
Cellulose-based electrospun fibers functionalized with polypyrrole and polyaniline for fully organic batteries†
A. C.
Baptista
 a,
I.
Ropio
a,
B.
Romba
a,
J. P.
Nobre
a,
C.
Henriques
b,
J. C.
Silva
b,
J. I.
Martins
cd,
J. P.
Borges
*a and
I.
Ferreira
*a
a,
I.
Ropio
a,
B.
Romba
a,
J. P.
Nobre
a,
C.
Henriques
b,
J. C.
Silva
b,
J. I.
Martins
cd,
J. P.
Borges
*a and
I.
Ferreira
*a
aCENIMAT/I3N, Departamento de Ciência dos Materiais, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal. E-mail: jpb@fct.unl.pt; imf@fct.unl.pt
bCENIMAT/I3N, Departamento de Física, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
cUniversidade do Porto, Faculdade de Engenharia, Departamento de Engenharia Química, 4200-465 Porto, Portugal
dLab2PT, Instituto de Ciências Sociais, Universidade do Minho, 4710 - 057 Braga, Portugal
First published on 30th November 2017
Abstract
A novel cellulose-based bio-battery made of electrospun fibers activated by biological fluids has been developed. This work reports a new concept for a fully organic bio-battery that takes advantage of the high surface to volume ratio achieved by an electrospun matrix composed of sub-micrometric fibers that acts simultaneously as the separator and the support of the electrodes. Polymer composites of polypyrrole (PPy) and polyaniline (PANI) with cellulose acetate (CA) electrospun matrix were produced by in situ chemical oxidation of pyrrole and aniline on the CA fibers. The structure (CA/PPy|CA|CA/PANI) generated a power density of 1.7 mW g−1 in the presence of simulated biological fluids, which is a new and significant contribution to the domain of medical batteries and fully organic devices for biomedical applications.
Introduction
Recently, cellulose paper has been (re)discovered as a smart material that can be used in electronics.1–4 Since cellulose is an environmentally friendly, cost effective and versatile material, a cellulose-based energy storage device will have significant advantages in comparison with many currently used batteries and supercapacitors. Therefore, the interest in cellulose-based bio-batteries for suppling power to implantable medical devices has increased and became a hot topic of research.5–8 In this context, a bio-battery can be defined as a biocompatible battery for in vivo usage.A. C. Baptista5 and co-workers proposed a new concept of a flexible and lightweight cellulose-based battery, so called bio-battery, activated by biological fluids. For concept demonstration, Al and Ag metallic thin films were deposited on each side of an ultrathin cellulose-based electrospun membrane as anode and cathode, respectively. This device, with an area of 2 cm2 and a thickness of 53 μm, placed in contact with simulated body fluids (<0.1 ml) displayed a power density of 3.38 μW cm−2. Later, Yong Kong9 and colleagues proposed a bio-battery composed of PPy doped with a biological polyelectrolyte (dextran sulfate, DS) as cathode and a bioresorbable Mg alloy as anode. This battery exhibited an energy density of 790 W h kg−1, using commonly biological media as electrolyte. Recently, Sha Li6 used cellulose-based composites as cathodes for zinc–air bio-batteries activated by simulated body fluids. PPy/CNTs composites were chemically synthetized and deposited on cellulose filter paper for the cathode while a zinc foil was used as anode. In the presence of simulated biological fluids, this device discharged in 24.5 hours at a current density of 60 μA cm−2. X. Jia8 and co-workers reported the development of a magnesium–air bio-battery using as cathode a silk fibroin–polypyrrole (SF–PPy) film, a bioresorbable Mg alloy as anode and a phosphate buffered saline (PBS) solution as electrolyte. This bio-battery exhibited a discharge capacity up to 3.79 mA h cm−2 for a current of 10 μA cm−2 at room temperature.
Electrically conductive polymers, such as PPy and PANI, are biocompatible and are therefore valuable materials that can be used as electrodes in lightweight and flexible biomedical batteries. The ability to customize conductive nanostructures to meet the requirements of specific applications gives electrospinning an advantage over other production methods. However, the electrospinning of continuous conductive fibers directly from conductive polymeric solutions is a great challenge.10 Some authors have proposed the addition of a carrier polymer to facilitate the electrospinning process; others reported on the combination of the electrospinning technique with a nanocoating procedure.11–13 The deposition of PPy or PANI on the surface of fabrics and yarns has been widely investigated in the last few years in order to incorporate these polymers in new and functional devices, such as sensors,14 biosensors15 and scaffolds for tissue engineering.16
Considering the growing need for power source miniaturization and the replacement, cost and risk inherent to conventional implantable medical devices, there is a need for the development of new electrical power source concepts. This work presents the development of flexible, lightweight non-toxic and conductive cellulose-based electrospun fibers functionalized with PPy and PANI. In order to obtain highly conductive fibers, the in situ polymerization of Py and Ani was carried out on the surface of cellulose acetate electrospun fibers. The polymerization conditions were extensively studied and lately the composite membranes were evaluated as electrodes for bio-batteries.
A fully polymeric bio-battery was constructed by assembling the CA/PPy and CA/PANI composite membranes separated by a CA electrospun membrane and tested with physiological simulated solution as electrolyte. The fully organic device reported here gives a new contribution towards the state of the art of bio-batteries taking advantage of the ionic content present in biological fluids, such as blood and sweat to supply low power consumption medical devices.
Results and discussion
A cellulose acetate membrane composed of sub-micrometric fibers with a smooth surface and an average diameter of 243 ± 58 nm was produced by electrospinning. Then, the CA electrospun membranes were coated with PPy or PANI through the in situ monomer oxidative polymerization as illustrated in Fig. 1a and b.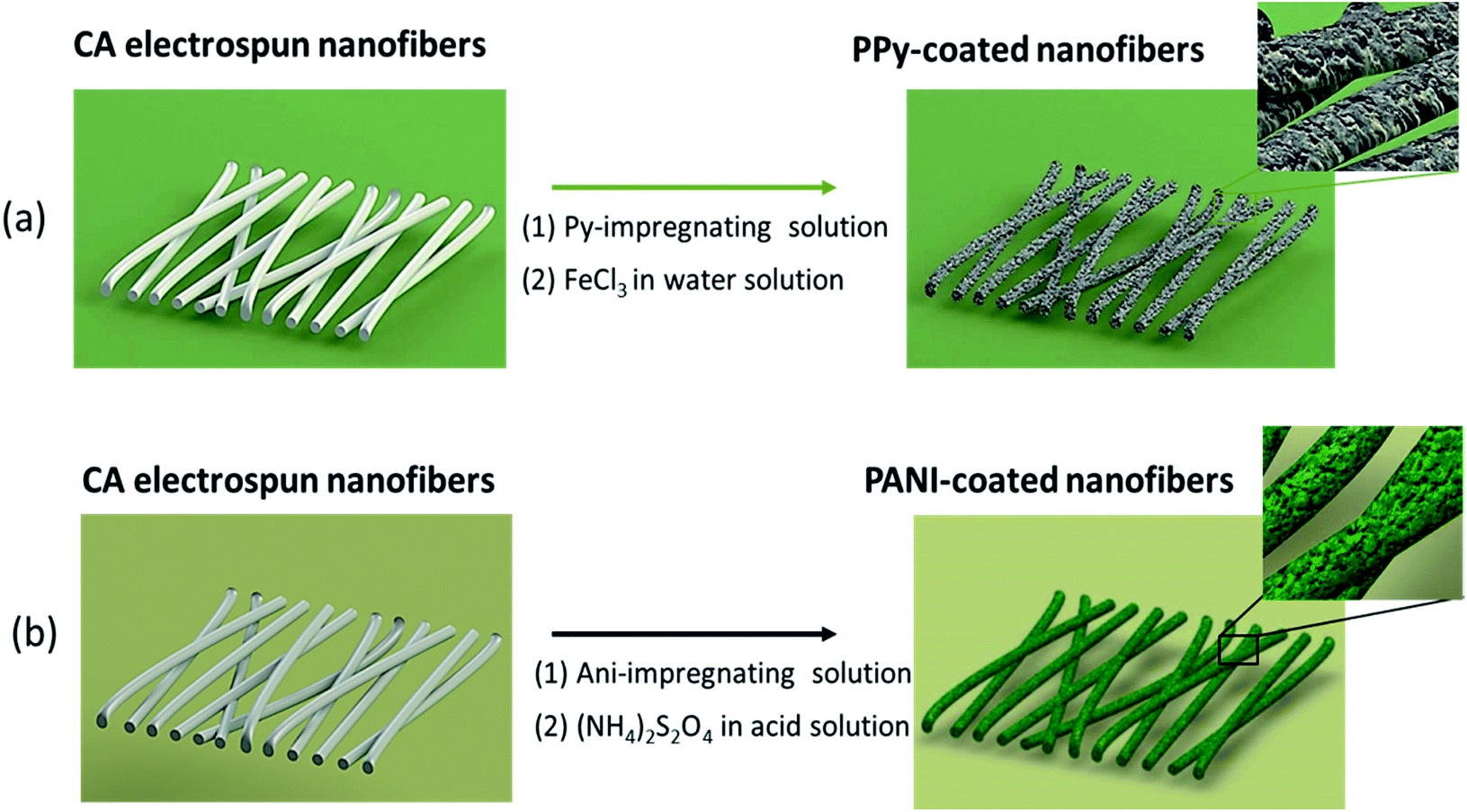 | ||
| Fig. 1 Illustration of the methodologies followed for the preparation of CA fibers coated with (a) PPy and (b) PANI by in situ chemical polymerization. | ||
The dependence of electrical conductivity on the Ox/Mon ratio of CA/PPy fibers obtained after 1 hour of polymerization using a pyrrole concentration of 0.025 M is shown in Fig. 2a. The electrical conductivity increased reaching maximum values around 5 × 10−3 S cm−1 as FeCl3/Py increased up to 2. Considering that the FeCl3 amount should be as small as possible due to its toxicity, the Ox/Mon ratio of 2 was chosen to carry out Py polymerization. Since cellulose can degrade under prolonged polymerization reaction or under aggressive reaction conditions,17 the influence of monomer concentration and polymerization time on the electrical conductivity was investigated (Fig. 2b). The conductivity increases remarkably up to values of 10−2 S cm−1 at 15 and 30 minutes of polymerization, for monomer concentrations of 0.075 and 0.05 M, respectively. This behavior is attributed to the high PPy yield and the formation of a continuous layer on CA nanofibers surface. However, for lower monomer concentrations, more than 90 min are required to achieve a conductivity close to 10−2 S cm−1. The morphology of CA/PPy composite fibers obtained using pyrrole concentrations of 0.025 M (Fig. 2c), 0.05 M (Fig. 2d) and 0.075 M (Fig. 2e) and a polymerization time of 45 min was observed by SEM. The PPy coating formed on CA fibers is not continuous for the lower monomer concentration 0.025 M, which explains the electrical conductivity measured. A continuous coverage of fibers and optimized conductivity values were obtained for monomer concentration of 0.05 M and a polymerization time of 30 min. PPy tends to form aggregates when the monomer concentration increases to 0.075 M, which explains the slight decrease in electrical properties. Some aggregates are also evident for the monomer concentration of 0.05 M but for a longer polymerization time (1440 min) – Fig. 2f. Both electrical and morphological results show that the most adequate conditions to obtain PPy-coated CA fibers with high electrical conductivity are the following: Py at a concentration of 0.05 M, an Ox/Mon ratio of 2 and a reaction time of 30 min.
 | ||
| Fig. 2 Influence of (a) oxidant/monomer ratios and (b) monomer concentration and reaction time on electrical conductivity of CA/PPy fibers prepared by in situ chemical oxidation; SEM images of CA/PPy fibers using different reaction conditions: pyrrole concentration of (c) 0.025 M, (d) 0.05 M and (e) 0.075 M and reaction time of 45 min, and (f) 0.05 M of pyrrole and 1440 min of reaction time. TEM images of CA/PPy fibers prepared by in situ chemical oxidation using 0.05 M of pyrrole concentration and 30 min of reaction time: (g) CA electrospun fibers with a uniform and continuous PPy coating and (h) a detailed image illustrating the PPy coating thickness. | ||
The TEM images of PPy coated CA nanofibers in the optimized synthesis conditions are shown in Fig. 2g and h. Nonwoven composite fibers have an average diameter of 290 ± 69 nm and a uniform and continuous PPy coating with a thickness in the range of 40–50 nm.
The electrical conductivity of polyaniline can change from 10−9 to 100 S cm−1 depending on its oxidation state, degree of protonation and type of dopant used. A higher conductivity is obtained for stronger protonic acids, such as HCl, as dopant agent. In this work, the synthesis of PANI using ammonium persulfate ((NH4)2S2O8), as the oxidant agent was investigated in detail to find the synthesis conditions that would lead to highly conductive fibers. Reports found in the literature have explained the nano-assembly of PANI on a cellulose-based network as a result of the interaction between the protonated nitrogen of PANI (in acidic medium) with the hydroxyl groups of cellulose through hydrogen bonds.18
The influence of monomer concentration, Ox/Mon molar ratio and reaction time on the electrical conductivity of CA/PANI fibers is shown in Fig. 3a. The pristine CA electrospun fibers have an electrical conductivity of (7.1 ± 0.8) × 10−11 S cm−1. During aniline polymerization, PANI gradually grows onto CA fibers forming a continuous sheath and the conductivity increases to values above 10−3 S cm−1 for a polymerization period of 30 min. Further increase in conductivity of the membranes up to 1 S cm−1 was achieved by adjusting polymerization time and Ox/Mon ratio.
 | ||
| Fig. 3 (a) Influence of polymerization time on the electrical conductivity of CA/PANI composite fibers using aniline concentration of 4 M and Ox/Mon molar ratios of 0.125, 0.25 and 0.5, respectively; and SEM images of CA/PANI fibers obtained after (b) 30 min, (c) 45 min and (d) 60 min of polymerization using a 4 M monomer concentration and an Ox/Mon molar ratio of 0.5; (e) influence of polymerization time on the electrical conductivity of CA/PANI composite fibers using an aniline concentration of 2 M and Ox/Mon molar ratios of 0.25 and 0.5, respectively; SEM images of CA/PANI composite fibers obtained after (f) 45 min and (g) 60 min of polymerization using a 2 M monomer concentration and an Ox/Mon molar ratio of 0.5. | ||
The granular aspect of the PANI formed on the fibers along the polymerization time is seen in Fig. 3b–d, for an Ox/Mon molar ratio of 0.5. Fig. 3b clearly shows a non-uniform coating for 30 min of polymerization and an increase of PANI aggregates on the fiber's surface with the polymerization time increase to 45 min and 60 min (Fig. 3c and d, respectively). For these polymerization periods the original structure of CA membrane, composed of individual cylindrical fibers, is not preserved due to aggregation of PANI particles forming a bulk-like material. Since the agglomeration of PANI particles took place on the surface of CA fibers for a monomer concentration of 4 M, a lower concentration, 2 M, was studied for two Ox/Mon ratios (0.25 and 0.5). A decrease of conductivity is observed in Fig. 3e for the Ox/Mon ratio of 0.25, indicating that the amount of monomer used was not enough to yield fibers completely covered by PANI even for prolonged polymerization times. Considering an Ox/Mon ratio of 0.5, the composites produced showed electrical conductivities of 10−1 S cm−1 for 45 and 60 min of polymerization. SEM images (Fig. 3f and g) confirmed the existence of complete fiber coverage by PANI aggregates on the surface of CA fibers.
Overall, we concluded that a continuous and uniform PANI coating of CA fibers with high electrical conductivity can be obtained with aniline at a concentration of 2 M, an Ox/Mon ratio of 0.5 and a 30–45 min reaction time. CA/PANI composite fibers produced under these conditions present an average diameter of 577 ± 59 nm while suggesting the formation of a PANI layer in the range of 160–170 nm.
The CA nonwoven membrane is composed of nanofibers that are loosely packed together without any chemical crosslinking point between fibers. Consequently, its tensile strength is relatively low. According to Fig. 4a, chemical polymerization of Py and Ani on the surface of CA electrospun fibers did not significantly affect the membranes' mechanical properties. The exception is the slight increase of the Young's modulus observed for CA/PPy membranes (42 ± 14 MPa for CA and 59 ± 13 MPa for CA/PPy) that may be due to the nanosized PPy particles on the surface of fibers increasing the cohesion of the membrane at the crossing points between fibers.
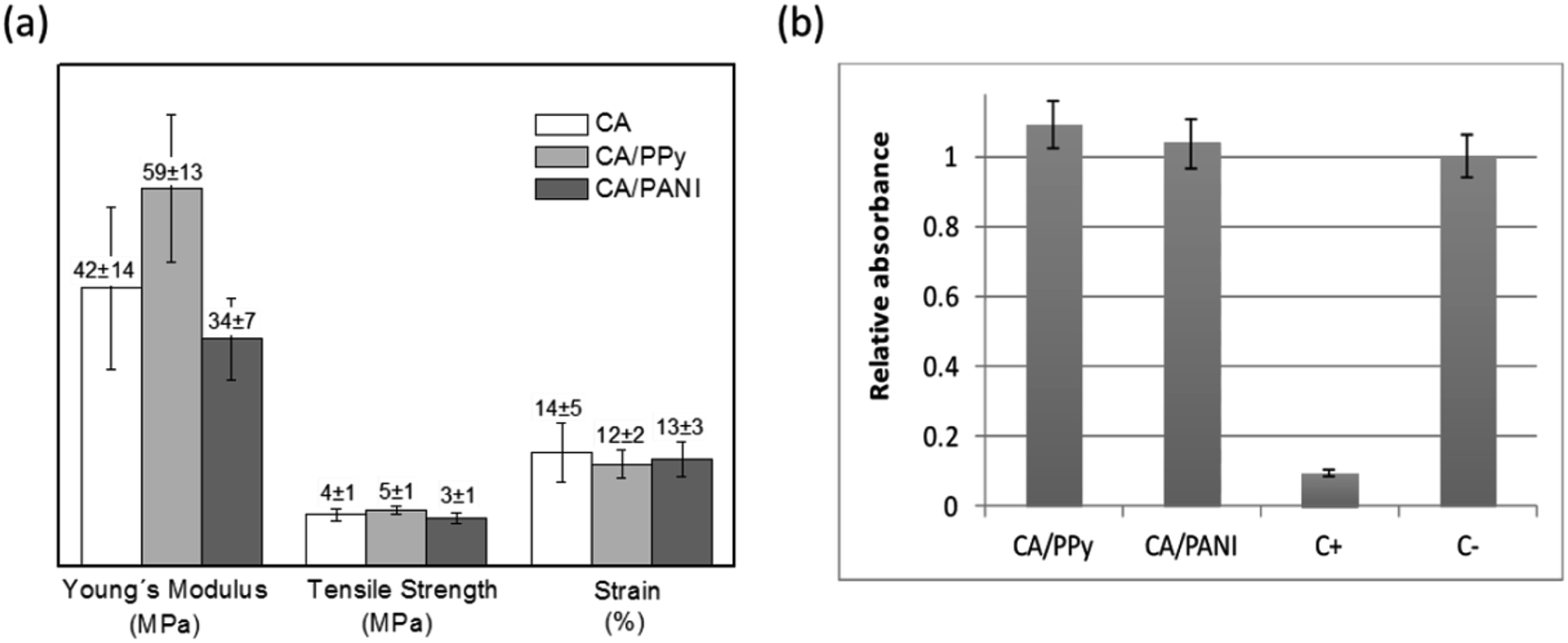 | ||
| Fig. 4 (a) Mechanical properties (Young's modulus, tensile strength and strain) obtained for CA, CA/PPy and CA/PANI membranes and (b) results of the cytotoxicity tests of the CA/PPy and CA/PANI composites using the extract method. Absorbance values are normalized to the average absorbance of the negative control wells and vertical lines represent the experimental standard deviation of the mean. No detrimental effects of extractables on cells were revealed. | ||
Although some authors have suggested that PPy and PANI can generally be regarded as a biocompatible synthetic polymer, it is very important to understand if the processing and morphology of the composites has affected the toxicity of the material.19,20 Following standard cytotoxicity test methods (ISO-10993-5), cells were exposed to extracts obtained by placing the CA/PPy and CA/PANI fibrous membranes in cell culture media. Cell viability was used as an indicator of toxicity and assessed using the resazurin reduction test – Fig. 4b. Both relative cell populations are above 90% indicating that the materials tested are free of harmful extractables or these are at a too low concentration to cause any acute cytotoxic effects capable of affecting cell proliferation and enzymatic activity.
To evaluate the composite materials described above as an electrode material for energy harvesting, CA/PPy and CA/PANI were assembled in the construction of a bio-battery. The CA/PPy and CA/PANI composite membranes were used as electrodes, separated by a cellulose acetate electrospun membrane that acts as the separator. The CA/PPy is the negative electrode and the CA/PANI is the positive electrode where the oxidation of PPy and the reduction of PANI occur, respectively, according to reactions (1) and (2) for discharge. Eventually, the O2 can also be reduced in the CA/PANI electrode configuring the system's hybrid operation.
The electrochemical behavior of the CA/PPy|CA|CA/PANI structure was evaluated by cyclic voltammetry in the presence of a physiological solution – 0.9% (w/v) NaCl solution.
The voltammogram obtained for CA/PPy as the working electrode shows that the anodic wave begins at −0.2 V with a shoulder around 0.08 V and a peak at 0.23 V, and the cathodic sweep shows two reduction peaks at −0.14 V and −0.25 V (Fig. 5a). The redox couples are associated with the oxidation of PPy to polaron and bipolaron states,21 as shown in Fig. 5b. Previous studies have reported that when potentials higher than 0.9–1.0 V vs. SCE are applied, PPy electrodes lose their electrochemical activity due to an irreversible redox reaction.22,23 This phenomenon was not observed during the electrochemical characterization. When CA/PANI is connected as the working electrode, the voltammograms show two redox pairs, with oxidation peaks at 0.23 V and 0.65 V, and reduction peaks at −0.25 V and −0.60 V (Fig. 5c). The first redox couple indicates the oxidation of leucoemeraldine form to protonated emeraldine, and the second couple to oxidation of emeraldine form to pernigraniline, as shown in Fig. 5d. The oxidation of emeraldine is generally considered to be less reversible than the well-known emeraldine–leucoemeraldine transition, and the oxidation products are easily hydrolyzed in acidified aqueous medium without aniline leading to the progressive degradation of the polymer.24–27 Therefore, the current density of the peaks decreases with the number of scanning cycles.
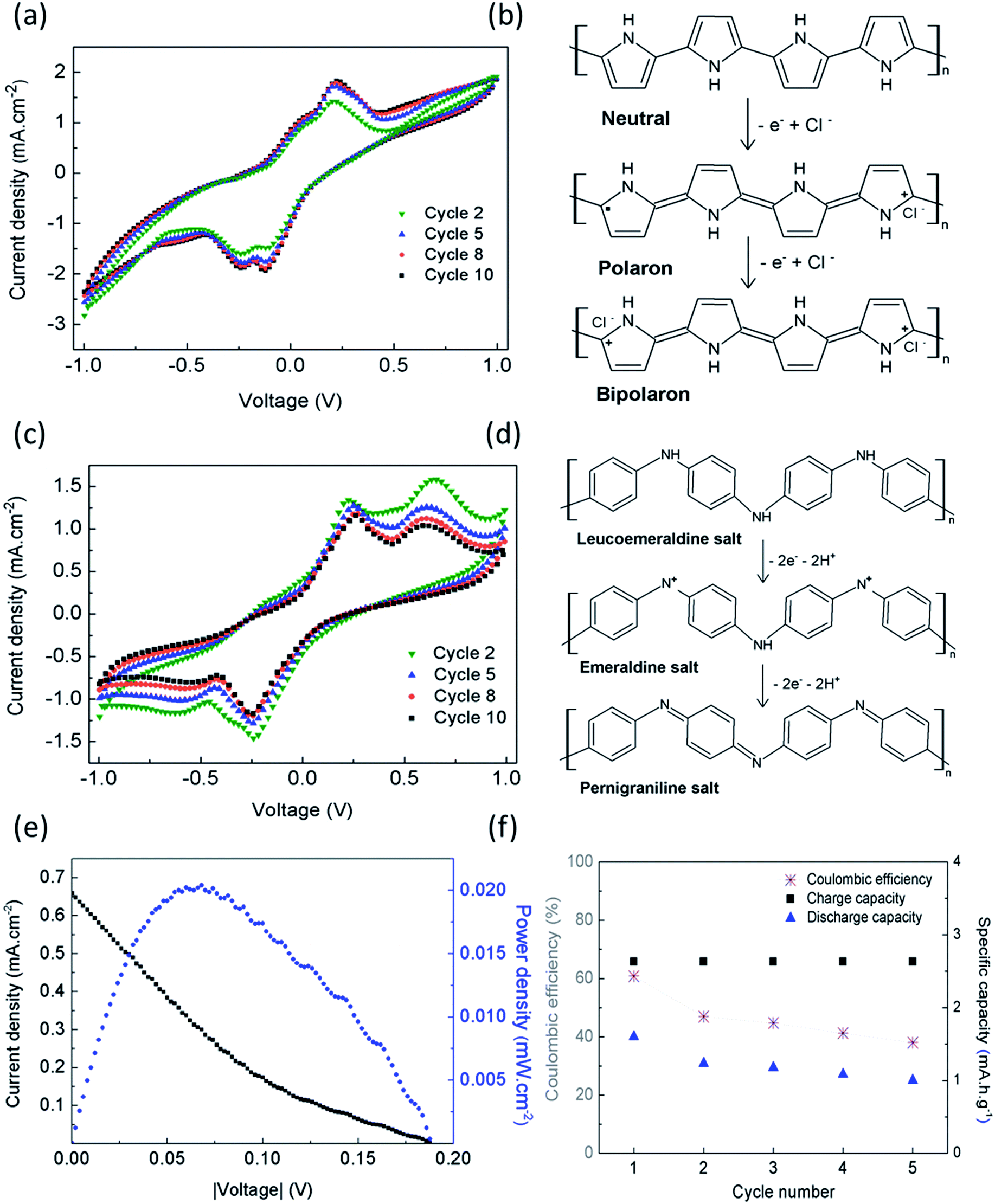 | ||
| Fig. 5 (a) Cyclic voltammogram obtained using CA/PPy as the WE and (b) mechanism suggested for the anodic oxidation of polypyrrole; (c) cyclic voltammogram obtained using CA/PANI as the WE; (d) mechanism suggested for the anodic oxidation of polyaniline; (e) I–V curve and power density determined for the bio-battery and (f) the charge and discharge capacity and coulombic efficiency obtained at a constant charge/discharge current density of 5.3 mA h g−1. | ||
The redox reactions occurring on the individual electrodes during charging under the prevailing experimental conditions can be summarized as follows (while the reverse reactions prevail for discharging):
Positive electrode
[–B–NH–B–NH+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) QNH+–B–NH–]2x → [–B–NQN–]4x + (8x)H+ + (4x)e− QNH+–B–NH–]2x → [–B–NQN–]4x + (8x)H+ + (4x)e− | (1) |
Negative electrode
| [–C4H3Ny+–]x + (xy)e− → [–C4H3N–]x | (2) |
The maximum power density (Pmax) of 1.7 mW g−1 (0.8 mW cm−3) was determined from I–V curves obtained from cyclic voltammetry (Fig. 5e). The power density of the devise matches the values found for typical pacemakers that require less than 10 μW to operate.
Short charge/discharge measurements (one cycle of 4 min) were carried out at different charge/discharge rates using a succession of decreasing charge/discharge current densities. Fig. S1 (in ESI section†) shows a decrease in ohmic losses with current density decreasing. The small discrepancy of the final charge potential for the same current density can be attributed to small current oscillations and/or structural modifications of polymers.
The device was submitted to charge/discharge cycles at a constant charge/discharge current density of 5.3 mA h g−1 for approximately 5 hours. The amount of electrolyte used was 20 μl, before measurements starts (1st cycle for t = 0 s), and no further addition has been performed during the following cycles. Fig. 5f shows the coulombic efficiency determined for the 5 cycles. The decrease of efficiency in 20% after the 5th cycle can be explained by the need of electrolyte addition. Since the device is not sealed, the small amount of electrolyte added when the measurement starts can be insufficient to guarantee the electrochemical stability of the charge/discharge experiments during prolong periods of time.
This hybrid device differs from conventional batteries that comprise alkali-metals-containing intercalation or insertion materials as electrodes in a closed system, since its concept beyond the redox characteristics of conductive polymers electrodes is also related with the active material of the circulating physiological fluid.
In addition to being flexible, lightweight and ultra-thin (<300 μm of thickness), this fully polymeric bio-battery has the advantage of having an economical production process making it a promising alternative to power implantable and portable microwatt electronic devices. Comparing the main properties of the proposed device with other ultralow power sources found in the literature (Table 1), we conclude that its power density per volume is competitive. The power density is 8 times higher than the first concept5 reported in 2011 using novel and non-toxic electrodes materials.
Conclusions
In this work, cellulose acetate electrospun membranes were evaluated as a polymer platform to produce fibrous conducting composites with high surface area. To produce cellulose-based composite fibers, the in situ polymerization of Py and Ani on the surface of CA electrospun fibers was investigated. CA/PPy and CA/PANI composites fibers were prepared through in situ chemical oxidative polymerization. During this study, chemical synthesis conditions, such as monomer concentration, the mass ratio oxidant/monomer and reaction time were investigated in detail. Morphologically uniform and electrically conductive cellulose-based fibers with high electrical conductivity – 10−2 and 10−1 S cm−1 for CA/PPy and CA/PANI, respectively – were obtained. Toxicity tests were performed for CA pristine membranes and CA/PPy composite fibers. Results indicate that the material tested is either free of harmful extractables or these are present at a low concentration which is insufficient to cause any acute cytotoxic effects capable of affecting cell proliferation and enzymatic activity. The electrochemical performance of cellulose-based bio-batteries was characterized by cyclic voltammetry. The composite materials, CA/PPy and CA/PANI, were used as electrodes (anode and cathode, respectively) for the construction of the bio-battery. This fully polymeric bio-battery showed a maximum power density of 1.7 mW g−1, which is a promising power value considering the power requirements of common implantable medical devices.In summary, conductive CA/PPy and CA/PANI composite fibers have been obtained with the advantage of preserving the main properties of electrospun membranes, such as the flexibility, porosity and large surface area, making them suitable electrodes for the bio-battery. A fully polymeric bio-battery was developed and validated demonstrating promising performance results, thus making it a new, economic alternative for supplying low-power consumption medical devices.
Experimental
Preparation of the CA electrospun membrane
Cellulose acetate (Mn 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with 40% acetyl groups, purchase from Sigma Aldrich) solutions – 12% wt – were prepared in a mixed-solvent system of acetone-dimethylacetamide (DMAc) with a solvent ratio of 2:1 (wt). To produce the fibers, the polymeric solution was loaded into a 1 ml syringe (B. Braun) connected to a blunt metallic needle with an internal diameter of 0.61 mm (21G from ITEC, Iberiana Technical). A syringe pump (100 series from Kd. Scientific) was used to eject out the polymer solution at a controllable feed rate (0.2 ml h−1) while a high voltage (20 kV) was applied (using a Glassman high voltage power supply) to the needle thus establishing an electric field between the needle and a grounded collector (an Al static plate). The environmental conditions were kept approximately constant at a relative humidity of 40% and a temperature of 22 °C. CA electrospun membranes were produced during 5 consecutive hours.
000 with 40% acetyl groups, purchase from Sigma Aldrich) solutions – 12% wt – were prepared in a mixed-solvent system of acetone-dimethylacetamide (DMAc) with a solvent ratio of 2:1 (wt). To produce the fibers, the polymeric solution was loaded into a 1 ml syringe (B. Braun) connected to a blunt metallic needle with an internal diameter of 0.61 mm (21G from ITEC, Iberiana Technical). A syringe pump (100 series from Kd. Scientific) was used to eject out the polymer solution at a controllable feed rate (0.2 ml h−1) while a high voltage (20 kV) was applied (using a Glassman high voltage power supply) to the needle thus establishing an electric field between the needle and a grounded collector (an Al static plate). The environmental conditions were kept approximately constant at a relative humidity of 40% and a temperature of 22 °C. CA electrospun membranes were produced during 5 consecutive hours.
Preparation of CA/PPy composite fibers by in situ chemical oxidation
Dried CA electrospun membranes with an area of approximately 3 cm × 2 cm were coated with PPy through in situ oxidative polymerization. The CA membranes were immersed in an aqueous solution of pyrrole with concentrations varying from 0.025 to 0.075 M for 10 min under magnetic stirring. The large amounts of hydroxyl groups in CA structure interact with the amine groups of pyrrole ensuring a uniform distribution of the monomer on the surface of nanofibers. Polymerization was carried out at room temperature by gently adding an aqueous solution of FeCl3·6H2O (Sigma Aldrich), as oxidant agent. Different Ox/Mon mass ratios – 1, 2, 3 and 4 – and polymerization time – 30, 45, 60, 90 and 1440 min – were evaluated. During pyrrole oxidative polymerization, the CA membranes turned from white to black within a few minutes, which confirmed the formation of PPy. After polymerization, the CA/PPy composites were thoroughly washed with distilled water and ethanol in order to extract the by-products and residues of the reaction and left to dry under normal ambient conditions.Preparation of CA/PANI composite fibers by in situ chemical oxidation
Dried CA electrospun membranes with an area of approximately 3 cm × 2 cm were coated with PANI through in situ oxidative polymerization. The CA membranes were immersed in 10 ml of an aqueous acid solution, HCl (1 M), containing aniline monomer under magnetic stirring and ice bath. The oxidant-containing solution was prepared by adding ammonium persulfate to 10 ml of an HCl solution (1 M). After an impregnating period of 60 min, the oxidant solution was added drop-wise to the monomer-containing solution. The mixture was stirred in an ice bath for 30, 45 and 60 min, respectively. Monomer concentrations of 2 and 4 M and Ox/Mon molar ratios of 0.125, 0.25 and 0.5 were evaluated for each reaction time. During aniline polymerization, the CA membranes turned from white to dark green color, which indicates the formation of PANI. At the end of each experiment, the obtained fibers were thoroughly washed with distilled water and ethanol in order to extract the by-products and residues of the reaction and dried in air at room temperature.Morphological characterization
Surface morphology of the electrospun coated fibers was obtained using a Focused Ion Beam Scanning Electron Microscope (SEM-FIB) from Zeiss (model Auriga). Double sided conductive carbon tape was used to attach the samples to a metallic holder. Samples were sputtered with gold (1 nm of thickness, approximately). Transmission Electron Microscopy (TEM, model H-8100 II from Hitachi), was carried out to estimate the thickness of PPy coating. Measurements on TEM images were made using the ImageJ software.30Electrical characterization
The electrical conductivity was measured along the fiber using an in-plane configuration. The fibers were placed on a rigid substrate and two parallel electrical contacts, separated by 1 mm, were made with silver glue. The I–V measurements were performed in a Cascade Microtech/Alessi REL-4500 probing platform connected to a HP 4145B Semiconductor Parameter Analyzer, at room temperature. The electrical conductivity was calculated using eqn (3):| σ = l/AR | (3) |
Mechanical characterization
Mechanical characterization of the electrospun membranes was carried out in a commercial mechanical testing system (Rheometric Scientific, Minimat Firmware 3.1) before electrochemical characterization.Ten samples of each membrane (CA, CA/PPy and CA/PANI) were analyzed to obtain mean values for the Young's modulus (E), the ultimate tensile strength (TS) and ultimate tensile strain (ε). The samples were kept in a desiccator under controlled humidity (40–45%) and room temperature (∼25 °C) before the stress–strain testing. The applied deformation rate was 1 mm min−1 and the temperature was kept constant at 25 °C.
Cytotoxicity assays
To evaluate if CA/PPy and CA/PANI composite fiber membranes could present toxicity once implanted in the body, cytotoxic effects were evaluated in vitro according to the International Standard (ISO 10993–5) using the extract method. Manipulations of cells and culture medium (DMEM supplemented with 10% FBS and 1% penicillin–streptomycin, from Invitrogen) were performed inside a biological safety cabinet (ESCO Labculture II). In order to obtain medium conditioned by extracts of the membranes, samples of CA/PPy and CA/PANI were sterilized by immersion in an aqueous solution of ethanol 70% v/v for 30 minutes and then rinsed 3 times in PBS. Each sample was then placed in a sterilized falcon centrifuge tube and culture medium was added in the proportion of 1 ml of medium to 4 mg of membrane. The membranes stood immersed and the tubes were placed inside an incubator at 37 °C for 48 h. Human fibroblasts, from the HFFF2 cell line (human fetal foreskin fibroblasts supplied by the European Collection of Authenticated Cell Cultures, Porton Down, UK) in their 10th passage, were seeded at a density of 16 k cells per cm2 in a 96 well microplate (Sarstedt) and incubated at 37 °C in a 5% CO2 humidified atmosphere incubator (Sanyo MCO19-AIC-UV) for 48 h. After that period, the medium was changed to proceed with the cytotoxicity test. Four test conditions were considered: culture medium (negative control), conditioned medium by CA/PPy, conditioned medium by CA/PANI, culture medium with 10% DMSO (positive control). For each test condition five replicas were prepared and 150 μl of the respective medium were used. After 24 h of incubating the cells in these conditions, a viability test was performed using resazurin (Alfa Aesar) as cell viability indicator. Resazurin is a blue and weakly fluorescent redox sensitive dye that is reduced by dehydrogenase enzymes in metabolically active cells. Resorufin, the oxidized form, is pink and fluorescent. The change in both absorbance and fluorescence correlates with the cell population in the sample. For this test, the media were changed by culture medium with 10% resazurin solution at a concentration of 0.2 mg ml−1 in PBS. This medium was also dispensed in five empty wells (without cells) to be used as a reference. The microplate was placed inside the CO2 incubator for three hours. Absorbance of the medium was then measured at 570 nm and 600 nm (Biotex ELX 800UV microplate reader) to evaluate the relative viability from the ratio of the absorbances measured for the tested conditions and for the negative control.Electrochemical characterization
The electrochemical performance of cellulose-based bio-batteries (PPy/CA|CA|CA/PANI structure) was characterized by cyclic voltammetry. The electrochemical experiments were carried out using a potentiostat Gamry Instruments-Reference 3000 and the batteries were tested in a homemade Teflon cell. The bio-battery cell was constructed using two identical pieces of the conductive materials (CA/PPy and CA/PANI) separated by a CA electrospun membrane. All the samples had an area of 0.25 cm2, a volume of 0.006 cm3 and weight of 2.9 mg, approximately. Two flexible carbon meshes were used as the collectors. The electrochemical cell (Fig. S2 in ESI†) has a reservoir in order to easily inject the electrolyte, 20 μl. Since the body fluids are mainly composed of water and ionic species such as Na+, Cl− and K+, a physiological solution of 0.9% (w/v) NaCl was used. Due to the low thickness (<0.1 mm) of the membranes, all measurements were carried out in a cell with two-electrode configuration. This configuration is the most appropriate to characterize solid-state electrochemical devices such as supercapacitors31 and batteries.32,33 The reference electrode (RE) and the counter electrode (CE) were merged into one electrode resulting in a two-electrode system. For cyclic voltammetry measurements, the potential was scanned in both directions between −1 and 1 V for 10 consecutive cycles with a scan rate of 80 mV s−1 under ambient conditions and room temperature.Charging/discharging tests
Charging/discharging experiments were carried out for PPy/CA|CA|CA/PANI structure at 25 °C using the potentiostat Gamry Instruments-Reference 3000.The cell was constructed using two identical pieces of the conductive materials (CA/PPy as the anode and CA/PANI as the cathode) separated by a CA electrospun membrane. The active materials have a total mass of 1.9 mg, approximately.
The setup (Fig. S2 – ESI†) was filled with 20 μl of electrolyte solution (0.9% NaCl) and no electrolyte refresh has been made during the measurement. It is important to note that the cell is not sealed.
Different charge/discharge rates were evaluated. For such analysis, short-time charge/discharge measurement (one cycle of 4 min) were tested at charge/discharge current densities of 20 mA g−1, 10.5 mA g−1, 5.3 mA g−1, 2.3 mA g−1, 1.3 mA g−1 and 0.7 mA g−1.
A cycling test of approximately 5 hours was performed by repeatedly charging (for 30 min) and discharging (also for 30 min) the device at a constant current density of 5.3 mA g−1. A lower cut-off potential of 0 V and an upper cut-off potential of 0.8 V was used for all experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Daniela Gomes from CENIMAT for the SEM images. This work was funded by H2020-ICT-2014-1, RIA, TransFlexTeg-645241; ERC-CoG-2014, CapTherPV, 647596, and also by FEDER funds through the COMPETE 2020 Programme and National Funds through the Portuguese Foundation for Science and Technology (FCT – MEC) under the project number POCI-01-0145-FEDER-007688, Reference ID/CTM/50025. Ana C. Baptista also acknowledges FCT-MEC for her postdoctoral grant with reference SFRH/BPD/104407/2014.References
- R. Martins, I. Ferreira and E. Fortunato, Phys. Status Solidi RRL, 2011, 5, 332–335 CrossRef CAS.
- I. Ferreira, B. Bras, N. Correia, P. Barquinha, E. Fortunato and R. Martins, J. Disp. Technol., 2010, 6, 332–335 CrossRef CAS.
- I. Ferreira, B. Brás, J. I. Martins, N. Correia, P. Barquinha, E. Fortunato and R. Martins, Electrochim. Acta, 2011, 56, 1099–1105 CrossRef CAS.
- R. Martins, P. Barquinha, L. Pereira, N. Correia, G. Gonçalves, I. Ferreira and E. Fortunato, Phys. Status Solidi RRL, 2009, 3, 308–310 CrossRef CAS.
- A. C. Baptista, J. I. Martins, E. Fortunato, R. Martins, J. P. Borges and I. Ferreira, Biosens. Bioelectron., 2011, 26, 2742–2745 CrossRef CAS PubMed.
- S. Li, Z. P. Guo, C. Y. Wang, G. G. Wallace and H. K. Liu, J. Mater. Chem. A, 2013, 1, 14300–14305 CAS.
- S. Li, K. Shu, C. Zhao, C. Wang, Z. Guo, G. Wallace and H. K. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 16679–16686 CAS.
- X. Jia, C. Wang, C. Zhao, Y. Ge and G. G. Wallace, Adv. Funct. Mater., 2016, 26, 1454–1462 CrossRef CAS.
- Y. Kong, C. Wang, Y. Yang, C. O. Too and G. G. Wallace, Synth. Met., 2012, 162, 584–589 CrossRef CAS.
- I. Ferreira, A. C. Baptista, J. P. Leitão, J. Soares, E. Fortunato, R. Martins and J. P. Borges, Macromol. Mater. Eng., 2013, 298, 174–180 CrossRef CAS.
- Y. Zhang and G. C. Rutledge, Macromolecules, 2012, 45, 4238–4246 CrossRef CAS.
- S. Nair, S. Natarajan and S. H. Kim, Macromol. Rapid Commun., 2005, 26, 1599–1603 CrossRef CAS.
- K. Ketpang and J. S. Park, Synth. Met., 2010, 160, 1603–1608 CrossRef CAS.
- Y. Chen, Y. Li, H. Wang and M. Yang, Carbon, 2007, 45, 357–363 CrossRef CAS.
- H. Yoon, Nanomaterials, 2013, 3, 524 CrossRef CAS PubMed.
- L. Ghasemi-Mobarakeh, M. P. Prabhakaran, M. Morshed, M. H. Nasr-Esfahani, H. Baharvand, S. Kiani, S. S. Al-Deyab and S. Ramakrishna, J. Tissue Eng. Regener. Med., 2011, 5, e17–e35 CrossRef CAS PubMed.
- D. Beneventi, S. Alila, S. Boufi, D. Chaussy and P. Nortier, Cellulose, 2006, 13, 725–734 CrossRef CAS.
- Z. Shi, S. Zang, F. Jiang, L. Huang, D. Lu, Y. Ma and G. Yang, RSC Adv., 2012, 2, 1040–1046 RSC.
- N. Ferraz, M. Strømme, B. Fellström, S. Pradhan, L. Nyholm and A. Mihranyan, J. Biomed. Mater. Res., Part A, 2012, 100, 2128–2138 CrossRef PubMed.
- P. M. George, A. W. Lyckman, D. A. LaVan, A. Hegde, Y. Leung, R. Avasare, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511–3519 CrossRef CAS PubMed.
- P. A. Kilmartin and G. A. Wright, Electrochim. Acta, 2001, 46, 2787–2794 CrossRef CAS.
- R. A. Khalkhali and G. G. Wallace, Iran. Polym. J., 2004, 13, 463–470 CAS.
- F. Beck, U. Barsch and R. Michaelis, J. Electroanal. Chem., 1993, 351, 169–184 CrossRef CAS.
- T. Kobayashi, H. Yoneyama and H. Tamura, J. Electroanal. Chem. Interfacial Electrochem., 1984, 177, 293–297 CrossRef CAS.
- A. G. MacDiarmid, L. S. Yang, W. S. Huang and B. D. Humphrey, Synth. Met., 1987, 18, 393–398 CrossRef CAS.
- A. Pron, F. Genoud, C. Menardo and M. Nechtschein, Synth. Met., 1988, 24(3), 193–201 CrossRef CAS.
- W.-S. Huang, B. D. Humphrey and A. G. MacDiarmid, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 2385–2400 RSC.
- S. J. Kim, J. H. We and B. J. Cho, Energy Environ. Sci., 2014, 7, 1959–1965 CAS.
- K. B. Lee, J. Micromech. Microeng., 2005, 15, S210–S214 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, J. Mater. Chem., 2011, 21, 16197–16204 RSC.
- A. I. Gopalan, K.-P. Lee, K. M. Manesh and P. Santhosh, J. Membr. Sci., 2008, 318, 422–428 CrossRef CAS.
- A. Gopalan, P. Santhosh, K. Manesh, J. Nho, S. Kim, C. Hwang and K. Lee, J. Membr. Sci., 2008, 325, 683–690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta06457h |
| This journal is © The Royal Society of Chemistry 2018 |