
Boosting pseudocapacitive charge storage in in situ functionalized carbons with a high surface area for high-energy asymmetric supercapacitors†
Hao
Zhang‡
a,
Mingjie
Lu‡
a,
Huanlei
Wang
 *a,
Yan
Lyu
a,
Dong
Li
a,
Shijiao
Sun
b,
Jing
Shi
a and
Wei
Liu
a
*a,
Yan
Lyu
a,
Dong
Li
a,
Shijiao
Sun
b,
Jing
Shi
a and
Wei
Liu
a
aSchool of Materials Science and Engineering, Ocean University of China, Qingdao 266100, People's Republic of China. E-mail: huanleiwang@ouc.edu.cn; huanleiwang@gmail.com
bCollege of Materials Science and Engineering, Nanjing Tech University, Nanjing 210009, People's Republic of China
First published on 8th August 2018
Abstract
Achieving both a high surface area and high heteroatom doping in carbon materials is a major challenge for aqueous-based supercapacitors. Herein, we choose an efficient carbonization–activation pathway to tune the porosity and heteroatom doping level of carbon materials by using jellyfish as the precursor and KOH as the activating agent. Highly Functionalized Jellyfish-Derived Activated Carbons (HFJDACs) possess a high surface area of up to 3300 m2 g−1 with nitrogen and oxygen heteroatom doping. Carbon activated at 500 °C displays a capacitance as high as 743 F g−1 and 639 F cm−3, while carbons activated at a temperature higher than 500 °C exhibit a favorable capacitance retention of >48.1% at 100 A g−1. These values are among the highest reported in the literature, and therefore these carbon materials can be used as the ideal negative electrode in asymmetric supercapacitors to circumvent the capacitance mismatch between oxide-based positive electrodes and carbon-based negative electrodes. The assembled asymmetric capacitor employing HFJDACs can achieve a high energy density of 43.4 W h kg−1 and an amazing cycle life with capacitance retention of 110% after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. These results demonstrate that adapting a scalable synthesis strategy for designing carbons with well-developed porosity and high level heteroatom doping is promising for advanced supercapacitors.
000 cycles. These results demonstrate that adapting a scalable synthesis strategy for designing carbons with well-developed porosity and high level heteroatom doping is promising for advanced supercapacitors.
Introduction
With the growth of the economy and the seriousness of environmental pollution, the demand for efficient, clean, and renewable energy storage has increased year by year.1–3 Supercapacitors, because of their high power density, high charge rate and good durability, have become very popular for their application in electric/hybrid vehicles, microgrids, and mobile electronic products.4–6 Supercapacitors can be divided into two categories, i.e. pseudocapacitors and electric double-layer capacitors. The former store charge through reversible surface/inside faradaic reactions on electrode materials (metal oxides are suitable) while the latter store charge based on reversible interfacial ion adsorption/desorption between the electrode and electrolyte (carbon materials are suitable).7 Nowadays, more and more occasions require supercapacitors to provide high energy density and superior cycle performance, which poses a great challenge for designing and preparing electrode materials to enhance the energy density while maintaining high power density.8 Traditional activated carbon has been commercialized for supercapacitors for a long time; however, activated carbons show low gravimetric capacitance (<300 F g−1 in aqueous electrolytes with 100–120 F g−1 in organic electrolytes) and inferior rate capability owing to the restricted ion transport kinetics by torturous pore channels.9–12 To enhance the pore access, a high surface area is required for improving the electrochemical kinetics.13 Besides, the capacitance of carbon materials is much lower than that of metal oxide materials. Therefore, finding an effective strategy to boost the charge storage of carbon materials is extremely desirable.In recent years, heteroatom-doped carbons have drawn much more interest as supercapacitor electrodes, since the introduction of heteroatoms can improve the conductivity, enhance the surface wettability, and provide additional pseudocapacitance.14–16 To date, the investigation of the roles of N/O dopants in carbons has been widely carried out, and different N/O configurations have diverse effects upon the performance of carbons. Taking N-containing functionalities as an example, pyridinic-N and pyrrolic-N can strengthen the surface-induced capacitive behavior and boost the diffusion of reactive electrolyte ions in carbon materials, while quaternary-N and pyridine-N-oxides with positive charge can ensure fast electron transfer to enhance the electroconductivity.17,18 However, the roles of different N/O configurations in different electrolytes are still debatable and unclear. There are two main strategies for obtaining heteroatom-doped carbon materials. The first is called the “heteroatom-introducing” strategy, which can be achieved by the heat-treatment of carbon precursors with heteroatom-containing gases or solid powders, such as ammonia, urea, and melamine for nitrogen-doped carbons.19–24 However, the introduction of heteroatoms in this way may lead to the destruction of the pore structure and low doping level. In particular, heteroatoms usually decorated on the surface rather than in the bulk of carbon materials and their corresponding surface functionalities may be decomposed during electrochemical processes, resulting in the decayed capacitance and poor cycling stability.25 The second is called the “heteroatom self-doping” strategy, which can be favorably achieved by the carbonization of heteroatom-containing precursors under an inert atmosphere. Since the heteroatoms are inherited from the precursors, the generated functional groups can be homogeneously distributed on the surface and inside the carbon matrix.16,26 This approach can lead to more stable energy storage.
Up to now, biomass, due to its advantages of low cost, sustainability, abundant resources, and versatility, has become strikingly attractive as a promising precursor for synthesizing carbon materials.12,27 Great efforts have been made in this field, and diverse precursors, such as bean shells,28 human hair,29 corn husks,12 eggshell membranes,30 and lignosulphonate-cellulose,31 have been utilized for carbon production. Among them, the capacitance ranges from 202 to 356 F g−1 in aqueous electrolytes, which is quite higher than that of commercial activated carbons. Selecting a precursor with abundant heteroatoms is beneficial for achieving carbons with developed porosity and functional groups by using the “heteroatom self-doping” strategy. Herein, we select jellyfish as a model precursor to develop carbons with an ultrahigh surface area and controllable heteroatom doping. As a new member of animal precursors, jellyfish contains rich proteins, minerals, and relatively low fats,32 and the main amino acids that make up the proteins are glycine, glutamate and proline. Therefore, the rich heteroatoms from the mentioned amino acids can be favorably inherited after carbonization, leading to self-doped carbon architectures. As illustrated in Scheme 1, the cleaned jellyfish was first pre-carbonized at 600 °C in N2. Then, the pre-carbonized product was uniformly mixed with KOH at a weight ratio of 1:3 for further activation at 500–800 °C. Finally, nitrogen- and oxygen-doped carbons (nitrogen: 0.65–6.26 at% and oxygen: 6.00–14.23 at%) with a high specific surface area up to 3291 m2 g−1 can be obtained. The carbon structure and the content of heteroatoms are adjusted by changing the activation temperature. Electrochemical evaluation indicates that the jellyfish-derived carbons exhibit superior capacitance, outstanding rate capability, and a long cycling life, making them ideal negative electrode materials for asymmetric supercapacitors in aqueous electrolytes. This work gives a case study to prepare heteroatom-doped porous carbons for high-performance energy storage devices by boosting pseudocapacitive charge storage.
 | ||
| Scheme 1 Schematic illustration of the synthesis process for the jellyfish derived carbons. | ||
Experimental
Synthesis of activated jellyfish-derived carbon
The salted jellyfish in vacuum packing was purchased from supermarket, which was collected from the Bohai Sea and processed by Penglailuning Seafood Co., Ltd (Yantai, China). The as-purchased salted jellyfish was thoroughly washed and dried before use. All experiments were performed in compliance with the Ocean University of China's policy on animal use and ethics. For direct carbonization, the dried jellyfish was annealed at 600 °C (heating rate: 3 °C min−1) for 2 h in N2. For the activation process, the pre-carbonized carbon was uniformly mixed with KOH at a weight ratio of 1:3 and then activated at 500–800 °C (heating rate: 3 °C min−1) for 2 h in N2. Finally, the obtained samples were repeatedly washed with 2 M HCl and distilled water until neutral and were then dried at 80 °C for 24 h in a vacuum oven. The resultant Highly Functionalized Jellyfish-Derived Activated Carbons are denoted as HFJDAC-T, where T refers to the activation temperature. For comparison, the non-activated Pre-carbonized Carbon is denoted as PC.
Synthesis of nickel cobaltite–graphene (NiCo2O4/GR) as the positive electrode
Nickel cobaltite–graphene was synthesized by following a previously reported process.33 Briefly, a mixture containing Ni(NO3)2·6H2O, Co(NO3)2·6H2O, urea ((urea/Ni2+/Co2+) = 30:1:2, by molar ratio), and graphene oxide (0.2 g, dispersed in 100 mL distilled water) was stirred for 20 min and then transferred into an autoclave, which was kept at 180 °C for 24 h. The resulting product was dried, collected, and calcined at 350 °C for 2 h in air.
Material characterization
For morphological and microstructural characterization, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed by using a Hitachi-4800 instrument and JEOL 2010 microscope equipped with an energy-dispersive X-ray (EDX) analyzer. The porous texture of the samples was determined by nitrogen adsorption–desorption at 77 K using a Micromeritics 3Flex™ surface characterization analyzer. Powder X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance powder diffractometer with monochromatized Cu Kα radiation. Raman spectra were recorded on a LabRAM HR800 spectroscope with a laser wavelength of 532 nm. Elemental characterization was conducted by X-ray photoelectron spectroscopy (XPS) using a multifunctional imaging electron spectrometer (Thermo ESCALAB 250XI).Electrochemical measurements
A slurry of an 80 wt% active material, 10 wt% conducting carbon (Super-P), and a 10 wt% polyvinylidene fluoride binder (PVDF) in ethanol was pressed onto nickel foam at a pressure of 10 MPa (the mass loading is about 2.0 mg cm−2) and then dried at 120 °C for 12 h. Electrochemical performance (including cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) cycling, and electrochemical impedance spectra (EIS) analysis) was evaluated using a Gamry Interface 1000 workstation in 2 M KOH solution at room temperature.For the three-electrode system, platinum foil and a Hg/HgO electrode were used as the counter and the reference electrode, respectively. The specific capacitance of the electrode (Cs) is calculated based on iΔt/mΔV, where i (A) is the constant discharge current, Δt (s) is the discharge time, m (g) is the weight of active carbon in the working electrode, and ΔV (V) is the potential window excluding the IR drop.
For the two-electrode system, an asymmetric supercapacitor device was assembled with the nickel cobaltite–graphene positive electrode and HFJDAC-600 negative electrode. The specific capacitance (Cd) is calculated from the discharge curves based on the equation Cd = iΔt/MΔV, where i (A) is the constant discharge current, Δt (s) is the discharge time, M (g) is the total mass of the positive and negative materials within the electrodes, and ΔV (V) is the potential window excluding the IR drop. The energy density E (W h kg−1) and power density P (W kg−1) of the asymmetric supercapacitor based on the mass of active materials were figured out by employing the following equations E = CdΔV2/2 and P = E/t, where t (h) represents the discharge time and ΔV (V) represents the cell potential excluding the ohmic drop.
Results and discussion
Morphology, structure and chemistry of HFJDACs
SEM analysis was used to examine the morphological evolution of jellyfish-derived carbons (Fig. 1 and S1†). All the samples are irregular monoliths on the micrometer scale. Activation at 500 °C gives rise to a relatively smooth surface for HFJDAC-500. As the activation temperature is increased to above 600 °C, a 3D honeycomb-like framework with a macroporous structure can be observed. TEM characterization was also carried out to obtain a more detailed investigation of the microstructure of HFJDACs. As seen from Fig. 2, the observed interconnected porous network consists of randomly distributed micropores and mesopores. It is visible that the graphitic ribbons become more and more apparent from HFJDAC-500 to HFJDAC-800, indicating the temperature-induced graphitization effect. Besides, the distortion degree of lattice fringes and the amount of structural defects tend to rise with the increase of activation temperature, suggesting the activation-induced structural disorder. In a word, the apparent interconnected hierarchical porous structure of HFJDACs can be beneficial for offering a large surface area (micropores), shortening the ion transport distance (mesopores), and serving as ion-buffering reservoirs (macropores),34,35 and the highly disordered structure with abundant defects of HFJDACs can introduce more active sites for ion adsorption. | ||
| Fig. 1 SEM images of (a) HFJDAC-500, (b) HFJDAC-600, (c) HFJDAC-700, and (d) HFJDAC-800. | ||
 | ||
| Fig. 2 TEM and high-resolution TEM micrographs of (a) HFJDAC-500, (b) HFJDAC-600, (c) HFJDAC-700, and (d) HFJDAC-800. (e) The energy-filtered TEM image of HFJDAC-600 and the corresponding elemental mapping images of (f) C, (g) N and (h) O atoms. | ||
The crystalline structure of jellyfish-derived carbons was examined by using XRD and Raman analysis. The XRD patterns of HFJDACs comprise two peaks centered at around 2θ = 24–25° and 2θ = 43–44° (Fig. 3a), representing the (002) and (100) planes of graphite. These two weak and broad peaks suggest that HFJDACs are X-ray amorphous. The interlayer spacing of graphitic layers for HFJDACs calculated based on the 2θ degree of the (002) peak increased with the rise of activation temperature, indicating the intercalation of potassium species during the activation process.36 The amorphous structure of HFJDACs was further investigated by Raman analysis (Fig. 3b and S2†). All HFJDACs exhibit two distinctive bands, positioned at ∼1350 and ∼1600 cm−1, which correspond to the D-band (defects and disorder) and G-band (graphitic). It is worth noting that the calculated values of ID/IG have obviously risen with the increase of activation temperature, indicating that the activation-induced structural disorder is dominant, which is consistent with the results of TEM characterization.25,37
 | ||
| Fig. 3 (a) XRD patterns of the HFJDAC samples. (b) Raman spectra of HFJDAC samples. (c) Nitrogen sorption analysis of the obtained PC and HFJDAC samples. (d) Pore size distribution calculated by using the DFT method. | ||
To further explore the porous texture of the HFJDAC samples, nitrogen adsorption–desorption measurements were employed, and the relevant textural parameters are summarized in Table 1. As shown in Fig. 3c, all the samples exhibit type I/IV isotherms with a specific surface area of <10, 505, 3289, 3291 and 2866 m2 g−1 for PC, HFJDAC-500, HFJDAC-600, HFJDAC-700 and HFJDAC-800, respectively, as calculated by the Brunauer–Emmett–Teller (BET) method. This clearly shows that the specific surface area and pore volume are significantly improved after activation, and the activation effect becomes more obvious when the activation temperature is higher than 500 °C. On activating at 800 °C, the total pore volume and specific surface area slightly decreased, which can be ascribed to the enhanced etching effect by KOH. In the low relative pressure range (P/Po < 0.01), the sharp adsorption indicates the existence of abundant micropores. An increasing uptake in the higher relative pressure range (0.01 < P/Po < 0.4) indicates the development of mesoporosity. Moreover, a slight rise in the pressure range from 0.95 to 1.0 (P/Po) suggests the existence of macropores. It can be concluded that HFJDACs display hierarchical porous structures with combined micropores, mesopores, and macropores. The pore size distributions calculated using the DFT method are shown in Fig. 3d, which can further confirm the presence of hierarchical porous structures. Such a high surface area and distinct characteristics of the porous structure of HFJDACs are beneficial for charge accumulation and ion diffusion kinetics.17,38
| Sample | S BET (m2 g−1) | V t (cm3 g−1) | ρ (g cm−3) | Pore volume (%) | d 002 (nm) | I D/IG | XPS composition (at%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V <2 nm | V >2 nm | C | N | O | S | ||||||
| a Surface area was calculated with the Brunauer–Emmett–Teller (BET) method. b The total pore volume was determined by the density functional theory (DFT) method. c ρ represents the packing density. | |||||||||||
| PC | <10 | 0.012 | 1.16 | 13.33 | 86.67 | — | — | 76.88 | 13.30 | 9.37 | 0.45 |
| HFJDAC-500 | 505 | 0.370 | 0.86 | 41.79 | 58.21 | 0.349 | 2.41 | 79.51 | 6.26 | 14.23 | — |
| HFJDAC-600 | 3289 | 1.565 | 0.67 | 69.99 | 30.01 | 0.351 | 3.68 | 89.11 | 3.54 | 7.35 | — |
| HFJDAC-700 | 3291 | 1.562 | 0.60 | 62.75 | 37.25 | 0.356 | 4.13 | 88.94 | 1.83 | 9.23 | — |
| HFJDAC-800 | 2866 | 1.257 | 0.73 | 77.37 | 22.63 | 0.358 | 4.62 | 93.35 | 0.65 | 6.00 | — |
Benefiting from the high intrinsic heteroatom content of jellyfish, the as-obtained PC and HFJDACs possess very high heteroatom-doping levels. The energy-filtered TEM image and the corresponding elemental mapping images of HFJDAC-600 manifest the homogeneous distribution of N and O atoms in the carbon framework (Fig. 2e–h). Besides, the XPS analysis confirms the coexistence of C, N and O in the HFJDAC samples (Fig. 4a), and the corresponding test results are given in Tables 1 and S1.† As shown in Fig. 4b and S3,† the high-resolution C 1s peak can be divided into four peaks with binding energies of 284.6 eV, 285–286 eV, 286–287 eV and 289–290 eV corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C, C–N/C–O, CO, and –COOH, respectively.2 For the PC sample, the nitrogen content is as high as 13.30 at% with a small amount of sulfur (0.45 at%). Remarkably, the N-content of HFJDACs is very susceptible to the activation temperature (Fig. 4c), decreasing from 6.26 at% for HFJDAC-500 to 3.54 at% for HFJDAC-600, 1.83 at% for HFJDAC-700, and 0.65 at% for HFJDAC-800, which suggests the decomposition and reconstruction of N-dopants.14 In the N 1s spectra (Fig. 4d and S4†), it can be found that the component of the N-dopants can be favorably tailored by the activation temperature, and the corresponding change of the relative content can be seen in Fig. 4e. The N configurations can be divided into pyridinic-N (N-6), pyrrolic-N (N-5), quaternary-N (N-Q) and pyridine-N-oxide (N-X).30,39 When the activation temperature increases, N-6 and N-X gradually disappeared, while the relative amount of N-5 and N-Q basically increased, indicating that N-6 and N-X functional groups are preferentially eliminated during the activation process. The high-resolution O 1s spectra of all the samples can be deconvoluted into three components (Fig. 4f and S5†), which are ascribed to –CO (531–532 eV, O-I, up to 84.56%), –C–OH/–C–O–C (532–533 eV, O-II, up to 67.57%), and –COOH (535–536 eV, O-III, up to 14.70%).40 These oxygen functionalities can further ameliorate the wettability of carbons to decrease the inert surface area and increase the amount of active sites, resulting in the enhancement of overall capacitance by pseudocapacitance contribution.17,41 In addition, the PC sample has a small amount of sulfur element (0.45 at%), but there is no detectable sulfur in HFJDACs, probably due to the gasification of sulfur species.
C/C–C, C–N/C–O, CO, and –COOH, respectively.2 For the PC sample, the nitrogen content is as high as 13.30 at% with a small amount of sulfur (0.45 at%). Remarkably, the N-content of HFJDACs is very susceptible to the activation temperature (Fig. 4c), decreasing from 6.26 at% for HFJDAC-500 to 3.54 at% for HFJDAC-600, 1.83 at% for HFJDAC-700, and 0.65 at% for HFJDAC-800, which suggests the decomposition and reconstruction of N-dopants.14 In the N 1s spectra (Fig. 4d and S4†), it can be found that the component of the N-dopants can be favorably tailored by the activation temperature, and the corresponding change of the relative content can be seen in Fig. 4e. The N configurations can be divided into pyridinic-N (N-6), pyrrolic-N (N-5), quaternary-N (N-Q) and pyridine-N-oxide (N-X).30,39 When the activation temperature increases, N-6 and N-X gradually disappeared, while the relative amount of N-5 and N-Q basically increased, indicating that N-6 and N-X functional groups are preferentially eliminated during the activation process. The high-resolution O 1s spectra of all the samples can be deconvoluted into three components (Fig. 4f and S5†), which are ascribed to –CO (531–532 eV, O-I, up to 84.56%), –C–OH/–C–O–C (532–533 eV, O-II, up to 67.57%), and –COOH (535–536 eV, O-III, up to 14.70%).40 These oxygen functionalities can further ameliorate the wettability of carbons to decrease the inert surface area and increase the amount of active sites, resulting in the enhancement of overall capacitance by pseudocapacitance contribution.17,41 In addition, the PC sample has a small amount of sulfur element (0.45 at%), but there is no detectable sulfur in HFJDACs, probably due to the gasification of sulfur species.
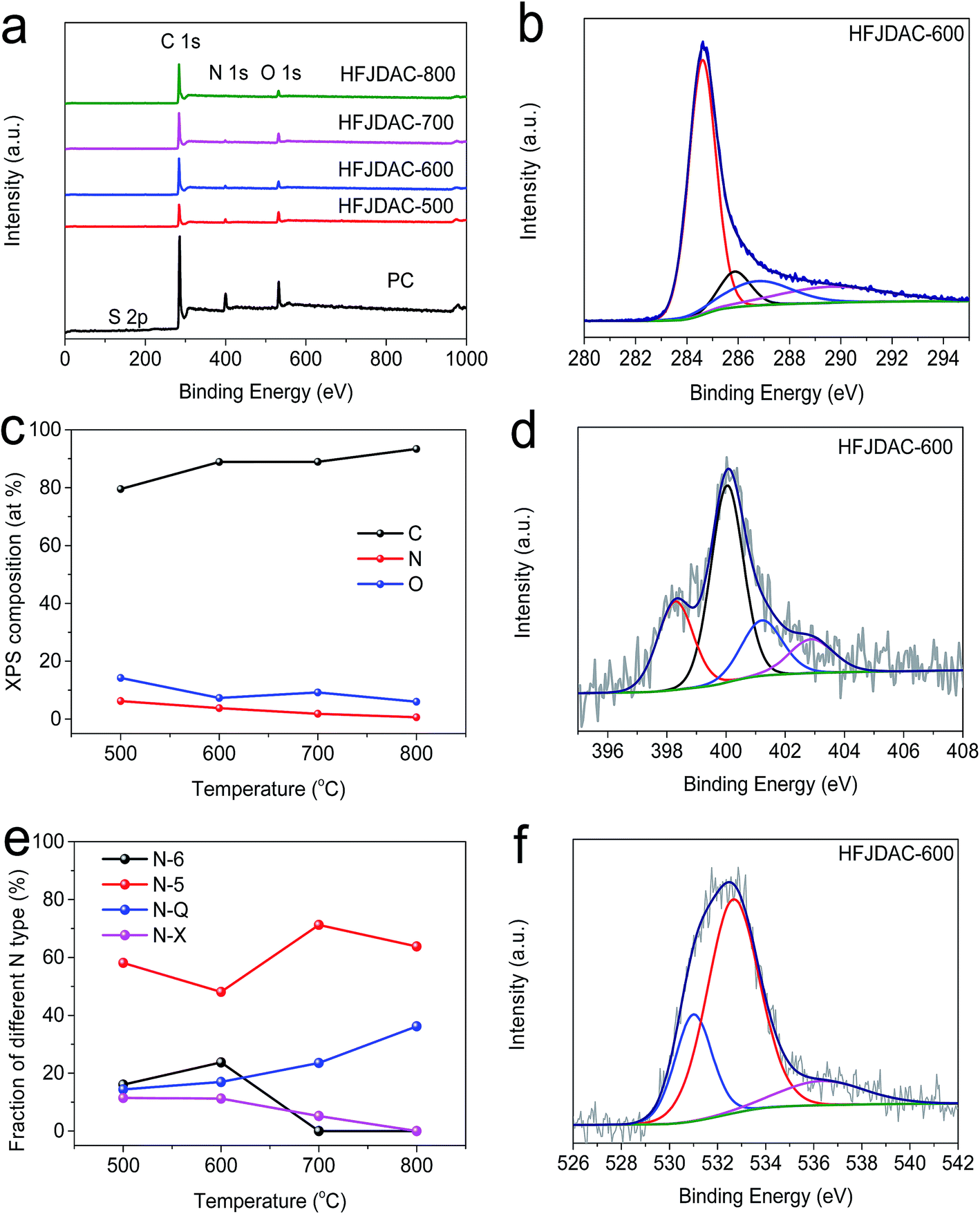 | ||
| Fig. 4 (a) XPS survey spectra of the PC and HFJDAC samples. (b) C 1s XPS spectra of the HFJDAC-600 sample. (c) XPS composition of the HFJDAC samples at various temperatures. (d) N 1s XPS spectra of the HFJDAC-600 sample. (e) Ratios of the different N types in the HFJDAC samples at various temperatures. (f) O 1s XPS spectra of the HFJDAC-600 sample. | ||
Electrochemical behavior of HFJDAC electrodes in a three-electrode setup
Benefiting from the high surface area, enriched heteroatoms (i.e. N and O), and combined macro/meso/microporous structure, HFJDACs could demonstrate excellent performance as electrodes for supercapacitors in alkaline electrolytes. Fig. 5a shows the CV curves of PC and HFJDACs collected at 10 mV s−1, and Fig. S6† exhibits the CV curves scanned from 5 to 200 mV s−1. It can be seen that the CV curves show a notable hump from −0.8 to −0.2 V. These distinct peaks offer a primary contribution of pseudocapacitance by surface redox reactions between nitrogenated/oxygenated functionalities and electrolyte ions.26 HFJDAC-500 demonstrates the highest distorted CV curve, linked to its low surface area (505 m2 g−1), high N-content (6.26 at%) and high O-content (14.23 at%). With the decrease of nitrogen amount, the shape of the CV curve becomes more and more rectangular, indicating the reduction of pseudocapacitance and the increase of electrical double-layer capacitance (EDLC). The integrated area of the CV curves of HFJDACs is much larger than that of PC, suggesting the significantly improved capacitance after KOH activation. As seen from Fig. 5b and S7,† the galvanostatic charge–discharge profiles of PC and HFJDACs show symmetrical and non-linear lines, which can prove the presence of both electrical double-layer capacitance and faradaic pseudocapacitance.42 Meanwhile, the IR drop for the discharge process is negligible, suggesting the excellent electrical conductivity of HFJDACs.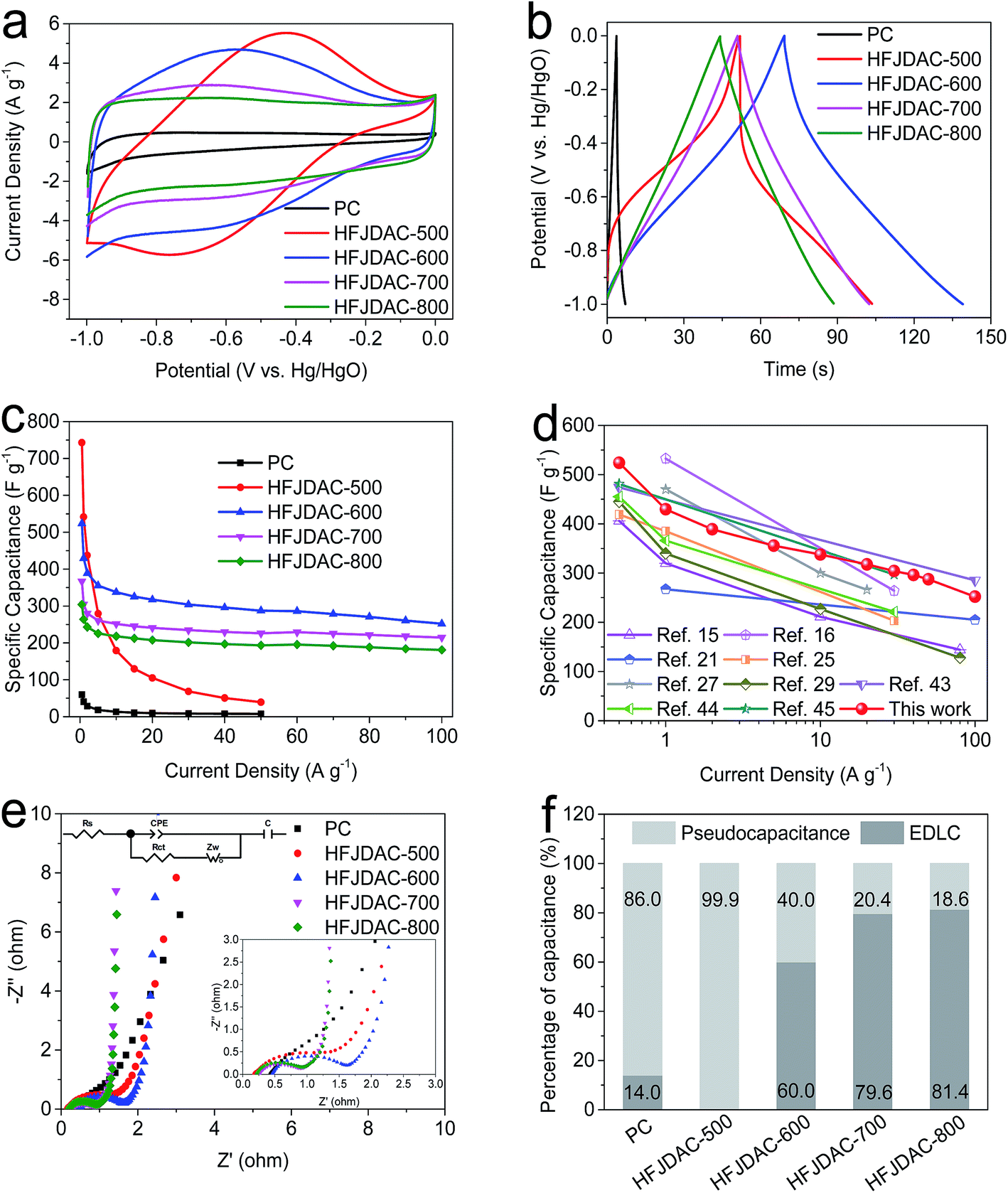 | ||
| Fig. 5 Electrochemical properties of the PC and HFJDAC samples in a three-electrode system tested in 2 M KOH: (a) CV curves at a scan rate of 10 mV s−1. (b) Galvanostatic charge–discharge curves at 5 A g−1. (c) Specific capacitances (F g−1) at different current densities. (d) Comparison of the rate capability between the HFJDAC-600 and state-of-the-art carbons reported in the literature. (e) Nyquist plots of the HFJDAC electrodes. (f) Percentage of the capacitance contribution evaluated for all the electrodes based on Trasatti analysis. | ||
Fig. 5c and S8† summarize the relationship between the gravimetric capacitances and the current densities (0.5–100 A g−1). The gravimetric capacitance is 743 F g−1 (196 mA h g−1), 524 F g−1 (144 mA h g−1), 367 F g−1 (102 mA h g−1), 304 F g−1 (84 mA h g−1), and 60 F g−1 (17 mA h g−1) at 0.5 A g−1 for HFJDAC-500, HFJDAC-600, HFJDAC-700, HFJDAC-800 and PC, respectively. The surface area-normalized capacitances are calculated to be 147.1, 15.9, 11.2, 10.6, and 1000.0 μF cm−2 for HFJDAC-500, HFJDAC-600, HFJDAC-700, HFJDAC-800, and PC, which are quite higher than 8.1 μF cm−2 for commercial activated carbon,43 confirming the contribution from pseudocapacitance. Besides, the measured packing densities of HFJDAC-500, HFJDAC-600, HFJDAC-700, HFJDAC-800, and PC are 0.86, 0.67, 0.60, 0.73, and 1.16 g cm−3. Consequently, the corresponding volumetric capacitance is 639, 351, 220, 222, and 70 F cm−3 for HFJDAC-500, HFJDAC-600, HFJDAC-700, HFJDAC-800, and PC, respectively. Although HFJDAC-500 shows the highest gravimetric and volumetric capacitances at low current density, HFJDAC-500 exhibits much lower capacitances of 39 F g−1 and 33.5 F cm−3 at 50 A g−1 (capacitance retention ratio is only 5.2%), indicating extremely poor rate capability. At 100 A g−1, carbons activated at temperatures higher than 500 °C offer a favorable capacitance retention ratio of 48.1–59.5%, manifesting that the high surface area and hierarchical porous structure can have a positive effect on rate capability. As shown in Table S2† and shown in Fig. 5d, it is noteworthy that the capacitance of HFJDAC-600 is quite competitive as compared with that of the state-of-art of reported carbons.15,16,21,25,27,29,43–45 Another significant performance metric is to characterize the relationship between gravimetric capacitance and the mass loading of active materials. As shown in Fig. S9,† when the mass loading rises from 2 mg cm−2 to 10 mg cm−2, HFJDAC-600 maintains a high capacitance of 382 F g−1 at 0.5 A g−1. As expected, the gravimetric capacitance gradually reduces as the mass loading increases, due to the enhanced ion and electron transport resistance/distance.46 A long-term cycling test was conducted to study the cycling stability of HFJDAC-600 (Fig. S10†). There is only 12% capacitance loss after 10000 cycles, indicating that the “heteroatom-self doping” strategy is more effective for stabilizing the functionalized carbons than the “heteroatom-introducing” strategy.
The EIS technique was applied to understand the electrochemical reaction process of the PC and HFJDAC electrodes (Fig. 5e). The equivalent series resistance (Rs) ranges from 0.17 to 0.45 Ω based on the intercept on the x-axis, which can explain the favorable conductivity in alkaline solutions.47 Moreover, the charge-transfer resistance (Rct) is 3.20 Ω for PC, 1.75 Ω for HFJDAC-500, 1.23 Ω for HFJDAC-600, 0.67 Ω for HFJDAC-700, and 0.72 Ω for HFJDAC-800. Among them, the PC and HFJDAC-500 show relatively high Rct values, further indicating that the low surface area and high content of N and O doping can impede ion diffusion.21 The Warburg diffusion line with an ∼45° slope illustrates the synergy of resistance and capacitance behaviors of the electrolyte-ions percolating into the carbon pores. The almost 90° line along the y-axis in the low frequency range for HFJDAC-700 and HFJDAC-800 represents that the electrical double-layer behavior is dominant,25,48 while the deviated vertical line for PC, HFJDAC-500, and HFJDAC-600 indicates the leading role of pesudocapacitive charge storage.
As shown in Fig. 5f and S11,† Trasatti method analysis was used to analyze the capacitance configuration of all the samples.49 It can be seen that these five samples can be divided into three “bins”: (PC and HFJDAC-500), HFJDAC-600, and (HFJDAC-700 and HFJDAC-800). The capacitance of PC and HFJDAC-500 mainly comes from pseudocapacitance. For HFJDAC-600, both EDLC and pseudocapacitance are equally important. The EDLC contribution becomes dominant in HFJDAC-700 and HFJDAC-800. With the activation temperature increasing from 500 °C to 800 °C, the pseudocapacitance contribution dramatically decreased owing to the largely decreased nitrogen species. The electrical double layer formed by ion adsorption on the interface between the electrode and electrolyte generally shows relatively faster kinetics than that formed by the pseudocapacitive reactions through heteroatom functional groups.50 Therefore, the more the pseudocapacitance contribution, the more capacitance loss will be produced at high current densities, which can explain the poor rate capability of HFJDAC-500. The optimized electrochemical performance of HFJDAC-600 indicates that the combined effects of the surface area, porous structure and heteroatom doping can affect the capacitance and rate capacity. It can be concluded that high heteroatom doping can boost pseudocapacitive charge storage of carbonaceous materials, while a high surface area with a hierarchical porous structure can ensure high electrical double-layer capacitance and smooth ion diffusion.
Electrochemical behavior of the asymmetric supercapacitor
Taking into account the win–win situation, the basic charge storage mechanisms behind carbon negative electrodes and metal oxide positive electrodes can cooperate to achieve high energy density and power density which are highly desired.51 Since extensive investigations have been conducted on battery-type faradaic electrodes (metal oxide electrodes) for enhancing the capacitive performance in asymmetric supercapacitors, it would be quite urgent to improve the properties of the capacitor-type electrode (carbon electrode) to match. Therefore, to meet the above purpose, HFJDAC-600 was selected as the negative electrode due to its high capacitance and excellent rate performance. For assessing the performance of HFJDAC-600, a NiCo2O4/graphene nanocomposite was used as the positive electrode. The NiCo2O4/GR possesses a mesoporous structure and high surface area of 115 m2 g−1, which are favorable for ion transport and charge accumulation. As shown in Fig. S12a and b,† the NiCo2O4/GR consists of rod-like nickel cobaltite (intimately anchored on the graphene surface) with a nanocrystallite size of around 10 nm. The NiCo2O4/GR shows high capacitances ranging from 821 F g−1 to 253 F g−1 at the current densities from 0.25 to 20 A g−1 (Fig. S13†). The charge storage mechanism of NiCo2O4/GR is based on near-surface redox reactions related to Ni2+/Ni3+ and Co2+/Co3+. Besides, the XRD pattern of the NiCo2O4/GR is seen in Fig. S12c,† with extra microstructural details being offered in our earlier report.33In the current study, an asymmetric supercapacitor based on the HFJDAC-600 negative electrode and NiCo2O4/GR positive electrode has been fabricated in 2 M KOH (Fig. 6a). According to the voltage window of HFJDAC-600 (−1 to 0 V) and NiCo2O4/GR (0–0.5 V), the operating cell voltage could be expanded to 1.5 V for the NiCo2O4/GR//HFJDAC-600 asymmetric supercapacitor. To balance the stored charge (q) of NiCo2O4/GR and HFJDAC-600, the mass ratio between the positive (m+) and negative (m−) electrodes can be calculated as m+/m− = (c− × ΔV−)/(c+ × ΔV+) (where c−/c+ represents the capacitance of the negative/positive electrode and ΔV−/ΔV+ represents the potential window of the negative/positive electrode during charge/discharge).52 Taking the capacitance at 2 A g−1 as an example, the mass ratio of NiCo2O4/GR to HFJDAC-600 is calculated to be about 1.1. For proving the superiority of the HFJDAC electrode, commercial activated carbon (called AC, Norit™) was also selected to assemble an asymmetric supercapacitor. Based on the capacitance of AC (Fig. S14†), the mass ratio of NiCo2O4/GR to AC is calculated to be about 0.5.
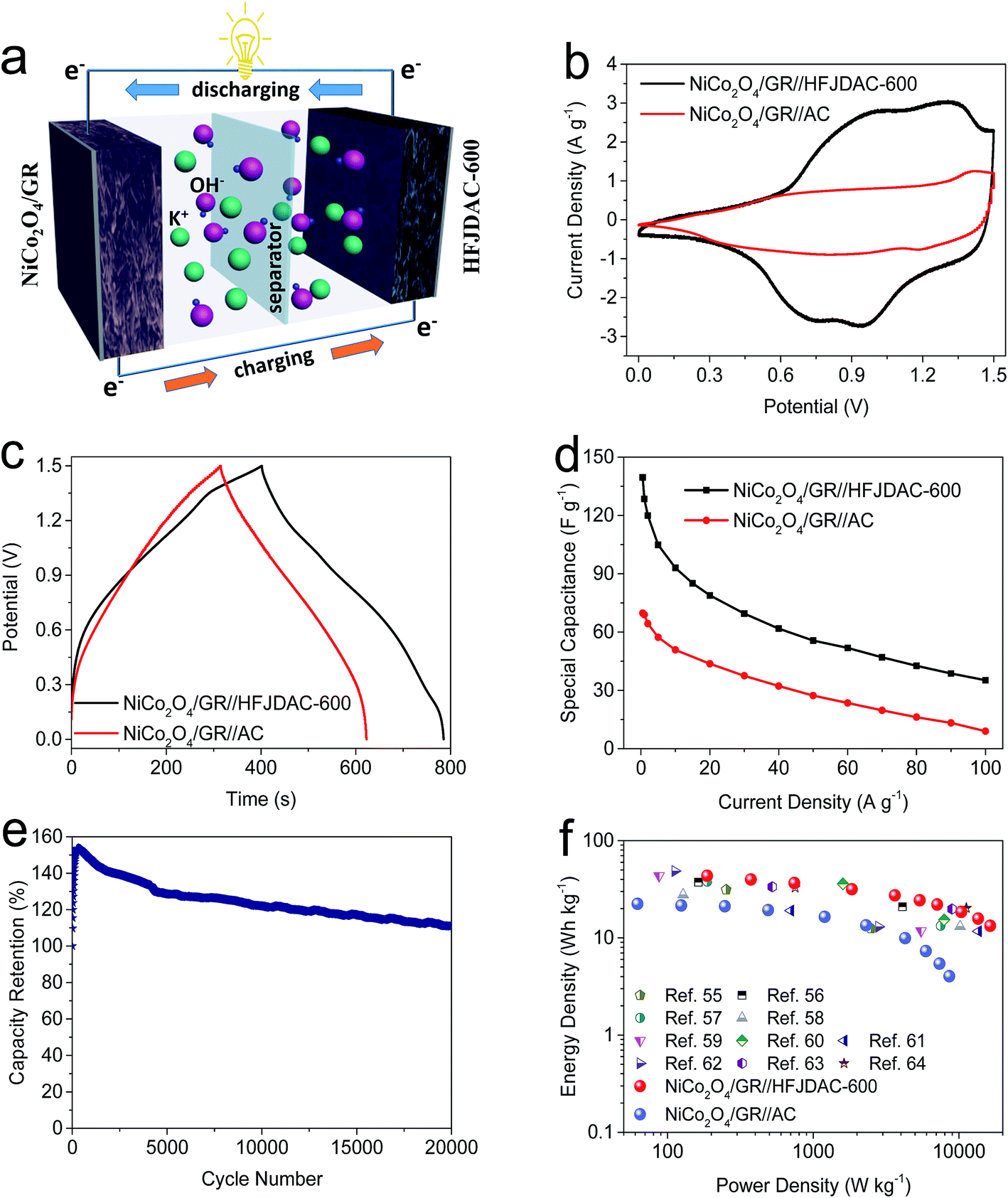 | ||
| Fig. 6 Electrochemical performance of asymmetric supercapacitors tested in 2 M KOH: (a) schematic illustration. (b) CV curves at 10 mV s−1. (c) Galvanostatic charge–discharge curves at 1 A g−1. (d) Specific capacitances at different current densities. (e) Cycling stability of NiCo2O4/GR//HFJDAC-600 tested at 20 A g−1 for 20000 cycles. (f) Comparison of energy-power density among the NiCo2O4/GR//HFJDAC-600 asymmetric supercapacitor, NiCo2O4/GR//AC asymmetric supercapacitor, and reported nickel/cobalt based asymmetric cells. | ||
The CV curves (Fig. 6b) of the two asymmetric capacitors at 10 mV s−1 are highly distorted (the prominent hump overlaid on top of the rectangular shape), suggesting the coexistence of EDLC and pseudocapacitance.53 Likewise, the coexistence of EDLC and pseudocapacitance can be confirmed from the nonlinear galvanostatic charge–discharge curves shown in Fig. 6c. The nearly symmetric shape also reveals excellent coulombic efficiency. The rate capability of the two asymmetric capacitors is further evaluated from 0.5 to 100 A g−1 (Fig. 6d). And the capacitance of the NiCo2O4/GR//HFJDAC-600 asymmetric cell is 140 F g−1 at 0.5 A g−1, while the capacitance reduces to 69 F g−1 for the NiCo2O4/GR//AC cell. At 100 A g−1, the capacitance of the NiCo2O4/GR//HFJDAC-600 cell is 35 F g−1, showing a capacitance retention ratio of 25%. However, the capacitance of the NiCo2O4/GR//AC cell is only 9 F g−1 (capacitance retention ratio: 13%). This phenomenon is caused by the excellent electrochemical performance of HFJDAC-600 and the total active mass reduction in the NiCo2O4/GR//HFJDAC-600 cell. Fig. 6e shows the cycling performance of the NiCo2O4/GR//HFJDAC-600 asymmetric cell at 20 A g−1 over 20000 cycles. The sharp increase of capacitance can be discovered during the first 300 cycles, which may be ascribed to the cycling-induced improvement in wettability (by heteroatoms) and the activation of electrodes (by the porous structure), leading to a more electroactive surface area.45,54,55 Then, the capacitance gradually decreases due to the pulverization of the electrodes and the wettability issues.8,33 After 20000 cycles, the capacitance still reaches 110% of its initial value, demonstrating an outstanding cycling stability. This result can also prove that the surface functionality in HFJDACs is pretty stable.
A comparison of energy-power densities among the NiCo2O4/GR//HFJDAC-600 asymmetric supercapacitor, NiCo2O4/GR//AC asymmetric supercapacitor and other reported nickel/cobalt based asymmetric cells is shown in Fig. 6f and Table S3.†55–64 At a power density of 187 W kg−1, the energy density of the NiCo2O4/GR//HFJDAC-600 cell can reach up to 43.4 W h kg−1, and a high energy density of 13.3 W h kg−1 can also be maintained at a power density of 16413 W kg−1. However, the maximal energy density for the NiCo2O4/GR//AC cell is only 22.4 W h kg−1. It is obvious that the electrochemical performance of the NiCo2O4/GR//HFJDAC-600 cell is competitive with previously reported state-of-the-art asymmetric devices.
Conclusions
To sum up, we have prepared jellyfish-derived N and O self-doped carbons, which possess a hierarchical architecture with pores of various sizes and an ultrahigh specific surface area of around 3300 m2 g−1, through a facile carbonization/activation approach. With the increasing activation temperature, the N-doping content gradually reduces, which causes a significant effect on determining the pseudocapacitance contribution in HFJDACs. Carbon activated at 500 °C with a high nitrogen content of 6.26 at% mainly displays pseudocapacitance generated by functional groups, resulting in a poor rate capability, while carbon activated at 700 and 800 °C with a low nitrogen content of 0.65–1.83 at% mainly shows electrical double-layer capacitance, leading to a relatively low specific capacitance. The optimized carbon activated at 600 °C with a moderate nitrogen content (3.54 at%) demonstrates a superior capacitance of 524 F g−1 at 0.5 A g−1 combined with a favorable capacitance retention ratio of 48.1% at 100 A g−1. By employing HFJDAC-600 as the negative electrode material in an asymmetric supercapacitor, a high specific energy of 43.4 W h kg−1 and an outstanding cycling performance with 110% capacitance retention over 20000 cycles can be achieved. These results indicate that carbons with a high surface area and hierarchical porous structure can boost pseudocapacitive charge storage in heteroatom self-doped structures. Moreover, functionalized carbons derived from biomass precursors can be extended to other energy storage systems for large-scale application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21471139 and 51602150), the Natural Science Foundation of Jiangsu Province (No. BK20161005), and the Fundamental Research Funds for the Central Universities (No. 201822008).Notes and references
- L.-F. Chen, Y. Feng, H.-W. Liang, Z.-Y. Wu and S.-H. Yu, Adv. Energy Mater., 2017, 7, 1700826 CrossRef.
- C. Wang, Y. V. Kaneti, Y. Bando, J. Lin, C. Liu, J. Li and Y. Yamauchi, Mater. Horiz., 2018, 5, 394–407 RSC.
- C. Li, J. Balamurugan, N. H. Kim and J. H. Lee, Adv. Energy Mater., 2018, 8, 1702014 CrossRef.
- R. R. Rosas, I. Fuentes, C. Vinas, F. Teixidor, E. Morallon and D. C. Amoros, Sustainable Energy Fuels, 2018, 2, 345–352 RSC.
- B. Zhao, L. Zhang, Q. Zhang, D. Chen, Y. Cheng, X. Deng, Y. Chen, R. Murphy, X. Xiong, B. Song, C.-P. Wong, M.-S. Wang and M. Liu, Adv. Energy Mater., 2018, 8, 1702247 CrossRef.
- J. Wu, Q. Zhang, J. Wang, X. Huang and H. Bai, Energy Environ. Sci., 2018, 11, 1280–1286 RSC.
- H. Duan, T. Yan, Z. Li, G. Chen, J. Zhang, L. Shi and D. Zhang, Sustainable Energy Fuels, 2017, 1, 1557–2156 RSC.
- H. Wang, C. M. B. Holt, Z. Li, X. Tan, B. S. Amirkhiz, Z. Xu, B. C. Olsen, T. Stephenson and D. Mitlin, Nano Res., 2012, 5, 605–617 CrossRef.
- B. Chang, W. Shi, S. Han, Y. Zhou, Y. Liu, S. Zhang and B. Yang, Chem. Eng. J., 2018, 350, 585–598 CrossRef.
- R. Thangavel, K. Kaliyappan, H. V. Ramasamy, X. Sun and Y.-S. Lee, ChemSusChem, 2017, 10, 2805–2815 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef PubMed.
- S. Song, F. Ma, G. Wu, D. Ma, W. Geng and J. Wan, J. Mater. Chem. A, 2015, 3, 18154–18162 RSC.
- J. Niu, R. Shao, J. Liang, M. Dou, Z. Li, Y. Huang and F. Wang, Nano Energy, 2017, 36, 322–330 CrossRef.
- S. Liu, J. Zhou and H. Song, Small, 2018, 14, 1703548 CrossRef PubMed.
- W. Qian, J. Zhu, Y. Zhang, X. Wu and F. Yan, Small, 2015, 11, 4959–4969 CrossRef PubMed.
- X. Wei, Y. Li and S. Gao, J. Mater. Chem. A, 2017, 5, 181–188 RSC.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef PubMed.
- D. H. Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef.
- B. Krüner, A. Schreiber, A. Tolosa, A. Quade, F. Badaczewski, T. Pfaff, B. M. Smarsly and V. Presser, Carbon, 2018, 132, 220–231 CrossRef.
- Z. Tang, Z. Pei, Z. Wang, H. Li, J. Zeng, Z. Ruan, Y. Huang, M. Zhu, Q. Xue, J. Yu and C. Zhi, Carbon, 2018, 130, 532–543 CrossRef.
- Y. Cui, H. Wang, X. Xu, Y. Lv, J. Shi, W. Liu, S. Chen and X. Wang, Sustainable Energy Fuels, 2018, 2, 381–391 RSC.
- C. Schneidermann, N. Jäckel, S. Oswald, L. Giebeler, V. Presser and L. Borchardt, ChemSusChem, 2017, 10, 2416–2424 CrossRef PubMed.
- H. Liu, L. Shi, P. Han, S. Ullah, J. Yu, B. Yang, C. Li, C. Zhu and J. Xu, Chem. Eng. J., 2018, 346, 143–150 CrossRef.
- S. Zhang, L. Sui, H. Kang, H. Dong, L. Dong and L. Yu, Small, 2018, 14, 1702570 CrossRef PubMed.
- L. Hu, J. Hou, Y. Ma, H. Li and T. Zhai, J. Mater. Chem. A, 2016, 4, 15006–15014 RSC.
- W. Yu, H. Wang, S. Liu, N. Mao, X. Liu, J. Shi, W. Liu, S. Chen and X. Wang, J. Mater. Chem. A, 2016, 4, 5973–5983 RSC.
- C. Chen, D. Yu, G. Zhao, B. Du, W. Tang, L. Sun, Y. Sun, F. Besenbacher and M. Yu, Nano Energy, 2016, 27, 377–389 CrossRef.
- G. Xu, J. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. Li and X. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- W. Qian, F. Sun, Y. Xu, L. Qiu, C. Liu, S. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379–386 RSC.
- Z. Li, L. Zhang, B. S. Amirkhiz, X. Tan, Z. Xu, H. Wang, B. C. Olsen, C. M. B. Holt and D. Mitlin, Adv. Energy Mater., 2012, 2, 431–437 CrossRef.
- Z. Zhao, S. Hao, P. Hao, Y. Sang, A. Manivannan, N. Wu and H. Liu, J. Mater. Chem. A, 2015, 3, 15049–15056 RSC.
- N. M. Khong, F. M. Yusoff, B. Jamilah, M. Basri, I. Maznah, K. W. Chan and J. Nishikawa, Food Chem., 2016, 196, 953–960 CrossRef PubMed.
- Y. Lv, H. Wang, X. Xu, J. Shi, W. Liu and X. Wang, Chem. Eng. J., 2017, 326, 401–410 CrossRef.
- L. Yao, Q. Wu, P. Zhang, J. Zhang, D. Wang, Y. Li, X. Ren, H. Mi, L. Deng and Z. Zheng, Adv. Mater., 2018, 30, 1706054 CrossRef PubMed.
- P. Lu, Y. Sun, H. Xiang, X. Liang and Y. Yu, Adv. Energy Mater., 2018, 8, 1702434 CrossRef.
- M. Lu, W. Yu, J. Shi, W. Liu, S. Chen, X. Wang and H. Wang, Electrochim. Acta, 2017, 251, 396–406 CrossRef.
- J. Yang, H. Wu, M. Zhu, W. Ren, Y. Lin, H. Chen and F. Pan, Nano Energy, 2017, 33, 453–461 CrossRef.
- N. Guo, M. Li, Y. Wang, X. Sun, F. Wang and R. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 33626–33634 CrossRef PubMed.
- Y. Cao, L. Xie, G. Sun, F. Su, Q.-Q. Kong, F. Li, W. Ma, J. Shi, D. Jiang, C. Lu and C.-M. Chen, Sustainable Energy Fuels, 2018, 2, 455–465 RSC.
- J. Ding, H. Wang, Z. Li, K. Cui, D. Karpuzov, X. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 941–955 RSC.
- N. He, S. Yoo, J. Meng, O. Yildiz, P. D. Bradford, S. Park and W. Gao, Carbon, 2017, 120, 304–312 CrossRef.
- C. Wang, F. Wang, Z. Liu, Y. Zhao, Y. Liu, Q. Yue, H. Zhu, Y. Deng, Y. Wu and D. Zhao, Nano Energy, 2017, 41, 674–680 CrossRef.
- N. Mao, H. Wang, Y. Sui, Y. Cui, J. Shi, W. Liu, S. Chen, X. Wang and D. Mitlin, Nano Res., 2017, 10, 1767–1783 CrossRef.
- X. Zhang, Y. Jiao, L. Sun, L. Wang, A. Wu, H. Yan, M. Meng, C. Tian, B. Jiang and H. Fu, Nanoscale, 2016, 8, 2418–2427 RSC.
- Z. Li, Z. Xu, H. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 RSC.
- J. Zhou, J. Lian, L. Hou, J. Zhang, H. Gou, M. Xia, Y. Zhao, T. A. Strobel, L. Tao and F. Gao, Nat. Commun., 2015, 6, 8503 CrossRef PubMed.
- C. Lu, D. Wang, J. Zhao, S. Han and W. Chen, Adv. Funct. Mater., 2017, 27, 1606219 CrossRef.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef PubMed.
- Z. H. Huang, T. Y. Liu, Y. Song, Y. Li and X. X. Liu, Nanoscale, 2017, 9, 13119–13127 RSC.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
- K. Ghosh, C. Y. Yue, M. M. Sk, R. K. Jena and S. Bi, Sustainable Energy Fuels, 2018, 2, 280–293 RSC.
- B. Li, F. Dai, Q. Xiao, L. Yang, J. Shen, C. Zhang and M. Cai, Adv. Energy Mater., 2016, 1600802 CrossRef.
- H. Tong, S. Yue, L. Lu, F. Jin, Q. Han, X. Zhang and J. Liu, Nanoscale, 2017, 9, 16826–16835 RSC.
- F. Zhu, M. Yan, Y. Liu, H. Shen, Y. Lei and W. Shi, J. Mater. Chem. A, 2017, 5, 22782–22789 RSC.
- X. Wang, C. Yan, A. Sumboja and P. S. Lee, Nano Energy, 2014, 3, 119–126 CrossRef.
- Y. Zhang, B. Wang, F. Liu, J. Cheng, X. Zhang and L. Zhang, Nano Energy, 2016, 27, 627–637 CrossRef.
- Y. Zhu, Z. Wu, M. Jing, H. Hou, Y. Yang, Y. Zhang, X. Yang, W. Song, X. Jia and X. Ji, J. Mater. Chem. A, 2015, 3, 866–877 RSC.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef PubMed.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef PubMed.
- H. Lai, Q. Wu, J. Zhao, L. Shang, H. Li, R. Che, Z. Lyu, J. Xiong, L. Yang, X. Wang and Z. Hu, Energy Environ. Sci., 2016, 9, 2053–2060 RSC.
- J. Hong, Y.-W. Lee, D. Ahn, S. Pak, J. Lee, A. R. Jang, S. Lee, B. Hou, Y. Cho, S. M. Morris, H. S. Shin, S. Cha, J. I. Sohn and J. M. Kim, Nano Energy, 2017, 39, 337–345 CrossRef.
- S. A. Rubaye, R. Rajagopalan, S. X. Dou and Z. Cheng, J. Mater. Chem. A, 2017, 5, 18989–18997 RSC.
- Z. Xiao, L. Fan, B. Xu, S. Zhang, W. Kang, Z. Kang, H. Lin, X. Liu, S. Zhang and D. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 41827–41836 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00348c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |