
Development of visible-light responsive and mechanically enhanced “smart” UCST interpenetrating network hydrogels
Yifei
Xu
 ,
Onkar
Ghag
,
Morgan
Reimann
,
Philip
Sitterle
,
Prithwish
Chatterjee
,
Elizabeth
Nofen
,
Hongyu
Yu
,
Hanqing
Jiang
and
Lenore L.
Dai
*
,
Onkar
Ghag
,
Morgan
Reimann
,
Philip
Sitterle
,
Prithwish
Chatterjee
,
Elizabeth
Nofen
,
Hongyu
Yu
,
Hanqing
Jiang
and
Lenore L.
Dai
*
School for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, Arizona 85281, USA. E-mail: Lenore.Dai@asu.edu; Tel: +1 480 965-4112
First published on 24th November 2017
Abstract
An interpenetrating polymer network (IPN), chlorophyllin-incorporated environmentally responsive hydrogel was synthesized and exhibited the following features: enhanced mechanical properties, upper critical solution temperature (UCST) swelling behavior, and promising visible-light responsiveness. Poor mechanical properties are known challenges for hydrogel-based materials. By forming an interpenetrating network between polyacrylamide (PAAm) and poly(acrylic acid) (PAAc) polymer networks, the mechanical properties of the synthesized IPN hydrogels were significantly improved compared to hydrogels made of a single network of each polymer. The formation of the interpenetrating network was confirmed by Fourier Transform Infrared Spectroscopy (FTIR), the analysis of glass transition temperature, and a unique UCST responsive swelling behavior, which is in contrast to the more prevalent lower critical solution temperature (LCST) behaviour of environmentally responsive hydrogels. The visible-light responsiveness of the synthesized hydrogel also demonstrated a positive swelling behavior, and the effect of incorporating chlorophyllin as the chromophore unit was observed to reduce the average pore size and further enhance the mechanical properties of the hydrogel. This interpenetrating network system shows potential to serve as a new route in developing “smart” hydrogels using visible-light as a simple, inexpensive, and remotely controllable stimulus.
Introduction
Environmentally responsive hydrogels are soft and “smart” polymeric networks that respond to external stimuli, such as temperature,1,2 pH,3,4 ions,5,6 electric fields,7,8 enzymes,9 and light,10,11 with significant volume transition. Owning to this unique feature, such functional materials uphold a variety of important applications including sensors,12 actuators,13 drug encapsulation and delivery,14–17 and oil cleanup.18,19 Temperature responsive hydrogels are the most commonly studied type of environmentally responsive hydrogels due to the ease of using temperature to trigger hydrogels’ “smart” responses. Among various temperature responsive hydrogels, crosslinked poly(N-isopropylacrylamide) (PNIPAAm) is well-known and extensively studied. The popularity of PNIPAAm can be attributed to its substantial volume change at its lower critical solution temperature (LCST) of approximately 33 °C. The hydrogels of LCST nature exhibit polymer coil and shrinkage above the LCST due to the reduced polymer–water interaction. The resulting polymer network then shows an overall hydrophobicity and causes de-swelling in water. With a transition temperature near both physiological and ambient room temperatures, PNIPAAm has been able to expand the application of hydrogels into diverse emerging fields such as flexible electronics,20,21 tissue engineering,22,23 artificial muscles,24,25 biomimetic robotics,26,27 3D printing,28,29 and other novel devices.Although PNIPAAm may represent the current state of the art of environmentally responsive hydrogels, it faces the following challenges. First, like typical hydrogels, PNIPAAm hydrogels have weak mechanical properties due to the porous structure filled with water. The use of a nanocrystalline clay as a crosslinker, such as LAPONITE®, has been reported to increase the toughness of hydrogels by lowering the crosslinking density to reduce the localized stress under deformation.30,31 However, LAPONITE® is highly hydrophilic and does not disperse in the majority of organic solvents which severely restricts its use to purely aqueous solvent systems.32 Second, the response behavior of PNIPAAm in water is limited to its LCST nature. When a material is desired to swell upon heating or shrink upon cooling, LCST-based polymer hydrogels are not capable of fulfilling this need. Contrary to that, certain unique polymers can demonstrate the property of UCST by swelling above a certain temperature. Ionic liquids as a type of novel solvent have been explored to reverse the phase transition of PNIPAAm into UCST behavior in replacement of water.33–36 However, their toxicity hinders the compatibility of the hydrogels in biomedical applications. Additionally, heat-associated problems such as the presence of a temperature gradient or dissipation of heat through the material can prevent PNIPAAm hydrogels from being used in applications that require fast and localized transitions. Another type of stimulus, light, is a less emphasized but appealing option to overcome this shortcoming. It can be easily focused into a beam of small size to aim at a specific area on a large hydrogel matrix and enable more precise responses. Hydrogels have been combined with chromophore materials to realize light responsiveness.37–40 Azobenzene, for example, has been employed to drive the volume transition of PNIPAAm-based hydrogels by its reversible photo-isomerization.37,41 However, the photoresponsive mechanism based on azobenzene groups as well as other prevailing chromophores requires that the wavelengths of the light stimuli are in either the ultraviolet or infra-red/near infra-red range, which can be harmful to living entities. Visible light is a safe, feasibly-controllable stimulus and can also be obtained from abundant sunlight. Therefore, visible-light responsive hydrogels are a better choice of environmentally responsive hydrogels in designing human-machine interfaced devices such as tactile displays for the visually-impaired.
In order to address the aforementioned challenges, a mechanically enhanced and visible-light responsive hydrogel with UCST characteristics was designed in this study by constructing an interpenetrating network (IPN) of PAAm and PAAc with chlorophyllin (Ch) incorporated as the chromophore. Interpenetrating network hydrogels have been known to demonstrate a greater degree of mechanical properties where traditional hydrogels fall short.42–44 For instance, ionically crosslinked alginate and covalently crosslinked PAAm can form a hybrid network via additional crosslinking between alginate and PAAm chains resulting in high toughness and strechability.43 In addition to ionic and covalent bonding, hydrogen bonding has also demonstrated a significant contribution to the mechanical enhancement of interpenetrating network hydrogels.42 As a weaker mode of bonding, hydrogen bonding can break before other types of bonding which facilitates the energy dissipation within the hydrogel network. In this study, a pair of two polymers, PAAm and PAAc, is selected because of their attractive intermolecular polymer–polymer interactions allowing complex formation by hydrogen bonding.45,46 We anticipate the formation of an IPN network between the PAAm and PAAc polymer chains and a resulting UCST swelling behavior via the formation and the dissociation of hydrogen bonding within the interpenetrating network. We also hypothesize that the chlorophyllin-incorporated interpenetrating network of PAAm–PAAc will represent a mechanically enhanced, positive-swelling, visible-light responsive hydrogel.
Experimental
Materials
Acrylamide (AAm), acrylic acid (AAc), N,N′-methylenebisacrylamide (MBAAm), azobisisobutyronitrile (AIBN), and chlorophyllin sodium copper salt (Ch) were purchased from Sigma-Aldrich Chemicals and used as supplied.Synthesis of the first phase PAAm chlorophyllin-containing hydrogel
The synthesis started with dissolving 2.35 mg AIBN as the initiator in 0.5 mL tetrahydrofuran (THF) solvent, followed by the addition of 4.5 mL deionized (DI) water, 670 mg AAm, 10 mg chlorophyllin, and 10 mg MBAA. After mixing and degassing, the solution was then heated at 68 °C for 8 hours to allow the polymerization reaction to proceed. Once the reaction was completed, the PAAm hydrogel was rinsed with DI water several times, cut into a disk-shaped geometry, and dried. The drying process included an air-drying step in a fume hood for 24 hours and then a vacuum-drying step in an oven at 40 °C for 24 hours.Synthesis of the second phase PAAc hydrogel for the preparation of a PAAm–PAAc chlorophyllin-containing interpenetrating network hydrogel
A monomer solution of AAc was prepared by mixing 0.345 mL (0.36 g) AAc, 2.35 mg AIBN, and 10 mg MBAA in 5 mL DI water followed by vortex mixing for 10 minutes. The previously obtained dry PAAm hydrogel was dropped into the AAc solution and allowed to soak for 24 hours at room temperature. After soaking, the above AAc solution was purged and then kept at 68 °C for 8 hours allowing the reaction to complete. During this process, the AAc monomer was polymerized and crosslinked within the first phase network of the PAAm gel matrix to form a PAAm–PAAc chlorophyllin-containing interpenetrating network hydrogel.Swelling tests
The swelling weights of the hydrogel samples were measured gravimetrically after wiping off the excessive water from the hydrogel surface. The hydrogel was incubated in water for 48 hours at room temperature prior to the weight measurement and any heat- or light-induced swelling test. The swelling ratio of the hydrogels, R, was calculated from the following equation: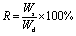 | (1) |
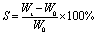 | (2) |
Fourier transform infrared spectroscopy (FTIR)
The ATR-FTIR spectra were taken on powders of ground dry interpenetrating network gel samples with a Bruker IFS 66V/S FTIR, with a wide-band Mercury Cadmium Telluride (MCT) detector, KBr mid-IR beamsplitter, and Pike diamond-ATR module.Scanning electron microscopy (SEM)
The morphology of the synthesized interpenetrating network hydrogels was studied using an SEM (FEI XL-30, Philips). Prior to SEM, the samples were dehydrated by a freeze-dryer (FreeZone1, Labconco Corporation) and glued onto the brass sample holders followed by a gold sputter coating process.Instron
The compressive tests were performed on synthesized hydrogels in their wet state by an Instron E3000 test system at room temperature. Three different strain rates, 0.5 mm min−1, 0.05 mm min−1, and 0.005 mm min−1, were run to study the effect of ramp rate on the corresponding mechanical properties of the prepared hydrogels. Prior to the test, all the samples were soaked in water for 48 h at room temperature to allow the hydrogel swelling to reach equilibrium.UV-Vis spectrophotometer
The temperature-dependent opacity transition of the interpenetrating network hydrogels was captured via a Cary 300 Bio UV-Vis spectrophotometer. The hydrogel sample was cut in the shape of a thin slice and was placed in a cuvette along with an adequate amount of water. The temperature control function was utilized for a temperature range from 15 °C to 50 °C, and a laser wavelength of 600 nm was used.Differential scanning calorimetry (DSC)
A TA differential scanning calorimeter (TA Instruments Q-20) was used to characterize the glass transition temperature of the prepared hydrogels in their dry state. Prior to the DSC measurement, the gel samples were first air dried at room temperature for 24 hours and then vacuum dried at 90 °C for 24 hours to further eliminate water retained in the network. During the measurement, the samples were subjected to cyclic heating and cooling at a ramp rate of 10 °C min−1.Results and discussion
Confirmation of interpenetrating network hydrogel synthesis
The functional groups of PAAm and PAAc were characterized by ATR-FTIR to confirm the synthesis. As shown in Fig. 1(a), the FITR spectra demonstrate the amide functional group of PAAm with N–H bending and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch which are indicated by the peaks at 1605 cm−1 and 1645 cm−1, respectively. Similarly, the presence of PAAc groups is proven by the C–O stretch and the CO stretch of the unsaturated carboxylic acid group as indicated by the peaks at 1240 cm−1 and 1695 cm−1, respectively. In the spectrum of the PAAm–PAAc interpenetrating polymer network (PAAm–PAAc IPN), the above signature peaks are all observed at 1238 cm−1, 1601 cm−1, 1645 cm−1 and 1708 cm−1, respectively, with minimal shifts compared to the individual spectrum of PAAm and PAAc polymer networks. The FTIR results demonstrate that the synthesis of the interpenetrating network hydrogel was able to successfully include both the PAAm and PAAc polymer networks. The peaks of N–H stretch of the amide group of PAAm and the O–H stretch of the carboxylic acid group of PAAc are broad and overlapping at the adjacent wavelength so they cannot be used to distinguish the functional groups of PAAm and PAAc. The PAAm and PAAc form the structure of the interpenetrating network hydrogel due to their intermolecular polymer–polymer interactions. PAAm and PAAc serve as the first phase and the second phase of the interpenetrating network, respectively. The intermolecular interaction is based on the dynamic mechanism of hydrogen bonding formation and dissociation between the amide group of PAAm and the carboxylic acid group of PAAc.46
O stretch which are indicated by the peaks at 1605 cm−1 and 1645 cm−1, respectively. Similarly, the presence of PAAc groups is proven by the C–O stretch and the CO stretch of the unsaturated carboxylic acid group as indicated by the peaks at 1240 cm−1 and 1695 cm−1, respectively. In the spectrum of the PAAm–PAAc interpenetrating polymer network (PAAm–PAAc IPN), the above signature peaks are all observed at 1238 cm−1, 1601 cm−1, 1645 cm−1 and 1708 cm−1, respectively, with minimal shifts compared to the individual spectrum of PAAm and PAAc polymer networks. The FTIR results demonstrate that the synthesis of the interpenetrating network hydrogel was able to successfully include both the PAAm and PAAc polymer networks. The peaks of N–H stretch of the amide group of PAAm and the O–H stretch of the carboxylic acid group of PAAc are broad and overlapping at the adjacent wavelength so they cannot be used to distinguish the functional groups of PAAm and PAAc. The PAAm and PAAc form the structure of the interpenetrating network hydrogel due to their intermolecular polymer–polymer interactions. PAAm and PAAc serve as the first phase and the second phase of the interpenetrating network, respectively. The intermolecular interaction is based on the dynamic mechanism of hydrogen bonding formation and dissociation between the amide group of PAAm and the carboxylic acid group of PAAc.46
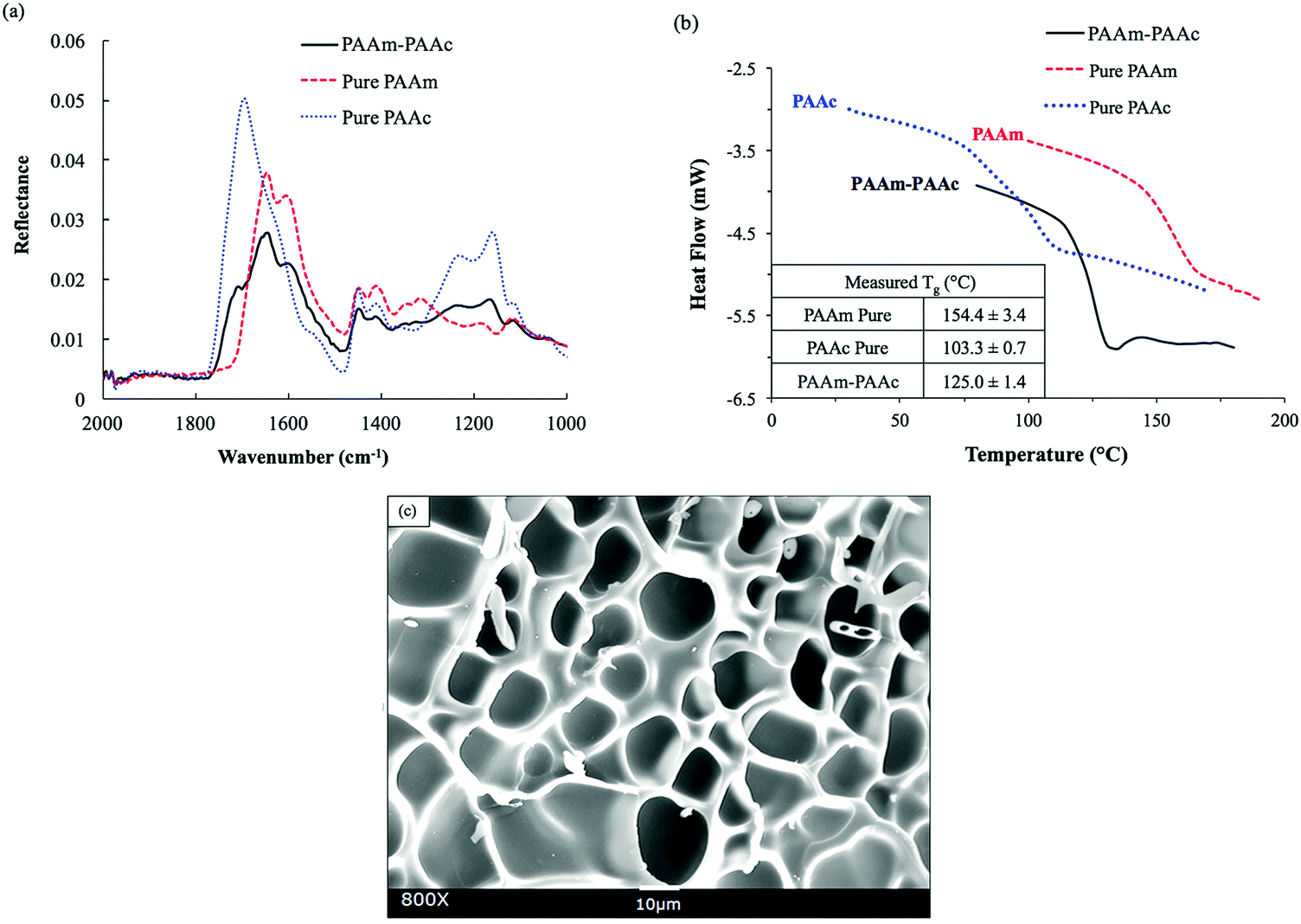 | ||
| Fig. 1 Conformation of the PAAm–PAAc IPN hydrogel: (a) comparison of the ATR-FTIR spectra between the interpenetrating network PAAm–PAAc hydrogel (black solid) and individual network hydrogels, pure PAAm (red dash) and pure PAAc (blue dot), in the wavenumber range from 1000 to 2000 cm−1. (b) Representative DSC thermograms showing the glass transition of PAAm–PAAc (black), PAAm (red), and PAAc (blue), with the resulting Tg values in the inset on the bottom left. (c) SEM image showing the morphology of the interpenetrating network PAAm–PAAc hydrogel (after freeze-drying) with an average pore size of 16.0 ± 4.4 μm. | ||
The formation of a PAAm–PAAc interpenetrating network was also confirmed by thermal evidence from DSC analysis. Previously, Ribellies et al.47 suggested that a well-mixed, highly cross-linked IPN is expected to have one single glass transition temperature (Tg) due to the close interaction between the two polymer networks that cannot express themselves independently. On the contrary, when the two polymer networks fail to interpenetrate or are only loosely cross-linked, two separate glass transitions should be observed in correspondence to a phase-separated system.47,48 As shown in Fig. 1(b), the single polymer networks of pure PAAm and PAAc show individual glass transitions at 154.4 ± 3.4 °C and 103.3 ± 0.7 °C, respectively. The interpenetrating network of PAAm–PAAc demonstrates a clear single glass transition at 125.0 ± 1.4 °C between the Tg values of pure PAAm and PAAc networks, which is evidence of the successful formation of the interpenetrating network between the PAAm and the PAAc. The morphological features of the PAAm–PAAc interpenetrating network after freeze-drying are shown by the SEM image in Fig. 1(c), and the macro-porous structures of the interpenetrating network hydrogel were revealed. The average pore size of the porous structures was calculated as 16.0 ± 4.4 μm based on 20 measurements randomly selected in the image using ImageJ.
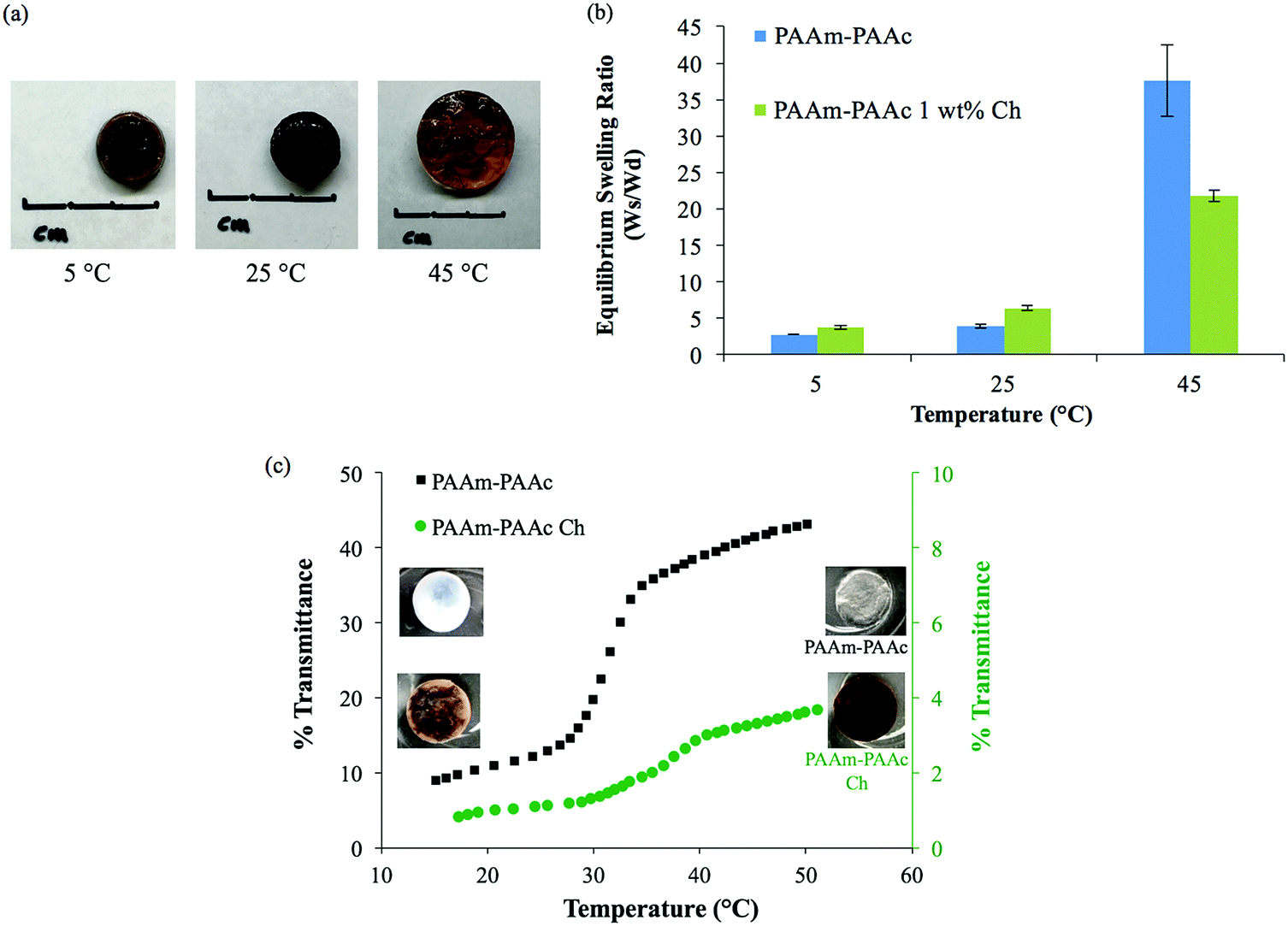
The intermolecular interaction between PAAm and PAAm within the interpenetrating network structure also facilitated a unique UCST swelling behavior of the synthesized PAAm–PAAc IPN hydrogel. Fig. 2(a) shows a pictorial depiction of the UCST characteristic of the synthesized PAAm–PAAc IPN hydrogel. At 5 °C, the PAAm–PAAc IPN hydrogel exhibited the most shrinkage. At both 25 °C and 45 °C, the PAAm–PAAc IPN hydrogel exhibited continuous volume expansion with a positive temperature dependence. In Fig. 2(b), the swelling ratios of the synthesized PAAm–PAAc IPN hydrogel were investigated at three different temperatures, 5 °C, 25 °C, and 45 °C, and a strong temperature dependence could be seen. As the media temperature rose from 5 °C to 45 °C, the swelling ratio increased over 13.6 ± 1.8 times. The volume transition of the PAAm–PAAc IPN hydrogel was much more dramatic over the tested temperature range between 25 °C and 45 °C than between 5 °C and 25 °C, which was visually observable by the increasing volume of the gel dimensions. Katono et al.49 also reported a similar positive swelling behavior for a poly(acrylamide-co-butyl methacrylate)-poly(acrylic acid) IPN system for application in drug delivery and attributed the phenomenon to the formation and the dissociation of an intermolecular complex. On a bulk scale, the overall interpenetrating network hydrogel exhibits de-swelling due to a more “tightly” held intermolecular complex by the stable formation of hydrogen bonding at a lower temperature. At a higher temperature, the hydrogel then shows enhanced swelling due to the dissociation of the intermolecular complex by the breakage of hydrogen bonding. However, Katono et al.'s work mainly focused on the swelling aspect and did not evaluate the formation of the interpenetrating network from other valuable angles, such as thermal and mechanical approaches, which will be discussed in the following sections of this study.
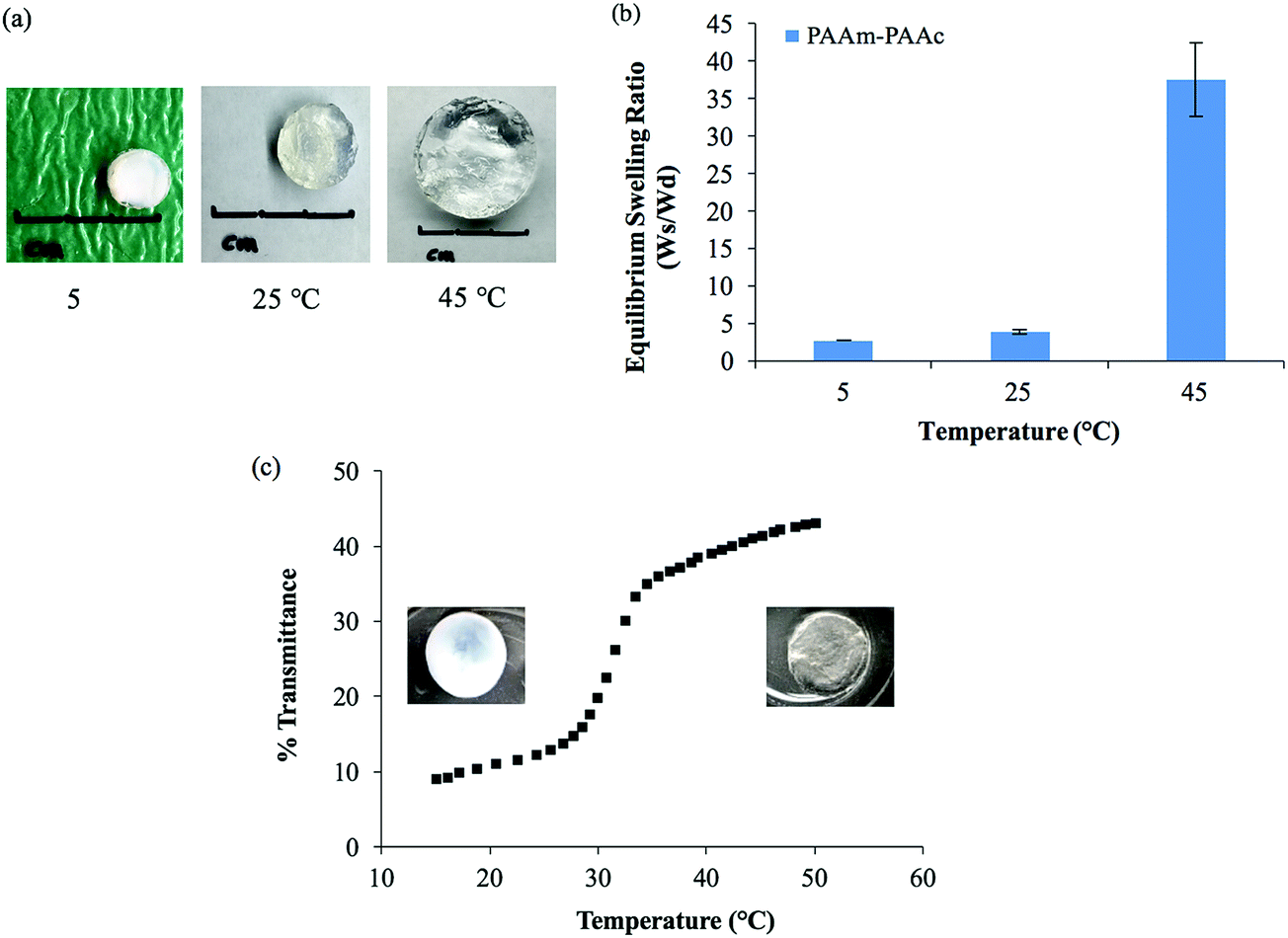 | ||
| Fig. 2 UCST characteristics of the PAAm–PAAc IPN hydrogel: (a) pictorial demonstration of the volume and opacity transition of the hydrogel. (b) Swelling ratio (Ws/Wd) at equilibrium at 5 °C, 25 °C, and 45 °C. (c) The change in optical transmittance of the hydrogel from 15 °C to 50 °C measured by UV-Vis spectrophotometer showing the opacity transition of the material; the left and right insets are representative screenshots of the hydrogel's opacity transition taken at the first 15 seconds after immersion in water at 15 °C and 50 °C from room temperature, respectively. | ||
In addition to the swelling behavior, the interpenetrating network hydrogel also shows temperature-dependent opacity transition. As also shown in Fig. 2(a), the color of the PAAm–PAAc IPN hydrogel appears as opaque at 5 °C and becomes clearly transparent at 45 °C. In order to further investigate this phenomenon, UV-Vis analysis is employed to quantify the opacity change of the PAAm–PAAc IPN hydrogel with respect to temperature. As shown in Fig. 2(c), the hydrogel has a low transmittance of only 8.9% with a white opaque color at 15 °C. As the temperature ramps up, the hydrogel responds to this environmental change by exhibiting a reduced opacity with the transmittance increasing to 43.0% measured at 50 °C. At this point, the hydrogel displays as almost transparent in the media instead of opaque. The most dramatic change of the percentage transmittance is observed to take place at approximately 32 °C while a more gradual transition occurs below 29 °C and above 35 °C. Unlike the volume transition described earlier, the opacity transition of the hydrogel is rather fast. When placing the hydrogel that is equilibrated in the media from a higher temperature (e.g. 50 °C) to a lower temperature (e.g. 15 °C), the opaqueness starts to form within the gel body immediately. Also, this opacity transition is observed as a reversible process. Zhang et al.50 reported a similar reversible opaque–transparent phase transition on a physically cross-linked poly(sulfobetaine methacrylate) double network hydrogel, and their work showed that the intramolecular and intermolecular associations were strong enough to drive a phase transition with sufficient water content. In our case, we hypothesize that the PAAm–PAAc IPN hydrogel at temperatures lower than UCST can be viewed as a collapsed coil structure due to intermolecular attractive forces between the acid and the amide group. Under this status, the interaction between the polymer chains was strong enough due to the predominant presence of hydrogen bonding formation that water was prevented from entering the polymer chain system. As a result, the separation of phases was pronounced and the solvent was minimized, giving the interpenetrating network hydrogel its opacity. At temperatures above UCST, the intermolecular forces subsided while the electrostatic bonding between water and the polymer chains became more dominant. The hydrophilicity of the network was then increased, which drove the transition from opaque to transparent. Overall, both the volume transition and the opacity transition contributed to the UCST characteristics of the PAAm–PAAc IPN hydrogel.
Another result of the formation of the PAAm–PAAc interpenetrating network was that the mechanical properties of the PAAm–PAAc IPN hydrogel were enhanced. As shown in Fig. 3, the compressive tests by Instron E3000 provided a quantitative comparison of the stress–strain profiles between the PAAm–PAAc IPN hydrogel and the hydrogels based on its individual polymer components. At a strain at 65%, for example, the PAAm–PAAc IPN hydrogel demonstrates a stress of approximately 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 Pa while the pure PAAm and PAAc hydrogels only show a stress of 14700 Pa and 6900 Pa, respectively. When compared to the PNIPAAm hydrogel, one of the most commonly studied environmentally responsive hydrogels, which was reported to show a stress response lower than 2000 Pa at approximately 25% strain,51 the PAAm–PAAc IPN gel showed a higher stress response of 3000 Pa under the same strain. Furthermore, the PAAm–PAAc IPN hydrogel can fully and repetitively recover to its original shape after being compressed up to above 65% strain without any observable fracture or material failure. In contrast, the pure PAAm hydrogel began to form fractures before reaching 35% strain and the pure PAAc hydrogel also failed to preserve the material integrity from the compressive test. The inset images in Fig. 3 vividly demonstrate that the PAAm–PAAc IPN hydrogel can withstand a large extent of compression while the pure PAAm and PAAc hydrogels suffer from severe deformation and permanent fractures from compression. Both the PAAm and PAAc hydrogels are very fragile in nature by their individual polymer network alone due to the high content of water. By forming the interpenetrated polymer network, the PAAm–PAAc IPN gel displays enhancement with a much greater toughness and a much better ability to stretch than the pure PAAm and PAAc hydrogels when the weight ratio of the monomer and the crosslinker remains the same. The enhanced mechanical properties of the PAAm–PAAc IPN hydrogel can be ascribed to the presence of hydrogen bonding between the amide groups of PAAm and the carboxylic acid groups of PAAc. Such hydrogen bonding serves to reinforce the intermolecular interaction and offers additional crosslinking between the polymer chains to strengthen the interpenetrated polymer networks. When subjected to an external mechanical load, some of the hydrogen bonds dissociate first and hence dissipate the energy, preventing further compromise of the material integrity. Upon the removal of the load, the hydrogen bonds can re-form, allowing the hydrogel to be applied in frequently operated devices. Similar mechanical enhancements through hydrogen bonding have also been observed on several other polymeric systems. Myung et al.52 reported the strain-hardening behavior of a poly(ethylene glycol)-poly(acrylic acid) IPN system with a significantly improved fracture strength and more than two times of modulus enhancement. Wang et al.53 also reported the role of hydrogen bonding in increasing the material strength in a n-acryloyl glycinamide-co-2-vinyl-4,6-diamino-1,3,5-triazine copolymer system. Over the change of pH, such a strengthening effect can be weakened and resumed reversibly based on the dissociation and re-construction of hydrogen bonding.
300 Pa while the pure PAAm and PAAc hydrogels only show a stress of 14700 Pa and 6900 Pa, respectively. When compared to the PNIPAAm hydrogel, one of the most commonly studied environmentally responsive hydrogels, which was reported to show a stress response lower than 2000 Pa at approximately 25% strain,51 the PAAm–PAAc IPN gel showed a higher stress response of 3000 Pa under the same strain. Furthermore, the PAAm–PAAc IPN hydrogel can fully and repetitively recover to its original shape after being compressed up to above 65% strain without any observable fracture or material failure. In contrast, the pure PAAm hydrogel began to form fractures before reaching 35% strain and the pure PAAc hydrogel also failed to preserve the material integrity from the compressive test. The inset images in Fig. 3 vividly demonstrate that the PAAm–PAAc IPN hydrogel can withstand a large extent of compression while the pure PAAm and PAAc hydrogels suffer from severe deformation and permanent fractures from compression. Both the PAAm and PAAc hydrogels are very fragile in nature by their individual polymer network alone due to the high content of water. By forming the interpenetrated polymer network, the PAAm–PAAc IPN gel displays enhancement with a much greater toughness and a much better ability to stretch than the pure PAAm and PAAc hydrogels when the weight ratio of the monomer and the crosslinker remains the same. The enhanced mechanical properties of the PAAm–PAAc IPN hydrogel can be ascribed to the presence of hydrogen bonding between the amide groups of PAAm and the carboxylic acid groups of PAAc. Such hydrogen bonding serves to reinforce the intermolecular interaction and offers additional crosslinking between the polymer chains to strengthen the interpenetrated polymer networks. When subjected to an external mechanical load, some of the hydrogen bonds dissociate first and hence dissipate the energy, preventing further compromise of the material integrity. Upon the removal of the load, the hydrogen bonds can re-form, allowing the hydrogel to be applied in frequently operated devices. Similar mechanical enhancements through hydrogen bonding have also been observed on several other polymeric systems. Myung et al.52 reported the strain-hardening behavior of a poly(ethylene glycol)-poly(acrylic acid) IPN system with a significantly improved fracture strength and more than two times of modulus enhancement. Wang et al.53 also reported the role of hydrogen bonding in increasing the material strength in a n-acryloyl glycinamide-co-2-vinyl-4,6-diamino-1,3,5-triazine copolymer system. Over the change of pH, such a strengthening effect can be weakened and resumed reversibly based on the dissociation and re-construction of hydrogen bonding.
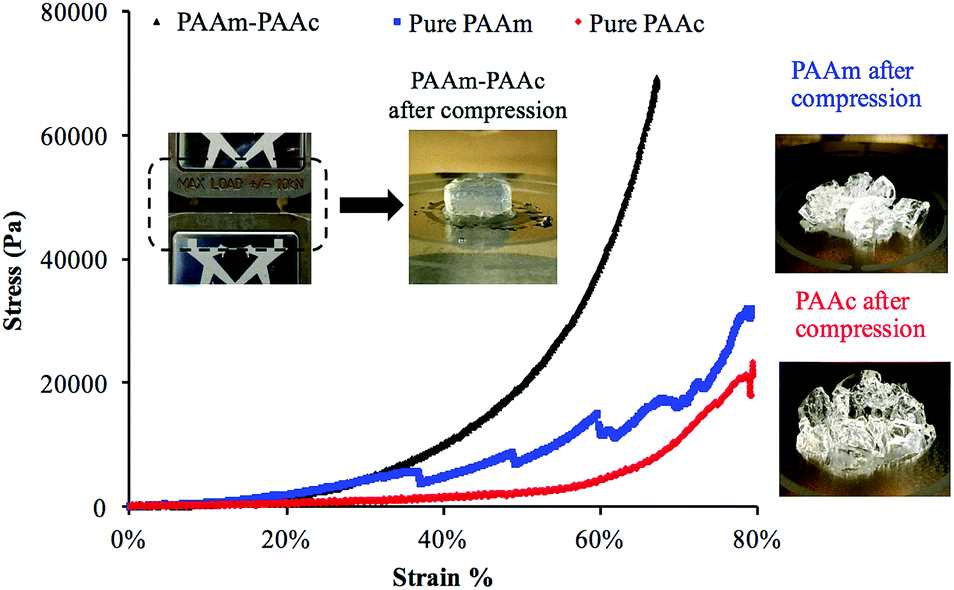 | ||
| Fig. 3 Instron E-3000 compressive analysis of the PAAm–PAAc IPN hydrogel (black) in comparison with the single phase of pure PAAm (blue) and PAAc (red) hydrogels, showing the enhanced mechanical properties. Left insets: the PAAm–PAAc IPN hydrogel being able to recover to its original shape after the compressive test; right insets: fractures and material failure of PAAm (top) and PAAc (bottom) after the compressive tests. | ||
Incorporation of chlorophyllin in the interpenetrating network hydrogel system
Chlorophyllin sodium copper salt (Ch) was selected as a suitable chromophore for the purpose of this study because its light responsiveness lies well within the wavelength range of visible-light. In Fig. 4(a), the UV-Vis spectroscopy of chlorophyllin salt aqueous solution indicates the absorption peaks at the wavelengths of approximately 404 nm and 625 nm. Farag also reported very similar absorption ranges between 350 and 480 nm and between 620 and 670 nm for chlorophyllin-based thin films.54 In the molecular structure shown in the inset image of Fig. 4(a), chlorophyllin has delocalized electrons that can easily be excited to orbitals at higher energy levels by light irradiation of appropriate wavelength. These electrons quickly fall back to their original state, releasing energy in the form of heat, which can induce the positive volume transition of the PAAc–PAAm interpenetrating network system. In addition, the excellent water solubility of chlorophyllin salt also allowed a facile process of its incorporation in the synthesis via simply mixing it into the precursor solution prior to polymerization.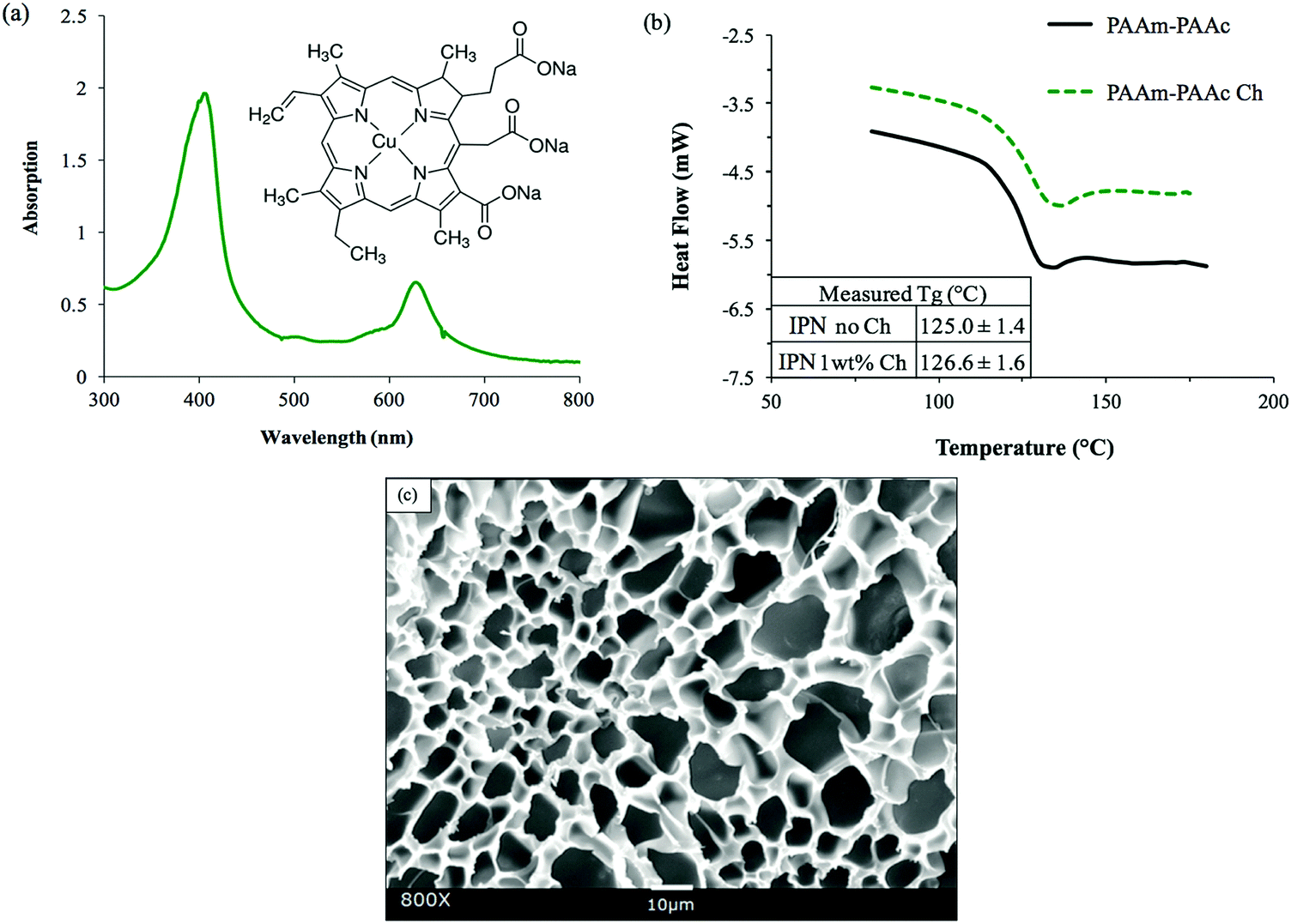 | ||
| Fig. 4 Incorporation of chlorophyllin into the interpenetrating-network hydrogel system: (a) UV-Vis spectrum of chlorophyllin salt aqueous solution between 300 and 800 nm wavelength. Inset: The molecular structure of chlorophyllin sodium copper salt. (b) Comparison of representative DSC thermograms showing the glass transition between non-chlorophyllin (black solid) and chlorophyllin-containing (green dash) interpenetrating network PAAm–PAAc hydrogels with the resulting Tg values (as a result of two runs) in the bottom left inset. (c) SEM images showing the morphology of the PAAm–PAAc Ch IPN hydrogel (after freeze-drying), with a pore size of 9.9 ± 4.3 μm. | ||
After the incorporation, as shown in Fig. 4(b), the DSC measurement detected a glass transition of the PAAm–PAAc chlorophyllin-containing interpenetrating polymer network (PAAm–PAAc Ch IPN) hydrogel to occur at 126.6 ± 1.6 °C. This was very close to the glass transition of the PAAm–PAAc IPN gel at 125.0 ± 1.4 °C, which suggested that the incorporation of chlorophyllin did not significantly affect the thermal properties of the PAAm–PAAc interpenetrating network system. Fig. 4(c) shows the SEM image of the PAAm–PAAc Ch IPN hydrogel sample after freeze-drying. Very similar to the PAAm–PAAc IPN hydrogel, macro-porous structures were also observed with no phase separation in the image. As the DSC analysis only reported Tg, the morphology of the PAAm–PAAc Ch IPN hydrogel additionally confirmed that the chlorophyllin as the chromophore was successfully incorporated into the matrix. Theoretically, the vinyl group on the chlorophyllin molecule can react with free radicals in the solution allowing the chlorophyllin to chemically bond onto the propagating polymer chain during polymerization.55–57 It is also worth noting that the average pore size of the PAAm–PAAc Ch IPN hydrogel is calculated as 9.92 ± 4.31 μm by ImageJ, which is 0.6 times smaller than the pore size of the PAAm–PAAc IPN hydrogel. Zhang et al.58 reported the use of different carbonyl group contents in the precursor solution to control the pore sizes of the synthesized hydrogel. In their work, the hydrogel was crosslinked by the carbonyl group of dialdehyde microfibrillated cellulose and the amino group of gelatin with a comparable range of pore sizes from 10 to 50 μm. The increase in the carbonyl group content of dialdehyde microfibrillated cellulose leads to a higher crosslinking degree between dialdehyde microfibrillated cellulose and gelatin, and therefore a decrease in the average pore sizes of the hydrogel. In our system, the multiple carbonyl groups on the chlorophyllin molecule can have a similar effect to increase the degree of crosslinking via hydrogen bonding, where the carbonyl group serves as the hydrogen bond acceptor with the amine group of the acrylamide as the donor. This enriched intermolecular interaction between the PAAm chains after the incorporation of chlorophyllin then causes a more compact porous structure of the polymeric matrix in the hydrogel with the reduction of the pore dimensions.
After introducing chlorophyllin, the PAAm–PAAc Ch IPN hydrogel still exhibited a clear UCST characteristic with an increasing swelling ratio as the temperature increases. A pictorial depiction of the UCST swelling behavior of the PAAm–PAAc Ch IPN hydrogel is shown in Fig. 5(a). Similar to the PAAm–PAA IPN hydrogel, a comparable positive volume expansion of the PAAm–PAAc Ch IPN hydrogel was observed as the media temperature was increased. In addition to the volume transition, the opacity transition of the PAAm–PAAc Ch IPN hydrogel was also observable as the color of the gel changed from opaque brown to transparent dark brown over the change of environmental temperature from 5 °C to 25 °C and above. Fig. 5(b) shows the comparison of the temperature-dependent swelling ratio between the PAAm–PAAc IPN and PAAm–PAAc Ch IPN hydrogels. The PAAm–PAAc Ch IPN hydrogel displays a slightly higher swelling ratio at both 5 °C and 25 °C yet a notably lower swelling ratio at 45 °C than the PAAm–PAAc IPN hydrogel. The total increase in the swelling ratio over the change of temperature for the PAAm–PAAc Ch IPN hydrogel, 5.70 ± 0.42 times, was considerably smaller than that for the PAAm–PAAc IPN hydrogel, 13.62 ± 1.75 times. This suggests that the incorporation of chlorophyllin may have an inhibitory effect on the swelling of the PAAm–PAAc Ch IPN hydrogel, which can be related to its smaller pore dimensions. The more compact porous structure can not only hold the polymer network more “tightly” together but also hinder the water diffusion through the network, resulting in less extent of expansion. A mimicking but much weaker behavior of opacity transition is also observed on the PAAm–PAAc Ch IPN hydrogel via UV-Vis analysis. As shown in Fig. 5(c), the opacity transition of the PAAm–PAAc Ch IPN hydrogel is captured increasing from only 0.6% to 4.3% transmittance over the range of the temperature increase. Since the PAAm–PAAc Ch IPN hydrogel has a dark color from the incorporation of chlorophyllin, the transmittance of the material is significantly lower than that of the PAAm–PAAc IPN hydrogel. The visual color of the PAAm–PAAc Ch IPN hydrogel also changes from an opaque brown to a transparent brown over the transition, and the most dramatic transition takes place at approximately 36 °C. This similar opacity transition behavior of the PAAm–PAAc Ch IPN hydrogel confirms that the temperature-dependent intramolecular and intermolecular interaction in the interpenetrating network is well sustained after the incorporation of chlorophyllin.
 | ||
| Fig. 5 UCST swelling behavior of the PAAm–PAAc Ch IPN hydrogel: (a) pictorial demonstration of the volume and the opacity transition of the hydrogel after incorporating cholorophyllin. (b) Comparison of the swelling ratio (Ws/Wd) at equilibrium between the non-chlorophyllin and chlorophyllin-containing hydrogel at 5 °C, 25 °C, and 45 °C. (c) The change in optical transmittance of the PAAm–PAAc Ch IPN hydrogel from 15 °C to 50 °C measured by UV-Vis spectrophotometer showing the opacity transition of the material; bottom left and bottom right insets are representative screenshots of the PAAm–PAAc Ch IPN hydrogel's opacity transition taken at the first 15 second after immersion in water at 15 °C and 50 °C from room temperature, respectively. | ||
Apart from the differences in the swelling ratio, the incorporation of chlorophyllin also has an effect on the mechanical properties of the interpenetrating network hydrogel system. The mechanical properties of the PAAm–PAAc Ch IPN hydrogel were also examined by a compressive test using Instron and compared to the PAAm–PAAc IPN hydrogel in Fig. 6(a). The PAAm–PAAc Ch IPN hydrogel demonstrates a significantly higher stress than the PAAm–PAAc IPN hydrogel at a given strain. For example, the PAAm–PAAc Ch IPN hydrogel shows a stress of approximately 120000 Pa at 60% strain in comparison with the PAAm–PAAc IPN hydrogel with a stress at approximately 38800 Pa. This suggests that the enhancement of the mechanical properties of the PAAm–PAAc interpenetrating network is further improved by incorporating chlorophyllin. This improvement can be related to the morphology of the PAAm–PAAc Ch IPN hydrogel again. The mechanical properties of hydrogels can be substantially affected by their porous structures. As discussed earlier, the smaller pore size, induced by the incorporation of chlorophyllin, can form a more compact packing of the polymer network which displays a stronger stress response toward increasing strain and better overall mechanical properties of the hydrogel in the bulk scale. A previous study by Chiu et al.59 reported a similar observation of an increase in compressive modulus with decreasing pore size for a poly(ethylene glycol)-co-(L-lactic acid) hydrogel. This consistent trend can be explained by the smaller pore size decreasing the water-to-hydrogel contact and hence reducing the plasticizing effect of water in the polymer network.60Fig. 6(b) shows the changing trend of the compressive tangent modulus of PAAm–PAAc IPN and PAAm–PAAc Ch IPN hydrogels calculated using the method described by Li et al.61 As the compression strain increases, the compressive tangent moduli of both the hydrogels demonstrate an exponential increase. Owing to the incorporation of chlorophyllin, the PAAm–PAAc Ch IPN hydrogel has a higher and more rapidly increasing modulus than the PAAm–PAAm IPN hydrogel as expected from the previous stress–strain comparison. Over the rise of the compression strain from 10% to 60%, the modulus of PAAm–PAAc Ch IPN significantly increases from 0.05 MPa to 0.67 MPa. When the applied strain is at a low level, the hydrogels display lower moduli because of the large amount of free water content contained in the highly three-dimensional porous structures in the network. At a high strain level, a strain-hardening effect on the hydrogels becomes more pronounced due to the release of free water from the hydrogel and the intensified physical entanglement from the re-orientation of the polymer network.52,61 The effect of strain rate on the responding mechanical property of the PAAm–PAAc Ch IPN hydrogel is also studied. As shown in Fig. 6(c), the stress–strain curves can clearly be differentiated by three different stain rates of 0.5 mm min−1, 0.05 mm min−1, and 0.005 mm min−1. At a given strain, the stress under a faster strain rate is found to be larger than that under a slower strain rate. This phenomenon is due to the fact that under a faster strain rate less relaxation is allowed to occur in the polymer network and thus a faster increasing stress takes place to respond to the strain.
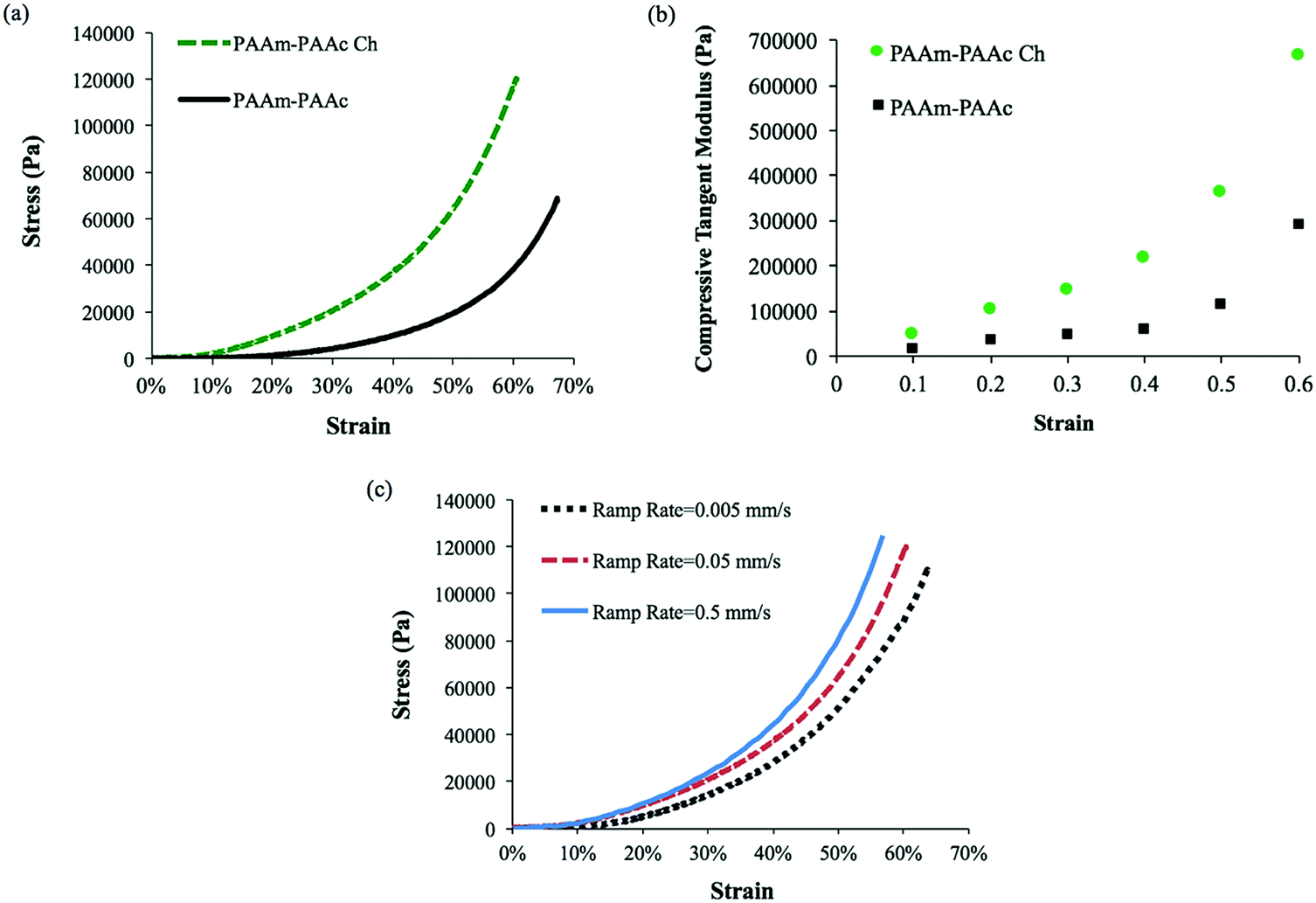 | ||
| Fig. 6 Comparison of compressive analysis: (a) stress–strain curves of PAAm–PAAc IPN and PAAm–PAAc Ch IPN hydrogels obtained by Instron showing further mechanical enhancement after incorporating chlorophyllin. (b) Calculated compressive tangent moduli of PAAm–PAAc IPN and PAAm–PAAc Ch IPN hydrogels with respect to strain ratio from 10% to 60% with an interval of 10%. (c) The effect of different strain rates on the PAAm–PAAc Ch IPN hydrogel. | ||
Fig. 7 demonstrates the visible-light responsiveness of the PAAm–PAAc Ch IPN hydrogel. The visible-light that comes from the LED lamp of a commercial projector (Epson) is used to mimic the illumination of the display screen from commonly used electronic devices, and white light is selected to maximize the optical output from the region of visible-light. All the light responsiveness tests were performed with the light intensity at 42.5 mW cm−2, which was measured at the height of the upper water surface. Upon the irradiation of the incident visible-light, the PAAm–PAAc Ch IPN hydrogel responds with a swelling weight increase of 18.3 ± 3.0% and 20.8 ± 4.5%, after 2 minutes and 10 minutes of exposure, respectively. This trend can be explained through a local heat effect which is caused by the time required for the light–heat conversion by chlorophyllin to equilibrate. Additionally, a more thorough diffusion process for water to transport from the solution to the polymer network will enhance the swelling weight increase in the chlorophyllin-containing hydrogel. The PAAm–PAAc IPN hydrogel as a control shows negligible responsiveness to visible light despite the exposure time, which is expected due to the inert nature of the PAAm–PAAc IPN hydrogel toward the stimulus without chlorphyllin converting light to heat. Owing to the fact that the intensity of light irradiation from the projector was not sufficient to raise the water temperature, the media remained at room temperature throughout each test.
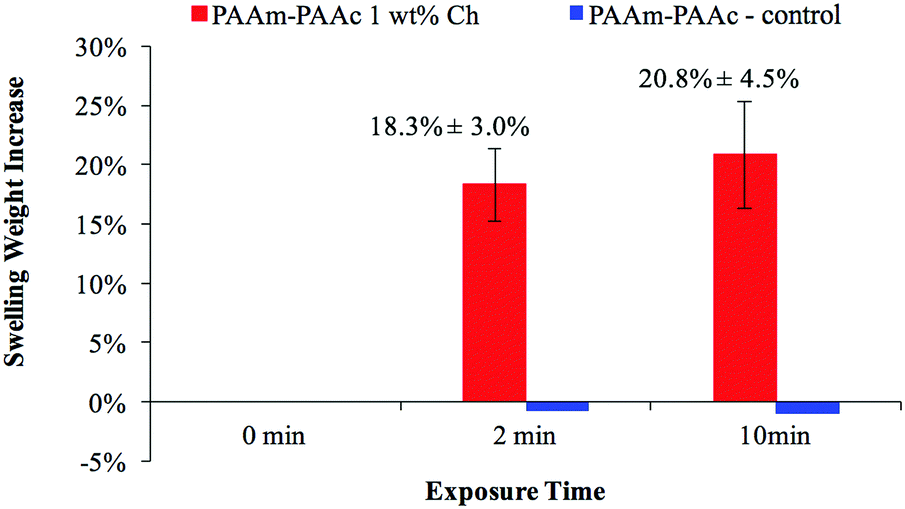 | ||
| Fig. 7 The visible-light responsiveness of the PAAm–PAAc Ch IPN network hydrogel measured by swelling weight with respect to exposure time (0 min, 2 min and 10 min). | ||
Conclusions
A novel visible-light responsive hydrogel system was synthesized and exhibited an enhancement of mechanical properties by the formation of an interpenetrating network between PAAm and PAAc. This formation of a PAAm–PAAc interpenetrating network was experimentally confirmed by the observation of positive swelling and the occurrence of a new Tg in between the Tg values of the pure PAAm and PAAc single networks. The visible light responsiveness of the synthesized hydrogel follows an UCST behavior, which is less studied compared to the LCST behavior prevalent in environmental responsive hydrogels. This interpenetrating network hydrogel system explored a new mechanism to overcome the aforementioned challenges in developing visible-light responsive “smart” materials, and further study is warranted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the financial support from the National Science Foundation CMMI 1462481 and the Fulton Undergraduate Research Initiative Program at Arizona State University. We would also like to acknowledge and thank Dr Matthew Green, Dr François Perreault, and Dr Konrad Rykaczewski at Arizona State University for the instrumental usage.References
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4, 22001 CrossRef PubMed.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS PubMed.
- A. Richter, G. Paschew, S. Klatt, J. Lienig, K.-F. Arndt and H.-J. P. Adler, Sensors, 2008, 8, 561–581 CrossRef CAS PubMed.
- R. Luo, H. Li and K. Y. Lam, Anal. Bioanal. Chem., 2007, 389, 863–873 CrossRef CAS PubMed.
- J. P. Gong, T. Nitta and Y. Osada, J. Chem. Inf. Model., 2013, 53, 1689–1699 CrossRef PubMed.
- T. Wallmersperger, D. Ballhause, B. Kroplin, M. Gunther and G. Gerlach, J. Intell. Mater. Syst. Struct., 2009, 20, 1483–1492 CrossRef CAS.
- T. Tanaka, N. Izumi, S.-T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CAS.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS PubMed.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- A. Suzuki, T. Ishii and Y. Maruyama, J. Appl. Phys., 1996, 80, 131 CrossRef CAS.
- G. Gerlach, M. Guenther, J. Sorber, G. Suchaneck, K. F. Arndt and A. Richter, Sens. Actuators, B, 2005, 111–112, 555–561 CrossRef CAS.
- J. Hoffmann, M. Plötner, D. Kuckling and W. J. Fischer, Sens. Actuators, A, 1999, 77, 139–144 CrossRef CAS.
- J. Iocozzia and Z. Lin, Macromolecules, 2017, 50, 4906–4912 CrossRef CAS.
- C. Feng, X. Pang, Y. He, Y. Chen, G. Zhang and Z. Lin, Polym. Chem., 2015, 6, 5190–5197 RSC.
- C. Feng, X. Pang, Y. He, B. Li and Z. Lin, Chem. Mater., 2014, 26, 6058–6067 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- A. M. Atta, R. A. M. El-Ghazawy, R. K. Farag and A. A. A. Abdel-Azim, React. Funct. Polym., 2006, 66, 931–943 CrossRef CAS.
- A. Ummadisingu and S. Gupta, Desalin. Water Treat., 2012, 44, 44–51 CrossRef CAS.
- H. Ko, R. Kapadia, K. Takei, T. Takahashi, X. Zhang and A. Javey, Nanotechnology, 2012, 23, 344001 CrossRef PubMed.
- V. T. Tran, H. Zhou, S. Hong, J. Youn, H. Chen, K. Koh and J. Lee, Curr. Appl. Phys., 2013, 13, 940–944 CrossRef.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS PubMed.
- T. Shimizu, M. Yamato, Y. Isoi, T. Akutsu, T. Setomaru, K. Abe, A. Kikuchi, M. Umezu and T. Okano, Circ. Res., 2002, 90, 40–48 CrossRef.
- K. L. Fujimoto, Z. Ma, D. M. Nelson, R. Hashizume, J. Guan, K. Tobita and W. R. Wagner, Biomaterials, 2009, 30, 4357–4368 CrossRef CAS PubMed.
- S.-H. Lee, T. H. Kim, M. D. Lima, R. H. Baughman and S. J. Kim, Nanoscale, 2016, 3248–3253 RSC.
- W. J. Zheng, N. An, J. H. Yang, J. Zhou and Y. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 1758–1764 CAS.
- Y. Zhu, N. A. Wood, K. Fok, T. Yoshizumi, D. W. Park, H. Jiang, D. S. Schwartzman, M. A. Zenati, T. Uchibori, W. R. Wagner and C. N. Riviere, Ann. Thorac. Surg., 2016, 102, 780–786 CrossRef PubMed.
- U. T. Hydrogel, M. Akashi, D. Ph, H. Yatani and D. Ph, Tissue Eng., Part A, 2010, 16, 2497–2504 CrossRef PubMed.
- X. Wang, Y. Sun, C. Peng, H. Luo, R. Wang and D. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 26131–26136 CAS.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 CAS.
- C. J. Wu, A. K. Gaharwar, B. K. Chan and G. Schmidt, Macromolecules, 2011, 44, 8215–8224 CrossRef CAS.
- M. Park, I. K. Shim, E. Y. Jung and J. H. Choy, J. Phys. Chem. Solids, 2004, 65, 499–501 CrossRef CAS.
- T. Ueki, Polym. J., 2014, 46, 1–10 CrossRef.
- T. Ueki, Y. Nakamura, A. Yamaguchi, K. Niitsuma, T. P. Lodge and M. Watanabe, Macromolecules, 2011, 44, 6908–6914 CrossRef CAS.
- R. Byrne, C. Ventura, F. Benito Lopez, A. Walther, A. Heise and D. Diamond, Biosens. Bioelectron., 2010, 26, 1392–1398 CrossRef CAS PubMed.
- M. W. Takeshi Ueki, Chem. Lett., 2006, 35, 964–965 CrossRef.
- C. Alvarez-Lorenzo, L. Bromberg and A. Concheiro, Photochem. Photobiol., 2009, 85, 848–860 CrossRef CAS PubMed.
- C.-W. Lo, D. Zhu and H. Jiang, Soft Matter, 2011, 7, 5604 RSC.
- L. Peng, M. You, Q. Yuan, C. Wu, D. Han, Y. Chen, Z. Zhong, J. Xue and W. Tan, J. Am. Chem. Soc., 2012, 134, 12302–12307 CrossRef CAS PubMed.
- B. Yan, J. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS PubMed.
- L. Li, B. Lu, Y. Zhang, X. Xing, X. Wu and Z. Liu, J. Polym. Res., 2015, 22, 176 CrossRef.
- Z. W. Low, P. L. Chee, D. Kai and X. J. Loh, RSC Adv., 2015, 5, 57678–57685 RSC.
- J. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. Nakajima, H. Furukawa, Y. Tanaka, T. Kurokawa, Y. Osada and J. P. Gong, Macromolecules, 2009, 42, 2184–2189 CrossRef CAS.
- X. C. Xiao, L. Y. Chu, W. M. Chen, S. Wang and Y. Li, Adv. Funct. Mater., 2003, 13, 847–852 CrossRef CAS.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef CAS.
- J. L. Gómez Ribelles, M. Monleón Pradas, J. M. Meseguer Dueas and C. Torregrosa Cabanilles, J. Non-Cryst. Solids, 2002, 307–310, 731–737 CrossRef.
- E. Bobo, Master thesis, University of Nebraska-Lincoln, 2013.
- H. Katono, A. Maruyama, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, J. Controlled Release, 1991, 16, 215–228 CrossRef CAS.
- Z. Zhang, T. Chao and S. Jiang, J. Phys. Chem. B, 2008, 112, 5327–5332 CrossRef CAS PubMed.
- R. E. Rivero, F. Alustiza, N. RodriGuez, P. Bosch, M. C. Miras, C. R. Rivarola and C. A. Barbero, React. Funct. Polym., 2015, 97, 77–85 CrossRef CAS.
- D. Myung, W. Koh, J. Ko, Y. Hu, M. Carrasco, J. Noolandi, C. N. Ta and C. W. Frank, Polymer, 2007, 48, 5376–5387 CrossRef CAS.
- Q. Wang, Y. Y. Zhang, X. Y. Dai, X. H. Shi and W. G. Liu, Sci. China: Technol. Sci., 2017, 60, 78–83 CrossRef CAS.
- A. A. M. Farag, Spectrochim. Acta, Part A, 2006, 65, 667–672 CrossRef CAS PubMed.
- L. Yuan and T. Kusuda, J. Appl. Polym. Sci., 2005, 96, 2367–2372 CrossRef CAS.
- J. Chen, S. Miao, J. Wan, J. Xia and X. Cao, Process Biochem., 2010, 45, 1928–1936 CrossRef CAS.
- L. Yuan, T. Kusuda and H. Satoh, J. Appl. Polym. Sci., 2007, 103, 681–686 CrossRef CAS.
- X. Zheng, Q. Zhang, J. Liu, Y. Pei and K. Tang, RSC Adv., 2016, 6, 71999–72007 RSC.
- Y.-C. Chiu, S. Kocagöz, J. C. Larson and E. M. Brey, PLoS One, 2013, 8, e60728 CAS.
- J. K. Carrow, P. Kerativitayanan, M. K. Jaiswal, G. Lokhande and A. K. Gaharwar, Polymers for bioprinting, Elsevier Inc., 2015 Search PubMed.
- W. Li, D. Wang, W. Yang and Y. Song, RSC Adv., 2016, 6, 20166–20172 RSC.
| This journal is © The Royal Society of Chemistry 2018 |