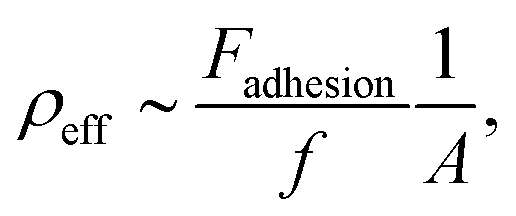DOI:
10.1039/C7SM01885A
(Paper)
Soft Matter, 2018,
14, 894-900
A modular approach for multifunctional polymersomes with controlled adhesive properties†
Received
19th September 2017
, Accepted 15th December 2017
First published on 15th December 2017
Abstract
The bottom-up approach in synthetic biology involves the engineering of synthetic cells by designing biological and chemical building blocks, which can be combined in order to mimic cellular functions. The first step for mimicking a living cell is the design of an appropriate compartment featuring a multifunctional membrane. This is of particular interest since it allows for the selective attachment of different groups or molecules to the membrane. In this context, we report on a modular approach for polymeric vesicles, so-called polymersomes, with a multifunctional surface, namely hydroxyl, alkyne and acrylate groups. We demonstrate that the surface of the polymersome can be functionalized to facilitate imaging, via fluorescent dyes, or to improve the specific adhesion to surfaces by using a biotin functionalization. This generally applicable multifunctionality allows for the covalent integration of various molecules in the membrane of a synthetic cell.
1 Introduction
“Omnis cellula e cellula.” – Every cell arises from another cell.1 This quote by Rudolf Virchow may cease to be true in the future, since the design and engineering of artificial cells has made significant progress during the last decades.2,3 The process leading to an artificial cell can be of two different ways: either the top-down4–6 or the bottom-up approach.7–10 The top-down approach relies on the concept of starting from an existing living cell in order to understand its basic constituents down to the minimal building blocks the cell is made of. A contrario, in the bottom-up approach, different building blocks are combined to more and more complex systems, until one creates a minimal system which mimics functionalities of living cells. Promising candidates for the construction of the membrane of an artificial cell system are amphiphilic polymers that are able to form stable vesicles, so-called polymersomes, composed of a polymeric bilayer.11,12 Compared to liposomes which are known as building blocks chosen by Nature, manmade polymersomes benefit from the versatility, stability, and tunability of synthetic polymer chemistry, especially regarding to chemical and biological functionalities.11 Polymersome-based compartments have been imbued with a number of different life-mimicking processes: enzymatic cascade reactions,13,14 sub-compartmentalization,15 reconstitution of transmembrane proteins,16,17 adenosine triphosphate (ATP) synthesis,18 response to external stimuli,19 gene expression,20 and adhesion properties of polymersomes.21–26 The latter has been of particular interest in the field of drug delivery research, where specific targeting moieties on the surface are desired to guide the synthetic entity to its place of action.27 A number of (bio)molecules were successfully linked to a polymersome surface, for example polyguanylic acid,28 enhanced green fluorescent protein (eGFP)29 and green fluorescent protein (GFP),30 horse radish peroxidase,31Candida antarctica Lipase B,32 biotin,29 peptides,33–35 sugars,36 antibodies,37,38 and drugs.39 Chemically, this is usually achieved by alkyne–azide click reactions,29,32N-hydroxysuccinimide (NHS),30,35 or maleimide chemistry37,39 complexation reactions, e.g. between biotin/streptavidin,28 or β-cyclodextrin/adamantane.31 However, examples of multifunctional polymersomes are sparse and so far limited to submicron dimensions.40 The functionalization is typically only achieved if several ligands are attached at the same functional groups24 or if one functional group is introduced by the initiator.25 To increase the chemical functionality of the membrane of compartments favorable for mimicking minimal cells, we present here a modular approach to multifunctional polymersomes by using block copolymers with orthogonal reactivity allowing for the selective binding of different molecules to the surface. We characterize the multifunctional polymersomes using different experimental techniques, such as fluorescence microscopy and micropipette force spectroscopy.
2 Materials and methods
2.1 Materials
All materials and solvents, unless stated otherwise, were purchased from Sigma-Aldrich. For the synthesis of the poly(butadiene)-block-poly(ethylene oxide) (PB-b-PEO) polymers, butadiene and ethylene oxide were supplied by GHC Gerling. The test of the functionalization of the polymersomes produced was realized by confocal laser scanning microscopy (CLSM) and fluorescence correlation spectroscopy (FCS). For this, Chromeo azide dye and BODIPY amine dye were used. Chromeo azide was purchased from Santa Cruz Biotechnology and 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethylenediamine hydrochloride (BODIPY amine dye) was supplied by ThermoFisher Scientific.
2.2 Instrumentations
CLSM measurements were performed on a TCS SP5 (Leica) using a 488 nm Ar+ laser at 15% power and a HCX PL APO CS 63× oil objective (N.A. 1.40). FCS measurements were carried out on a commercial setup (Zeiss, Germany) consisting of a module ConfoCor 2 and an inverted microscope model Axiovert 200 with a Zeiss C-Apochromat 40×/1.2 W water immersion objective. The Chromeo azide dye was excited using a He–Ne laser (λex = 543 nm) and the emission was detected in the range λem = 560–615 nm. BODIPY amine dye was excited using an Ar+ laser (λex = 488 nm) and its emission was detected in the range λem = 505–550 nm. Dynamic light scattering (DLS) was measured with a Nicomp™ 380 Submicron Particle Sizer (PSS-Nicomp) at an angle of 90° or on an ALV spectrometer consisting of a goniometer and an ALV-5004 multiple-tau correlator (320 channels) which allows measurements over an angular range from 20° to 150°. A He–Ne laser (λ0 = 632.8 nm) was used as light source.
2.3 Micropipette force spectroscopy
The functionalization of polymersomes was probed via the measurement of the specific adhesion of such polymersomes on dedicated surfaces. This experimental investigation was performed via the micropipette force spectroscopy technique, which involves the high-resolution optical imaging of the deflection of a double-L-shape micropipette. Here, the cantilever was a double-L-shape micropipette made from borosilicate glass capillaries with an initial diameter of 1 mm (WPI, Borosilicate Glass Capillaries TW 100-6).41 The cantilever was about 2 cm long and featured an outer diameter of the order of 10 μm. The micropipette was first pulled by means of a pipette puller (Sutter Instrument, P-97 Flaming/Brown Micropipette Puller) and, subsequently, bent in a characteristic double-L-shape using a microforge (Narishige, microforge MF-900). The deflection of the micropipette was observed with an inverted microscope (Olympus IX-83) using long distance objectives of 20× and 40× magnification and a camera recording at 10 fps (Grashopper, GS3-U3-41C6M-C). A multifunctional polymersome was aspirated at the tip of the micropipette by applying a precise negative pressure using a pressure-driven pump (MFCS-8C, Fluigent). The micropipette was linked to 3-axis piezo-driven manual micromanipulators (Burleigh, PCS-5400) in order to precisely control its position. The tested substrates were a normal microscope glass slide and a neutravidin-coated glass slide (PolyAn). The surfaces were carefully cleaned with ethanol, isopropanol and dried under a clean nitrogen stream prior to any experiments. The surfaces were fixed side-by-side on a substrate holder, which was connected to high-precision motorized linear stages (Newport, Conex LTA-HS). The whole setup was placed on an active anti-vibration table (Accurion, Halcyonics i4-large). The deflection of the micropipette from the zero-force position was determined by an image auto-correlation analysis (wrote with MATLAB, R2014b, MathWorks Inc.) of the intensity patterns of the micropipette. The spring constant of the micropipette was 2.30 ± 0.03 nN μm−1, resulting in a force resolution of about 200 pN with this configuration. The reader will find more details on the setup and on the technical aspects of the method in ref. 41.
2.4 PB-b-PEO synthesis
Three different types of PB-b-PEO were synthesised: one with an alkyne functionalization, another with an acrylate functionalization and one featuring both functionalizations. The reactions were carried out in flamed-out glassware under an argon atmosphere. Freshly prepared cumylpotassium was used as an initiator. To do so, potassium (2.5 g) was washed with tetrahydrofuran (THF) and petroleum ether and dried under vacuum. Subsequently, THF and cumyl methyl ether (5.2 mL) were added (ratio potassium to cumyl methyl ether 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume). The reaction was allowed to take place for 48 h, after which the mixture was filtered. The concentration of cumylpotassium in the filtrate was determined by an exemplary polymerization of 1,3-butadiene and determination of the molecular weight. 1,3-Butadiene was polymerized anionically in THF at −65 °C to yield primarily 1,2-addition. Cumylpotassium was added to 1,3-butadiene (10 g). The polymerization proceeded for 72 h in THF (200 mL). After that, ethylene oxide (4.3 g) was added while cooling and polymerized for 72 h. The reaction was finished by the addition of degassed methanol. The polymer was obtained by precipitation in cold acetone and dried at reduced pressure (yields: 95–100%).
:1 in volume). The reaction was allowed to take place for 48 h, after which the mixture was filtered. The concentration of cumylpotassium in the filtrate was determined by an exemplary polymerization of 1,3-butadiene and determination of the molecular weight. 1,3-Butadiene was polymerized anionically in THF at −65 °C to yield primarily 1,2-addition. Cumylpotassium was added to 1,3-butadiene (10 g). The polymerization proceeded for 72 h in THF (200 mL). After that, ethylene oxide (4.3 g) was added while cooling and polymerized for 72 h. The reaction was finished by the addition of degassed methanol. The polymer was obtained by precipitation in cold acetone and dried at reduced pressure (yields: 95–100%).
2.5 PB-b-PEO functionalization
2.5.1 Alkyne functionalization.
The reaction was carried out in flamed-out glassware. Dichloromethane (DCM) was freshly distilled and PB-b-PEO was dried from benzene prior to the reaction. PB-b-PEO (0.2 g, 26.32 μmol) and propiolic acid (0.42 g, 6.03 mmol) were dissolved in DCM (10 mL, dry). The solution was stirred and cooled at −20 °C. A solution of N,N′-dicyclohexylcarbodiimide (DCC) (5.40 mg, 26.21 μmol) and 4-dimethylaminopyridine (DMAP) (1.8 mg, 14.73 μmol) in DCM (3 mL, dry) was added dropwise over a period of 20 min. The cooling bath was removed and the reaction mixture was stirred for an additional 24 h at room temperature. The crude reaction mixture was washed twice with HCl (1 M, 5 mL) and brine (5 mL). The combined aqueous phase was extracted with DCM (3 mL). The combined organic phase was dried using MgSO4, concentrated under reduced pressure and dialysed overnight against DCM (200 mL, 1000 MWCO dialysis tube). The product was dried under vacuum and the yield was at least 47%.
2.5.2 Acrylate functionalization.
The reaction was carried out in flamed-out glassware. As previously mentioned, DCM was freshly distilled and PB-b-PEO was dried from benzene prior to the reaction. PB-b-PEO (0.2 g, 26.32 μmol) and triethyl amine (1.34 mg, 13.24 μmol, dry) were dissolved in DCM (10 mL, dry). The solution was stirred and cooled at 0 °C. Acryoyl chloride (54.5 mg, 0.60 mmol) was added dropwise. After 30 min, the cooling bath was removed and the reaction mixture was stirred for an additional 72 h at room temperature. The crude reaction mixture was washed thrice with NaHCO3 (saturated, 5 mL). The combined aqueous phase was extracted with DCM (5 mL). The combined organic phase was washed with water until the aqueous phase was neutral (thrice, 5 mL) and the combined organic phase was extracted again with DCM (5 mL). The organic phase was dried using MgSO4 and concentrated under reduced pressure. The obtained solution was dialysed overnight against DCM (200 mL, 1000 MWCO dialysis tube). The product was dried under vacuum and the yield was at least 97%.
3 Results and discussion
Polymersomes based on PB-b-PEO polymers with orthogonal reactivity on their surface were produced from end-functionalized block copolymers by self-assembly or by using a microfluidic platform.42 These complementary approaches allowed for the generation of trifunctional polymersomes on different size ranges: either submicron (from self-assembly in aqueous environment) or with diameters of several tens of micrometers (via microfluidics). First, we characterize the synthesized PB-b-PEO polymers featuring different functionalizations and later on we probe the pertinence of such functionalized block copolymers after self-assembly in polymersomes by means of fluorescence spectroscopy and by adhesion measurements.
3.1 Characterization of functionalized PB-b-PEO polymers
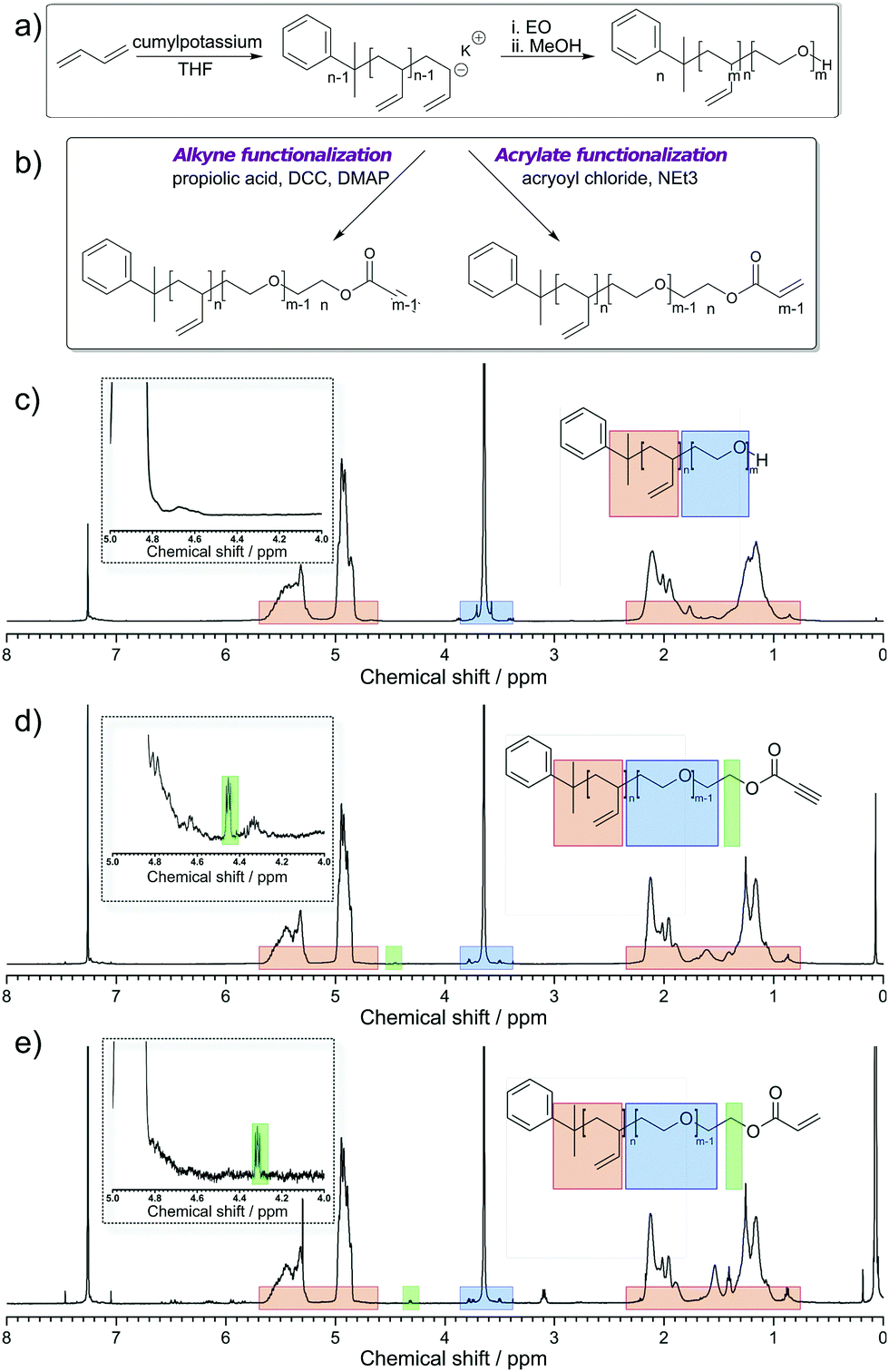
PB-b-PEO was synthesized via the sequential living anionic polymerization of 1,3-butadiene and ethylene oxide (Fig. 1a). The block copolymer was obtained with a hydroxyl end group after the synthesis. Starting from this material, an activated alkyne end group was introduced via Steglich esterification with propiolic acid, mediated by DCC and DMAP (Fig. 1b – left side). Similarly, an acrylate group was attached to the polymer's chain end by esterification with acryloyl chloride (Fig. 1b – right side). The characterization data for all polymers are summarized in Table 1. Fig. 1c–e summarize the 1H NMR spectra of the starting PB-b-PEO-OH block copolymer and the reactive products. The end group modifications do not alter the signals of the polymeric backbone (5.7–4.6 ppm and 2.3–0.8 ppm PB, 3.6 ppm PEO). The degree of functionalization was determined from the integral of the methylene group adjacent to the end group at ca. 4.4 ppm (highlighted in green). In case of the acrylate-functionalized block copolymer, the resonances of the double bond group are visible from 6.5 to 5.8 ppm. Even if not a quantitative functionalization was achieved in all cases, with the degree of functionalization, the overall density of the functional groups on the polymersome can be adjusted.
 |
| | Fig. 1 (a) Synthesis of PB-b-PEO using 1,3 butadiene and ethylene oxide (EO) in THF (for clarity, only the 1,2 addition is shown) and (b) post-polymerization modification using propiolic acid, DCC and DMAP or acryloyl chloride and trimethylamine (NEt3); (c) 1H NMR spectrum of PB-b-PEO-OH in CDCl3; (d) 1H NMR spectrum of PB-b-PEO-alkyne in CDCl3. (e) 1H NMR spectrum of PB-b-PEO-acrylate in CDCl3. | |
Table 1 Characterization data of PB-b-PEO and its derivatives with a low polydispersity index
| Polymer |
M
n (kg mol−1) |
Đ
|
Degree of functionalization (%) |
| PB237-b-PEO101-OH |
17.2 |
1.16 |
100 |
| PB237-b-PEO101-alkyne |
17.2 |
1.11 |
28 |
| PB237-b-PEO101-acrylate |
17.2 |
1.17 |
62 |
3.2 Production of functionalized PB-b-PEO based polymersomes in microfluidic environment and characterization via fluorescence microscopy
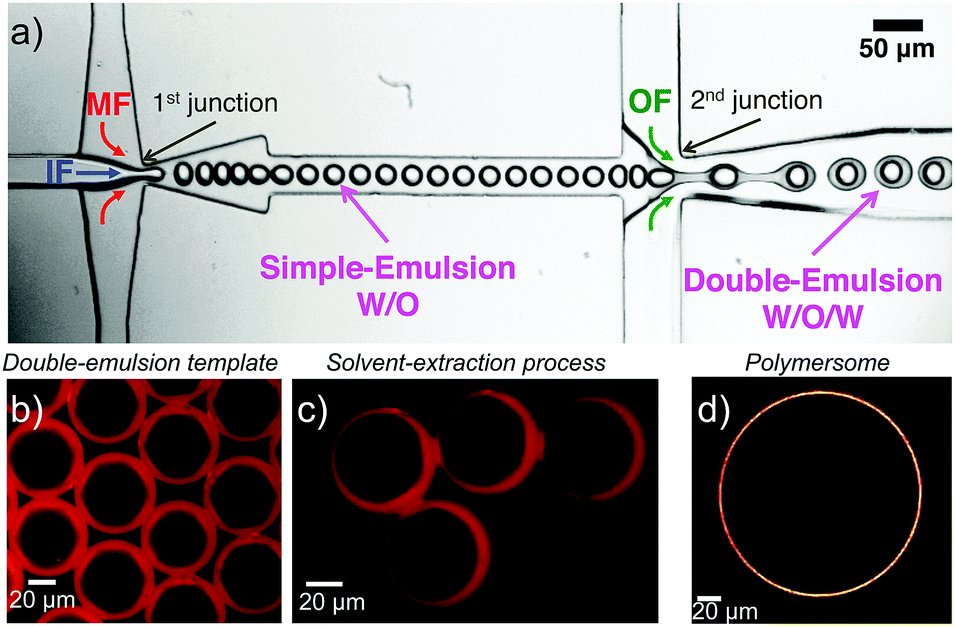
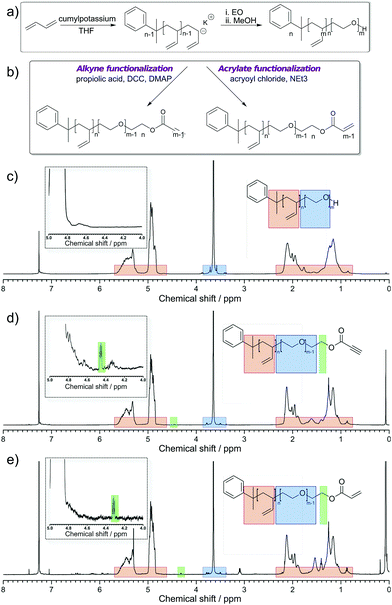
In order to produce micrometer-sized polymersomes, the previously described amphiphilic PB-b-PEO block copolymers were dissolved at a concentration of 1 wt% in oleic acid and flushed through a special microfluidic chip design in combination with two other aqueous phases. The microfluidic design used here is presented in Fig. 2a, and similar to the one reported in ref. 42. It consisted of two consecutive cross-junctions featuring a flow-focusing configuration. At the first junction, the inner aqueous fluid (IF), consisting of a mixture of water and the surfactant Synperonic F108 (Sigma-Aldrich) at 1 wt% was sheared off by the middle oil fluid (MF), in which the polymer was dissolved, generating a water-in-oil emulsion. These droplets were then sheared off by the outer aqueous fluid (OF) at the second junction of the microfluidic design. The OF consisted of a mixture of water, surfactant Synperonic F108 (1 wt%), glycerol (15 wt%, Sigma Aldrich), poly(diallyldimethylammonium chloride) solution (2 wt%, Sigma Aldrich) and ethanol (14 wt%, Roth). The double-emulsion was produced using the typical flow rates for the different fluid phases of 50 μL h−1, 100 μL h−1 and 500 μL h−1 for the IF, the MF and the OF, respectively. This generated double emulsion served as a precursor for polymersome formation (Fig. 2b).
 |
| | Fig. 2 (a) Microfluidic setup to generate double emulsions. (b) Double emulsion template obtained at the outlet of the microfluidic chip. The double emulsions are observed under fluorescence microscopy (the dye Nile Red is incorporating in the stock solution of MF prior to the experiment). (c) Solvent-extraction process resulting from the presence of ethanol in the continuous OF. (d) Confocal micrograph of a polymersome obtained after solvent-extraction. | |
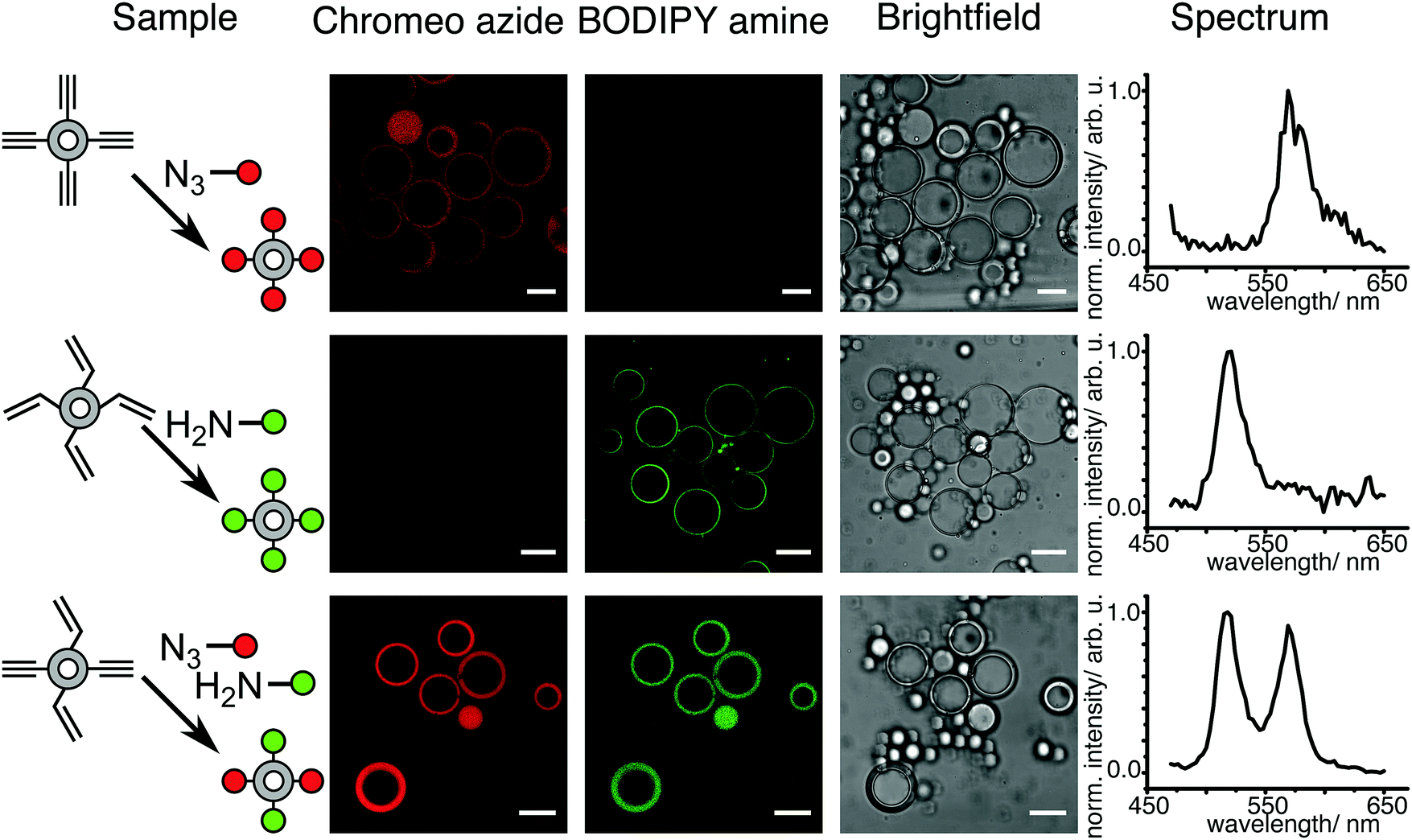
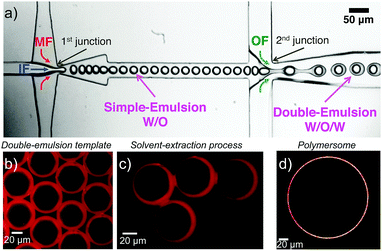
Monodisperse polymersomes were finally obtained after extraction of the oil (oleic acid) from the MF (Fig. 2c and d). This solvent-extraction process was initiated by the presence of ethanol in the OF, which is an appropriate solvent for oleic acid. During this extraction process, the amphiphilic block copolymer (in the MF) diffused at the water–oil interface. There, it self-assembled with the hydrophilic PEO blocks pointing towards the in- and outside of the membrane layer resulting in the formation of the polymersomes with a homogeneous size distribution of about 29 ± 7 μm (inner diameter) for the chosen parameters. Furthermore, the polymersomes produced with this method featured alkyne, acrylate and OH-groups on their surface and were very stable for a long period of time (at least three months). The polymersomes were labeled with specific dyes after the extraction process, which enabled both the imaging and validation of the functionality of the polymers self-assembled in polymersome configuration (Fig. 3). Alkyne functionality was labeled using the water-soluble azide-functionalized Chromeo azide dye. Chromeo azide is known to react with the activated alkyne in a 1,3-dipolar click reaction without the addition of a catalyst.43 The acrylate functionality was labeled with the water-soluble amine-bearing boron-dipyrromethene (BODIPY) amine by nucleophilic addition. The final functionalized polymersomes were imaged by CLSM (Fig. 3). The labeled alkyne polymersomes (Fig. 3 – top row) showed only fluorescence of the Chromeo azide at 560 nm, while the modified acrylate polymersomes (Fig. 3 – middle row) showed fluorescence only for the BODIPY amine at 520 nm. Polymersomes with both functional groups in their membrane (Fig. 3 – bottom row) and subsequent labelling with both dyes exhibited fluorescence for both chromophores, proving the attachment of both dyes to the polymersomes.
 |
| | Fig. 3 CLSM micrographs of polymersomes with alkyne or/and acrylate functionality labeled with Chromeo azide or/and BODIPY amine and spectral detection of the dyes (scale bar 20 μm). | |
3.3 Demonstration of the covalent linkage of the different functionalizations to the terminal groups of PB-b-PEO polymers
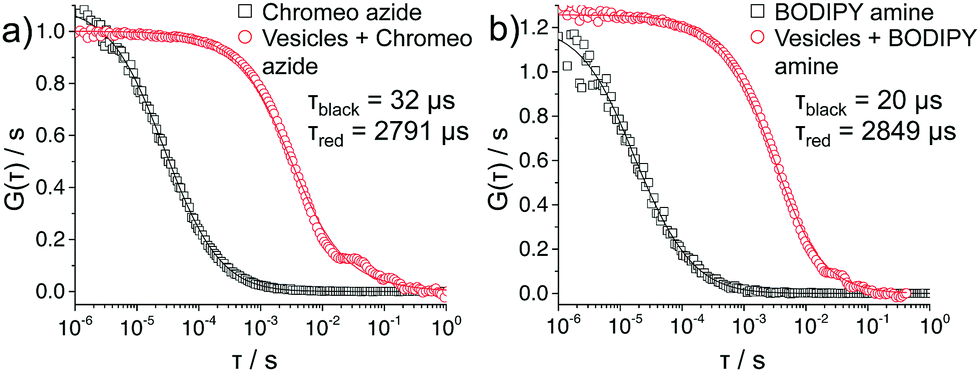
The covalent linkage to the terminal groups was demonstrated by FCS measurements on polymersomes prepared by aqueous self-assembly (Fig. 4). Indeed, the polymersomes prepared in microfluidic environment were too large for this technique, thus the polymersomes were produced via the film hydration method44 leading to diameters of about 120 nm. They were reacted under the same conditions with the two dyes as for the CLSM experiments presented above. As both dyes were used in under stoichiometric amounts, they fully react with the polymersomes and no excess of dye is present (note: if an excess of dye is used, dialysis can be used to remove any excess). For both Chromeo azide- and BODIPY amine-labeled polymersomes, the decline of the autocorrelation curve G(τ) was delayed compared to that of the freely diffusing dyes confirming that the dyes are attached to larger species and thus diffuse significantly slower. A fit (solid lines in Fig. 4) of the experimental autocorrelation curves with a single component model function45 yielded the diffusion time τ, the diffusion coefficient D and through the Stokes Einstein equation46 the hydrodynamic radius RH of the respective fluorescent species. The free dyes showed a diffusion time of either 32 or 20 μs, which corresponded to a RH of 0.6 and 0.5 nm, a typical value for small, freely diffusing molecules.47 In contrast, the labeled polymersomes exhibited slower diffusion times of about 2800 μs, corresponding to RH of about 60 nm, providing evidence of the attachment of the dye to the polymersome.
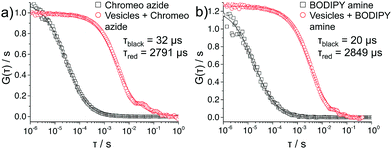 |
| | Fig. 4 FCS autocorrelation curves of functionalized polymersomes labeled with (a) Chromeo azide, and (b) BODIPY amine. | |
3.4 Surface functionalization of PB-b-PEO-acrylate based polymersomes characterized via adhesion experiments
In order to confirm the successful covalent linkage of the acrylate functionalization to the terminal groups of PB-b-PEO polymers, we further modified the acrylate-functionalized polymersomes with amine-functionalized biotin. We realized this step in order to study the specific adhesion of such prepared polymersomes on neutravidin-coated surfaces (Fig. 5a). Using the micropipette force spectroscopy technique,41,48,49 we measured the adhesion force between PB-b-PEO polymersomes (functionalized or not) and dedicated surfaces. A single PB-b-PEO polymersome was immobilized by partial aspiration at the opening of the micropipette (see Fig. 5b – left side). As the surface approached the polymersome and established contact, the soft micropipette was deflected (see Fig. 5b – right side). From the optical detection of the micropipette deflection, and taking into account its spring constant (2.30 ± 0.03 nN μm−1), we obtained force–displacement curves (see Fig. 5c) for reiteratively recorded approach and retraction cycles.
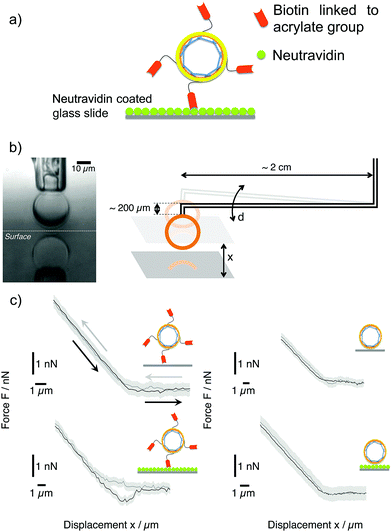 |
| | Fig. 5 (a) Sketch of biotinylated PB-b-PEO-acrylate polymersomes interacting with a neutravidin-coated surface. (b) Left: Optical micrograph showing a polymersome aspired at the tip of a micropipette in proximity to a reflecting surface. Right: Sketch of the micropipette force spectroscopy technique (not to scale), which is based on the deflection d of a double-L-shaped micropipette. (c) Left: Force–displacement curves obtained for biotinylated PB-b-PEO-acrylate polymersomes on neutravidin-coated glass (bottom) and uncoated glass substrates (top). Grey curves indicate the approach of the surface to the polymersome; black curves represent the retraction of the surface. Solid lines are represent the average of 4 consecutive force–displacement cycles (the background represents the standard deviation). The configuration of biotinylated PB-b-PEO-acrylate polymersomes on neutravidin-coated glass (bottom) exclusively displays a significant adhesion peak of 0.9 ± 0.2 nN. Right: Force–displacement curves obtained for non-biotinylated PB-b-PEO-acrylate polymersomes on neutravidin-coated glass (bottom) and uncoated glass (top) do not exhibit any adhesion peak. | |
We studied the adhesion of PB-b-PEO-acrylate polymersomes in 4 different experimental configurations: biotinylated PB-b-PEO-acrylate polymersomes on normal glass and on neutravidin-coated glass surfaces (Fig. 5c – left side) as well as non-biotinylated PB-b-PEO-acrylate polymersomes on normal glass and on neutravidin-coated glass surfaces (Fig. 5c – right side). We probed the adhesion of a polymersome by performing 4 cycles of approach and retraction and, subsequently, repeated the force–displacement measurements on the other surface for the same polymersome. Each force–displacement curve shown Fig. 5c represents the average over all approach and retraction cycles; the background denotes the standard deviation.
Due to the strong affinity between biotin and neutravidin,50–52 a significant adhesion peak during retraction (about 1 nN, see Fig. 5c – bottom left) was exclusively observed for the biotinylated PB-b-PEO-acrylate polymersomes on the neutravidin-coated glass surface. For all the other configurations, no adhesion peak was observed, which provides evidence for the successful biotin functionalization of the PB-b-PEO-acrylate based polymersomes. In the following quantitative analysis, we take advantage of the optical detection of the contact area A between the functionalized polymersome and the substrate during a force–displacement measurement. As reported in the literature, the elementary force quantum of a single biotin–avidin pair is about f = 160 pN.53 We can therefore estimate the effective density of biotin–neutravidin bonds, ρeff, from
| | 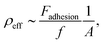 | (1) |
where
Fadhesion denotes the measured adhesion force of the polymersome. The contact area
A between the polymersome and the substrate is on the order of 80 μm
2, as inferred from the optical image sequences that were obtained during the force–displacement measurements. Based on the aforementioned strength of a single bond, we find that the measured adhesion forces correspond to an effective density of one biotin–neutravidin bond per ∼14 μm
2.
4 Conclusion
In conclusion, functional block copolymers with orthogonal chemical functionality were synthesized and self-assembled into polymersomes of different size regimes, either by film hydration or by employing a microfluidic approach. These polymersomes exhibit a multifunctional surface carrying hydroxyl, alkyne and acrylate groups. By changing the stoichiometry, the chemical functionality can be controlled and the surface of the polymersome can be functionalized with various groups to facilitate imaging or improve specific adhesion to surfaces, i.e. mimicking the adhesive properties of real cells. Biotin was successfully linked to the polymersomes and the adhesion to avidin-modified substrates was demonstrated by measuring the adhesion forces using a micropipette force spectroscopy technique. These multifunctional polymersomes are a step towards the design of multicompartimentalized protocells that might be used as efficient nano- or micro-reactors in synthetic biology.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is part of the MaxSynBio consortium which is jointly funded by the Federal Ministry of Education and Research of Germany and the Max Planck Society. The authors thank Christian Kreis for technical assistance and discussions. Open Access funding provided by the Max Planck Society.
References
-
R. Virchow, Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre: zwanzig Vorlesungen, gehalten während der Monate Februar, März und April 1858 im pathologischen Institute zu Berlin, Hirschwald, 1858 Search PubMed.
- D. Deamer, Trends Biotechnol., 2005, 23, 336–338 CrossRef CAS PubMed.
- P. Schwille and S. Diez, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 223–242 CrossRef CAS PubMed.
- A. Meyer, R. Pellaux and S. Panke, Curr. Opin. Microbiol., 2007, 10, 246–253 CrossRef CAS PubMed.
- S. K. Lee, H. Chou, T. S. Ham, T. S. Lee and J. D. Keasling, Curr. Opin. Biotechnol., 2008, 19, 556–563 CrossRef CAS PubMed.
- K. Brenner, L. You and F. H. Arnold, Trends Biotechnol., 2008, 26, 483–489 CrossRef CAS PubMed.
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387 CrossRef CAS PubMed.
- P. Schwille, Science, 2011, 333, 1252–1254 CrossRef CAS PubMed.
- A. D. Griffiths and D. S. Tawfik, Trends Biotechnol., 2006, 24, 395–402 CrossRef CAS PubMed.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- S. M. Kuiper, M. Nallani, D. M. Vriezema, J. J. Cornelissen, J. C. van Hest, R. J. Nolte and A. E. Rowan, Org. Biomol. Chem., 2008, 6, 4315–4318 CAS.
- S. F. van Dongen, M. Nallani, J. J. Cornelissen, R. J. Nolte and J. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- H. C. Shum, Y.-j. Zhao, S.-H. Kim and D. A. Weitz, Angew. Chem., 2011, 123, 1686–1689 CrossRef.
- C. Nardin, J. Widmer, M. Winterhalter and W. Meier, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 4, 403–410 CrossRef CAS.
- D. L. Richmond, E. M. Schmid, S. Martens, J. C. Stachowiak, N. Liska and D. A. Fletcher, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9431–9436 CrossRef CAS PubMed.
- H.-J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS PubMed.
- N. P. Kamat, G. P. Robbins, J. Rawson, M. J. Therien, I. J. Dmochowski and D. A. Hammer, Adv. Funct. Mater., 2010, 20, 2588–2596 CrossRef CAS PubMed.
- C. Martino, S.-H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420 CrossRef CAS PubMed.
- T. O. Pangburn, F. S. Bates and E. Kokkoli, Soft Matter, 2012, 8, 4449–4461 RSC.
- J. J. Lin, F. S. Bates, D. A. Hammer and J. A. Silas, Phys. Rev. Lett., 2005, 95, 026101 CrossRef PubMed.
- J. J. Lin, J. A. Silas, H. Bermudez, V. T. Milam, F. S. Bates and D. A. Hammer, Langmuir, 2004, 20, 5493–5500 CrossRef CAS PubMed.
- G. P. Robbins, R. L. Saunders, J. B. Haun, J. Rawson, M. J. Therien and D. A. Hammer, Langmuir, 2010, 26, 14089–14096 CrossRef CAS PubMed.
- B. Iyisan, A. Janke, P. Reichenbach, L. M. Eng, D. Appelhans and B. Voit, ACS Appl. Mater. Interfaces, 2016, 8, 15788–15801 CAS.
- N. P. Kamat, S. J. Henry, D. Lee and D. A. Hammer, Small, 2013, 9, 2272–2276 CrossRef CAS PubMed.
- V. Balasubramanian, B. Herranz-Blanco, P. V. Almeida, J. Hirvonen and H. A. Santos, Prog. Polym. Sci., 2016, 60, 51–85 CrossRef CAS.
- P. Brož, S. M. Benito, C. Saw, P. Burger, H. Heider, M. Pfisterer, S. Marsch, W. Meier and P. Hunziker, J. Controlled Release, 2005, 102, 475–488 CrossRef PubMed.
- J. A. Opsteen, R. P. Brinkhuis, R. L. Teeuwen, D. W. Löwik and J. C. van Hest, Chem. Commun., 2007, 3136–3138 RSC.
- R. Nehring, C. G. Palivan, S. Moreno-Flores, A. Mantion, P. Tanner, J. L. Toca-Herrera, A. Thünemann and W. Meier, Soft Matter, 2010, 6, 2815–2824 RSC.
- M. Felici, M. Marzá-Pérez, N. S. Hatzakis, R. J. Nolte and M. C. Feiters, Chem. – Eur. J., 2008, 14, 9914–9920 CrossRef CAS PubMed.
- S. F. Van Dongen, M. Nallani, S. Schoffelen, J. J. Cornelissen, R. J. Nolte and J. van Hest, Macromol. Rapid Commun., 2008, 29, 321–325 CrossRef CAS.
- S. F. van Dongen, W. P. Verdurmen, R. J. Peters, R. J. Nolte, R. Brock and J. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef PubMed.
- M. A. Petersen, L. Yin, E. Kokkoli and M. A. Hillmyer, Polym. Chem., 2010, 1, 1281–1290 RSC.
- D. Demirgöz, T. O. Pangburn, K. P. Davis, S. Lee, F. S. Bates and E. Kokkoli, Soft Matter, 2009, 5, 2011–2019 RSC.
- L. You and H. Schlaad, J. Am. Chem. Soc., 2006, 128, 13336–13337 CrossRef CAS PubMed.
- Z. Pang, W. Lu, H. Gao, K. Hu, J. Chen, C. Zhang, X. Gao, X. Jiang and C. Zhu, J. Controlled Release, 2008, 128, 120–127 CrossRef CAS PubMed.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
- B. Iyisan, J. Kluge, P. Formanek, B. Voit and D. Appelhans, Chem. Mater., 2016, 28, 1513–1525 CrossRef CAS.
- C. T. Kreis, M. Le Blay, C. Linne, M. M. Makowski and O. Bäumchen, Nat. Phys., 2017 DOI:10.1038/nphys4258.
- J. Petit, I. Polenz, J.-C. Baret, S. Herminghaus and O. Bäumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 1–6 CrossRef CAS PubMed.
- H. Elamari, I. Jlalia, C. Louet, J. Herscovici, F. Meganem and C. Girard, Tetrahedron: Asymmetry, 2010, 21, 1179–1183 CrossRef CAS.
- J. P. Reeves and R. M. Dowben, J. Cell. Physiol., 1969, 73, 49–60 CrossRef CAS PubMed.
- K. Koynov and H.-J. Butt, Curr. Opin. Colloid Interface Sci., 2012, 17, 377–387 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- Z. Petrášek and P. Schwille, Biophys. J., 2008, 94, 1437–1448 CrossRef PubMed.
- E. Evans, K. Ritchie and R. Merkel, Biophys. J., 1995, 68, 2580–2587 CrossRef CAS PubMed.
- M.-J. Colbert, A. Raegen, C. Fradin and K. Dalnoki-Veress, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 30, 117 CrossRef CAS PubMed.
- P. Vermette, T. Gengenbach, U. Divisekera, P. A. Kambouris, H. J. Griesser and L. Meagher, J. Colloid Interface Sci., 2003, 259, 13–26 CrossRef CAS PubMed.
- P. Vermette, H. J. Griesser, P. Kambouris and L. Meagher, Biomacromolecules, 2004, 5, 1496–1502 CrossRef CAS PubMed.
- C. A. Helm, W. Knoll and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8169–8173 CrossRef CAS.
- E.-L. Florin, V. T. Moy and H. E. Gaub,
et al.
, Science, 1994, 264, 415–417 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sm01885a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Oliver
Bäumchen
*a and
Katharina
Landfester
*b
*b,
Oliver
Bäumchen
*a and
Katharina
Landfester
*b