
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-electrochemical hydrogen production from neutral phosphate buffer and seawater using micro-structured p-Si photo-electrodes functionalized by solution-based methods†
Anurag
Kawde
 ab,
Alagappan
Annamalai
c,
Lucia
Amidani
b,
Manuel
Boniolo
d,
Wai Ling
Kwong
d,
Anita
Sellstedt
e,
Pieter
Glatzel
b,
Thomas
Wågberg
c and
Johannes
Messinger
*ad
ab,
Alagappan
Annamalai
c,
Lucia
Amidani
b,
Manuel
Boniolo
d,
Wai Ling
Kwong
d,
Anita
Sellstedt
e,
Pieter
Glatzel
b,
Thomas
Wågberg
c and
Johannes
Messinger
*ad
aUmeå University, Department of Chemistry, Sweden. E-mail: johannes.messinger@kemi.uu.se
bEuropean Synchrotron Radiation Facility (ESRF), Grenoble, France
cUmea University, Department of Physics, Sweden
dMolecular Biomimetics, Department of Chemistry – Ångström Laboratory, Uppsala University, Sweden
eUmeå University, Department of Plant Physiology, Umeå Plant Science Centre (UPSC), Umeå, Sweden
First published on 16th August 2018
Abstract
Solar fuels such as H2 generated from sunlight and seawater using earth-abundant materials are expected to be a crucial component of a next generation renewable energy mix. We herein report a systematic analysis of the photo-electrochemical performance of TiO2 coated, microstructured p-Si photo-electrodes (p-Si/TiO2) that were functionalized with CoOx and NiOx for H2 generation. These photocathodes were synthesized from commercial p-Si wafers employing wet chemical methods. In neutral phosphate buffer and standard 1 sun illumination, the p-Si/TiO2/NiOx photoelectrode showed a photocurrent density of −1.48 mA cm−2 at zero bias (0 VRHE), which was three times and 15 times better than the photocurrent densities of p-Si/TiO2/CoOx and p-Si/TiO2, respectively. No decline in activity was observed over a five hour test period, yielding a Faradaic efficiency of 96% for H2 production. Based on the electrochemical characterizations and the high energy resolution fluorescence detected X-ray absorption near edge structure (HERFD-XANES) and emission spectroscopy measurements performed at the Ti Kα1 fluorescence line, the superior performance of the p-Si/TiO2/NiOx photoelectrode was attributed to improved charge transfer properties induced by the NiOx coating on the protective TiO2 layer, in combination with a higher catalytic activity of NiOx for H2-evolution. Moreover, we report here an excellent photo-electrochemical performance of p-Si/TiO2/NiOx photoelectrode in corrosive artificial seawater (pH 8.4) with an unprecedented photocurrent density of 10 mA cm−2 at an applied potential of −0.7 VRHE, and of 20 mA cm−2 at −0.9 VRHE. The applied bias photon-to-current conversion efficiency (ABPE) at −0.7 VRHE and 10 mA cm−2 was found to be 5.1%.
Introduction
The CO2 released by burning fossil fuels for transportation, production of electricity and heating is a major contributor to global warming.1–3 In order to limit the greenhouse effect, it is necessary to replace fossil fuels with renewable and carbon-neutral or ‘green’ fuels.1 On Earth's surface, sunlight is the most abundant source of free energy, and seawater can provide a plentiful supply of electrons and protons as ingredients for making solar fuels.4–8 Natural photosynthesis and recent lab-scale artificial photosynthesis devices have demonstrated high efficiencies in solar-driven water splitting into molecular hydrogen and oxygen.9–12 However, enabling global-scale solar-fuel generation requires the fabrication of devices, in particularly of photoelectrodes, from earth-abundant materials. To obtain the maximal device performance, the interplay between light-harvesting, charge separation and electron/proton transport, as well as water oxidation and reduction catalysis needs to be carefully optimized2,3,12–20The intrinsic band gap (1.1 eV) of silicon photo-electrodes make them capable of absorbing a broad spectrum of the solar light. Over the past decades, they have thus gathered much attention towards photo-electrochemical (PEC) water splitting.21–26 The band structure of n-type Si promotes the photo-anodic oxygen evolution reaction (OER),27,28 while p-type Si facilitates the photo-cathodic hydrogen evolution reaction (HER).22 However, the difference between the potentials of the valence band edge of p-Si and that of the H+/H2 redox couple is relatively small which limits the photo-voltage and leads to slow HER kinetics.29 The surface area of the Si photo-electrodes can be significantly increased via nano or micro structuring.30 This not only increases light absorption of Si but also improves HER kinetics by shortening charge carrier transport pathways.24,31
When Si is placed in an aqueous environment, it forms an insulating oxide layer, which significantly reduces its HER efficiency.32 In addition, Si is only stable at strongly acidic33 and alkaline34 pH if protected. Both processes can be suppressed highly efficiently by applying a thin TiO2 over coating.35–37 In most studies the fabrication of the TiO2 protective layer involves the use of relatively expensive high vacuum technology such as atomic layer deposition (ALD).34,38–40 While ALD is used industrially for applying thin metal layers during the production of electronic components for computers and other electronic devices, it may still be useful to explore cheap and scalable wet-chemical methods for producing large areas of coatings of 20–50 nm thickness.41,42 The performance of Si/TiO2 photo-electrodes can be improved by decoration with suitable co-catalysts.38,43 Decoration with NiOx has been studied for improving OER catalysis,27,44,45 but there are only a few studies on the application of NiOx as HER catalysts at neutral (or near neutral) pH.46–49 To the best of our knowledge, NiOx has not yet been studied as HER co-catalyst on the surface of p-Si/TiO2 for photoelectrochemical seawater reduction to H2.
Herein, we report the fabrication of efficient HER photo-electrodes consisting of CoOx or NiOx decorated p-Si/TiO2. The photo-electrodes were prepared by micro-structuring commercial p-Si wafers using a well-documented electroless wet etching technique.50,51 The resulting microwire surface was coated with a protective TiO2 layer and subsequently functionalized with CoOx or NiOx co-catalysts using simple solution-based techniques. The electrodes obtained by the various fabrication steps were systematically studied, and the photo-electrochemical responses of the final p-Si/TiO2/NiOx and p-Si/TiO2/CoOx electrodes were measured in neutral pH phosphate buffer and in artificial seawater at pH 8.4. The PEC performance of functionalized p-Si photoelectrodes were also studied without a TiO2 layer to establish the effect of TiO2 as efficient charge separation layer between p-Si and the co-catalysts. Detailed electrochemical and X-ray spectroscopic studies were performed to elucidate the origin of the significantly better performance of p-Si/TiO2/NiOx as compared to p-Si/TiO2/CoOx.
Results and discussion
Preparation and structural characterization of HER photo-electrodes
The experimental steps involved in the synthesis of our p-Si photo-electrodes are summarized schematically in Fig. 1 (see ESI† for experimental methods). After a reductive cleaning of the commercial p-Si wafers to remove the surface SiO2, wet etching was employed to obtain high-aspect ratio micro-structured p-Si electrodes.50,51 These ‘as-synthesized’ p-Si photo-electrodes (Fig. 2(a) and (b)) were subsequently spin-coated in two steps, first with a TiO2 sol and subsequently with either a NiOx or CoOx sol. Each spin-coating step was followed by drying at room temperature and calcination at 380 °C. The scanning electron microscopy (SEM) top and side views of the as-synthesized p-Si (Fig. 2(a) and (b)) show the microstructure morphology. Fig. 2(c)–(f) show that the sol–gel method in combination with spin coating resulted in the formation of a protective TiO2 overcoating and a co-catalysts film that fully covers the p-Si. The energy dispersive X-ray spectroscopy (EDX) cross-sections of the functionalized p-Si photo-electrodes are shown in ESI Fig. S1–S3.† On the basis of the SEM and TEM data shown in ESI Fig. S4† we conservatively estimate that the TiO2 layer has a thickness ranging between 20–50 nm. The elemental composition and the presence of CoOx and NiOx were confirmed using XPS, for details see ESI Fig. S5.†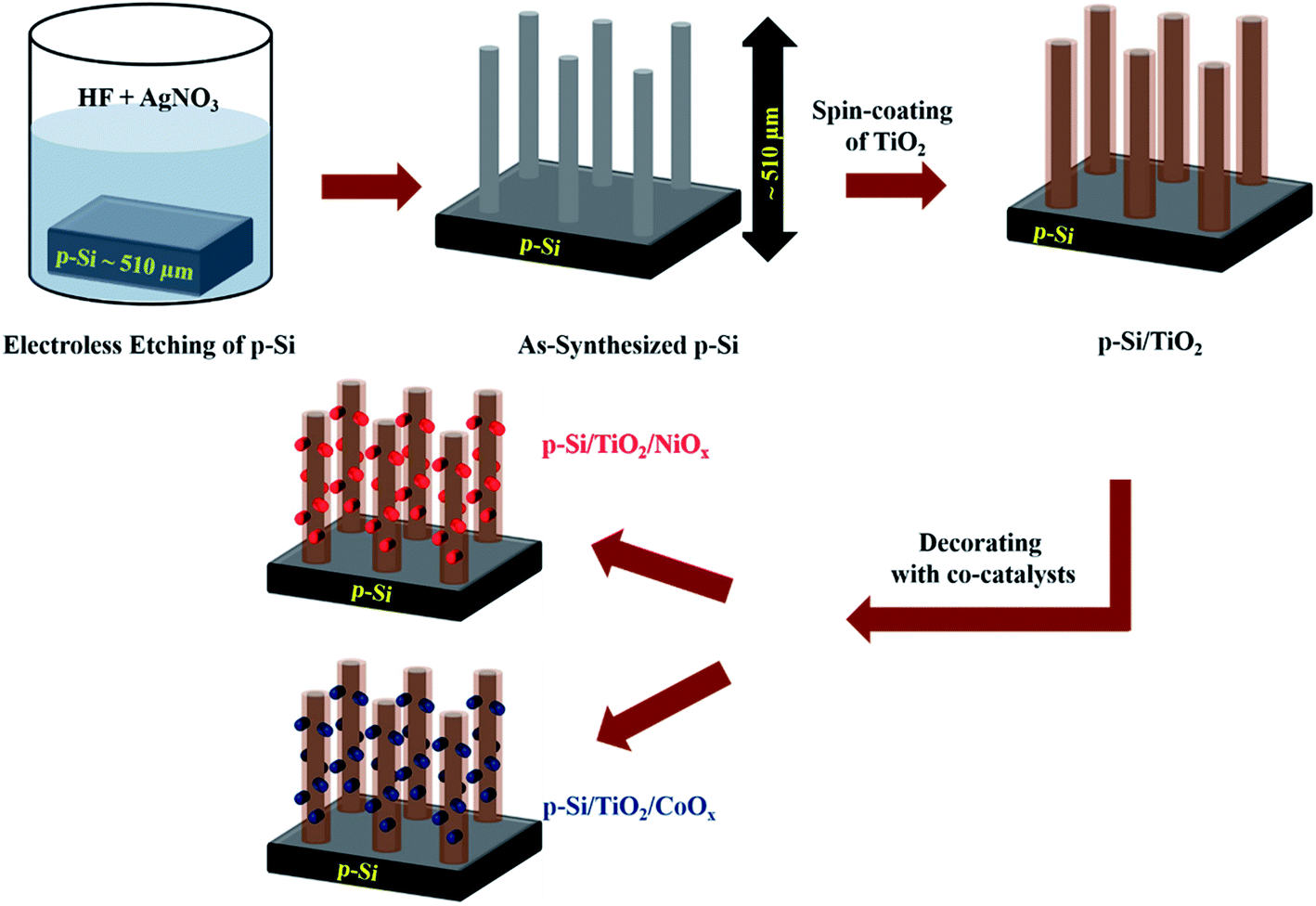 | ||
| Fig. 1 Schematic representation (not to scale) of the preparation steps of p-Si micro-wires by wet-etching, their coating with TiO2 and subsequent functionalization with NiOx or CoOxvia spin coating and sintering. The procedure is described in detail in the ESI.† | ||
 | ||
| Fig. 2 SEM Images showing the planar top views (a), (c) and (e) and the cross-sectional view (b), (d) and (f), of as-synthesized and functionalized p-Si. | ||
Photo-electrochemical characterization of the HER electrodes in neutral phosphate buffer
The photo-electrochemical performances of the p-Si photo-electrodes obtained after each preparation step were carefully studied at 1 sun illumination in neutral phosphate buffer. The linear sweep voltammogram (LSV) traces in Fig. 3a show that the photocurrent density of untreated planar p-Si stays negligible over the entire potential range studied (dark currents are shown in ESI Fig. S6†). The slightly higher photocurrent performance of the as-synthesized, micro-structured p-Si photoelectrode (purple trace, Fig. 3a) as compared to planar p-Si (green trace, Fig. 3a) is in line with an earlier report34 and can be explained by the better light absorption and charge transport owing to the one dimensional morphology and improved surface area. This advantage of micro-structuring52 is even more obvious for the fully functionalized photo-electrodes as shown in Fig. 3b (LSVs for planar Si and planar Si/TiO2 are shown in Fig. S7†). Coverage of microstructured p-Si with a protective TiO2 layer leads to a further minor increase in the photocurrent (black trace in Fig. 3a), likely due to the absence of an insulating Si-oxide surface layer and/or the better catalytic HER performance of TiO2vs. p-Si. The nearly 6-fold increase in photocurrent after functionalization with CoOx (J = −0.50 mA cm−2@0 VRHE) and the 15-fold increase in case of NiOx (J = −1.40 mA cm−2@0 VRHE) shows that the photo-charge carriers of our control sample (J = −0.09 mA cm−2@0 VRHE) can be used much more efficiently for proton reduction if suitable co-catalysts are provided (blue and red traces in Fig. 3a). Interestingly, Fig. 3c shows that the current densities obtained are much smaller in absence of the intermittent TiO2 layer, indicating that function of the TiO2 layer goes far beyond a simple protective function. The magnitude of the photo current is illustrated for the fully assembled photo-electrodes in Fig. 3d by comparing the corresponding light and dark traces.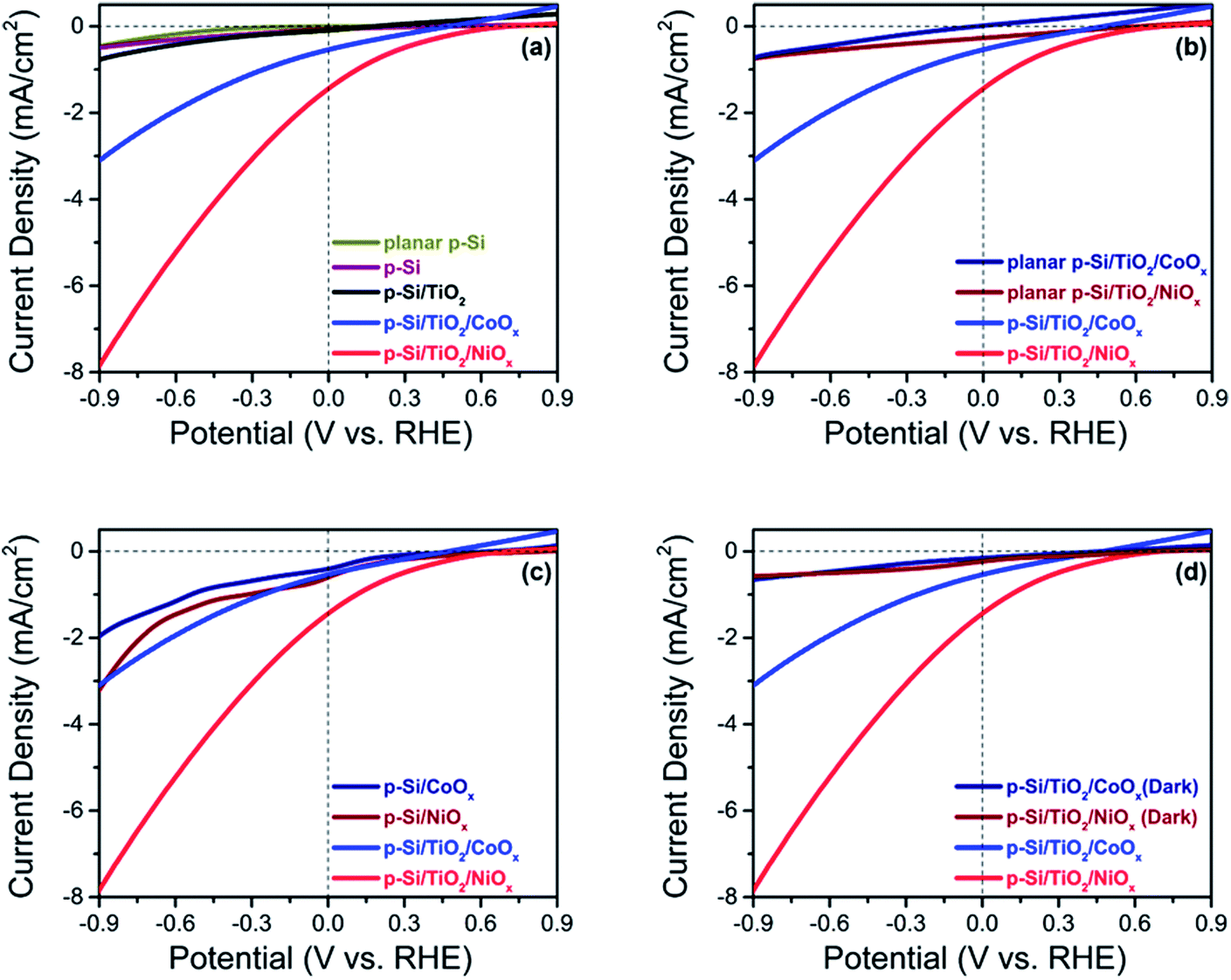 | ||
| Fig. 3 Linear sweep voltammograms recorded under 1 sun illumination in potassium phosphate buffer (0.2 M; pH 7.00) (a) planar p-Si, a surface etched p-Si with microwires (p-Si), and functionalized p-Si photo-electrodes (b) fully functionalized planar Si and functionalized p-Si (c) functionalized p-Si with and without the intermittent TiO2 layer (d) functionalized p-Si in dark and in illumination. | ||
The same trends as for the photo currents are also seen in the respective onset potentials (Vonset) for H2-evolution, here defined as the potential needed for reaching a net photocurrent density of −0.1 mA cm−2.53 A positive (anodic) shift of the onset potential (Vonset) was observed for the co-catalyst functionalized electrodes as compared to as-synthesized and p-Si/TiO2: the Vonset of the control p-Si/TiO2 sample was observed at −0.66 VRHE, whereas those for p-Si/TiO2/CoOx and p-Si/TiO2/NiOx were shifted to +0.27 VRHE and +0.42 VRHE, respectively (ESI Table S1†). The Vonset value for our p-Si/TiO2/NiOx photoelectrode is remarkable when compared to a recent report by Zhang and coworkers,33 where a Vonset = +0.25 VRHE (J = −0.1 mA cm−2) was obtained for p-Si/NiCoSex nanopillars in 0.5 M H2SO4 where the concentration of protons is 106 times higher as compared to the neutral pH electrolyte employed here.33,43 The excellent performance of NiOx at neutral pH condition, which is further manifested by the detailed comparison to further co-catalysts functionalized p-Si based photo-electrodes reported in the literature (ESI Table S1†), may be attributed either to an intrinsically better HER performance of NiOx compared to CoOx, and/or to a superior charge transfer between p-Si and the NiOxvia the protective TiO2 underlayer. This will be analysed in subsequent sections.
Photochemical H2 generation in neutral phosphate buffer
The photo-generation of H2 was measured using gas chromatography (GC) at a constant potential of 0 VRHE. Fig. 4a shows the results for p-Si/TiO2/CoOx (blue) and p-Si/TiO2/NiOx (red). Over a period of 5 hours of continuous illumination, a near linear production of H2 was observed yielding ∼128 μmol cm−2 and ∼44 μmol cm−2 of H2 for p-Si/TiO2/NiOx and p-Si/TiO2/CoOx respectively. The simultaneously recorded photocurrent density (Fig. 4b) was essentially stable at −1.48 mA cm−2 (p-Si/TiO2/NiOx) and −0.48 mA cm−2 (p-Si/TiO2/CoOx) over the five hours of the measurements, indicating a total lifetime extending by far the chosen duration of this experiment. XES scans taken after these experiments demonstrate that Pt from the counter electrode was not deposited onto our working electrodes during these experiments (ESI Fig. S8†). This results in a Faradaic efficiency of 96% (p-Si/TiO2/NiOx) and 93% (p-Si/TiO2/CoOx) and in applied bias photon to current conversion efficiencies (ABPE) of 1.74% for p-Si/TiO2/NiOx and 0.54% for p-Si/TiO2/CoOx at 0 VRHE. The excellent performance of NiOx may in part also be attributed to its conversion into Ni(OH)2 upon contact with water.48,49,54 Ni(OH)2 formation is in agreement with the XPS spectra taken after the 5 hour performance test (ESI Fig. S5†). While we cannot exclude that a transient light-induced formation of NiOOH contributes to the excellent performance of NiOx, as suggested recently for NiOx nano-particles,54 we do not observe a stable NiOOH formation in the above mentioned XPS spectra, as expected under negative bias.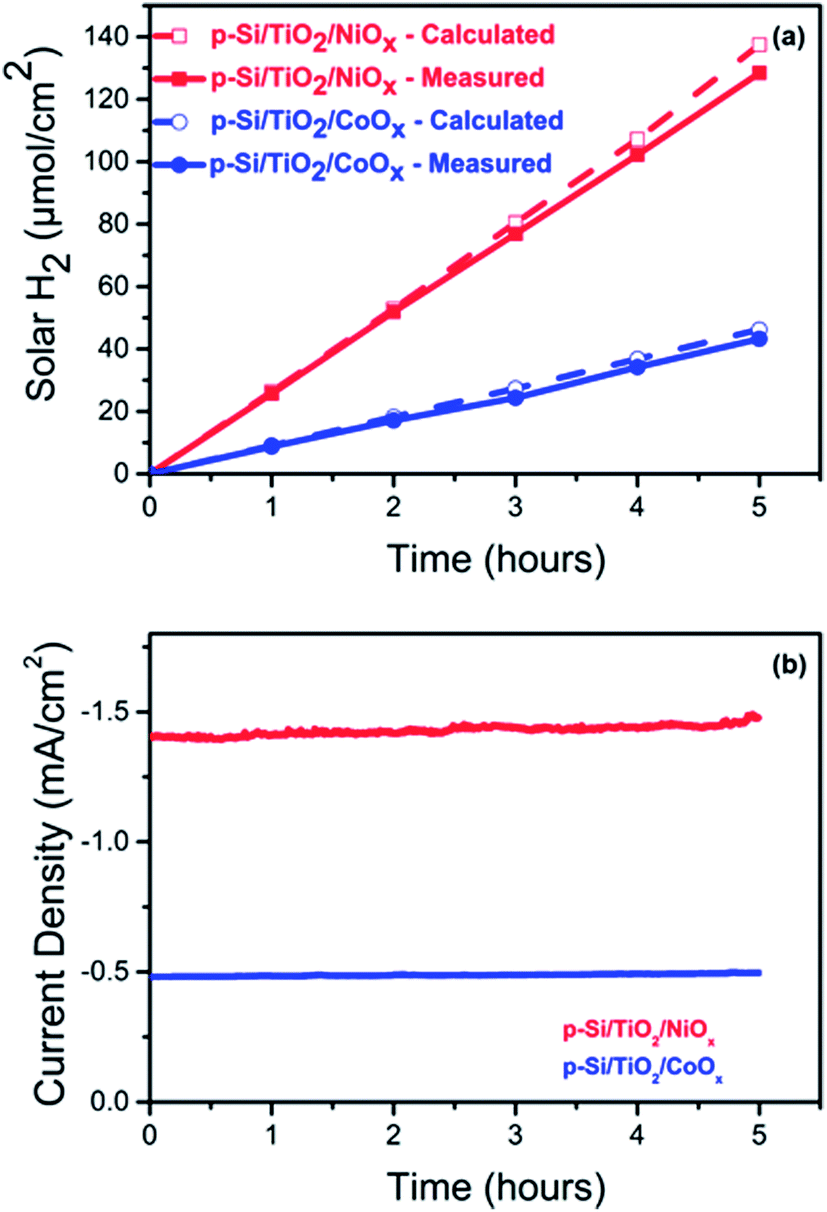 | ||
| Fig. 4 Comparison of (a) the calculated and measured solar hydrogen production per geometric surface area of the photoelectrode and (b) of the current density of NiOx (red) and CoOx (blue) functionalized p-Si photo-electrodes over 5 hours of continuous operation. The data displayed in (a) and (b) were obtained simultaneously at 0.0 VRHE and 1 sun illumination. | ||
Charge transport kinetics
To understand the differences between the photoelectrochemical performance of non-functionalized and NiOx- or CoOx-functionalized p-Si/TiO2 photo-electrodes, the transport of photoelectrons generated in p-Si to the surface and further to the protons in the phosphate buffer (pH 7.0) were studied by using electrochemical impedance spectroscopy (EIS). The Nyquist plots of the EIS data measured at 1 sun illumination are shown in Fig. 5 along with the equivalent circuit diagram employed for their analysis. The smaller semi-circle (high frequency region) corresponds to the resistance for the charge transfer from p-Si through TiO2 to the surface (RCT1, CPE1), whereas the larger semicircle (low frequency region) corresponds to the photoelectrode/electrolyte interfacial charge transfer resistance described by RCT2 and CPE2. The data in Fig. 5 and ESI Table S2† show that the charge transport resistance for both processes is smallest for NiOx, and increases for p-Si/TiO2/CoOx and is largest for the non-functionalized p-Si/TiO2. RCT1 increases from 807 Ω to 1672 Ω and 1763 Ω, while RCT2 from 2519 Ω to 6646 Ω and 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 840 Ω. The effective Rs also increases in the same order, indicating that p-Si/TiO2/NiOx has the best charge transfer kinetics.55
840 Ω. The effective Rs also increases in the same order, indicating that p-Si/TiO2/NiOx has the best charge transfer kinetics.55
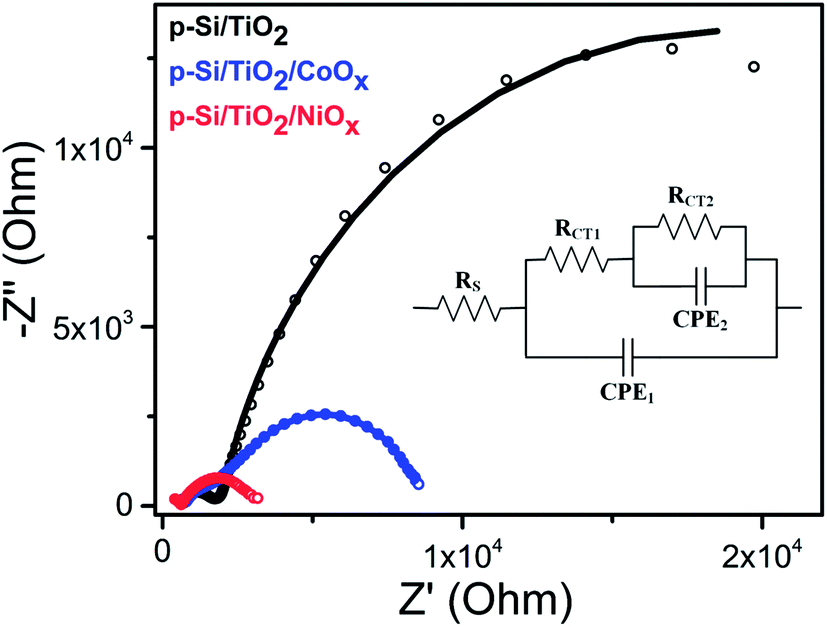 | ||
| Fig. 5 Nyquist plots of p-Si/TiO2 (black), p-Si/TiO2/CoOx (blue), p-Si/TiO2/NiOx (red) measured at 0.0 VRHE under 1 sun illumination. The inset shows the equivalent circuit employed for analyzing the Nyquist plots. The fit data are provided in ESI Table S2.† | ||
To further investigate the electronic and interfacial properties, the flat band potential (VFB) and the carrier concentration (ND) were studied by Mott–Schottky measurements. Fig. 6(a–c) show that all photo-electrodes display a negative slope in the Mott–Schottky plots, consistent with the p-type doping of the Si substrate. The carrier concentrations (ND) of the photo-electrodes were calculated from the slopes of Mott–Schottky plots, yielding values of 1.52 × 1014 cm−3, 1.98 × 1015 cm−3 and 3.73 × 1015 cm−3 for p-Si/TiO2 and CoOx or NiOx functionalized photo-electrodes, respectively. Thus, both CoOx and NiOx functionalized photo-electrodes show one-order higher donor densities compared to the p-Si/TiO2 control sample. This is in line with the observed H2 generation rates at neutral pH, as well as with the order of the current densities and the charge transfer properties described above.
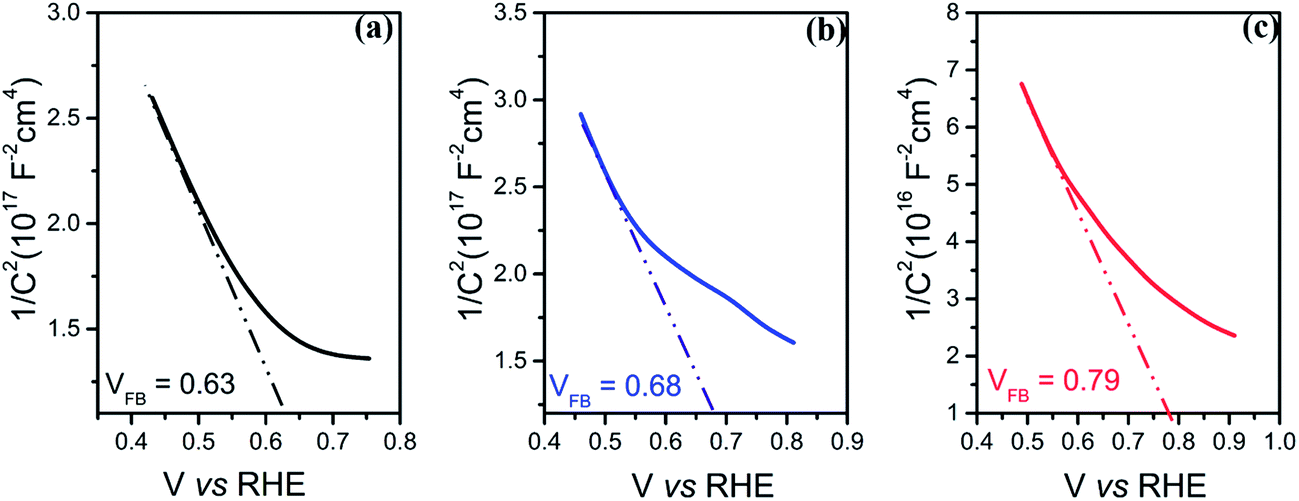 | ||
| Fig. 6 Mott–Schottky measurements of (a) p-Si/TiO2 (b) p-Si/TiO2/CoOx and (c) p-Si/TiO2/NiOx. | ||
By fitting the linear regions (minimum range of 200 mV)56 of the Mott–Schottky plots and extrapolating the line to the X-axis, the flat band potential VFB can be deduced. Significant differences were observed: the VFB of the Si/TiO2 control sample was 0.63 VRHE, whereas VFB of the CoOx and NiOx modified photo-electrodes were 0.68 VRHE and 0.79 VRHE, respectively. The anodic shifts of 0.05 V and 0.16 V compared to the control indicate that p-Si/TiO2/NiOx has the least recombination losses,57 suggesting that NiOx passivates the surface defects more efficiently than CoOx. The more positive anodic shifts of the VFB for the p-Si/TiO2/NiOx may be attributed to dissipation of potential drop at the Helmholtz layer and space charge region at the p-Si/TiO2/NiOx – electrolyte interface.45 An anodically shifted VFB enhances the HER kinetics by lowering the potential for transporting electrons to the surface.46,58–62
Effects of co-catalysts on the local geometry of TiO2
The electrochemical characterization of our photo-electrodes shows that the charge transfer properties of the interfacial TiO2 layer are improved to various degrees by applying the CoOx or NiOx co-catalyst layers and sintering at 380 °C for two hours, indicating a modification of the TiO2 coating by this procedure. In addition, the significant and different performance increases of p-Si/CoOxvs. p-Si/TiO2/CoOx and p-Si/NiOxvs. p-Si/TiO2/NiOx indicate an important role of the TiO2 layer for the overall performance of the photo-electrodes. Thus, K-edge HERFD-XANES measurements were performed at the Ti K-edge (Fig. 7a and b) to elucidate changes in the atomic and electronic structure of TiO2 induced by co-catalyst decoration. A schematic view of the optics and the experimental set up is presented elsewhere.63,64 The pre-edge region of the XAS spectra (4967 to 4976 eV) is particularly sensitive to the symmetry at the Ti sites.65,66 The pre-edge of crystalline TiO2 has three characteristic peaks, denoted as A1, A2 and A3.67 The pre-edge peaks A2 and A3 mainly arise from dipolar transitions from the Ti 1s shell and are thus affected by p–d mixing,66,68 which in turn depends on the local bonding geometry. In contrast, the quadrupolar 1s to 3d transition gives rise to A1 and the low energy shoulder in A2.69,70 Thus the number and position of the peaks in the pre-edge region contain information about the local coordination of Ti in TiO2.67 In anatase and amorphous TiO2, the intensity of this shoulder is particularly sensitive to the presence of low coordination Ti sites as demonstrated by investigations of TiO2 nanoparticles.71,72 Additional structural information can be obtained from the spectral shape of the post-edge region (B and C in Fig. 7a), which derives from the excitation of a 1s electron to one of the unoccupied states with p-symmetry with respect to Ti. Such transitions are highly sensitive to the local structure around the absorber.67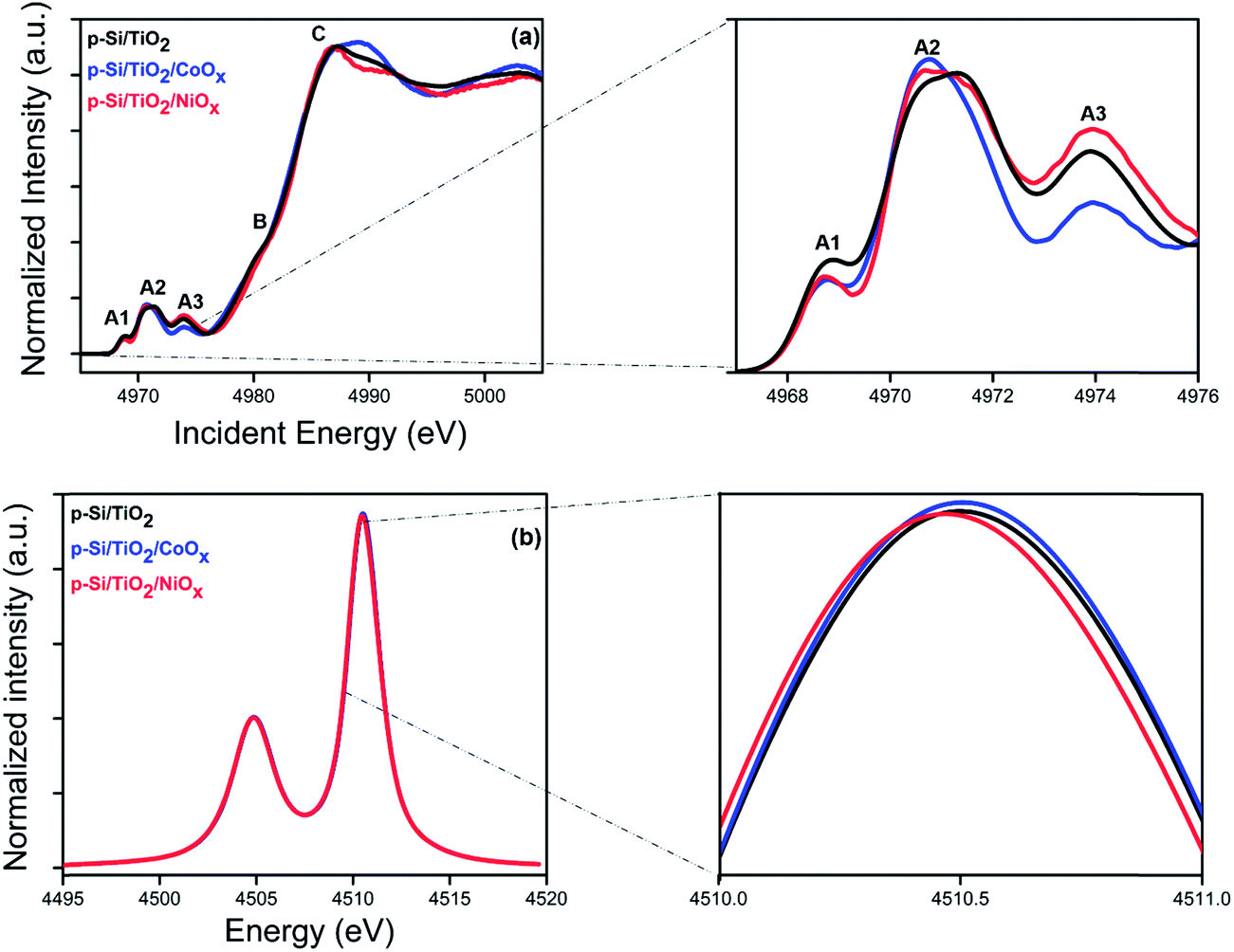 | ||
| Fig. 7 X-ray spectroscopy characterization of p-Si/TiO2 (black), p-Si/TiO2/CoOx (blue), p-Si/TiO2/NiOx (red): (a) HERFD-XANES spectra acquired at the Ti K-edge. The magnified view on the right shows the pre-edge peaks. (b) Kα emission lines. The magnified view on the right shows the Kα peak maximum. | ||
Fig. 7a shows the HERFD-XANES spectra of p-Si/TiO2, p-Si/TiO2/CoOx, and p-Si/TiO2/NiOx, while the reference spectra of TiO2 anatase, TiO2 rutile, and sol–gel processed protective TiO2 are displayed in ESI Fig. S9.† The overall spectral shape with three strong pre-edge features is preserved in all three samples. Together with the largely invariant Kα lines (Fig. 7b) this finding confirms that the crystallinity of the protective TiO2 layer remains unchanged, i.e. that in all cases Ti is in oxidation state +IV with a six-fold coordination. However, a detailed comparison revealed clear spectral differences between the pre-edge peaks of the three samples that are indicative of variously distorted TiO2 octahedra. In addition, the absorption peak C is also different for all the samples.71 The precise nature of these changes could possibly be addressed using computational modeling, which is beyond the scope of this study. Nevertheless, the present data demonstrate that an intricate interplay between all components of the photo-electrode, including the TiO2 layer, exists that contributes to the overall catalytic performance.
Functional characterisation of HER activity in artificial seawater (pH 8.4)
Because of the vast seawater resources on Earth, we tested also the photo-catalytic performance of our best electrode, p-Si/TiO2/NiOx, in artificial seawater. Fig. 8 displays the photo-electrochemical performance of p-Si/TiO2/NiOx in neutral phosphate electrolyte and artificial seawater (pH 8.4). A shift of Vonset by −0.12 VRHE is observed for artificial seawater compared to neutral phosphate buffer. This cathodic shift is expected due to the lower proton concentration at the higher pH. However, despite the less favourable onset potential, the p-Si/TiO2/NiOx photoelectrode was highly efficient in H2 generation at increasing potentials. A photocurrent density of 10 mA cm−2 was achieved at 1 sun illumination and an applied potential of −0.7 VRHE, and 20 mA cm−2 at about −0.9 VRHE.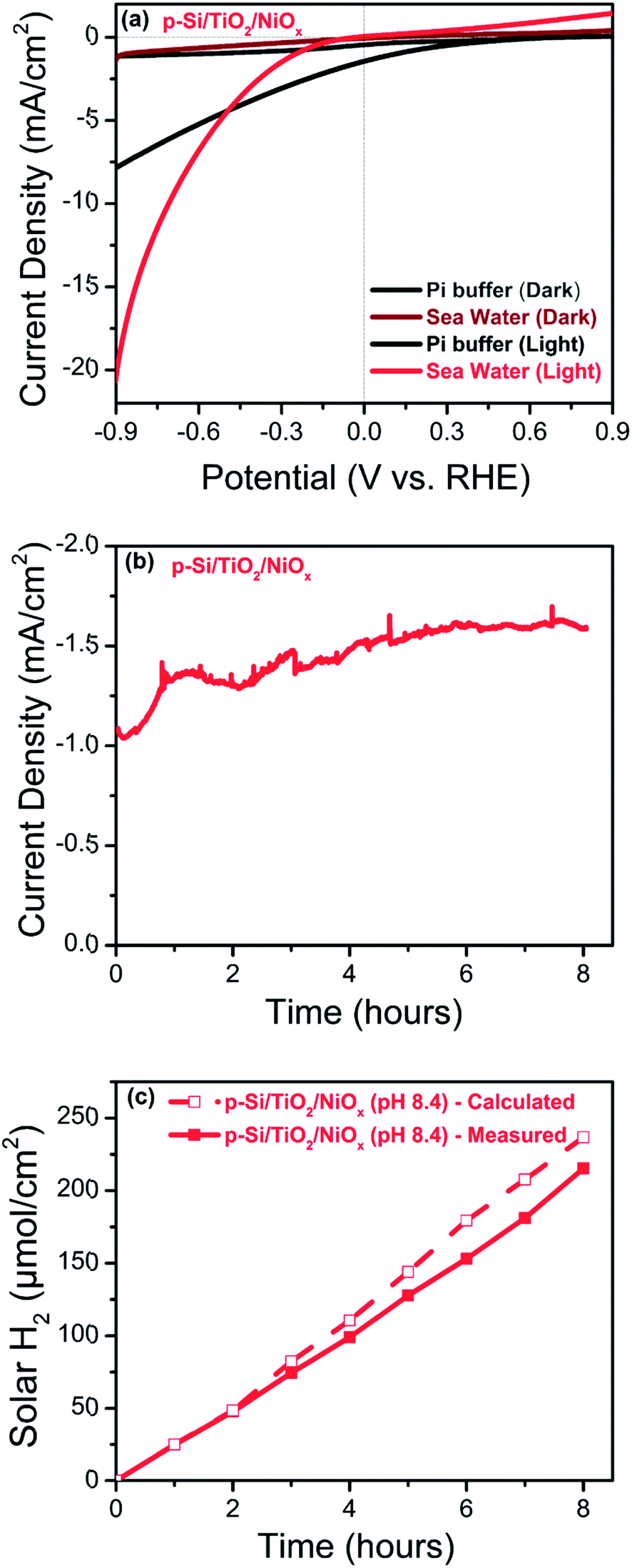 | ||
| Fig. 8 Photo-electrochemical performance of the p-Si/TiO2/NiOx photoelectrode recorded under 1 sun illumination in artificial seawater at pH 8.4 (red) or pH 7.0 (black; 0.2 M phosphate buffer): (a) linear sweep voltammograms, (b) photo-current density measured at −0.3 VRHE and (c) comparison of calculated (open squares) and measured (closed squares) photo generated hydrogen per geometric surface area of the photo-electrode at −0.3 VRHE and 1 sun. | ||
To the best of our knowledge, there are only a small number of studies describing the photo-electrochemical hydrogen generation by seawater splitting.4,6–8 In a recent study, Mase et al.73 reported the photocatalytic production of hydrogen peroxide (H2O2) from seawater at pH 1.3 with a solar-to-electricity efficiency of 0.28%, and a photocatalytic production efficiency of H2O2 of 0.55%. Ji et al.6 found that their NiO/Ni/La2Ti2O7 photo catalysts were suitable for solar hydrogen generation from both natural and artificial seawater, but that Mg2+ and Ca2+ reduce the rate of H2-evolution. Fig. 8(b) and (c) show that our photoelectrode when operated in artificial seawater at an applied potential of −0.3 VRHE and 1 sun illumination, produces H2 at a nearly constant rate of about −1.5 mA cm−2 over 8 hours. The possibility of photo-oxidation of Cl− to Cl2 has been discussed for the photo-electrochemical seawater splitting4,7 however, we did not detect chlorine gas formation employing gas chromatography. The ABPE in the seawater splitting was calculated to be 1.26% at −0.3 VRHE, which corresponds to a current density of −1.5 mA cm−2, after 8 hours of illumination with a Faradaic efficiency close to 91%, further indicating that Cl− oxidation and Mg2+/Ca2+ deposition are at best minor side reactions. Importantly, an ABPE of 5.1% was observed at −0.7 VRHE. These surprisingly good results highlight the importance of the TiO2 layer in combination with NiOx as an efficient HER catalysts, which can likely be further optimized in future for even more efficient solar seawater splitting.
Conclusions
We demonstrate here a simple solution-based approach for the synthesis of photo-electrodes with earth abundant materials. The photo-electrodes presented in this study can harvest solar energy and utilize it for splitting corrosive seawater, making it an attractive system for large-scale application. Our finding of sustained H2 evolution with >1.2% ABPE in artificial seawater using a p-Si/TiO2/NiOx at a bias of −0.3 VRHE is highly encouraging, but further significant improvements to optimize the presented photo-electrodes are needed before practical applications become feasible. Importantly, the dual effect of NiOx, on the charge transfer through the protective TiO2 layer and for HER catalysis, points towards new ways for photoelectrode manufacturing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Andrey Shchukarev collected the XPS data and provided valuable input for their interpretation. The SEM and EDX data were obtained at the Umeå Core Facility for Electron Microscopy (UCEM). The authors thank European Synchrotron Radiation Facility for granting beamtime and the ID 26 beamline staff for the assistance during the experiments. The Knut and Alice Wallenberg's Foundation via the Artificial Leaf Project Umeå provided financial support (KAW 2011.0055). T. W. acknowledges support from Vetenskapsrådet (2017-04862), Energimyndigheten (45419-1), and Ångpanneföreningen (15-483).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef PubMed.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, M. Fontecave and D. R. MacFarlane, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- W. Luo, Z. Yang, Z. Li, J. Zhang, J. Liu, Z. Zhao, Z. Wang, S. Yan, T. Yu and Z. Zou, Energy Environ. Sci., 2011, 4, 4046–4051 RSC.
- J. Augustynski, R. Solarska, H. Hagemann and C. Santato, Proc. SPIE, 2006, 6340, U140–U148 CrossRef.
- S. M. Ji, H. Jun, J. S. Jang, H. C. Son, P. H. Borse and J. S. Lee, J. Photochem. Photobiol., A, 2007, 189, 141–144 CrossRef.
- X. Guan, F. A. Chowdhury, N. Pant, L. Guo, L. Vayssieres and Z. Mi, J. Phys. Chem. C, 2018, 125, 13797–13802 CrossRef.
- L. Zhang, I. Álvarez-Martos, A. Vakurov and E. E. Ferapontova, Sustainable Energy Fuels, 2017, 1, 842–850 RSC.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef PubMed.
- B. Wang, J. Zhang and F. Huang, Appl. Surf. Sci., 2017, 391, 449–456 CrossRef.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- T. Sharifi, C. Larsen, J. Wang, W. L. Kwong, E. Gracia-Espino, G. Mercier, J. Messinger, T. Wågberg and L. Edman, Adv. Energy Mater., 2016, 6, 1600738 CrossRef.
- A. Annamalai, R. Sandström, E. Gracia-Espino, N. Boulanger, J.-F. Boily, I. Mühlbacher, A. Shchukarev and T. Wågberg, ACS Appl. Mater. Interfaces, 2018, 10, 16467–16473 CrossRef PubMed.
- D. Bae, B. Seger, P. C. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef PubMed.
- J. Barber, Sustainable Energy Fuels, 2018, 2, 927–935 RSC.
- X. Cheng, Y. Zhang, H. Hu, M. Shang and Y. Bi, Nanoscale, 2018, 10, 3644–3649 RSC.
- S. K. Matta, C. Zhang, Y. Jiao, A. O'Mullane and A. Du, Nanoscale, 2018, 10, 6369–6374 RSC.
- E. L. Warren, J. R. McKone, H. A. Atwater, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 9653–9661 RSC.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig and H. A. Atwater, J. Am. Chem. Soc., 2011, 133, 1216–1219 CrossRef PubMed.
- X. Wang, K.-Q. Peng, Y. Hu, F.-Q. Zhang, B. Hu, L. Li, M. Wang, X.-M. Meng and S.-T. Lee, Nano Lett., 2013, 14, 18–23 CrossRef PubMed.
- K.-Q. Peng, X. Wang, L. Li, Y. Hu and S.-T. Lee, Nano Today, 2013, 8, 75–97 CrossRef.
- A. Sartori, M. Orlandi, S. Berardi, A. Mazzi, N. Bazzanella, S. Caramori, R. Boaretto, M. Natali, R. Fernandes and N. Patel, Electrochim. Acta, 2018, 271, 472–480 CrossRef.
- H. Meng, K. Fan, J. Low and J. Yu, Dalton Trans., 2016, 45, 13717–13725 RSC.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef PubMed.
- Z. Chen, G. Ma, Z. Chen, Y. Zhang, Z. Zhang, J. Gao, Q. Meng, M. Yuan, X. Wang and J.-M. Liu, Appl. Surf. Sci., 2017, 396, 609–615 CrossRef.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Science, 2010, 327, 185–187 CrossRef PubMed.
- C. Liu, N. P. Dasgupta and P. Yang, Chem. Mater., 2013, 26, 415–422 CrossRef.
- K. Sun, S. Shen, Y. Liang, P. E. Burrows, S. S. Mao and D. Wang, Chem. Rev., 2014, 114, 8662–8719 CrossRef PubMed.
- E. L. Warren, H. A. Atwater and N. S. Lewis, J. Phys. Chem. C, 2013, 118, 747–759 CrossRef.
- H. Zhang, Q. Ding, D. He, H. Liu, W. Liu, Z. Li, B. Yang, X. Zhang, L. Lei and S. Jin, Energy Environ. Sci., 2016, 9, 3113–3119 RSC.
- Y. J. Hwang, A. Boukai and P. Yang, Nano Lett., 2008, 9, 410–415 CrossRef PubMed.
- M. R. Shaner, S. Hu, K. Sun and N. S. Lewis, Energy Environ. Sci., 2015, 8, 203–207 RSC.
- S. Ida, K. Kearney, T. Futagami, H. Hagiwara, T. Sakai, M. Watanabe, A. Rockett and T. Ishihara, Sustainable Energy Fuels, 2017, 1, 280–287 RSC.
- Y. Yu, Z. Zhang, X. Yin, A. Kvit, Q. Liao, Z. Kang, X. Yan, Y. Zhang and X. Wang, Nat. Energy, 2017, 2, 17045 CrossRef.
- Y. W. Chen, J. D. Prange, S. Dühnen, Y. Park, M. Gunji, C. E. Chidsey and P. C. McIntyre, Nat. Mater., 2011, 10, 539 CrossRef PubMed.
- M. T. McDowell, M. F. Lichterman, A. I. Carim, R. Liu, S. Hu, B. S. Brunschwig and N. S. Lewis, ACS Appl. Mater. Interfaces, 2015, 7, 15189–15199 CrossRef PubMed.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef PubMed.
- J. Yu, X. Zhao and Q. Zhao, Thin Solid Films, 2000, 379, 7–14 CrossRef.
- S.-Y. Lien, D.-S. Wuu, W.-C. Yeh and J.-C. Liu, Sol. Energy Mater. Sol. Cells, 2006, 90, 2710–2719 CrossRef.
- J. R. McKone, E. L. Warren, M. J. Bierman, S. W. Boettcher, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Energy Environ. Sci., 2011, 4, 3573–3583 RSC.
- K. Sun, N. Park, Z. Sun, J. Zhou, J. Wang, X. Pang, S. Shen, S. Y. Noh, Y. Jing and S. Jin, Energy Environ. Sci., 2012, 5, 7872–7877 RSC.
- W. Zhen, X. Ning, B. Yang, Y. Wu, Z. Li and G. Lu, Appl. Catal., B, 2018, 221, 243–257 CrossRef.
- J.-Y. Jung, J.-Y. Yu and J.-H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7955–7962 CrossRef PubMed.
- M. A. Melo, S. A. Carminati, J. Bettini and A. F. Nogueira, Sustainable Energy Fuels, 2018, 2, 958–967 RSC.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543–9550 RSC.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- K. Peng, Y. Xu, Y. Wu, Y. Yan, S. T. Lee and J. Zhu, Small, 2005, 1, 1062–1067 CrossRef PubMed.
- K. Q. Peng, Y. J. Yan, S. P. Gao and J. Zhu, Adv. Mater., 2002, 14, 1164–1167 CrossRef.
- K. Q. Peng and S. T. Lee, Adv. Mater., 2011, 23, 198–215 CrossRef PubMed.
- A. Kargar, J. Khamwannah, C. H. Liu, N. Park, D. Wang, S. A. Dayeh and S. Jin, Nano Energy, 2016, 19, 289–296 CrossRef.
- K. Han, T. Kreuger, B. Mei and G. Mul, ACS Catal., 2017, 7, 1610–1614 CrossRef PubMed.
- A. Annamalai, A. Subramanian, U. Kang, H. Park, S. H. Choi and J. S. Jang, J. Phys. Chem. C, 2015, 119, 3810–3817 CrossRef.
- Y. Zhao, N. Anderson, K. Zhu, J. Aguiar, J. Seabold, J. V. D. Lagemaat, H. Branz, N. Neale and J. Oh, Nano Lett., 2015, 15, 2517–2525 CrossRef PubMed.
- A. W. Bott, Curr. Sep., 1998, 17, 87–92 Search PubMed.
- A. J. Bard, J. Phys. Chem., 1982, 86, 172–177 CrossRef.
- H. Gerischer, Surf. Sci., 1969, 18, 97–122 CrossRef.
- K. Rajeshwar, Encycl. Electrochem., 2007, 6, 1–53 Search PubMed.
- W. Gomes and F. Cardon, Prog. Surf. Sci., 1982, 12, 155–215 CrossRef.
- W. Schmickler and E. Santos, Interfacial electrochemistry, Springer Science & Business Media, 2010 Search PubMed.
- M. Rovezzi and P. Glatzel, Semicond. Sci. Technol., 2014, 29, 023002 CrossRef.
- L. Amidani, A. Naldoni, M. Malvestuto, M. Marelli, P. Glatzel, V. Dal Santo and F. Boscherini, Angew. Chem., Int. Ed., 2015, 54, 5413–5416 CrossRef PubMed.
- G. A. WlvcnuNls, Am. Mineral., 1987, 72, 89–101 Search PubMed.
- F. Farges, G. E. Brown and J. Rehr, Phys. Rev. B: Solid State, 1997, 56, 1809 CrossRef.
- R. Asahi, Y. Taga, W. Mannstadt and A. J. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 7459 CrossRef.
- G. Fronzoni, R. De Francesco, M. Stener and M. Causa, J. Phys. Chem. B, 2006, 110, 9899–9907 CrossRef PubMed.
- D. M. Pickup, E. A. A. Neel, R. M. Moss, K. M. Wetherall, P. Guerry, M. E. Smith, J. C. Knowles and R. J. Newport, J. Mater. Sci.: Mater. Med., 2008, 19, 1681–1685 CrossRef PubMed.
- W. B. Kim and J. S. Lee, J. Catal., 1999, 185, 307–313 CrossRef.
- V. Luca, J. Phys. Chem. C, 2009, 113, 6367–6380 CrossRef.
- V. Luca, S. Djajanti and R. F. Howe, J. Phys. Chem. B, 1998, 102, 10650–10657 CrossRef.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00291f |
| This journal is © The Royal Society of Chemistry 2018 |