in the carbon electrode as it gets intercalated and deintercalated with lithium, the spectra often suffer from poor resolution due to a number of overlapping resonances. We have shown that a simple signal processing method of analyzing the operando NMR signal by taking the derivative of the signal with respect to the operando time, referred to as dOp NMR (derivative operando NMR), results in significantly more detailed information about the changes that occur during the cycling of the cell. By using this method we have identified the formation and removal of six stages of lithium intercalation and deintercalation from LiC72 to LiC6 and back. In addition, the dOp NMR method allows for the detection of additional gas like stages of Li in graphite, which were previously not observed experimentally. In this work, using slow rates of potential perturbation at 2 μV s−1, we have identified the electrochemical potentials at which LiC72 is formed from gas like Li precursors.A partial projection over specific regions of the dOp NMR spectrum as a function of the operando time or voltage can be presented in a cyclic fashion what is termed a dOp NMR CV. Through the dOp NMR CV method we have found that the rates of formation and removal of lithium dendrites 7Li resonances closely follow the electrochemical current as long as dendrites are present in the cell. Similarly, the rates of formation and removal of 7Li resonances associated with the lithium intercalated graphite also follow the electrochemical current flow with slight deviations due to an experimental artifact from incomplete 7Li relaxation of Li stages as well as possible inhomogeneity of the cell voltage within the electrode in the electrochemical reactions after stage 3.
The dOp methodology could be applied to other operando analyses such as in situ XRD or in situ IR or Raman spectroscopy, where time independent processes can be removed to enhance the signals of relevant time dependent chemical transitions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported in part by the National Science Foundation under Seed Grant NSF-DMR-1420451. JLLL acknowledges the support of the GAANN fellowship from the Department of Education (P200A150267) and the Ohio State Department of Chemistry and Biochemistry.References
- T. Christen and M. W. Carlen, J. Power Sources, 2000, 91, 210–216 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- B. Y. Liaw and R. Kostecki, Electrochem. Soc. Interface, 2011, 20, 41–42 CrossRef CAS.
- J. R. Dahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 9170–9177 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS.
- T. Ohzuku, J. Electrochem. Soc., 1993, 140, 2490 CrossRef CAS.
- L. Y. Beaulieu, S. D. Beattie, T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A419 CrossRef CAS.
- S. Whitney, S. R. Biegalski, Y. H. Huang and J. B. Goodenough, J. Electrochem. Soc., 2009, 156, A886 CrossRef CAS.
- R. Downing, G. Lamaze, J. Langland and S. Hwang, J. Res. Natl. Inst. Stand. Technol., 1993, 98, 109 CrossRef CAS PubMed.
- D. X. Liu, L. R. Cao and A. C. Co, Chem. Mater., 2016, 28, 556–563 CrossRef CAS.
- D. X. Liu and A. C. Co, J. Am. Chem. Soc., 2016, 138, 231–238 CrossRef CAS PubMed.
- D. X. Liu, J. Wang, K. Pan, J. Qiu, M. Canova, L. R. Cao and A. C. Co, Angew. Chem., Int. Ed., 2014, 53, 9498–9502 CrossRef CAS PubMed.
- R. E. Gerald, J. Sanchez, C. S. Johnson, R. J. Klingler and J. W. Rathke, J. Phys.: Condens. Matter, 2001, 13, 8269–8285 CrossRef CAS.
- M. Letellier, F. Chevallier and F. Béguin, J. Phys. Chem. Solids, 2006, 67, 1228–1232 CrossRef CAS.
- F. Chevallier, F. Poli, B. Montigny and M. Letellier, Carbon, 2013, 61, 140–153 CrossRef CAS.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- S. A. Krachkovskiy, J. D. Bazak, P. Werhun, B. J. Balcom, I. C. Halalay and G. R. Goward, J. Am. Chem. Soc., 2016, 138, 7992–7999 CrossRef CAS PubMed.
- H. J. Chang, A. J. Ilott, N. M. Trease, M. Mohammadi, A. Jerschow and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 15209–15216 CrossRef CAS PubMed.
- J.-M. Tarascon, A. Gozdz, C. Schmutz, F. Shokoohi and P. Warren, Solid State Ionics, 1996, 86–88, 49–54 CrossRef CAS.
- M. Letellier, F. Chevallier and M. Morcrette, Carbon, 2007, 45, 1025–1034 CrossRef CAS.
- J. L. Markley, W. J. Horsley and M. P. Klein, J. Chem. Phys., 1971, 55, 3604–3605 CrossRef CAS.
- P. J. Grandinetti, J. T. Ash and N. M. Trease, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 121–196 CrossRef CAS PubMed.
- C. P. Slichter, Principles of Magnetic Resonance, Springer-Verlag, Berlin, 1980 Search PubMed.
- A. J. Ilott, S. Chandrashekar, A. Klöckner, H. J. Chang, N. M. Trease, C. P. Grey, L. Greengard and A. Jerschow, J. Magn. Reson., 2014, 245, 143–149 CrossRef CAS PubMed.
- R. R. Ernst, Rev. Sci. Instrum., 1966, 37, 93 CrossRef CAS.
- RMN 1.1, PhySy Ltd., 2017 Search PubMed.
- M. D. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 79–88 CrossRef CAS.
- A. J. Smith, J. C. Burns, X. Zhao, D. Xiong and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A447 CrossRef CAS.
- M. A. McArthur, S. Trussler and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A198–A207 CrossRef CAS.
- M. Hara, A. Satoh, N. Takami and T. Ohsaki, J. Phys. Chem., 1995, 99, 16338–16343 CrossRef CAS.
- K. Tatsumi, J. Electrochem. Soc., 1996, 143, 1923 CrossRef CAS.
- N. M. Caffrey, L. I. Johansson, C. Xia, R. Armiento, I. A. Abrikosov and C. Jacobi, Phys. Rev. B, 2016, 93, 195421 CrossRef.
- J.-C. Charlier, X. Gonze and J.-P. Michenaud, Europhys. Lett., 1994, 28, 403–408 CrossRef CAS.
- K. R. Kganyago and P. E. Ngoepe, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 205111 CrossRef.
- G. Volpilhac and J. Hoarau, Synth. Met., 1981, 4, 77–79 CrossRef CAS.
- O. Pecher, D. M. Halat, J. Lee, Z. Liu, K. J. Griffith, M. Braun and C. P. Grey, J. Magn. Reson., 2017, 275, 127–136 CrossRef CAS PubMed.
- B. J. Walder, K. K. Dey, M. C. Davis, J. H. Baltisberger and P. J. Grandinetti, J. Chem. Phys., 2015, 142, 014201 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta07521a |
| This journal is © The Royal Society of Chemistry 2018 |