
Cellulose solvent-based pretreatment for enhanced second-generation biofuel production: a review
Behzad
Satari
 a,
Keikhosro
Karimi
ab and
Rajeev
Kumar
*cde
a,
Keikhosro
Karimi
ab and
Rajeev
Kumar
*cde
aDepartment of Chemical Engineering, Isfahan University of Technology, Isfahan 84156-83111, Iran
bIndustrial Biotechnology Group, Research Institute for Biotechnology and Bioengineering, Isfahan University of Technology, Isfahan 84156-83111, Iran
cCenter for Environmental Research and Technology (CE-CERT), Bourns College of Engineering, University of California Riverside (UCR), Riverside, CA 92507, USA. E-mail: rkumar@cert.ucr.edu; rajeev.dartmouth@gmail.com
dBioEnergy Science Center (BESC), Oak Ridge National Laboratory (ORNL), Oak Ridge, TN, USA
eCenter for Bioenergy Innovation (CBI), Oak Ridge National Laboratory (ORNL), Oak Ridge, TN, USA
First published on 28th September 2018
Abstract
Cellulose, in addition to hemicellulose and lignin, makes the major fraction of lignocellulosic biomass – the only sustainable feedstock to meet the long-term sustainable energy need of the world. Cellulose is soluble in a number of solvents, e.g., concentrated phosphoric acid (CPA), N-methylmorpholine-N-oxide (NMMO), and ionic liquids (ILs), and can be regenerated by an anti-solvent without major derivatization for various applications. For one, the regenerated and much less crystalline cellulose is highly reactive for its biological conversion to sugars, fuels, and chemicals mediated by enzymes and/or microbes. This ability can be used as a core pretreatment step for improved bioprocessing of lignocelluloses. In this comprehensive review, cellulose solvent-based lignocellulosic fractionation technologies for enhanced enzymatic hydrolysis to improve biofuel and renewable chemical production are reviewed. The first part is focused on the background information of lignocellulosic biomass, lignocellulosic derived biogas, biohydrogen, and ethanol as well as acetone, butanol, and ethanol (ABE) production, and enzymatic hydrolysis. In the second part, the conditions for pretreatments applying CPA, NMMO, and IL solvents, improvements in enzymatic hydrolysis rates and yields for solids resulting from application of these pretreatments, and the features of the lignocellulosic structure affecting the improved bioprocessing have been thoroughly reviewed.
1 Different generations and types of biofuels
Recent concerns about climate change due to greenhouse gas emissions and energy crisis have prompted the need for transition from unsustainable fossil-derived energies to sustainable and renewable energies.1Development of sustainable and economically viable biorefinery processes for biofuel production needs to use renewable carbon sources.2 Biofuels produced from food-based crops like sugar- and starch-based substrates, e.g., sugarcane and corn, are considered as first-generation biofuels.3,4 Nevertheless, there is a food-versus-fuel debate in using feedstocks for first-generation fuels. Therefore, next-generation biofuels were introduced and are considered as essential for meeting the world's energy demand in the transportation sector.5–7
Second-generation biofuels are produced from lignocellulosic biomass, which can reduce the carbon emission, increase energy efficiency, and reduce nations' energy dependency.3,7–9 Non-food lignocellulosic substrates are abundant and potentially low-cost organic sources for renewable chemical and fuel production. Lignocellulosic wastes can be originated from industrial wastes (e.g., sawdust, paper mill discards, and food industry wastes), forestry wastes (i.e., hardwoods and softwoods), agricultural residues (e.g., straws, stover, and non-food seeds), domestic wastes (e.g., kitchen wastes, sewage, and waste papers), and municipal solid wastes.10–12
Third-generation biofuels are produced from algae.13,14 Biofuel production from algal species, including Botryococcus braunii, Chaetoceros calcitrans, several Chlorella species, Isochrysis galbana, Nannochloropsis, Schizochytrium limacinum, and Scenedesmus species, is a promising technology since algae are fast growing, compared to many terrestrial plants, with no soil needed, while they have high capturing ability for CO2 and other greenhouse gases.15 Algae contain substantial amounts of carbohydrates and lipids (up to 70%), making them promising feedstocks for conversion to biofuels, e.g., by simple hydrolysis followed by fermentation or consolidated bioprocessing.16 A comprehensive overview of the composition, properties, and challenges of algal biomass for biofuel application was recently presented by Vassilev and Vassileva.17 Biodiesel, bioethanol, biohydrogen, and biogas were reported to be produced from micro- and macro-algae via different technologies.13,14
Fourth-generation biofuels use engineered algae for biofuel production from oxygenic photosynthetic organisms.18 Gaseous biofuels, algal ethanol, algal butanol, four carbon alcohols, and algal biodiesel were reported to be possible to produce by using this technology.18
Nonetheless, the production cost of biofuels is extremely sensitive to the feedstock cost.19 Although algae do not need freshwater and can grow on wastewater streams (e.g., saline/brackish water/coastal seawater), harvesting and carbon supply are the major factors of algal biomass production cost.20 Harvesting microalgae usually needs flocculation to aggregate small algal cells followed by filtration, centrifugation, and sedimentation to separate the algae from the liquid medium. Besides, advanced and cheaper technologies are required for the extraction of algal oil. Although the land use is low for algal cultivation, infrastructure requirements, and mixing and separation costs are still high. Moreover, the high cost of edible crops and land requirements to meet the demand make them unsustainable. Therefore, lignocellulosic biomass is the only sustainable and low-cost feedstock to meet the near future growing energy needs and mitigate environmental problems.20,21
Regarding environmental impacts, all types of biofuels reduce greenhouse gas (GHG) emissions22 (Fig. 1). Life cycle assessment for biofuel production from different sources was performed and the net GHG emission for different fuels, e.g., fossil fuel, soya oil biodiesel, palm biodiesel, sugarcane ethanol, wheat ethanol, corn ethanol, corn stover ethanol, and algal biodiesel, was compared.21,23 It was shown that among them corn stover derived ethanol released the lowest net GHG emission.
 | ||
| Fig. 1 A simplified diagram showing a neutral carbon cycle for biofuel production from plants. | ||
Less (or negligible) competition to food, production of value-added byproducts, and energy security are among the advantages of second-generation biofuels. As shown schematically in Fig. 2, the main steps for second-generation biofuel and chemical production are usually substrate preparation, including size reduction and pretreatment, carbohydrate saccharification, fermentation, and product separation and purification.24 The processing cost for second-generation ethanol is approximately two to three times higher than that for gasoline on an energy equivalent basis;25 therefore, substantial attention has been recently focused on the improvement of process economy and technology development to make second-generation biofuels economically viable.
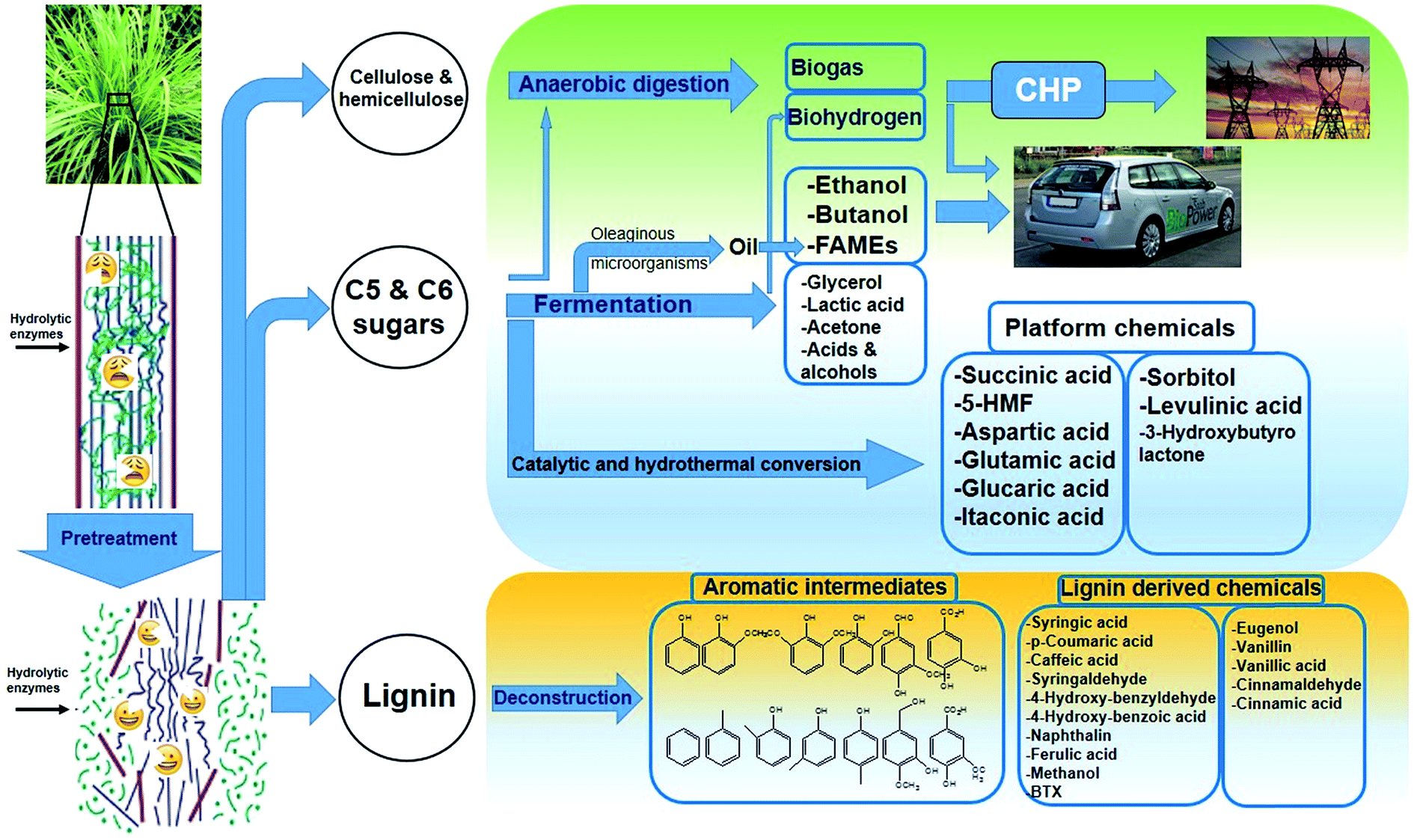 | ||
| Fig. 2 Schematic of the production of various chemicals from lignocellulosic feedstocks (second-generation biofuels and chemicals). | ||
1.1 Bioethanol
Ethanol, blended with gasoline or as a neat fuel in vehicles, is an attractive transportation fuel, giving a high octane number and heat of vaporization.26 Currently, ethanol mainly produced by fermentation routes using sugar- and starch-based feedstocks, e.g., sugarcane and maize, is called first-generation ethanol27 (Fig. 3). Following fermentation, ethanol is separated and purified from the fermentation broth via distillation and molecular sieves, respectively.28 The industrial technology for the fermentation of glucose to ethanol is quite robust and high concentrations of ethanol (12–15%) can be achieved.29 In the production of ethanol from starch, an extra step of liquefaction and saccharification by α-amylases and glucoamylases, respectively, is necessary for converting starch to sugar30 (Fig. 3). Since the production capability of the first-generation ethanol is limited and is unsustainable on a large scale, second-generation ethanol was then introduced, which utilizes a variety of lignocelluloses as substrates.27,31 (Fig. 3).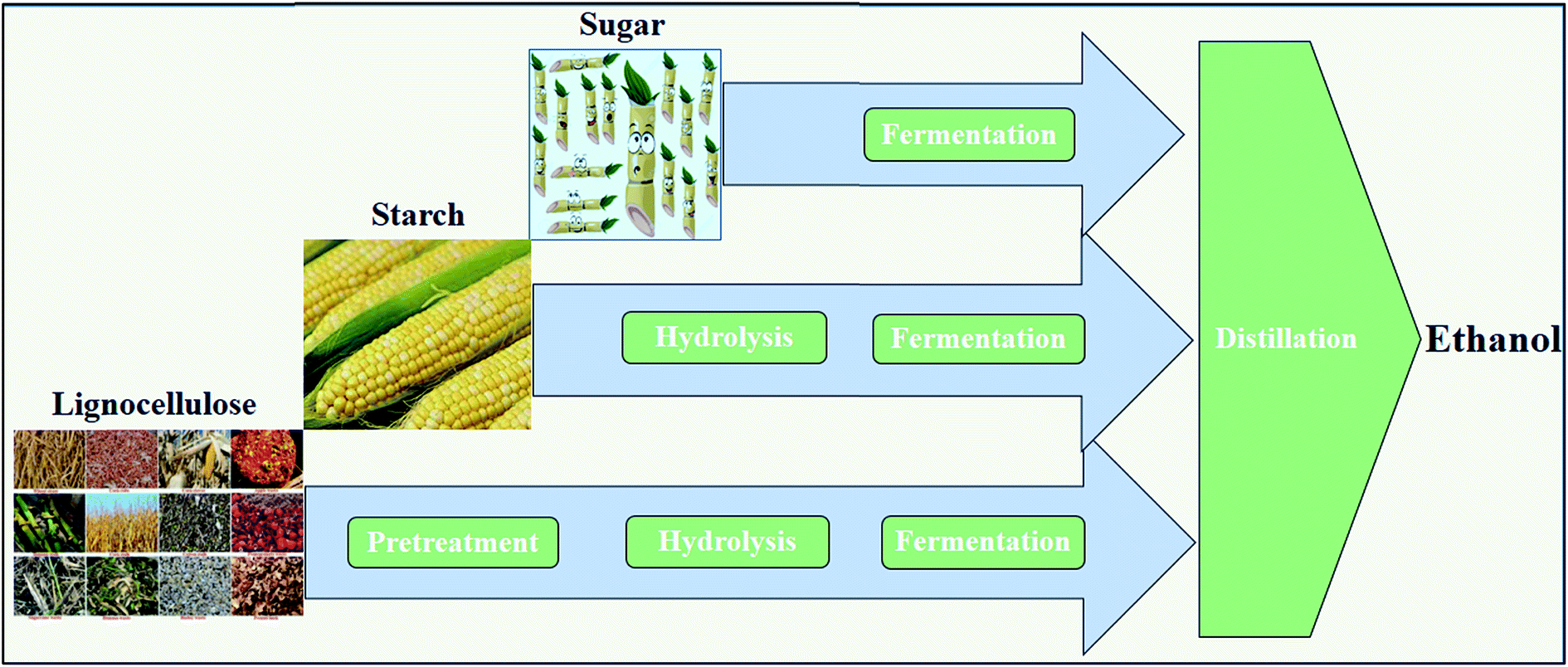 | ||
| Fig. 3 Conversion of different feedstocks to ethanol via the fermentation route. The conversion from sugar- and starchy-based materials to ethanol is called first-generation and production from lignocelluloses is called second-generation. | ||
1.2 Biobutanol
For gasoline blending, butanol, a four-carbon alcohol, is more desirable than ethanol due to its higher energy density, lower hygroscopicity, lower Reid vapor pressure, better blending ability, and use in conventional combustion engines without modification.32 Besides the fuel extender, biobutanol can be used as a feedstock for the synthesis of a variety of commercial products.33,34 The fermentative route of production, e.g., by microorganisms that belong to the genus Clostridium, is more sustainably and environmentally attractive than the petrochemical route.35 These microorganisms typically produce a mixture of different solvents, mainly including acetone, ethanol, and butanol; thus, the process is referred to as acetone–butanol–ethanol (ABE) fermentation.36,37 However, the major challenge in the microbial production of butanol is the low butanol titer due to product inhibition.38,39 Several strategies have been reported to address these issues40 such as genetic and metabolic engineering of microorganisms40 and promising integrated continuous culture technology with efficient product recovery techniques, e.g., using metal–organic frameworks,41 liquid–liquid extraction,42–44 pervaporation technique,45 and gas stripping.46Butanol can be synthesized via different metabolic and engineered pathways from different substrates. Starch/sugars can be converted to butanol via the clostridial route that includes glycolysis, pyruvate:ferredoxin oxidoreductase, thiolase, 3-hydroxybutyryl-CoA dehydrogenase, crotonase, butyryl-CoA dehydrogenase, and butyraldehyde/butanol dehydrogenase. The conversion of lignocellulosic feedstocks to biobutanol also follows the same route after being converted to C5 and C6 sugars in the preceding pretreatment and/or enzymatic saccharification steps. Lignocellulosic biobutanol production has received a lot of attention, and it has recently been the focus of vast studies.47,48 However, the low butanol titers and yields and requirement of extra pretreatment and enzymatic saccharification steps are some of the challenges in butanol production from lignocellulosic biomass. Moreover, syngas or CO2/H2 can also be fermented to butanol via the clostridial pathway.49 For starch and sugars, there is another non-fermentative pathway based on amino acid metabolism plus 2-keto acid decarboxylase and alcohol dehydrogenase.36 Aerobic butane-utilizing bacteria use monooxygenase to oxidize butane to a butanol mixture (95% butanol, 5% iso-butanol).36
1.3 Biodiesel
Biodiesel, a mixture of fatty acid methyl esters (FAMEs), can be produced by transesterification of vegetable oil or animal fat. It recently received much attention as a renewable source of energy.50,51 However, these resources for biodiesel production do not meet the large-scale demands for transportation fuel and a sustainable renewable source is required.Nonetheless, some microorganisms, called oleaginous microorganisms, can store intracellular lipids, usually referred to as single cell oil (SCO), especially triacylglycerols (TAGs).51 Microbial oil, as a raw feedstock for biodiesel production, is advantageous compared to vegetable oil because it has a short life cycle, requires less labor, is less affected by venue, season, and climate, and is easier to scale up.52 Different oleaginous microorganisms, including microalgae, yeasts, fungi, and bacteria, were reported to produce substantial amounts of SCO (e.g., 20–50% dry cell weight).53–55 However, it is possible to increase lipid accumulation in oleaginous microorganisms via metabolic engineering technology, involving the enhancement of fatty acid synthesis approaches, enhancement of TAG synthesis approaches, regulation of related TAG biosynthesis bypass approaches, blocking of competing pathways, and multigene approaches.56
A variety of carbon sources from lignocellulose-based carbohydrates and other low-cost industrial wastes, e.g., glycerol, food processing waste, and even wastewater, have been reported to be assimilated by oleaginous microorganisms to produce lipids.57–61 Auxiliary nutrients such as phosphorous and nitrogen are available from the waste streams. However, lipid accumulation in oleaginous microorganisms is usually triggered by nutrient starvation, e.g., nitrogen or phosphorus, relative to the carbon source.62
Lipid production from lignocellulosic biomass has attracted substantial attention in recent years and many research studies have focused on its commercialization; however, substantial process improvements and reduction in the production cost are required.63–66
1.4 Biogas
Besides liquid biofuels, biomass with high organic content can be converted to another form of energy, biogas, via anaerobic digestion (AD). In this process, the organic matter is biologically decomposed by an assortment of microbes under oxygen-free conditions, producing biogas (about 50–75% CH4 and 25–50% CO2).67,68 The AD process can be divided into four steps: (i) hydrolysis of proteins and lipids to amino acids and long-chain fatty acids and carbohydrates into sugars, (ii) conversion of hydrolysis products and monomers to volatile fatty acids (VFAs) and other minor products such as alcohol by acidogenic bacteria, (iii) conversion of VFAs to acetate, carbon dioxide, and/or hydrogen by acetogenic bacteria, and (iv) methane formation from other stage products by methanogenesis69 (Fig. 4). Although methanogenesis is usually considered as the rate-limiting step in the AD process for a number of substrates, the hydrolysis step is believed to be the limiting step for lignocelluloses. Sawatdeenarunat et al.70 classified the current technologies in the AD process of lignocellulosic biomass into anaerobic co-digestion, solid-state anaerobic digestion (SS-AD) (more than 15% TS) and using alternative biological pretreatment of feedstock for further biological conversion to sugars, e.g., by using rumen microorganisms.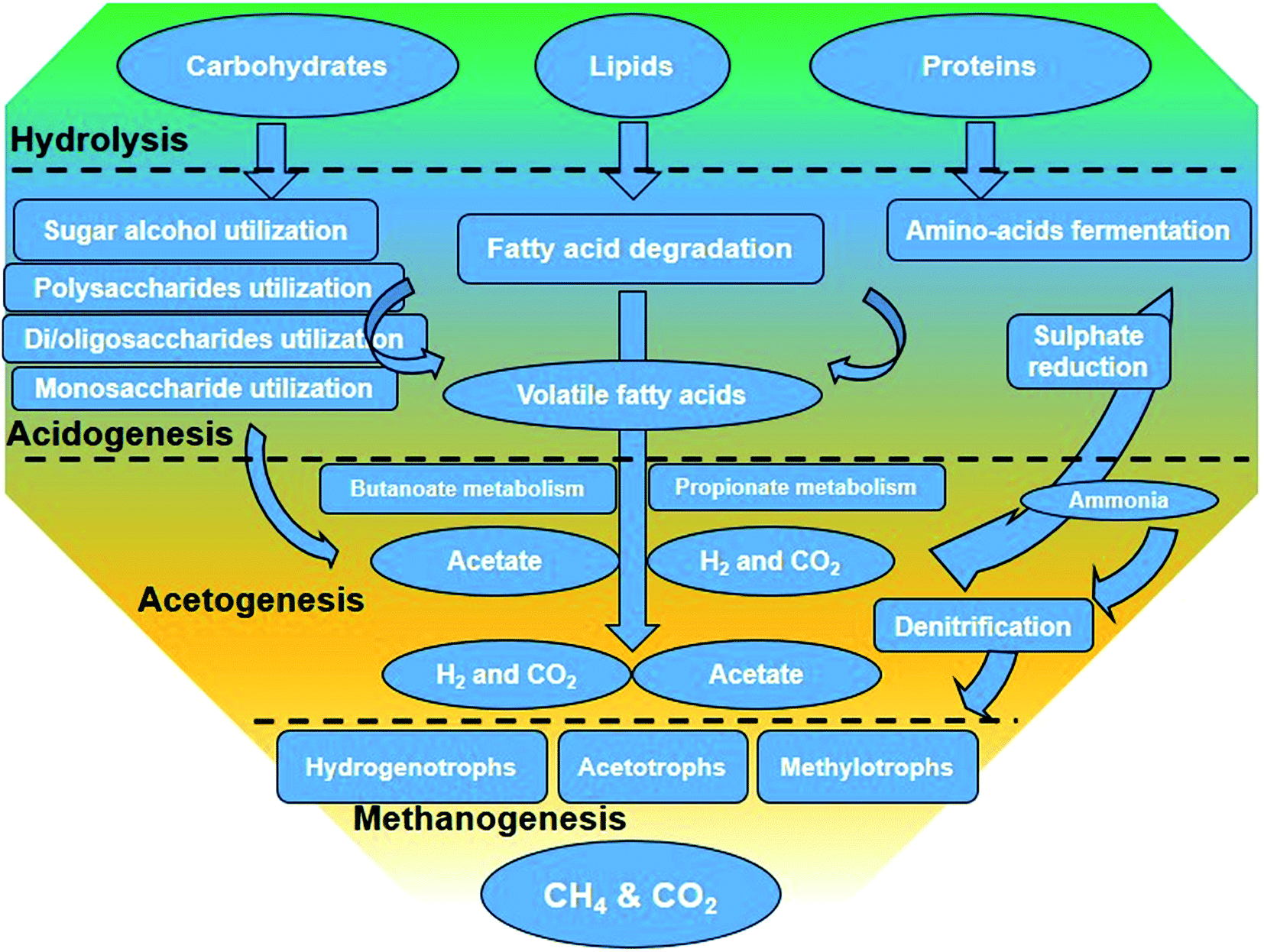 | ||
| Fig. 4 The main steps in the degradation of organic matter through the anaerobic digestion process.71 | ||
1.5 Biohydrogen
Biologically produced hydrogen, biohydrogen, is recently becoming of great interest as a renewable energy carrier, because hydrogen utilization for combustion, in fuel cells, and/or electricity production produces no carbon byproducts.72 Biological pathways for hydrogen production are primarily divided into photobiological processes and light independent methods.73,74 Green algae from the genera Chlamydomonas, Scenedesmus, Lobochlamys, and Chlorella can reduce protons of water in the presence of light to produce mixed oxygen and hydrogen gases.75 Some photosynthetic bacteria were also reported to produce hydrogen by the same mechanism of biophotolysis as that by green algae. Fermentative biohydrogen production, classified as photofermentation and dark fermentation, can be performed by a wide variety of microorganisms, e.g., strict anaerobes, facultative anaerobes, and aerobes kept under anoxic conditions.73,76 Fermentative hydrogen production is more advantageous over the photosynthetic method since various organic feedstocks can be converted to hydrogen with high production rates and simple operations.77 Several factors, including inoculum, i.e., mixed and pure cultures, substrates, reactor type, availability of nitrogen and phosphate micro-nutrients and metal ions, temperature, and pH, were reported to influence fermentative hydrogen production.78,79 Because of the higher hydrogen evolution rate, dark fermentation hydrogen production is more commercially feasible than photofermentation. In dark fermentation, organic substrates like glucose are converted by facultative and obligate anaerobes to hydrogen, volatile fatty acids, and carbon dioxide operated at mesophilic, thermophilic, or hyperthermophilic temperatures in the absence of light.80 The knowledge of biological pathways for dark fermentation hydrogen production is quite mature and is comprehensively presented in the literature.73,75,76,80–85 Here a brief discussion on the strategies to enhance biological hydrogen production and the feedstocks is presented.Different carbon sources, e.g., agricultural residues, industrial waste, organic fraction of municipal waste, and pure sugars, were reported as feedstocks for biohydrogen production.72,86,87 Lignocellulosic feedstocks are promising raw materials for biohydrogen production and recently have been the focus of a number of studies.72 Different approaches for bioconversion of lignocellulosic biomass to H2, i.e., separate hydrolysis and fermentation, simultaneous saccharification and fermentation, and consolidated bioprocessing of lignocellulosic biomass to H2, have been discussed by Cheng et al.72 and Ren et al.88 Application of various pretreatment technologies for enhanced lignocellulosic bioconversion to biohydrogen has also been the topic of several studies.89–93
While the theoretical hydrogen yield is 12 mole H2 per mole of glucose, natural and genetically modified microorganisms can produce hydrogen at a maximum yield of 4 mol mol−1 glucose when acetic acid is the only VFA product.85 The strategies for biohydrogen production improvement include microbial culture immobilization, bioreactor modifications, optimization of operating parameters (i.e., temperature, pH, organic loading rate, hydrolytic retention time, and H2 partial pressure), substrate type and inorganic nutrients, metabolic engineering of microbes, and cogeneration of biohydrogen and biomethane.73,78,81,94,95
The inoculum for dark fermentation biohydrogen production can be either pure cultures or anaerobic microbial consortia. Mixed culture is generally preferable because of the ease of operation, no need for sterilization, and, especially for lignocelluloses, the presence of hydrolytic activities.96 In such systems, methanogenesis activity can be easily eradicated by a heat shock or pH control, and the hydrogen-producing bacteria can sporulate.74,97
Another noteworthy approach based on cell-free hydrogen production was originally proposed by Dr Jonathan Woodward at Oak Ridge National Laboratory,98,99 and then has recently been revived by Ye et al.100 and Zhang et al.101
2 Lignocellulosic biomass structure
Lignocelluloses typically contain lignin, carbohydrate polymers (∼75%; i.e., cellulose, hemicellulose, and pectin), acetate, proteins, salt, ash, and minerals.102Table 1 summarizes the major composition (carbohydrates and lignin) of some lignocelluloses used for second-generation biofuel production. Being the nature's most abundant organic substance after cellulose, lignin comprises 28–30% of woody gymnosperm stems and 20–24% of woody angiosperms.103 The lignin composition varies between hardwoods and softwoods. Lignin has a heterogeneous three dimensional β-O-4, β-5, β-1, β-β, 5–5, and 4-O-5 linked structure of phenylpropane units, e.g., p-hydroxycinnamyl, p-coumaryl, coniferyl, guaiacyl, syringyl, and sinapyl alcohol.102,104 Lignin acts as a cement to hold the cell components together and provides the biomass integrity.105| Biomass type | Substrate | Glucan | Xylan | Mannan | Galactan | Arabinan | Lignin | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Acid insoluble | Acid soluble | ||||||||
| a The carbohydrate contents were measured by analyzing the sugars released during a concentrated sulfuric acid (72%) hydrolysis at 30 °C followed by a dilute acid treatment at 121 °C to cleave the carbohydrates to monomeric sugars. Acid-insoluble lignin was measured gravimetrically after subtracting the ash content of final acid-insoluble materials.128 | ||||||||||
| Hardwood |

|
41.7 | 14.3 | 2.6 | 3.2 | 2.0 | 30.2 | 114 | ||

|
45.2 | 20.3 | 4.2 | — | — | 21.0 | 3.3 | 115 | ||

|
39.2 | 13.1 | 1.8 | 0.9 | — | 14.7 | 116 | |||

|
40.3 | 16.9 | 1.7 | 0.6 | 0.3 | 20.3 | 117 | |||

|
49.0 | 14.9 | 2.0 | 0.5 | 0.8 | 24.6 | 1.0 | 118 | ||

|
43.6 | 20.3 | 1.5 | 1.2 | — | 26.3 | 119 | |||
| Softwood |

|
47.3 | 4.4 | 11.7 | 2.2 | 1.2 | 29.8 | 0.5 | 120 | |

|
38.2 | 8.5 | 11.3 | 4.3 | — | 29.5 | 4.9 | 121 | ||

|
38.5 | 5.0 | 11.7 | 1.9 | 1.3 | 28.5 | 117 | |||
| Agricultural residues and grasses |

|
32.0 | 17.9 | — | 1.73 | 1.78 | 21.4 | 122 | ||

|
37.4 | 22.4 | — | 0.51 | 6.2 | 13.2 | 1.9 | 123 | ||

|
38.8 | 22.2 | 1.7 | 2.7 | 1.4 | 18.5 | 124 | |||

|
40.87 | 20.82 | — | — | 1.53 | 24.81 | 125 | |||

|
35.3 | 23.9 | — | 1.9 | 4 | 19.2 | 0.7 | 126 | ||

|
41.33 | 17.96 | 0.85 | 1.26 | 1.96 | 16.4 | 1.78 | 127 | ||
Cellulose, with over 1011 metric tons production per year, is composed of linear chains of several hundreds to over tens of thousands of β-D-glucopyranose residues linked by β-1,4 glycosidic bonds with numerous inter- and intra-molecular hydrogen bonds.106 It is a ubiquitous polysaccharide of the plant cell wall (Fig. 5), which makes it insoluble in water and common organic solvents.104,107,108 Aggregation of cellulose chains forms nanofibrils and a 5–10 nm microfibril, hypothesized to be composed of 36 chains of cellulose, is used to define the next level of aggregation, which is observable via high magnification microscopy, e.g., electron microscopy, and atomic force microscopy109–112 (Fig. 5). Cellulose is the dominant component of the primary cell wall (20–40% of cell wall dry matter).113 The research on cellulose revealed that native celluloses are crystalline and are composites of two forms, Iα (with one-chain triclinic structure) and Iβ (a two-chain monoclinic structure), which coexist in all native forms.107
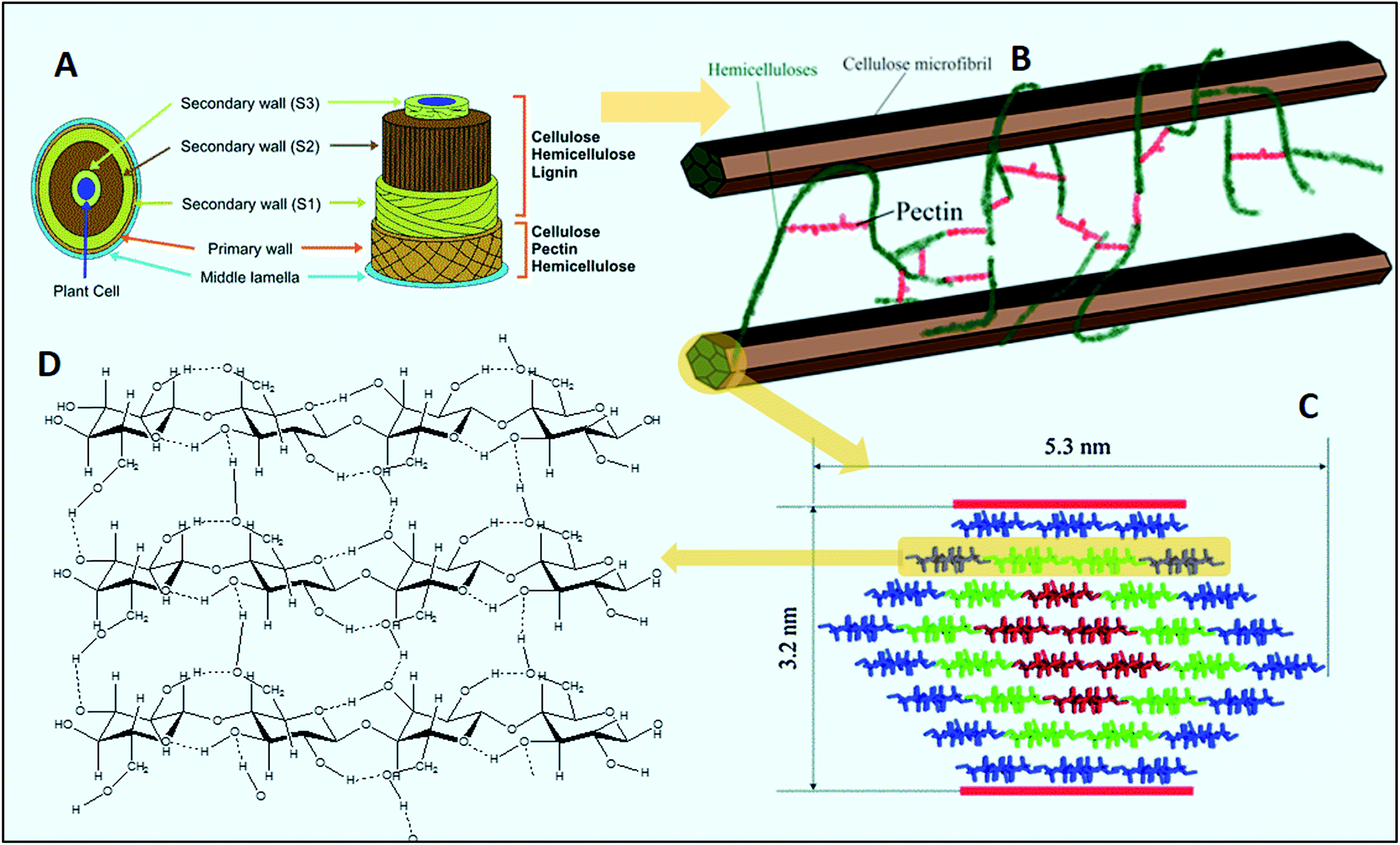 | ||
| Fig. 5 (A) Pictorial illustration of the lignocellulosic biomass framework (modified from Menon and Rao137 with permission), (B) a simplified model showing the interaction of carbohydrate polymers present in the cell wall, modified from Himmel et al.,138 (C) structure of a 36-chain model for the cellulose Iα or Iβ elementary fibril (the reds show six true crystalline chains; greens are 12 subcrystalline chains with a small degree of disorder; the blues are 18 surface chains that are subcrystalline with a large degree of disorder, taken from ref. 111), and (D) a model of inter- and intra-chain hydrogen-bonding patterns in cellulose, taken from ref. 139 with permission. | ||
Hemicellulose, a stereo-irregular polysaccharide, is a heterogeneous plant cell wall polymer composed of linear β(1,4)-D-glycan backbones branched with one monosaccharide and/or small oligosaccharides, with an approximate degree of polymerization of 200.129–131 Unlike cellulose, hemicellulose has an amorphous, random, and branched structure, which is more susceptible to thermal, biological, and acid hydrolysis.132–135 Xylan, mainly in the form of heteroxylan, is usually substituted with acetate and arabinose residues. It is the most abundant hemicellulose in nature, which dominantly contains β-D-xylopyranosyl residues linked by 1,4 glycosidic bonds.102,104 The xylan content of the plant cell wall may vary depending on the biomass type, ranging between 15 and 35% of total dry weight.102 Hemicellulose interacts with cellulose and lignin and build a rigid network structure which is a barrier to enzyme-catalyzed deconstruction of cellulose.136
Pectin (pectic polysaccharides) is a heterogeneous polysaccharide with a dominantly methyl esterified or de-esterified homogalacturonan (HG) backbone. Located in the cell wall and middle lamella of plants, pectin is the major component of the primary walls of several non-woody plant cells.140,141 After cellulose, pectin acts as a major plant load-bearing component and plays a “glue” role in holding cell-wall components together.138,142–145
3 Biomass recalcitrance and pretreatment
Lynd et al.146 first defined the “biomass recalcitrance” as the natural resistance of lignocelluloses and their components to microbial and enzymatic deconstruction. Later, Himmel et al.138 summarized the factors contributing to the biomass recalcitrance as “(i) epidermal tissue of plant body, especially cuticle and epicuticular waxes, (ii) the arrangement and density of the vascular bundles, (iii) the relative amount of sclerenchymatous (thick wall) tissue, (iv) the degree of lignification, (v) the structural heterogeneity and complexity of cell-wall constituents such as microfibrils and matrix polymers, (vi) the challenges for enzymes acting on an insoluble substrate, and (vii) the inhibitors to subsequent fermentations that exist naturally in cell walls or are generated during conversion processes”. Due to the biomass inherent recalcitrance, the release of fermentable sugars via appropriate enzymatic hydrolysis as well as microbial hydrolysis is the bottleneck of the industrial lignocellulosic biorefineries.147,148Therefore, an efficient pretreatment step is required to obtain renewable chemicals and fuels from lignocelluloses.149 A suitable enzymatic or acid hydrolysis can then be applied to the pretreated substrates to convert them to fermentable sugars or the AD process can be applied to obtain biogas. There are many reviews in the literature on pretreatment methods to enhance the enzymatic digestibility of lignocellulosic feedstocks.11,69,136,150–157 Pretreatment is a “physical”, “chemical”, “physico-chemical”, or “biological” process, which can open up the lignocellulosic recalcitrance structure and make it amenable for subsequent enzymatic/microbial degradation. Physical pretreatments are divided into mechanical comminution and pyrolysis, whereas physicochemical pretreatments are steam explosion, ammonia fiber expansion (AFEX), and carbon dioxide explosion, and chemical pretreatments can be categorized into ozonolysis, acid hydrolysis, alkaline hydrolysis, oxidative delignification, and organosolv processes.11,158,638,639 The two most commonly used technologies for pretreatment of lignocelluloses are dilute acid and alkaline pretreatments.159 Dilute acid and alkaline pretreatments mainly target hemicellulose and lignin fractions, respectively, in lignocellulosic biomass. Acids like HCl and H2SO4 and bases like sodium hydroxide and sodium carbonate are mostly employed, and the pretreatment temperature, time, and acid/base concentration are among the main factors determining the effectiveness of pretreatment. An additional process and/or additional chemicals are required for recovering and neutralizing the hydrolysates and removing the inhibitory compounds for downstream processes. Hydrothermal pretreatment with only hot water, which is performed by using saturated steam at temperatures and pressures below the water critical point (subcritical water) or supercritical water, has the advantages of a low amount of biological inhibitor production, minimal chemical cost, and relatively low cost of reactors compared with using acid or alkali solutions. A technology used for hydrothermal pretreatment, called steam explosion, is a pretreatment in which the lignocellulosic biomass is heated up by high-pressure steam (160–240 °C and pressures 0.7–4.8 MPa) followed by an explosive decompression. Hemicelluloses are mostly hydrolyzed in this pretreatment via the reaction called “autohydrolysis”.160,161
For an advanced and low-cost pretreatment, several key criteria should be considered. It should be effective for a variety of lignocellulosic types with different characteristics. Significant sugar degradation products, formation of inhibitory byproducts for subsequent sugar fermentation, and production of waste residues should not occur during the pretreatment. Moreover, the pretreatment should need minimum heat and power requirement and reasonable size and moderate cost reactors.150,153,162
An efficient biomass pretreatment strategy should, therefore, be capable of effectively disrupting and removing the linkages among cellulose, hemicellulose, and lignin present in the plant cell walls. Furthermore, reordering or removing highly ordered hydrogen bonds in cellulose fibers and subsequently increasing the porosity and surface area, resulting in cellulose accessibility to cellulase, are highly desirable traits of an effective pretreatment.150,153,162
Recently, a new pretreatment category based on cellulose solvent lignocellulosic fractionation, meeting the desired criteria, was added to the traditional biomass pretreatments. A number of low toxicity and mostly environmentally friendly solvents, including N-methyl-morpholine-N-oxide (NMMO), ionic liquids (ILs), LiCl/N,N-dimethylacetamide (LiCl/DMAc), aqueous NaOH solution, alkali/urea and NaOH/thiourea aqueous solutions, tetra butyl ammonium fluoride/dimethyl sulfoxide system, metal complex solutions, concentrated phosphoric acid, and molten inorganic salt hydrates, have been introduced as cellulose solvents for regenerating cellulosic materials.163–165 The cellulose solvents can be classified into (i) derivatizing, (ii) non-derivatizing, and (iii) aqueous and non-aqueous systems having the ability to eliminate the inter- and intra-molecular hydrogen bonds among cellulose molecules.166 The cellulose can then be recovered using an anti-solvent such as water, ethanol, or acetone. The parallel arrangement of cellulose I, in most regenerated celluloses, is irreversibly converted into an anti-parallel orientation, cellulose II, which is much easier to hydrolyze using cellulases.167 Cellulose II is thermodynamically more stable and has a more dense packing structure than cellulose I.168 However, as examined by Wada et al.,169 the hydrolysis of cellulose II (and especially its hydrate form) proceeds faster than the hydrolysis of cellulose I. Changes in polarity, crystallinity, and ultrastructure of cellulose I to cellulose II have been reported to be the factors responsible for cellulose II faster hydrolysis.167
While some of the traditional pretreatments suffer from relatively low sugar yields, require severe reaction conditions (high temperature and/or high pressure), and result in the formation of fermentation inhibitory compounds, cellulose solvent-based pretreatments can be performed under relatively mild conditions (100–160 °C), resulting in an insignificant amount of inhibitors from degradation of cellulose and hemicelluloses.170,171 Cellulose solvent-based fractionations are regarded as biomass-independent or feedstock agnostic pretreatments, which can break the recalcitrant structure of biomass by increasing the cellulose accessibility more than the traditional pretreatments.172 The recovery of non-fermentable co-products, e.g., pure and unaltered lignin, in these methods, adds revenue streams to the fermentation products.173,174 The use of cellulose solvents over traditional solvent systems, which are typically (e.g., ethanol) volatile, for biomass pretreatment is promising for the future of lignocellulosic biorefineries.
This review paper has mainly focused on the most promising cellulose solvent-based pretreatment, i.e., concentrated phosphoric acid (CPA), N-methyl-morpholine-N-oxide (NMMO, or NMO), and ionic liquids (ILs). Although a few other reviews are available in the literature,172,175–186 this review is intended to be a comprehensive review, with the focus on recent research on cellulose solvent-based pretreatments to improve the reactivity of lignocelluloses for biogas, ethanol, and renewable chemical production. Furthermore, as the pretreatment is a preceding step to microbial conversion mediated by enzymes, the basic concepts and the limiting factors in the enzymatic hydrolysis of lignocelluloses are also briefly reviewed.
4 Hydrolysis of pretreated lignocellulosic substrates
The hydrolysis of lignocelluloses has long been done by dilute and concentrated acids, e.g., sulfuric acid.187,188 The main drawback of acid hydrolysis is degradation of sugars and formation of byproducts that showed severe inhibition to fermentation microorganisms. High investments and maintenance cost, high utility and disposal costs, high energy consumption for acid recovery, and environmental impacts are among the major disadvantages of acid hydrolysis.189 Hydrolysis of lignocellulosic materials by “enzymatic” processes has emerged as a prominent process for the production of monomeric sugars, e.g., for subsequent production of fuel ethanol.190,191 Cellulases and hemicellulases are the two enzymes typically used for the depolymerization of lignocellulosic carbohydrates to fermentable sugars for second-generation biofuel production. Although a lot of efforts have been made to reduce the production costs, the enzymes are still expensive.192,193Cellulose can be hydrolyzed by three glycoside hydrolases: endo-1,4-β-D-glucanases (EG) (EC 3.2.1.4), which randomly hydrolyze internal β-1,4-glucosidic bonds in the cellulose microfibril; exo-1,4-β-D-glucanases or cellobiohydrolases I and II (CBH) (EC 3.2.1.91), which progressively convert cellulose into cellodextrins; and 1,4-β-D-glucosidases (EC 3.2.1.21), which hydrolyze cellobiose and cellodextrins to glucose.139,194–196 In a synergistic mixture, cellulases have higher combined activities than the sum of their individual activities.197 Cellulases typically have two separate domains: a catalytic domain (CD) and a cellulose binding module (CBM), comprised of approximately 35 amino acids, linked by a flexible linker region.198 Over the years, several kinetic models for lignocellulosic biomass hydrolysis by cellulase have been proposed and developed199–201 to understand the mechanisms. For example, recently a comprehensive model was developed by Bansal et al.202 that included the following steps: (i) adsorption of cellulases onto the substrate via the binding domain, (ii) direction of cellulases to a bond (located on the chain end or cleavable bond) susceptible to hydrolysis on the substrate surface, (iii) formation of the enzyme–substrate complex, (iv) hydrolysis of the β-glycosidic bond and simultaneous direction of the enzyme to the cellulose chain, (v) desorption of cellulases from the substrate or repetition of step iv or steps ii/iii if only the catalytic domain detaches from the chain, and (vi) hydrolysis of cellobiose to glucose by β-glucosidase (if available in the enzyme mixture). However, the exact mechanism of cellulose hydrolysis mediated by fungal cellulases is still unknown as the binding mechanism of the binding module to cellulose, catalytic action of cellulase, and stimulation of cellulose hydrolysis by CBMs are still not clearly understood.203
The catalytic domains in cellulase are connected to one or more CBMs by peptide linkers of varying length and structure.139,204 CBMs, with high binding affinity, increase the interaction between cellulase and the cellulose surface and enhance enzyme penetration into the substrates.139,205,206 Several synergistic proteins, e.g., plant expansins and expansin-like proteins such as swollenin,207 and auxiliary activity family 9 (formerly GH61) proteins,208,209 are able to enhance the enzymatic hydrolysis of cellulose by cellulase in ways that are not yet clearly understood.210
Hemicellulases refer to a diverse combination of enzymes that can synergistically hydrolyze hemicellulose from mixed sources and are divided into two major categories: depolymerases and debranching enzymes (accessory enzymes).195,211 The former group is either endo-acting enzymes, that attack polymer chains internally, or exo-acting enzymes that act processively212 from the reducing or non-reducing terminals.213 Depolymerases mainly include xylanases, mannanases, β-glucanases, and xyloglucanases, and debranching enzymes are α-glucuronidase, α-arabinofuranosidase, α-D-galactosidase, acetyl xylan esterase, and ferulic acid esterase.211,214
Várnai et al.215 reported that the synergistic action of xylanase and mannanase can improve the total hydrolysis of pretreated softwood. Synergism is defined as “the ratio of the rate or yield of product released by enzymes when used together to the sum of the rate or yield of these products when the enzymes are used separately in the same amounts as they were employed in the mixture”.216 It depends on both the ratio of the enzymes involved and the characteristics of the enzymes and substrate.102 Synergism, as reviewed by Van Dyk and Pletschke,102 can be grouped into cellulase component interaction, as mentioned earlier, hemicellulase interaction, and combined enzymes on complex substrates.
For degradation of lignocelluloses, many aerobic bacteria and fungi, e.g., Acidothermus cellulolyticus, Trichoderma reesei, and Aspergillus niger, produce free enzymes. Nonetheless, some anaerobic bacteria from genera of Clostridium, Acetivibrio, Bacteriodes, and Ruminicoccus are capable of producing multi-enzyme extracellular protein complexes, called cellulosomes, which can degrade cellulose, hemicellulose, and pectin.102,139,198,205,217 The most important characteristic difference between cellulosomes and free enzyme is cohesion-containing scaffoldin(s) and dockerin-containing enzymes (hemicellulases, cellulases, and pectinases).218,219 Besides, free non-cellulosomal enzymes usually contain a CBM that attach to the substrate. The structure and function of cellulosomes and their differences with free enzymes have been reviewed by Bayer et al.220
The enzymatic hydrolysis and fermentation can be conventionally performed by separate enzymatic hydrolysis and fermentation (SHF) or via an integrated process, i.e., simultaneous saccharification and fermentation (SSF), non-isothermal simultaneous saccharification and fermentation (NSSF), simultaneous saccharification, filtration, and fermentation (SSFF), or simultaneous saccharification and co-fermentation (SSCF)221 (Fig. 6). Although a recent study reported a higher ethanol yield by SHF over SSF at very high solid concentration by using new preparations of a cellulolytic enzyme, Cellic® CTec2,222 integrated approaches were developed to enhance the overall ethanol yield by reducing the inhibitory effect of sugar released during the hydrolysis process on enzymes.223 Another approach, called consolidated bioprocessing (CBP), can convert biomass to biofuel by using anaerobic bacteria capable of producing cellulosome enzymes with high activity and ferment the resulting sugars to, e.g., ethanol, in a single step.192,203,219 Although CBP is a more effective process than the others, it is in the developing stage and further developments in metabolic and genetic engineering are required to meet the industrial requirements.
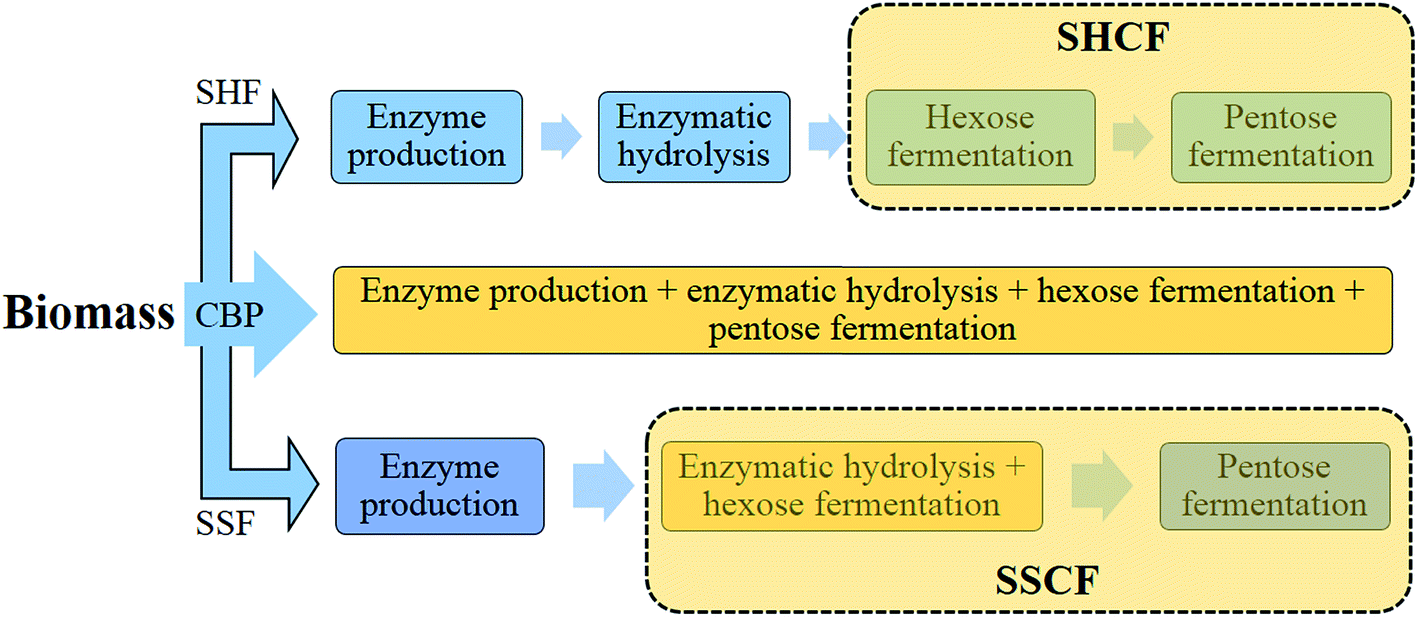 | ||
| Fig. 6 Different strategies for hydrolysis and fermentation of lignocellulosic substrates (SHF: separate hydrolysis and fermentation; SSF: simultaneous saccharification and fermentation; CBP: consolidated bioprocessing; SHCF: separate hydrolysis and co-fermentation; and SSCF: simultaneous saccharification and co-fermentation). | ||
Commercial enzymes are usually a cellulase mixture derived from fungi such as T. reesei supplemented with β-glucosidase and contain more than 80 proteins.195,224 Novozymes is one of the companies that provide enzymes for process optimization and commercialization of cellulosic ethanol. In this regard, the company started a dedicated work in 2000, under a national renewable energy laboratory (NREL) subcontract funded by the United States Department of Energy (DOE), to reduce the cost of cellulases.203 In 2007, the company estimated 40–100 times higher cost for the hydrolytic cellulase enzyme than the cost of enzymes for starch hydrolysis to glucose on a per gallon ethanol basis.157 The outcome of the work was Cellic CTec & Cellic HTec enzyme cocktails in March 2009 followed by an improved and cost-effective product, Cellic® CTec2, in February 2010, and the company reported a 35% lower enzyme price. The company then developed a new generation of enzyme, called Cellic® CTec3, with 1.5 times better performance than the previous best product in the market. Cellic® CTec3 has been shown to work across a variety of feedstocks with a consumption of approximately 50 kg of Cellic® CTec3 to produce 1 ton of ethanol (https://www.novozymes.com/). The cellulase assays usually measure the production of reducing sugars from high molecular weight cellulose,225 like Whatman 1 filter paper, as first developed by Ghose226 and later adopted and modified by NREL.227 The protein content of the enzymes is also of great interest, which is usually measured by the Bradford assay,228 Pierce BCA assay,229 and total crude protein by Kjeldahl nitrogen analysis.230
Equivalent glucose yield, proposed by the NREL, as % of theoretical yield (% cellulose or glucan digestibility) is usually calculated by using eqn (1):
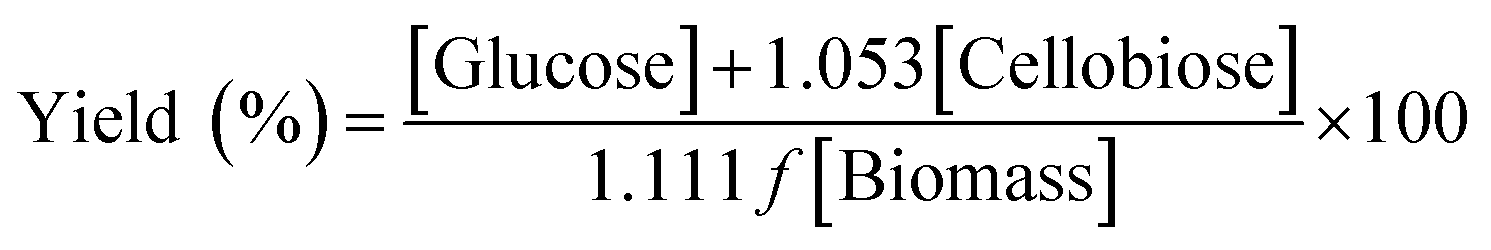 | (1) |
5 Obstacles in the enzymatic hydrolysis of lignocelluloses and the role of pretreatment
The enzymatic hydrolysis performance of lignocelluloses is affected by not only cellulolytic enzyme-related factors (discussed in Section 4) but also by the physical, chemical, and morphological characteristics of the lignocellulosic materials.151,232–234 Cellulose crystallinity, structure, degree of polymerization (DP), and accessibility, as well as hemicellulose and lignin contents are among the main structural and physicochemical features of cellulosic substrates that control the rate and extent of enzymatic hydrolysis.113,235–238 Among all the factors that control cellulose hydrolysis mediated by fungal enzymes and to an extent, by cellulolytic organisms such as Clostridium thermocellum and other microbes, cellulose accessibility to enzymes/microbes is believed to be the main factor affecting cellulose deconstruction.113,172,239,240 However, tracking only one factor governing biological conversion is practically impossible because an increase in cellulose accessibility in biomass is usually accompanied by hemicellulose and lignin removal and/or reduction in cellulose crystallinity.5.1 Cellulose crystallinity and degree of polymerization (DP)
Cellulose microfibrils exist in different polymorphs, i.e., crystalline, paracrystalline (disordered), and amorphous structures. Amorphous cellulose is much easier to hydrolyze than crystalline cellulose.241 One of the major obstacles for efficient hydrolysis of cellulose mediated by fungal enzymes is the cellulose crystalline structure since lignin- and hemicellulose-free substrates, e.g., cotton fibers, still show resistance to enzymatic degradation.242 However, based on findings in the literature, the correlation between the cellulose crystallinity and enzymatic hydrolysis rate and yield is still debatable.243–247Although cellulose accessibility and enzyme adsorption can be affected by cellulose crystallinity, lignin/hemicellulose contents and distribution, biomass porosity, and biomass particle size can also affect the accessibility.243 Besides, some reports have stated a constant crystallinity for cellulose during the course of hydrolysis;244 while others reported a decrease in cellulose crystallinity during hydrolysis.245Reported by Hall et al.,246 at constant adsorbed enzyme concentration, crystallinity was found to be a more influencing factor for enzymatic hydrolysis rates than enzyme adsorption. Mittal et al.247 found a strong correlation between the initial rate of digestion (up to 24 hours) and the amorphous content for four cellulose samples with different degrees of polymerization and crystallinity indexes, which were subjected to aqueous sodium hydroxide and anhydrous liquid ammonia treatments. Besides, they reported a weak correlation of allomorph type with initial digestibility; however, a strong correlation with cellulose conversion was found at later hydrolysis times. Cui et al.248 prepared four types of cellulose allomorphs from α-cellulose and concluded that the amorphous content had a strong positive influence on cellulose digestibility. The allomorph digestibility was reported to be in the following order: cellulose III > cellulose II > cellulose Iα > cellulose Iβ. In contrast, the crystalline polymorph of cellulose was reported to have a negligible influence on the conversion degree of non-dried and dried cellulose samples into glucose.249 Finally, cellulose crystallinity can affect the synergism among cellulase components and the cellulase processivity, which has a notable effect on the hydrolysis.241
The crystallinity index measurements are highly dependent on the technique applied, i.e., Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), Nuclear Magnetic Resonance (NMR) Spectroscopy, and Raman spectroscopy, and also the methods used for calculating the crystallinity index from the raw spectrographic data.204,242 The cellulose crystallinity index (CrI) from XRD spectra has long been calculated by different calculation approaches.243 The most frequent and simple calculation technique is based on the peak height according to the empirical method of Segal et al.250 for native cellulose:
| CrI (%) = [(I002 − Iam)/I002] × 100 | (2) |
The degree of polymerization (DP) of cellulose is the number of glucose units in the cellulose molecule chain and varies between 6000 in the primary cell wall and up to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in the secondary cell wall.252 The DP of cellulose is believed to contribute to the enzymatic hydrolysis of lignocelluloses since a long cellulose chain has more hydrogen bonds, while a shorter chain has more cellulose ends available to the exoglucanases.253
000 in the secondary cell wall.252 The DP of cellulose is believed to contribute to the enzymatic hydrolysis of lignocelluloses since a long cellulose chain has more hydrogen bonds, while a shorter chain has more cellulose ends available to the exoglucanases.253
However, tracking changes in the DP of cellulose, especially for complex lignocelluloses, during the course of pretreatment cannot be easily assayed. A method developed by Zhang and Lynd254 is only applicable to pure cellulosic substrates. Besides, DP is not typically an independent factor influencing cellulose digestibility because altering DP is always accompanied by crystallinity changes.148,255
5.2 Cellulose accessibility to cellulases
One of the primary barriers for cellulase enzymes in the hydrolysis of lignocellulose is their limited access to much of the cellulose confined in a highly packed structure.256 The presence of lignin significantly decreases the swelling/accessibility of cellulose resulting in low sugar yields at commercially viable low enzyme loading.120 Arantes and Saddler257 found that the required protein loading to achieve efficient hydrolysis of lignocellulosic substrates, regardless of their source, structure, and type of pretreatment, had a strong linear dependency on the cellulose accessibility for each substrate. Biomass porosity is considered as the lignocellulosic interior surface area and exterior surface area and is largely determined by the particle size.258 The accessible pore sizes required for anaerobes and cellulase and hemicellulase enzymes were reported to be at least 0.2–20 μm and 40–60 nm in width, respectively, to allow sufficient penetration.253 Wiman et al.259 correlated the higher rate of enzymatic hydrolysis, in spite of the negative effect of lignin accumulation on the particle surface, with the increase in specific surface area. Rollin et al.240 also showed that increasing cellulose accessibility is more important than removing lignin in the enzymatic hydrolysis of pretreated substrates, while removing lignin increases the accessibility of hemicelluloses which in turn affects cellulose accessibility.260 Similar to cellulose crystallinity, a strong relationship was observed between the accessible cellulose surface and the degree of synergistic action of cellulase components, which is crucial to enhance the hydrolysis efficiency.261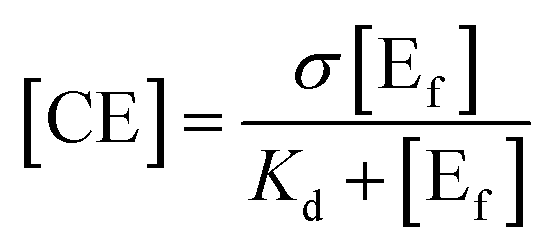 | (3) |
The concentration of free enzymes is measured either directly by analyzing the adsorbed protein on the substrate or calculated as the difference between the total amount of protein initially added and the amount left in aqueous solution at any time.262,265–267 The enzymes were reported to adsorb quickly in the initial stage and remain attached throughout hydrolysis.268 For instance, the equilibrium time for cellulase on pretreated sugarcane bagasse was approximately 120 min and was even shorter for Avicel (10 min), while β-glucosidase (from A. niger) was not significantly adsorbed.269
5.3 Hemicellulose content
Hemicelluloses, a physical barrier around cellulose, can retard the enzymatic hydrolysis by precluding the access of enzymes to cellulose (Section 5.2) and inhibiting the endoglucanase and cellobiohydrolase activity.270,271 The presence of xylan is believed to limit the cellulose hydrolysability, as evident by slow digestion of delignified substrates compared to pure cellulose.272,273 Although it is commonly found in the pulp and paper industry that xylan and other hemicelluloses adsorb on cellulose and enhance pulp strength, Kumar et al.274,640 recently showed that hemicellulose adsorption and their strong association with cellulose during pretreatments can retard cellulose digestion significantly; however, supplementation of xylanase to cellulase was shown to relieve the inhibition. In another report, Wang et al.275 also reported that the re-adsorption of dissolved xylan, produced during the pretreatment, on cellulose can inhibit the cellulose hydrolysis by cellulases. The supplementation of cellulases by xylanase was suggested to hydrolyze the xylan adsorbed on cellulose and potentially improve the hydrolysis efficiency of lignocelluloses. As discussed earlier (Section 4), the supplementation of xylanase has also been reported to synergistically improve the performance of cellulases in the hydrolysis of lignocelluloses.216,276–278 Nonetheless, hemicellulase supplementation to cellulase not only enhances cellulose accessibility to cellulase by simultaneously removing structural/non-structural hemicelluloses but also depolymerizes shorter hemicellulose oligomers in the solution that have been shown to be strongly inhibitory to cellulases by Kumar and Wyman and others.279–284 On the other hand, a negative effect of xylose accumulation on cellulase cocktails was also observed.285 Partial removal of hemicelluloses by concentrated NaOH was reported to be more effective than complete removal for poplar, and a maximum enzymatic hydrolysis of 94.6% was achieved.285 More information on the inhibitory effects of sugars and oligomers on the enzymatic hydrolysis is provided in Section 5.5.5.4 Lignin content
In general, lignin plays a negative role in the biochemical processes for producing lignocellulosic biofuels.286–289 Nonetheless, Nakagame et al.290 concluded that an increase in the carboxylic content of lignin resulted in a decrease in non-productive binding of cellulase and consequently an increase in hydrolysis yield. A slight enhancement in enzymatic hydrolysis was also reported by Wang et al.291 by adding Kraft lignin to the enzymatic hydrolysates. Lai et al.292 reported contrasting results for the effect of ethanol organosolv lignin on enzymatic hydrolysis. They found that the addition of 8 g L−1 hardwood organosolv lignin significantly improved the enzymatic yield of organosolv pretreated sweetgum and loblolly pine, while addition of softwood organosolv lignin was shown to decrease the yields.Lignin can retard the enzymatic hydrolysis of lignocelluloses via three mechanisms: (1) enzymes can be adsorbed on lignin through hydrophobic interactions, electrostatic interactions, and/or hydrogen-bonding interactions, (2) lignin in lignocellulosic materials acts as a surface barrier to block the accessible surface of carbohydrates through physical blockage on the surface and chemical blockage through the lignin–carbohydrate complex, and (3) enzyme deactivation by soluble lignin.293,294
Öhgren et al.278 evaluated the effects of partial delignification of corn stover by acid-catalyzed or autocatalysis pretreatment to increase the enzymatic hydrolysis yield. Due to the delignification, a slight increase in glucose yield and a decrease in xylose yield due to hemicellulose loss were observed. Várnai et al.272 concluded that the limitation in the enzymatic hydrolysis of spruce was mainly due to the presence of lignin, since the removal of lignin with chlorite delignification doubled the hydrolysis yield with near theoretical yield within 2 days. Nlewem et al.295 performed alkali, dilute acid, and hot water pretreatments on switchgrass and compared its enzymatic hydrolysability. Although it was not only due to delignification, the alkali pretreatment generally produced glucose at higher concentrations than the others, since it caused higher reduction in lignin content and lots of pores were formed by the pretreatment. In another study, fungal delignification of wet milled rice straw by Trichoderma viride in the presence of a surfactant for 30 days resulted in 74% of lignin removal and 56% of enzymatic saccharification.296
5.5 Formation of inhibitory byproducts
Besides hemicellulose and lignin-derived compounds, some inhibitory byproducts produced during pretreatment, e.g., furan aldehydes, weak acids, and hydrolysis-derived substances like soluble mono/oligomeric sugars (Section 5.3), hamper the performance of cellulases and fermentable organisms.307,310,311 Furan aldehydes, i.e., furfural and 5-hydroxymethylfurfural (HMF), are formed by dehydration of pentose and hexose sugars, respectively312,313 (Fig. 7). By the release of acetic acid during pretreatment, mainly by hydrolysis of the acetyl group, or by re-hydrolysis, furan aldehydes can be converted to weak acids such as levulinic acid and formic acid (eqn (4)).314–316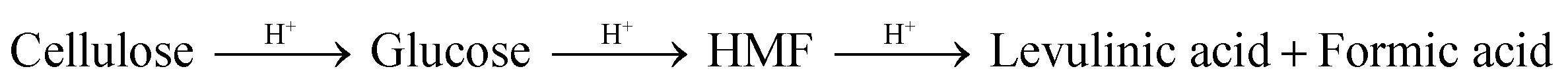 | (4) |
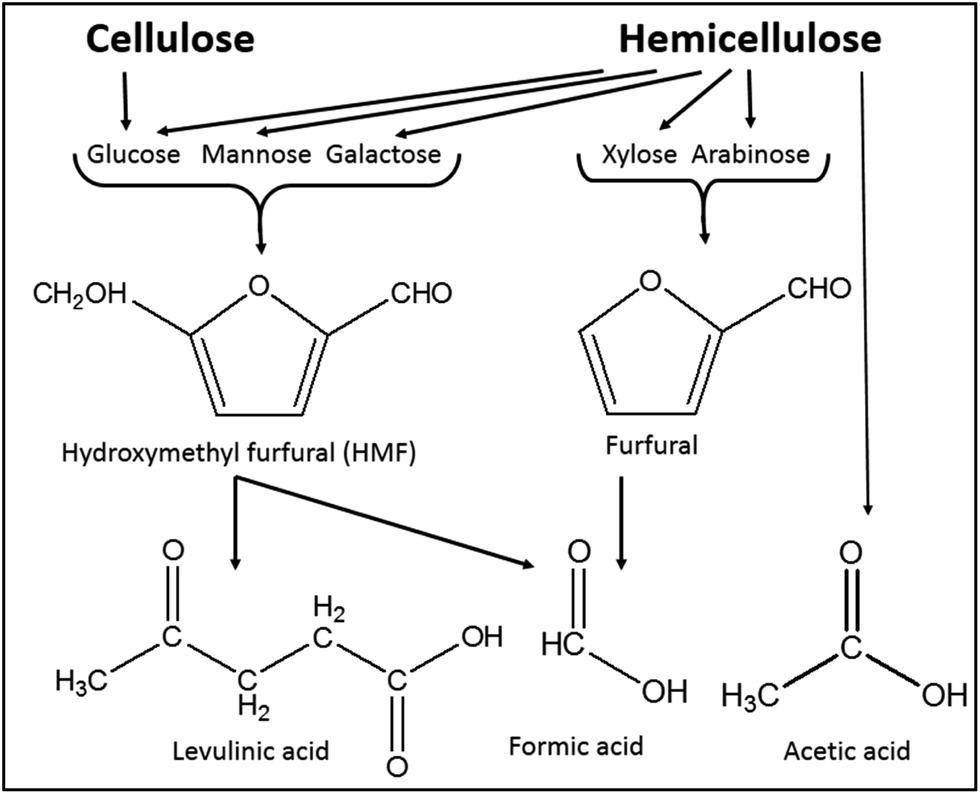 | ||
| Fig. 7 Formation of major inhibitory by-products from main carbohydrates present in lignocelluloses (modified from Reginatto et al.317). | ||
Formation of inhibitory byproducts during pretreatment is strongly dependent on feedstock and pretreatment type applied. For example, agricultural residues and hardwoods with higher amounts of acetylated xylan generate a higher concentration of acetic acid during pretreatment. Most of the pretreatments under severe conditions, such as a long reaction time and high temperature, result in the formation of inhibitory by-products. In acid-catalyzed thermochemical pretreatment processes, dehydration of pentose sugars and uronic acid results in inhibitory byproducts (Fig. 7). In addition, the splitting of the lignin's β-O-4 ether and other acid labile linkages forms phenolic and non-phenolic aromatic inhibitory compounds. However, the formation of carboxylic acids by the peeling-off reaction takes place under alkaline conditions.307,311
Jing et al.318 compared the inhibitory effect of the major lignocellulose degradation products on Spezyme®CP cellulase and found it be in the following order: lignin derivatives > furan derivatives > organic acids > ethanol. Arora et al.319 reported a severe inhibition by formic acid (5 or 10 mg mL−1) on the enzymatic hydrolysis of cellulose powder as well as dilute acid-pretreated poplar.
Xiao et al.320 quantitatively calculated the inhibitory effect of sugars on cellulase and β-glucosidase during the enzymatic hydrolysis of softwood substrates and showed a dramatic increase in the inhibition of both enzymes by increasing the glucose concentration. They also reported the significant inhibitory effect of mannose, xylose, and galactose during the hydrolysis on cellulase activity but not on β-glucosidase activity. Xylooligomers (XOs), especially at high concentrations, were reported to have a greater inhibitory effect than xylan and xylose, resulting in the decrease of the initial hydrolysis rate and final glucose yield of Avicel.280,284 Addition of xylanase and β-xylosidase was recommended to reduce xylooligomer and xylan inhibition of enzymatic hydrolysis of pretreated corn stover.321 In a recent study, Kumar and Wyman279 revealed that mannan polysaccharides and their enzymatically derived oligomers were more inhibitory to cellulase than XOs and cellobiose. They also showed that cellulase inhibition dramatically increased with mannan backbone substitution with galactose. However, the amount of mannan re-adsorption on cellulose after pretreatment was reported to be higher than that of glucomannan and galactomannan at the same concentrations.322 In a recent study, the Cellic® CTec3 enzyme mixture was reported to be more resistant than Celluclast 1.5L cellulase to the inhibitory compounds produced during steam pretreatment of poplar and lodgepole pine.323 Furthermore, monomeric sugars were shown to have greater inhibitory effects than phenolics, depending upon their types, and oligomeric sugars.
It is notable that the discussed byproducts also have inhibitory effects on the bioconversion routes leading to biofuel and renewable chemical production. For example, the concentrations of furfural and HMF in the range of 0.5–1 g L−1 and formic and acetic acids at more than 4 g L−1 were reported to be toxic in batch lactic acid fermentation by Rhizopus oryzae.324 For a recombinant S. cerevisiae strain, initial furfural concentrations below 5 g L−1 were reported to have a negligible effect on ethanolic fermentation in a xylose and glucose containing medium, while xylose consumption rates were affected at initial furfural concentrations of 10–15 g L−1.325
6 Concentrated phosphoric acid pretreatment
Phosphoric acid (85%) was first recognized as a swelling agent to produce reactive cellulose from air dried cellulose by Walseth326 in the 1950s. Since then, phosphoric acid swollen cellulose (PASC) has been the subject of vast studies as a cellulose substrate for cellulase activity assays and preparation of microcrystalline cellulose.327–329 Bellamy and Holub330 patented a process using CPA (80–85%) for decrystallization of cellulose to improve its hydrolysis. The process included the formation of a gel by mixing cotton and wood pulp with CPA at room temperature followed by acid removal from the cellulosic substrate by water washing. Zhang et al.,331 however, observed cellulose dissolution behavior when the phosphoric acid concentration reached greater than 80.5%, the critical concentration value for the dissolution of Avicel. During the first stage of the dissolution, an esterification reaction between the hydroxyl group of cellulose and phosphoric acid occurs and cellulose phosphate (cellulose-O-PO3H2) is formed. In the second stage, a competitive hydrogen-bond reaction between the cellulose hydroxyl groups and the solvent molecules or hydrogen ions happens and regenerated cellulose and phosphoric acid without major substitution are recovered.332,333 Meanwhile, cellulose hydrolysis remains minimum since the reaction temperature is kept low enough (30–70 °C) to retard the depolymerization and side reactions.334Conte et al.335 by applying high- and low-field NMR confirmed that direct bonding between phosphoric acid and cellulose is formed. Zhang et al.333 particularly investigated the structural changes of microcrystalline cellulose (MCC) dissolution in 83% phosphoric acid (at temperatures of 30–70 °C) with X-ray diffraction, solid-state cross-polarization magic angle spinning 13C-NMR, and X-ray photoelectron spectroscopy (XPS). The XRD pattern demonstrated a decrease in χc (crystallinity index) with increasing temperature (from 30 to 70 °C) or time (from 2 to 6 h). χc was calculated according to the following equation,
| χc=Fc/(Fa + Fc) × 100% | (5) |
Besides, the crystallinity characteristic peaks for both cellulose I and II diminished or greatly decreased after cellulose regeneration from concentrated phosphoric acid (CPA). In the spectra of CP/MAS and 13C solid-state NMR, distinct peaks of C4 verified a transition from crystalline to amorphous cellulose after CPA treatment.333 The XRD patterns of MCC treated with 85% CPA at 323 K also demonstrated that more cellulose I was converted to cellulose II by increasing the reaction time from 0 to 6 h.336 Jia et al.337 chemically modified MCC with phosphoric acid in order to enhance its processing for applications in gelling materials and emulsion stabilizers. Regenerated cellulose at some angles corresponding to crystallographic planes of cellulose II exhibited less crystallinity compared to intact MCC. Besides, the crystallinity index was reduced by 48% after regeneration.
The dissolution was also capable of fractionating lignocellulose components under the modest reaction conditions, and the cellulose can be regenerated by an organic solvent, e.g., ethanol and acetone, or water.240,334,641 Addition of an antisolvent, e.g., acetone, causes the dissolved cellulose and hemicellulose to precipitate and partial dissolution of lignin in acetone also takes place. Besides, hemicellulose oligomers are fractionated from cellulose due to higher solubility in water and poor solubility in the water/acetone mixture.334 The regenerated amorphous cellulose, precipitated from the dissolved cellulose, demonstrated extremely high reactivity for enzymatic digestibility, suggesting the dissolution technique as a new approach for the pretreatment of lignocelluloses.331 Recently, a new cellulose solvent- and organic solvent-based lignocellulosic fractionation (COSLIF) method using concentrated phosphoric acid, as a cellulose solvent, and an organic solvent (e.g., acetone or ethanol) for the solute precipitation, under modest reaction conditions was developed.334 This novel pretreatment was able to effectively disrupt the lignocellulosic structure of switchgrass,340 bamboo,338 common reed,339 and miscanthus and hybrid poplar.340Table 2 summarizes the results of glucan digestibility improvement after COSLIF, as well as the applied conditions, for different lignocelluloses. As can be seen in Table 2, COSLIF pretreatment is performed under mild conditions, e.g., temperatures of ca. 50–60 °C, atmospheric pressure, and short pretreatment time (∼1 h), using acetone, ethanol, and water as anti-solvents. Compared with other most commonly used pretreatment methods, such as dilute acid, alkali, and hydrothermal, the sugar yields for CPA pretreatment for a variety of hardwoods and agricultural residues are very high. For instance, over 90% glucan digestibility was achieved after 72 h hydrolysis even at low enzyme loadings. Moreover, some studies reported ethanol yield enhancement by the CPA pretreatment (Table 3).
| Substrate | COSLIF condition | Enzymatic hydrolysis | Glucan digestibility | Ref. |
|---|---|---|---|---|
| Sesbania grandiflora (L.) Pers. | H3PO4 (85%), 50 °C for 45 min, 95% (v/v) ethanol as an organic solvent | 1 FPU cellulase from Sigma | 86% glucose in 72 h | 346 |
| Achyranthes aspera and Sida acuta weed | 70%, 75%, and 80% phosphoric acid (1.0 g/8.0 mL), and 60 °C for 1 h, and acetone as an organic solvent | 30 FPU g−1 dry biomass Celluclast 1.5 L and 60 U g−1 dry biomass β-glucosidase | Up to 86.2% and 82.2% glucan conversion yields, respectively | 347 |
| Alamo switchgrass (Panicum virgatum) | 85% H3PO4, 60 °C, 1 atm, for 45 min, 95% (v/v) ethanol as an organic solvent | Novozymes 50013, 15 and 3 FPU g−1 glucan, supplemented with 10 IU g−1 β-glucosidase | 90% and 85%, respectively, in 72 h | 240 |
| Moso bamboo | 85% H3PO4, 50 °C, 1 atm, for 60 min, 95% (v/v) ethanol as an organic solvent | Novozymes 50013 and β -glucosidase (Novozymes 50010), 1, 2, 5, and 15 FPU of cellulase per g glucan supplemented with 10 β-glucosidase IU g−1 | 88.2%, 89.8%, 93.3%, and 94.9%, respectively, in 72 h | 338 |
| Common reed (Phragmites australis) | 85% H3PO4, 50 °C, 1 atm, and 60 min, 95% (v/v) ethanol as an organic solvent | 15, 10, and 5 FPU and 30 units of β-glucosidase per gram of glucan, (Novozymes 50013 and Novozyme 50010) | 94%, 93%, and 90%, respectively, 24 h | 339 |
| Miscanthus and poplar | 85% H3PO4, 50 °C, 1 atm, and 60 min, 95% (v/v) ethanol as an organic solvent | 5 FPU of cellulase and 10 units of β-glucosidase per gram of glucan, (Novozymes 50013 and Novozyme 50010) | ∼93% in 72 h | 340 |
| Microcrystalline cellulose | 83% H3PO4 and ice-cold distilled water as an anti-solvent | 15 FPU g−1 cellulose and 60 IU β-glucosidase per g cellulose | 100% cellulose conversion after 3 h | 331 |
| Avicel and α-cellulose | 81.7% phosphoric acid at room temperature for a half-hour, and acetone as an organic solvent | 15 FPU per g glucan of Genencor Spezyme®CP cellulase and 60 IU g−1 glucan of Novozymes 188 β-glucosidase | 100% conversion within 3 h | 334 |
| Corn stover and switchgrass | 84% phosphoric acid at 50 °C for 45 min, and acetone as an organic solvent | 15 FPU g−1 glucan of Genencor Spezyme®CP cellulase and 60 IU g−1 glucan of Novozymes 188 β-glucosidase | ∼96–97% in 24 h | 334,341 |
| Hybrid poplar and douglas fir | 85% phosphoric acid at 50 °C for 60 min, and acetone as an organic solvent | 15 FPU g−1 glucan of Genencor Spezyme®CP cellulase and 60 IU g−1 glucan of Novozymes 188 β-glucosidase | ∼97% and ∼75% in 24 h for hybrid poplar and douglas fir, respectively | 334 |
| Oriented strand board, chipboard, plywood, and wallpaper | 85.9% phosphoric acid at 50 °C for 30 min, and acetone as an organic solvent | 20 FPU cellulase (Sigma, C2730) and 50 IU β-glucosidase (Sigma, G0395) per gram of substrate | 87.0–93.5% in 96 h | 348 |
| Hybrid poplar (P. tormentosa Carr.) | 85% phosphoric acid and room temperature until complete dissolution, and water as solvent | 50 FPU 1:1 blend of Celluclast 1.5 L and Novozyme 188/g substrate |
92%, 72 h | 349 |
| Industrial hemp stalks | 85.9% H3PO4 at 50 °C for 1 h, and organic solvent, acetone | 15 FPU cellulase (Spezyme CP), and 60 IU β-glucosidase per gram of glucan | 95.9%, 24 h | 350 |
| Bermudagrass, reed, and rapeseed | 85% phosphoric acid at 50 °C for 60 min, and acetone as an organic solvent | 25 FPU of Celluclast® 1.5 L per gram of cellulose | 97.5–99.4% (24 h) | 351 |
| Eastern gamagrass (Trypsacum dactyloides) and switchgrass | The pretreatment method reported by Zhang et al.334 and modified by Ge et al.352 | 100 μL of Novozymes 188, or 600 μL of cellulase and 200 μL of Novozymes 1800 for high solid-loading | 80.5–99.8% and 73.5–87.1%, for eastern gamagrass and switchgrass, respectively, 36 h | 353 |
| Giant reed, elephantgrass, and sugarcane clone | 85% phosphoric acid at 50 °C for 60 min, and organic solvent, acetone | 300 μL of cellulase (Sigma C2730) and 100 μL of Novozymes 188 (Sigma C6105) | Glucose yields from biomass: 0.306, 0.309, 0.331, 0.317, and 0.290 g g−1 for giant miscanthus, giant reed, giant miscanthus (Q42641), elephantgrass, and sugarcane, respectively | 352 |
| Corn stover and Avicel | 85%(w/w) phosphoric acid, 2% (w/v) solid loading, described by Zhang et al.331 | 5 FPU g−1 of glucan (Novozymes 50013) and 10 units of β-glucosidase (Novozymes 50010) per gram of glucan | ∼90% (72 h) for corn stover and 100% (6 h) for Avicel | 354 |
| Substrate | CPA pretreatment condition | Method and microorganism | Ethanol yield | Ref. |
|---|---|---|---|---|
| a SHF: separate enzymatic hydrolysis and fermentation; SSF: simultaneous saccharification and fermentation; NSSF: non-isothermal simultaneous saccharification and fermentation. | ||||
| Dedicated energy crops and crop residues | The same as reported by Zhang et al.331 | SHF, three self-flocculating Saccharomyces cerevisiae strains: SPSC01, ATCC24859, ATCC4126 | 0.375 to 0.396 g g−1 (SPSC01), 0.380 to 0.394 g g−1 (ATCC24859), and 0.384 to 0.405 g ethanol per g (ATCC4126) glucose | 352 |
| Oil palm empty fruit bunches (OPEFB) | The same as reported by Zhang et al.334 | SSF, S. cerevisiae | 89.4% of theoretical maximum ethanol yield | 355 |
| Aspen wood (Populus tremula) | Phosphoric acid (85%), 12.5% solid loading, 50 °C, 90 rpm, 30 min, and acetone as an organic solvent | NSSF, Mucor hiemalis | 72.4% of theoretical maximum ethanol yield | 356 |
| Rice straw, elmwood, and pinewood | The same as of Rollin et al.240 | SHF, Mucor indicus | Over 78–92% ethanol yield based on glucose consumed | 342 |
| Tripsacum dactyloides | The same as of Zhang et al.334 | SHF, a self-flocculating yeast strain SPSC01 | Up to 0.496 g ethanol per g glucose | 353 |
COSLIF was observed to follow a different mechanism than alkali or acid pretreatment with respect to changes in lignocellulosic components. Zhu et al.341 compared glucan, hemicellulose, and lignin contents of the COSLIF and dilute acid (DA) pretreated corn stover. They reported that COSLIF removed more lignin compared to DA pretreatment. Siripong et al.347 reported removal of all xylan and ca. half of the acid-insoluble lignin from two wood species as a result of CPA (80%) pretreatment. Similarly, Rollin et al.240 reported a 67% and 34% hemicellulose and lignin removal, respectively, from switchgrass by CPA pretreatment. They reported a greater increase in cellulose susceptibility to hydrolysis in COSLIF pretreatment than soaking in aqueous ammonia (SAA, 10% w/w ammonia, 140 °C, 20:1 liquid/solid ratio, 14 h) for Alamo switchgrass (Fig. 8). However, the cellulose content remained almost constant after both the pretreatments. Another action of CPA pretreatment is to hydrolyze hemicellulose acetyl groups to acetic acid.334,339 The remaining hemicellulose can be enzymatically depolymerized and used as a co-substrate for fermentation.342,343
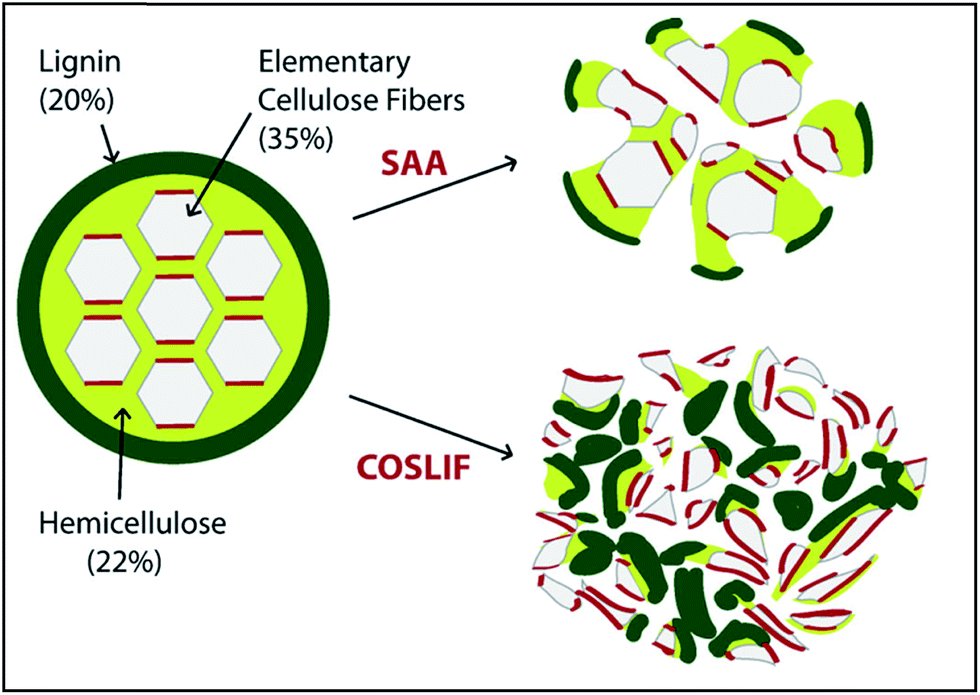 | ||
| Fig. 8 Conceptual image of alteration in the lignocellulose structure as a result of cellulose solvent- (concentrated phosphoric acid) and organic solvent-based lignocellulose fractionation (COSLIF) and soaking in aqueous ammonia (SAA) pretreatments (taken from ref. 235 with permission). | ||
There are few studies in the literature that showed biogas production improvement by CPA pretreatment. A study showed 40% improvement in the methane yield obtained after CPA pretreatment (85.7% CPA at 50 °C for 30 min) compared with that of the untreated oil palm empty fruit bunches.344 Conversely, CPA pretreatment did not improve the methane yield for berry and poplar woods.345 This is presumably due to the repelling interaction of anaerobic bacteria and biomass surface after CPA pretreatment. In addition, the pores generated following CPA pretreatment may not be large enough for anaerobic bacteria to penetrate into the biomass structure.
6.1 Criteria for efficient phosphoric acid pretreatment
A narrow range of phosphoric acid concentrations is required for the cellulose phase transition from swelling to dissolution to occur. Only phosphoric acid above its critical concentration is able to disrupt the lignocellulose recalcitrant structure.331 The critical phosphoric acid concentration is in the range of 77–83 wt%, depending on substrate type and its moisture content. Fig. 9 shows the SEM images of pretreated cotton fibers with a range of o-phosphoric acid concentrations.357 As shown in this figure, amorphogenesis begins to develop at the surface of the cotton fibers when the acid concentration was increased to near its critical values of cellulose dissolution. At 74% acid concentration, splitting, roughening, fibrillation, and peeling/delamination were observed, indicating that amorphogenesis started at the surface of the cotton fibers. Amorphogenesis is developed by increasing acid concentration, and destruction and diminishing of fiber structure were observed at 76% and 78% acids, respectively.351 Moxley et al.350 also found that a minimum phosphoric acid concentration of 81% is required to obtain a very rapid hydrolysis rate and high digestibility of hemp stalks. Jia et al.337 discovered minimum 77.8 wt% CPA for significant solubilization of MCC powder. Zhang et al.331 showed that 77 wt% of CPA caused only cellulose swelling while ice-cold phosphoric acid (≥83%) completely dissolved MCC.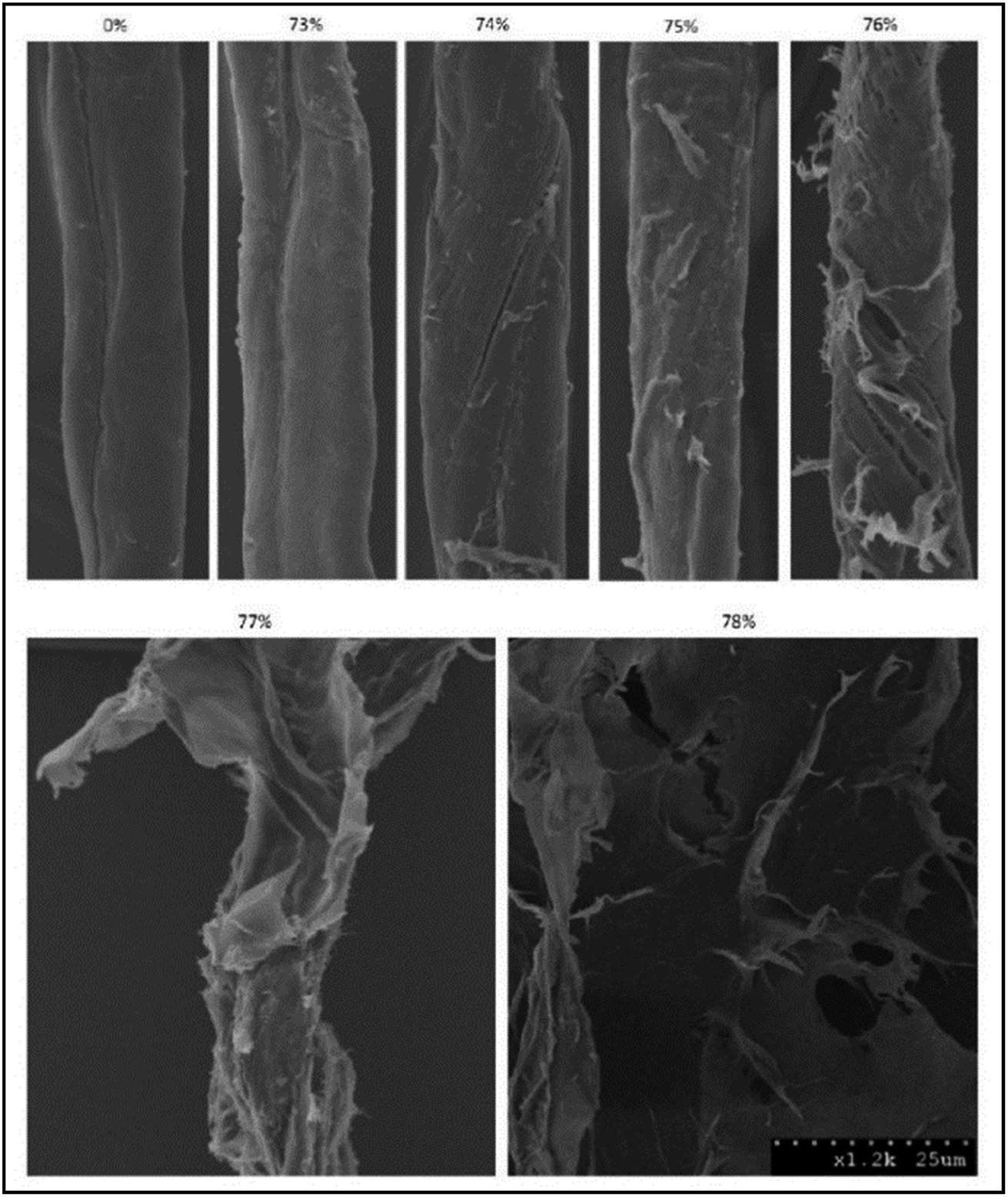 | ||
| Fig. 9 SEM images of cotton linter pretreated with different concentrations of O-phosphoric acid (0–78% w/w). Pretreatment conditions were: ice-cold temperature, one hour with occasional mixing, and water as an antisolvent (taken from ref. 357). | ||
The dissolution of (ligno)celluloses in CPA also depends on the reaction temperature and time. Cellulose dissolution by CPA usually occurs at modest reaction temperatures.334 Moxley et al.350 investigated the effect of 84.0% H3PO4 pretreatment at different reaction times (from 30 to 120 min) at 50 °C and pretreatment temperatures (from 40 to 60 °C) for 60 min on the enzymatic glucan digestibility of hemp stalks. Higher reaction temperatures and time resulted in faster fiber dissolution; however, significant hydrolysis of cellulose and hemicellulose or sugar degradation occurred under these conditions. In terms of enhanced MCC processing ability by CPA, however, a decreasing trend in solubility was observed by increasing the temperature from 5 to 75 °C.337 Sathitsuksanoh et al.339 optimized the COSLIF conditions for enhanced saccharification at decreased cellulase loadings by response surface methodology (RSM). The optimal conditions were 85% (w/v) CPA, 50 °C, and 60 min, regardless of the biomass moisture contents from 5–15% (w/w). These modest reaction conditions can minimize sugar degradation, inhibitor formation, and capital investment of industrial plants.
Addition of volatile organic solvents is used for regenerating amorphous cellulose and hemicellulose, dissolving the organic solvent lignin soluble fraction, and for the recycling and re-concentration of PA.358 Recently, replacement of acetone by ethanol was presented and widely used in a modified version of the COSLIF. This modification is advantageous because ethanol is more chemically stable than acetone for solid/liquid separation and less corrosive. Besides, a very high yield of acetone recovery (e.g., >99.99%) is required for having an economically viable COSLIF implementation, whereas lower ethanol recycling/recovery after pretreatment (e.g., 98–99%) is acceptable,338–340 since the remaining ethanol can be separated in an ethanol distillation process. Moreover, a 40% decrease in organic solvent consumption was achieved in the replacement of acetone by ethanol.339
6.2 Why is phosphoric acid so effective in enhancing enzymatic hydrolysis?
Many studies reported that substrate accessibility to cellulase determines the susceptibility of lignocellulosic substrates to enzymatic hydrolysis.257,260,261,297,359–361 Cellulose accessibility to cellulase (CAC) is usually quantified by cellulase adsorption Langmuir kinetics, as discussed in Section 5.2.261,362 Recently, a quantitative assay for CAC, based on the adsorption of a nonhydrolytic fusion protein containing CBM and GFP, was developed by Hong et al.363 and applied for pretreated substrate characterization. CAC (m2 g−1 of cellulose) was calculated by multiplying a constant to maximum cellulase adsorption capacity obtained from the Langmuir equation (eqn (3)).363 For pretreated lignocellulosic biomass, total substrate accessibility to cellulase (TSAC) represented the cellulase adsorption capacity for the whole biomass and was calculated by adding CAC and non-cellulose accessibility to cellulase (NCAC).341 TSAC was equal to CAC for protein thioredoxin-GFP-CBM (TGC) adsorption to biomass. Similarly, CAC and TSAC (m2 g−1 biomass) can be calculated by TGC adsorption after BSA blocking of the lignin fraction. Therefore, NCAC (m2 g−1 biomass) can be calculated as the difference between TSAC and CAC.341 TSAC, CAC, and NCAC (m2 g−1 biomass) measurements of intact lignocelluloses were reported to be approximately 1 m2 g−1 biomass.240,339–341 Untreated Alamo switchgrass (Panicum virgatum), for example, had 1.27, 0.49, and 0.77 m2 g−1-biomass TSAC, CAC, and NCAC, respectively.364 SAA slightly improved all the accessibilities, while COSLIF resulted in a considerable increase of 9.6 and 8.0 for TSAC and CAC (m2 g−1 biomass), respectively.240 Similarly, an almost 2-fold increase in the accessibilities was observed for COSLIF-treated corn stover compared to DA pretreatment.341 TSAC (m2 g−1-biomass) of miscanthus and poplar also increased after COSLIF pretreatment but more radically from 0.18 to 20.7 and 0.23 to 18.2, respectively.340 Common reed followed the same pattern as miscanthus and poplar after the pretreatment.339Breaking or even restructuring the highly ordered intra- and inter-molecular hydrogen-bond network of crystalline cellulose is believed to enhance its depolymerization rate.106,337,365 The evidence of breaking hydrogen-bonding networks in cellulose fibers of switchgrass after COSLIF was confirmed by CP/MAS 13C-NMR and FTIR.365 Other analytical techniques, e.g., microscopy and X-ray diffraction, also showed the disruption of the hydrogen-bond network of cellulose for MCC regenerated from CPA.337 John et al.366 investigated the structures of native and regenerated celluloses by X-ray methods and proposed the same lattice plane location of the inter-molecular hydrogen bonds between adjacent cellulose molecules. The empty space between adjacent cellulose chains could be occupied by the hydrogen ion from phosphoric acid; therefore, inter-molecular hydrogen bond formation is destroyed during the regeneration process.349
Recently, computer simulations have been employed to study the biomass recalcitrance at the molecular level that otherwise cannot be analyzed with available experimental techniques.367,368 Molecular dynamics simulation (MDS) and quantum chemical calculations, e.g., density functional theory (DFT) methods, are the tools of molecular simulation. These techniques have been used for the simulation of lignin biosynthesis and degradation,369,370 cellulose insolubility,371 and recently for the simulation of the effect of ammonia pretreatment on cellulose Iβ.106 Although models of secondary plant cell walls incorporating cellulose, xylan, water, and lignin by MD simulations were generated,372 the molecular simulation studies on lignocelluloses are scarce. This is due to the complex lignocellulose biomass structure and also the intricate relationship between enzymes, chemicals, and biomass. Molecular simulation for lignocelluloses is still in its early stage of development and needs further investigation to fill the gap of advancing analytical methods in pretreatment.
6.3 Summary and future perspectives of phosphoric acid pretreatment
Taking all into consideration, COSLIF was successful with a number of agricultural residues and hardwoods342,373 and demonstrated the advantages of high glucan digestibility even at low cellulase loadings, high hydrolysis rates, modest reaction conditions, higher revenues from co-products (acetic acid, lignin, and hemicelluloses), and less inhibitor formation. Besides, the remaining CPA on treated biomass did not show inhibitory effects for enzymatic hydrolysis or fermentation processes. However, it is still in its early stage of development and its commercialization is a far promising priority that needs pervasive consideration. Although there are only a few studies in the literature, CPA pretreatment does not seem to be very effective in improving biogas production from lignocelluloses. Substantial reduction in the use of chemicals (both CPA and organic solvent) is required in order to have an economically competitive process. Improvement of ethanol production process economy was suggested by the production of two major value-added byproducts, i.e., unaltered and purified lignins by the COSLIF and byproducts from fermentation.342 Although CPA pretreatment seems to be very promising given the high end-product and by-products yields, a detailed techno-economic analysis of CPA pretreatment is required in order to study the feasibility of this pretreatment for a large-scale operation.7 N-Methylmorpholine-N-oxide (NMMO) pretreatment
N-Methylmorpholine-N-oxide (NMMO or NMO) is categorized as a family of cyclic, aliphatic, tertiary amine oxides.374,375 Tertiary amine oxide systems were first patented by Graenacher and Sallmann376 in 1939 to dissolve cellulose for enhanced chemical processing. However, Johnson,377 for the first time in 1969, introduced a cyclic mono(N-methylamine-N-oxide) compound to interact with inter-molecular hydrogen bonding networks and dissolve cellulose, wool, silk, hair, and feather, which are insoluble in commonly used solvents. Since the late 1970s, research on the dissolution of cellulose in NMMO was initiated when McCorsley and Varga378 produced a highly concentrated, yet economical, cellulose solution by dissolving cellulose in a NMMO–water system. At that time, research on NMMO–cellulose tertiary systems was mainly focused on producing regenerated cellulose fibers that have applications in textiles and nonwovens, the lyocell process, strengthening paper films, and paper coatings.374,377,379–381 However, this technology has been recently introduced as a pretreatment method of lignocelluloses, e.g., for the improvement of either second-generation bioethanol115,382–388 or biogas production.388–397Having a strong N–O dipole, which acts as either an ionic or donative and single bond, NMMO is capable of disrupting the hydrogen-bond networks of cellulose and building new hydrogen bonds between the polymer and the solvent375,379,398 (Fig. 10). Cellulose dissolution in NMMO leads to a tertiary phase of the cellulose–NMMO–water system.379,399 Hydration with 1–1.2 water molecules per NMMO (water content 13.3–17 wt%) significantly improves its interaction with a solute and boosts its solvation ability, while increasing the water content to 19–24% and 25–30% results in heterogeneous swelling by forming balloons and ballooning, respectively.400 Higher water contents (above 35%) make fibers swell homogeneously and precipitate, because in the tertiary system water is further preferred to form hydrogen bonds with NMMO than cellulose.375,379,400 Ballooning and swelling modes of cellulose dissolution are more efficient for biogas production, while for ethanol production pretreatment with 85% NMMO leads to better lignocellulose bioprocessing.384Fig. 11 shows a microscopic image of wood fiber swollen by ballooning in NMMO solution, where the three zones of the membrane of the balloons, the inside of the balloons, and the nonswollen crystalline regions are easily identified.
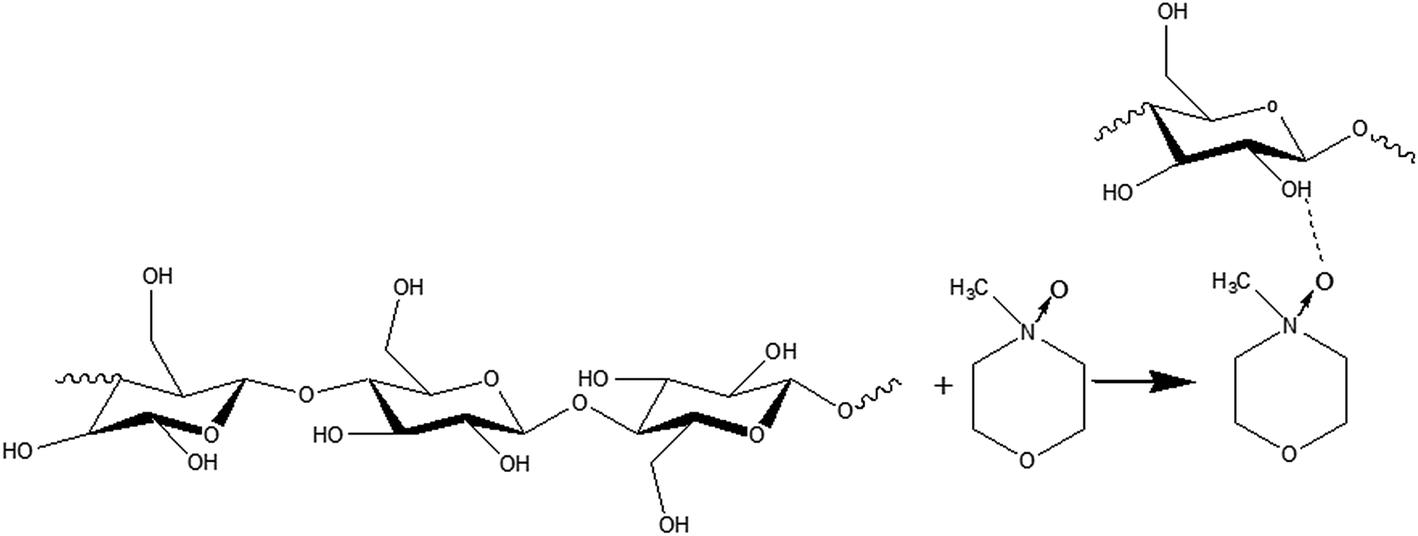 | ||
| Fig. 10 The mechanism of cellulose dissolution in NMMO, adapted from Wang et al.163 with permission. | ||
 | ||
| Fig. 11 Wood fiber swollen by ballooning in a 78 wt% NMMO solution in water, taken from ref. 404. | ||
Lignocelluloses are directly dissolved in the solvent at moderate temperatures (90–130 °C) under atmospheric pressure for 20 min to 5 h with negligible derivatization. Cellulose is subsequently regenerated by adding water as an anti-solvent to the slurry. The regenerated cellulose (cellulose precipitated from the NMMO solution) is converted from cellulose I to cellulose II structure, which is much more reactive for cellulase adsorption and subsequent hydrolysis.401,402 The solvent is washed away from the regenerated solids with distilled boiling water, and the excess water can be easily vaporized due to the low vapor pressure of NMMO, allowing approximately 99% of NMMO recycling.403
7.1 Effect of NMMO pretreatment on the superstructure of lignocelluloses
The enhancement in the digestibility of regenerated lignocellulosic biomass by NMMO pretreatment is mainly due to reduced cellulose crystallinity. The crystalline structure of regenerated lignocellulose from the NMMO solution as well as the untreated one are usually expressed by the Total Crystallinity Index (TCI) and Lateral Order Index (LOI) using FTIR.405 The FTIR spectra of lignocelluloses can also give some valuable information on the structure and the variation in characteristic bands by the pretreatment. Table 4 summarizes the characteristics of bands, their corresponding functional groups, and assignments to the major biomass constituents.| Wavenumber (cm−1) | Functional group | Assignment |
|---|---|---|
| 3175 | –OH stretching (inter-molecular hydrogen-bonds) | Cellulose II |
| 2900 | C–H stretching | Cellulose |
| 1740 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (acetyl or carboxylic acid) O stretching (acetyl or carboxylic acid) |
Hemicellulose and lignin |
| 1510, 1610 | C=C stretching (aromatic ring) | Lignin |
| 1465 | C–H3 (bending) | Lignin |
| 1420, 1430 | C–H2 (bending) | Cellulose |
| 1375 | C–H (bending) | Cellulose |
| 1335 | –OH (bending) | Cellulose |
| 1315 | C–H2 (wagging) | Cellulose |
| 1158 | C–O–C (stretching) | Cellulose |
Purwandari et al.396 reported that the TCI (the absorbance ratio A1427/A898 calculated from FTIR spectra) of oil palm empty fruit bunch (OPEFB) reduced by up to 78% following the pretreatment in 85% NMMO at 120 °C for 3 h. In addition, ballooning and swelling modes of NMMO result in a lower TCI at 120 °C than at 90 °C. This finding is in contrast to ballooning and swelling modes of NMMO pretreated cotton that result in lower crystallinity indexes at 90 °C than 120 °C.384 However, compared to the untreated cotton, crystallinity indexes decrease slightly for different modes of dissolution, ballooning, and swelling.121 Besides, the intra-molecular hydrogen-bonding OH stretching at about 3350 cm−l (FTIR spectra) in pretreated cotton is broadened and shifted to a higher wave number,384 which is an indication of transforming cellulose I to cellulose II.406,407 This finding is in accordance with another report on NMMO pretreatment of straw389 and also confirms that the pretreatment reduced the structural lignin content. NMMO pretreatment of bagasse at 130 °C for 1 h transformed the crystalline structure into an amorphous form, since the TCI and LOI decreased from 1.39 and 1.44 to 1.18 and 1.10, respectively.385 The LOI, a criterion for the estimation of the amorphous to crystalline portion of the structure, considerably decreased from 2.68 to 0.88 when the straw fraction of manure was pretreated for 5 h at 120 °C using 85% NMMO and decreased more with increase in pretreatment time to 15 h.389 Moreover, the LOI and TCI of rice straw pretreated with 85 wt% NMMO for 5 h at 120 °C decreased from 0.46 to 0.40 and 1.69 to 1.62, respectively.387 Likewise, Khodaverdi et al.408 reported that NMMO (85%) treatment of cotton linter at 120 °C for 2 h resulted in a TCI and LOI decrease from 7.1 and 2.7 for untreated cotton to 3.3 and 1.1, respectively.
The FTIR analysis also indicated that lignin and acetyl groups from the hemicellulose backbone were partially removed by the pretreatment, while the cellulose content increased. Liu et al.362 qualitatively studied the abundance and distribution of lignin and cellulose in NMMO-pretreated pine flour using the FTIR technique. Diminishing of the absorbance peaks at 1270 cm−1 and 1596 cm−1, corresponding to lignin,409 indicated a reduction in lignin content on the surface of pine flour after NMMO pretreatment.362 Furthermore, crystallinity measurement of the biomass by X-ray diffraction confirmed a linear correlation (R2 = 0.91) between cellulose crystallinity and initial hydrolysis rates of the pine flour samples. Virtanen and Maunu410 investigated the dissolution process of softwood pulp fibers in NMMO at 110 °C for 15, 30, and 90 min by employing different NMR spectroscopic methods: solid state cross polarization magic angle spinning (CP-MAS), 13C and 15N spectroscopies, and 1H high resolution MAS NMR spectroscopy. The cellulose crystallinity of the NMMO pretreatment sample for 90 min decreased by 15%, and the C4 signal appeared different from the untreated pulp, while it remained almost constant for the first 30 min of treatment with a broadening C4 signal.
7.2 Changes in composition and microstructure during NMMO pretreatment
In general, carbohydrate contents of lignocelluloses do not undergo significant changes and high solid recoveries are achieved after NMMO pretreatments.115,394,396,397,411 This is an advantage of NMMO pretreatment over conventional pretreatment methods, since carbohydrate loss is a major problem in most chemical, physicochemical, and biological pretreatments.136,187 However, a longer pretreatment time and/or temperature lead to partial removal of acid-insoluble lignin and xylan (or mannan in softwoods) and enrichment of the glucan constituent.382,383,386,387,389,393,411 Furthermore, structural studies confirmed the liberation of acetic acid from the acetyl groups of biomass during NMMO pretreatment, especially at longer pretreatment times and higher temperatures.115 The ash content was also reported to decrease from 5.4% up to 1.3% as a result of NMMO pretreatment of OPEFB,396 while no considerable change was reported for rice straw.387Cellulose accessibility for NMMO-treated substrates was then evaluated by comparing the maximum adsorption capacity (by Langmuir adsorption isotherm) of pretreated samples and enzyme lignin (EnzL),362 prepared by complete hydrolysis of carbohydrates in the pretreated biomass with excessive cellulase loadings.261 The maximum adsorption capacity of cellulase onto pine flour samples as well as cellulose accessibility considerably increased with increasing NMMO pretreatment time from 30 to 120 min at 120 °C. Moreover, a nearly good linear correlation between cellulose accessibility and overall glucan conversion rate was also reported for pine flour.362
The other rapid specific surface area assessment technique is the water retention value (WRV) or water swelling capacity, which has been used to quantify the swelling potential of paper pulps.417 WRV is the ability of water adsorption or the swelling capacity of the substrate and reflects the accessibility of the substrate to subsequent hydrolysis by enzymes.412 Besides, since substrate swelling and water adsorption occur mainly in the amorphous regions, the WRV can be used as a criterion to assess changes in the crystalline structure after pretreatment.418 The water swelling capacity of birch hardwood after pretreatment with 85% NMMO at 130 °C for 3 h substantially increased by 46.6–119.9% depending on the applied drying method.383 The WRV of triticale straw also slightly increased by 10%, 10%, and 20% at NMMO pretreatment times of 1, 3, and 15 hours, respectively, and a smaller increase of 10% for rice straw was realized.416 However, significant reduction in the WRV of cellulose was reported by NMMO pretreatment in dissolution mode at either 90 or 120 °C.384 This behavior was observed less at lower concentrations of NMMO (than 85%), but it still had lower WRV values compared to the untreated one.384
7.3 Advantages and disadvantages of NMMO pretreatment
NMMO is able to dissolve up to 15 wt% of cellulose419 with no/less chemical modification under relatively mild conditions (low/moderate temperatures and atmospheric pressures). High bioprocess efficiency, high solvent recovery, and formation of low carbohydrate degradation and inhibitory products are also among the favorable characteristics of NMMO pretreatment. Tables 5 and 6 summarize an overview of treatment conditions along with improvements in saccharification/fermentation and biogas production from different lignocelluloses after NMMO pretreatment. These tables show that NMMO pretreatment is conducted under relatively mild conditions, i.e., temperature 90–130 °C for a few hours, using ∼85% NMMO. As can be seen in these tables, NMMO pretreatment causes significant improvement in ethanol, biogas, and enzymatic hydrolysis yields for different types of lignocelluloses including hardwoods, softwoods, agricultural residues, and other cellulosic substrates. By applying NMMO pretreatment, ethanol can be produced by S. cerevisiae, M. indicus, and Z. mobilis via different strategies, e.g., SSF, SHF, and NSSF (Table 5). The pretreatment resulted in up to 100% conversion of cellulose in enzymatic hydrolysis and 93.3% ethanol yields of theoretical maximum for rice straw (Table 5). An improvement of about 100% in the methane yield was also reported after NMMO pretreatment of cotton linter.392 At pilot scale, maximum hydrolysis sugar yields of 195 and 175 mg sugar per g wood for spruce and birch wood chips, respectively, in NSSF with Mucor indicus were also achieved.386| Substrate | NMMO condition | Method and fermentation microorganism | Glucan conversion and/or ethanol yield | Ref. |
|---|---|---|---|---|
| a Non-isothermal simultaneous saccharification and fermentation. | ||||
| Spruce and oak | 90, 110, and 130 °C, 1–3 h | NSSFa, Saccharomyces cerevisiae | Up to 85.4% and 89% improvement in ethanol yield for spruce and oak, respectively | 115 |
| Rice straw | 85 wt% NMMO, 120 °C, 1, 3, and 5 h, 5% loading | SSF, S. cerevisiae | Hydrolysis yield of glucan 96%, 93.3% of theoretical maximum ethanol yield | 387 |
| Cotton linter | 90 and 120 °C, 0.5–15 h using 85%, 79%, and 73% NMO | SSF, S. cerevisiae | Improvement of up to 100% yield in enzymatic hydrolysis and 83.75% ethanol yield | 384 |
| Spruce and birch | 85% NMMO, 130 °C, 1–5 h | Bench-scale and airlift cultivations, Mucor indicus | Maximum ethanol yields of 195 and 175 mg g−1 wood for spruce and birch, respectively | 386 |
| Birch | 85% NMMO, 130 °C, 3 h | SHF, S. cerevisiae, | Maximum 76.8% ethanol of theoretical yield, 9-fold increase in ethanol yield compared to untreated | 383 |
| Wheat straw | 85% NMMO 120 °C for 1–5 h | Anaerobic cultivations, M. indicus | Up to 92.1% of theoretical maximum ethanol yield | 382 |
| Sugarcane bagasse | NMMO monohydrate, 130 °C, 1 h | SSF, Zymomonas mobilis | Approximately 0.15 g ethanol per g bagasse (86% of the theoretical maximum ethanol yield) | 385 |
| Substrate | Pretreatment condition | (Improvement in) methane yield | Ref. |
|---|---|---|---|
| a Volatile solid. | |||
| Oil palm empty fruit bunch (OPEFB) | 90 and 120 °C, 1, 3, and 5 h, 85%, 79%, and 73% NMMO | Methane yield up to 0.408 Nm3 kg−1-VS, improvement by 167% compared to untreated | 396 |
| 73, 79, and 85% NNMO, 90 and 120 °C, 1, 3, and 7 h | Maximum 0.408 Nm3 CH4 per kg-VS | 395 | |
| Softwood spruce, rice straw, and triticale straw | 130 °C, 1–15 h, 85% NMMO | Up to 245, 157, and 203 Nml CH4 per g raw material, respectively, 400–1200% improvement compared to the raw materials | 397 |
| Forest residues | 120 °C, 3, 7, and 15 h, 75% and 85% NMMO | Up to 0.17 Nm3 kg−1-VSa methane yield (83% of theoretical maximum yield) | 393 |
| Straw fraction of cattle and horse manure | 5 h and 15 h, 120 °C, 85% NMMO | Maximum methane yield increase by 53% and 51% for cattle and horse manure, respectively, after 15 h pretreatment | 389 |
| Barley straw and forest residues | 85% NMMO, 3–30 h, 90 °C | 0.23 and 0.15 Nm3 CH4 per kg-VS from barley straw and forest residues, respectively; corresponding to 88% and 83% of the theoretical maximum yields | 394 |
| Blended-fiber waste textiles | 85% w/w NMMO, 120 °C, 2 h | Up to 62.18% of theoretical maximum methane yield (after 6 days) | 391 |
| Forest residues | NMMO concentrations of 75% and 85%, 120 and 90 °C, 3 and 15 h | Maximum 141% increase in methane production (75% NMMO at 120 °C for 15 h) | 388 |
| Jeans textiles | 85% NMMO, 120 °C, 3 h | Two-stage semi-continuous process, 400 mL methane per g-VS per day | 390 |
| Cotton linter | 85% NMMO, 5% w/w solid loading, 120 °C, 3 h | Approximately 100% methane yield (% of maximum theoretical) for 5 g L−1 cellulose concentration after 30 days | 392 |
The solvent is recycled by treating the solution with ion-exchange resins to remove contaminants and subsequently dewatering the solvent.420 Due to the low vapor pressure of NMMO, excess water can be easily vaporized from the recycled solvent leaving the monohydrate form of NMMO.403 However, the water evaporation unit demands high-energy input which has considerable negative effects on the economy of the whole process.421,422 Besides, in order to have an economically feasible process of bioethanol and biogas production by NMMO pretreatment of lignocelluloses, more than 99 percent of NMMO recovery is required.421,422 Some side reactions and/or NMMO ring cleavage can occur in cellulose–NMMO solutions, especially at elevated process temperatures,423,424 which hamper efficient solvent recovery. A study showed that a smaller amount of reducing sugars was liberated from NMMO-pretreated sugarcane bagasse at 130 °C rather than 100 °C, possibly due to NMMO or cellulose degradation.385 In some studies, recycled NMMO showed the same performance in hydrolysis improvement and biogas production of pretreated sugarcane bagasse and barley straw, respectively,385,394 as compared with fresh NMMO. However, in contrast, forest residues with high lignin and bark content resulted in 55% reduction in methane yield after pretreatment with recycled NMMO in comparison with those pretreated with the fresh NMMO.394
The remaining NMMO in the regenerated solids may prove to have inhibitory effects on fermenting organisms and/or hydrolytic enzymes. NMMO concentrations of 5 and 100 g L−1 have been shown to reduce the enzymatic hydrolysis yields by 12% and 76%, respectively, after 12 h of hydrolysis for cotton linter.384 Although NMMO decreased the glucose uptake rate by S. cerevisiae a little, it had negligible impact on the final ethanol yield even at a concentration of 100 g L−1.115,384 However, the ethanol yield and productivity decreased at concentrations above 2% NMMO for M. indicus, while the total production of metabolites was not significantly changed.386 This was because some glucose shunted from the ethanol to the glycerol pathway as the glycerol yield and production increased in proportion to NMMO concentration. Recently, He et al.425 introduced a NMMO-tolerant cellulase-producing strain from a newly isolated Galactomyces sp. CCZU11-1. The results showed that up to 25% (w/v) NMMO had no significant effect on the saccharification of NMMO-pretreated sugarcane bagasse prepared at 130 °C for 1 h or fermentation by S. cerevisiae. On the other hand, NMMO remaining in the pretreated substrate at concentrations higher than 0.002% was reported to considerably decrease the methane yield.393
Once the solvent was washed away from the substrate, NMMO leaving the process ends up in the wastewater stream. Nevertheless, it is not of great concern, since NMMO is an environmentally friendly solvent.426,427
Techno-economic analysis of NMMO pretreatment of spruce for bioethanol and biogas421 and forest residues for biogas production elucidated high process energy efficiency.422 In the case of bioethanol production, a biogas plant in parallel to valorize pentoses can improve the process economy.421 This is because most ethanol-producing organisms cannot assimilate pentoses efficiently.343 When forest residues were co-digested with two-thirds of organic fraction of municipal solid waste in order to avoid nitrogen deficiency, the process of biogas production was evaluated to be financially feasible at 15% internal rate of return or higher for a minimum plant capacity of 50000 tons per year.422 Generally, large amounts of water need to be vaporized in order to efficiently recover NMMO, and this is among the barriers for its commercialization.421
8 Ionic liquid pretreatment
8.1 Ionic liquids: historical evolution and general properties
Ionic liquids (ILs) are usually defined as large organic salts, composed entirely of an organic cation and an organic or inorganic anion, which exist in liquid form at or below 100 °C.428 The field of ILs was first discovered in 1914 by Walden,429 who synthesized and characterized ethyl-ammonium nitrate ([EtNH3][NO3]) by neutralizing ethylamine with concentrated nitric acid. Organic based chloroaluminates ILs were first developed by Hurley et al.430 in 1951. A new class of ILs with melting point lower than ambient temperature based on 1-alkyl-3-methylimidazolium cation, called room-temperature ionic liquids (RTILs) and considered as the first generation ILs, has emerged since 1982 after the study by Wilkes et al.431 The replacement of the moisture-sensitive anion in the first generation ILs by the tetrafluoroborate ion ([BF4]−) and other anions resulted in more water-stable ILs in 1992, known as second-generation ILs.432 Third generation ILs, known as “task-specific” ionic liquids (TSIL), which covalently incorporate either anions or cations or both as functional groups, were introduced by Davis433 in 2004.ILs have negligible vapor pressures, high viscosity, and reasonable thermal and chemical stability, compared with typical organic solvents.434,435 These properties can be changed and controlled by selection of cations and anions developed for a special application. This is why ILs are usually defined by the term “designer solvents”.436 ILs, due to their unique properties, have received significant attention for vast applications in chemical and biochemical industries.428,435,437–441
8.2 Solvation in ILs
A simulation and vibration spectroscopy study of water–IL mixtures suggested the concentration dependent solubility of water in ILs. At low concentrations, the dissolution mechanism of water is molecular dispersion, while water aggregation takes place at higher concentrations. Dissolution of benzene in [DMIM](1,3-dimethylimidazolium)[PF6], however, makes an expansion in the IL structure, while the long-range charge ordering pattern in the IL still exists.442 One of the promising solvation features of ILs is their ability to dissolve monosaccharides, which are barely soluble in common solvents, except water.443,444 Like benzene, a simulation understanding of glucose dissolution in the ionic liquid [DMIM][Cl] has been established.445,446 The nature of the solute–solvent interaction in the system is mainly the hydrogen bond with a high chloride content of the IL. Youngs et al.446 suggested that the dominant coordinate of glucose dissolution in excess IL is the formation of three hydrogen-bonds between the OH groups of glucose and three anions, and an OH⋯Cl⋯HO bridge between the last two OH groups and the fourth chloride. The RTILs that contain dicyanamide anions were also reported to dissolve significant amounts of glucose, sucrose, lactose, and cyclodextrin.447 Other monosaccharides, including arabinose, fructose, mannose, and xylose, seem to have partial to high solubility in different ILs.448 Surprisingly, not only monosaccharides but also oligosaccharides and even polysaccharides are soluble in ILs. α-Cyclodextrin and starch, for example, were shown to have 30% and 10% solubility, respectively, in [BMIM](1-butyl-3-methylimidazolium)[Cl].448 Unlike dissolving saccharides in classic solvents, e.g., DMF and DMSO, the derivatization of native carbohydrates in ILs is of great importance since it is a green process.443 However, the aim of most studies on the carbohydrate–IL interaction is to produce non-derivatized cellulose, which has demonstrated vast applications in fiber and composite fiber production,449 as monoliths and films,443 and more recently, lignocellulosic biomass pretreatment.173The selection of cations and specially anions in ILs plays a crucial role in cellulose dissolution.108 Since the cellulose–IL bond, in nature, is the hydrogen-bond,446 it seems that anions with more hydrogen-bond-acceptor capability, e.g., OAc−, HCOO−, (MeO)2PO2−, and Cl−, are the suitable candidates for the solubility, while ILs with low-basicity anions, such as dicyanamide-based ILs, are not that efficient in dissolving cellulose.108 ILs containing ‘noncoordinating’ anions, including [BF4]− or [PF6]−, on the other hand, display no cellulose solubility.452 Unlike anions, cations in ILs play an unclear, but effective role, in the cellulose dissolution.108Table 7 lists the structure of some well-known cations of ILs for cellulose dissolution. Li et al.454 performed a simulation study and concluded that ILs with unsaturated heterocyclic cations can dissolve cellulose, whereas ILs with saturated ring cations can hardly dissolve cellulose. The reason for that was reported to be related to the structure factor and dynamic effect of the cations. Zhang et al.455 synthesized and used 1-allyl-3-methylimidazolium chloride as a non-derivatizing solvent for molecular dissolution of cellulose at room temperature. Although intra- and inter-molecular hydrogen-bond disruption were mainly due to the formation of a chloride hydrogen-bond network, it was suggested that the small polarized cation, [AMIM]+, also helped the attack on oxygen atoms of cellulose hydroxyl in this case.455,456 The 13C NMR spectrum of MCC dissolved in [AMIM][Cl] clearly resolved the six signals of carbon atoms of unmodified anhydroglucose similar to cellulose dissolved in sodium hydroxide solution or [BMIM][Cl].455 However, the dissolution mechanism was not dominated by hydrogen bond formation between cellulose and chloride. Thus, first, it is important to compare cellulose solubility in chloride alkali metal salts. Chloride in LiCl, for example, perfectly interacts with cellulose hydroxyl groups and dissolves cellulose in the presence of N,N-dimethylacetamide (DMAc).455 Nevertheless, other chloride salts, e.g., sodium, potassium, barium, and calcium chloride, are unable to dissolve cellulose.455 Although concentrated zinc chloride is able to dissolve cellulose, its solvation behavior is due to the formation of the zinc–cellulose complex.457 Second, a unique anion with solubility potential, when combining with all range of cations, was not found. Vitz et al.458 conducted a thorough study on cellulose dissolution in imidazolium-based ILs with particularly bromide and chloride anions. An odd-even effect for imidazolium chloride ILs with more cellulose solubility in even-numbered alkyl chains of cations compared to the odd-numbered was observed, whereas, this pattern was not generalized for imidazolium-based ILs containing bromide anions. 1-Ethyl-3-methylimidazolium diethyl phosphate, however, demonstrated the maximum solubility of cellulose among all the imidazolium-based ILs. Although the role of cation is not yet well-clarified, it was reported that the size and polarizability and attached functional groups of the cation, e.g., hydroxyl end-group, or basic oxygen atoms affected its solubility.108
| Cation structure | Name |
|---|---|
|
R = CH3: 1,3-dimethylimidazolium |
| R = C2H5: 1-ethyl-3-methylimidazolium | |
| R = C3H7: 1-propyl-3-methylimidazolium | |
| R = C4H9: 1-butyl-3-methylimidazolium | |
| R = C5H11: 1-pentyl-3-methylimidazolium | |
| R = C6H13: 1-hexyl-3-methylimidazolium | |
| R = C7H15: 1-octyl-3-methylimidazolium | |
| R = C8H17: 1-nonyl-3-methylimidazolium | |
| R = C9H19: 1-decyl-3-methylimidazolium | |
|
1-Cyano-3-methylimidazolium |
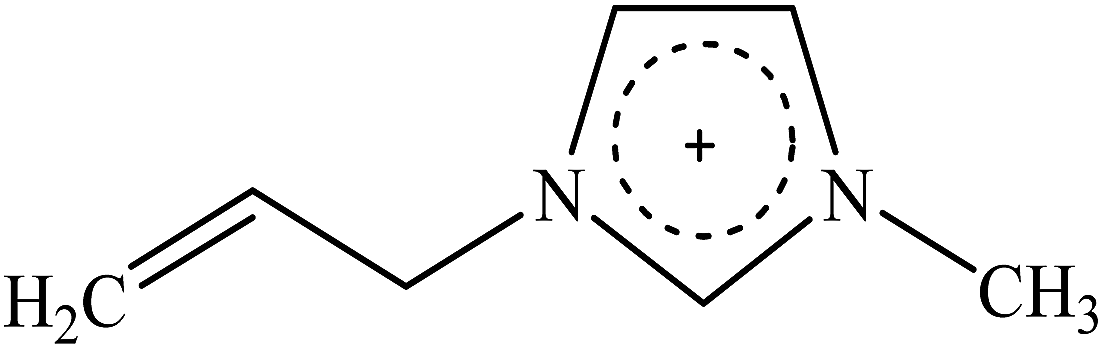
|
1-Allyl-3-methylimidazolium |
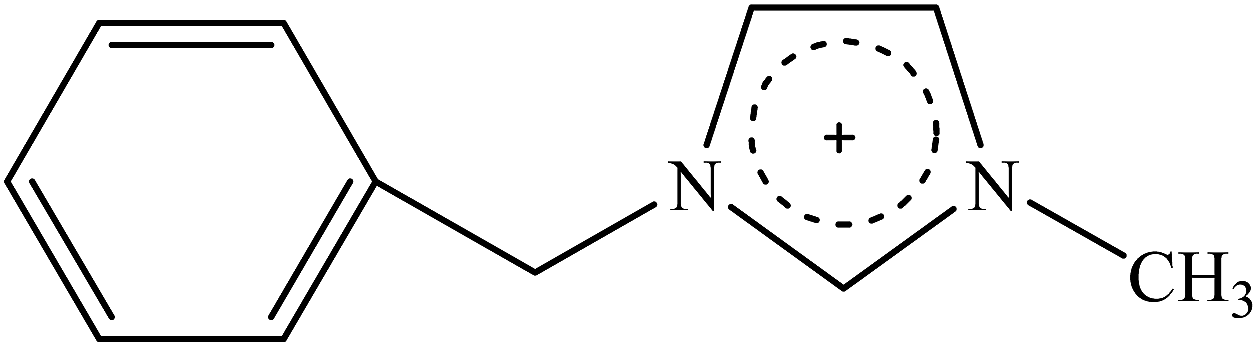
|
1-Benzyl-3-methylimidazolium |
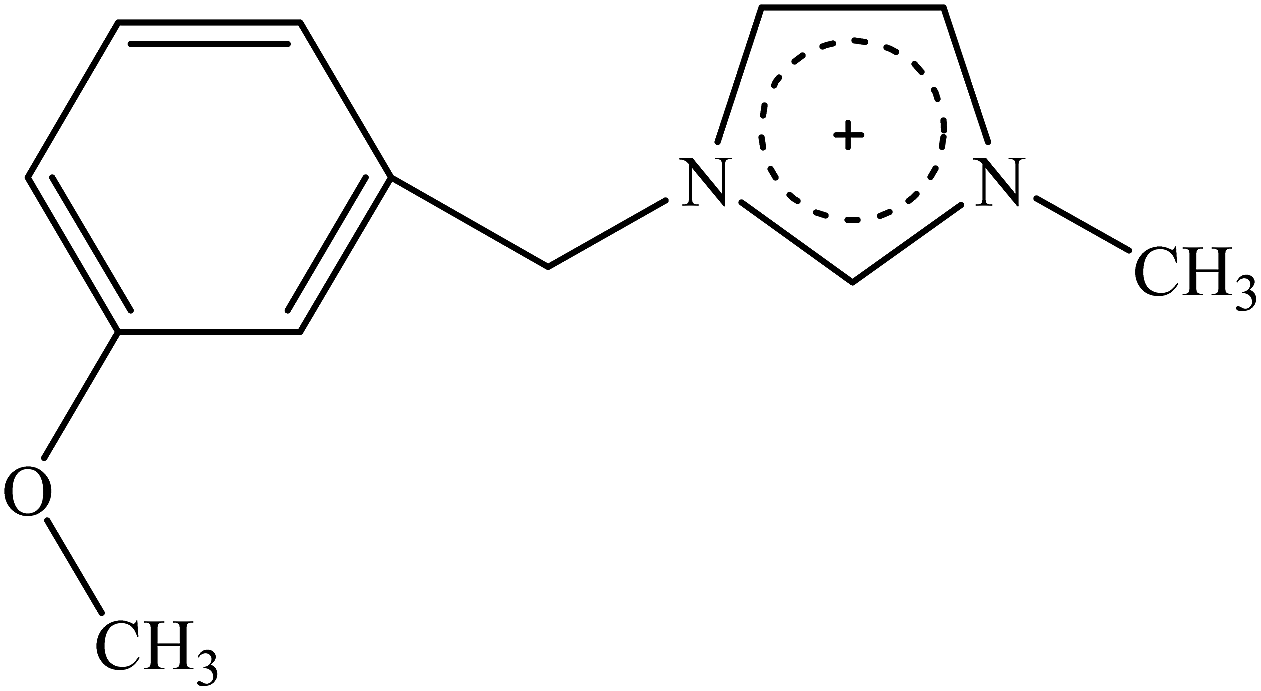
|
1-(3-Methoxybenzyl)-3-methylimidazolium |
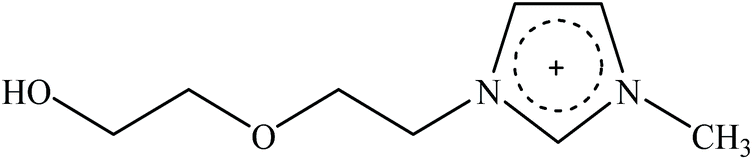
|
1-(3,6-Dioxahexyl)-3-methylimidazolium |
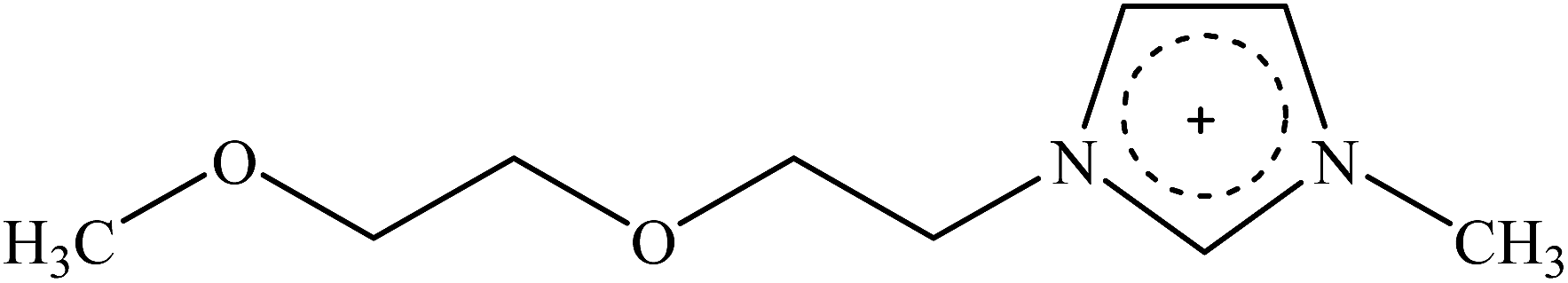
|
1-Ethyl-3-(3,6-dioxaheptyl)imidazolium |
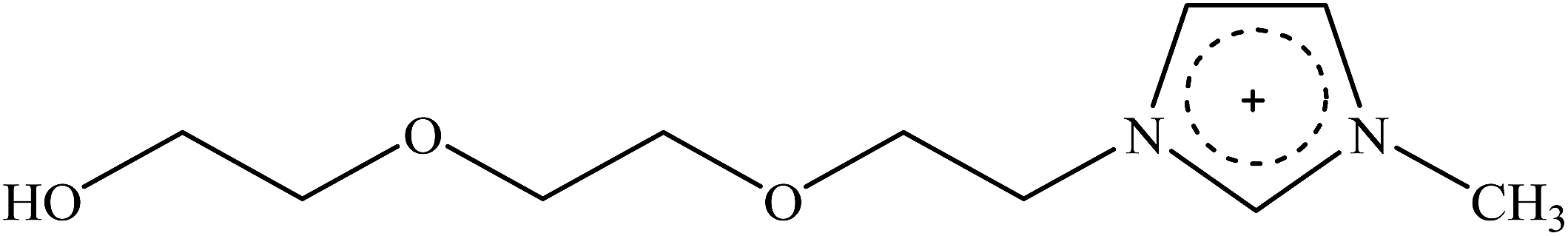
|
1-(3,6,9-Trioxanonyl)-3-methylimidazolium |
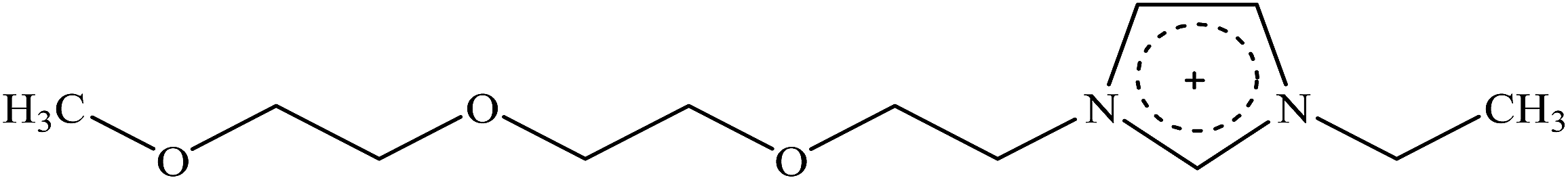
|
1-Ethyl-3-(3,6,9-trioxadecyl)-imidazolium |

|
1-Butyl-3-(3,6,9-trioxadecyl)-imidazolium |
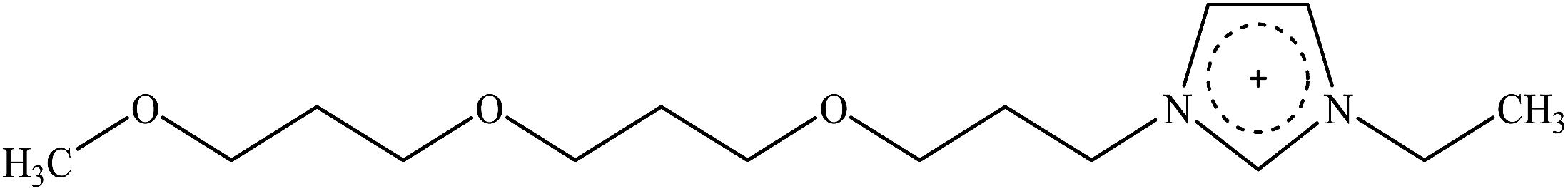
|
1-Ethyl-3-(4,8,12-trioxatridecyl)-imidazolium |

|
3,3-Ethane-1,2-diylbis(1-methyl-1H-imidazole-3-ium) |
|
1-Butyl-3-methylpyridinium |
|
1-Butyl-1-methylpyrrolidinium |
|
1-Ehyl-3-(3,6,9,12,15,18,21-heptaoxadococyl)-imidazolium |
|
1-(3,6-Dioxaheptyl)-3-(3,6,9-trioxadecyl)-imidazolium |
|
N-Benzyl-N,N-dimethylammonium |
|
Tetrabutylphosphonium |
|
Trihexyltetradecylphosphonium |
|
R = CH3: 8-metyl-1,8-diazabicyclo[5.4.0]undec-7-enium |
| R = C4H9: 8-butyl-1,8-diazabicyclo[5.4.0]undec-7-enium | |
| R = C8H17: 8-octyl-1,8-diazabicyclo[5.4.0]undec-7-enium | |
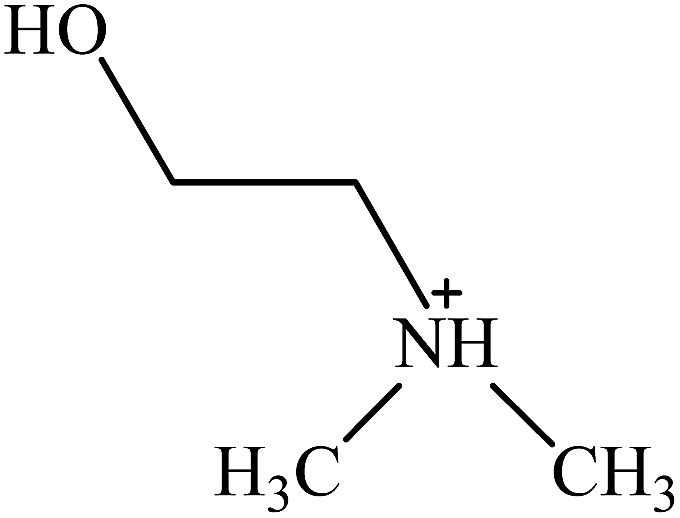
|
N,N-Dimethylathanolammonium |

|
Choline bis[(trifluoromethane)sulfonyl]imide |
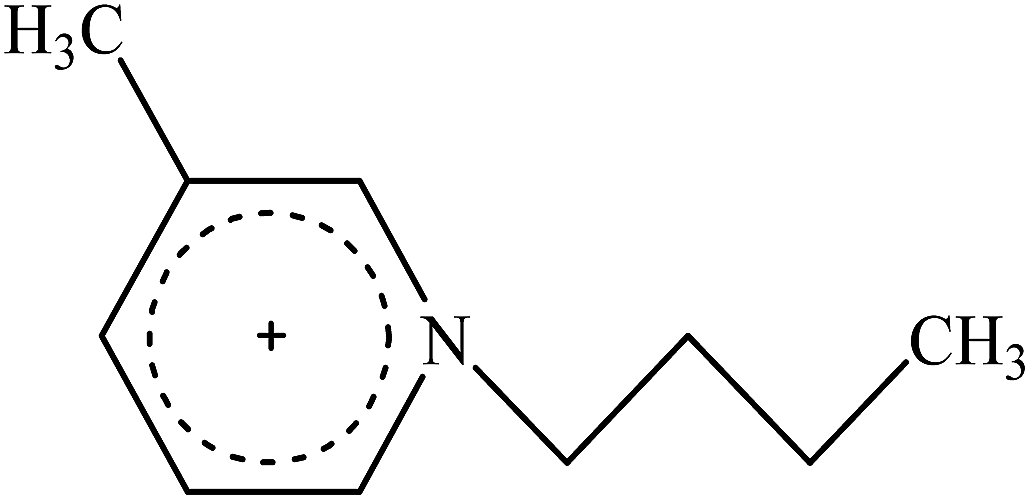
|
1-Butyl-3-methylpyridinium |
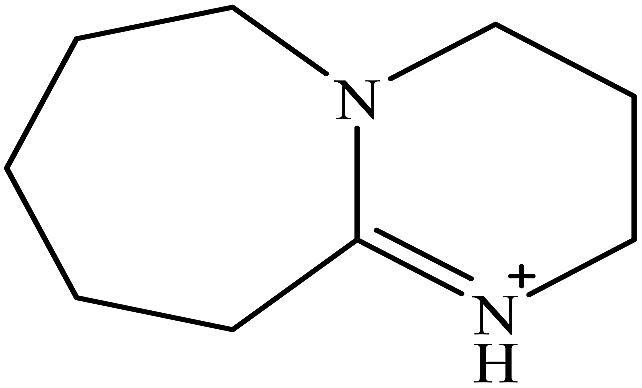
|
1,8-Diazabicyclo[5.4.0]undec-7-enium |
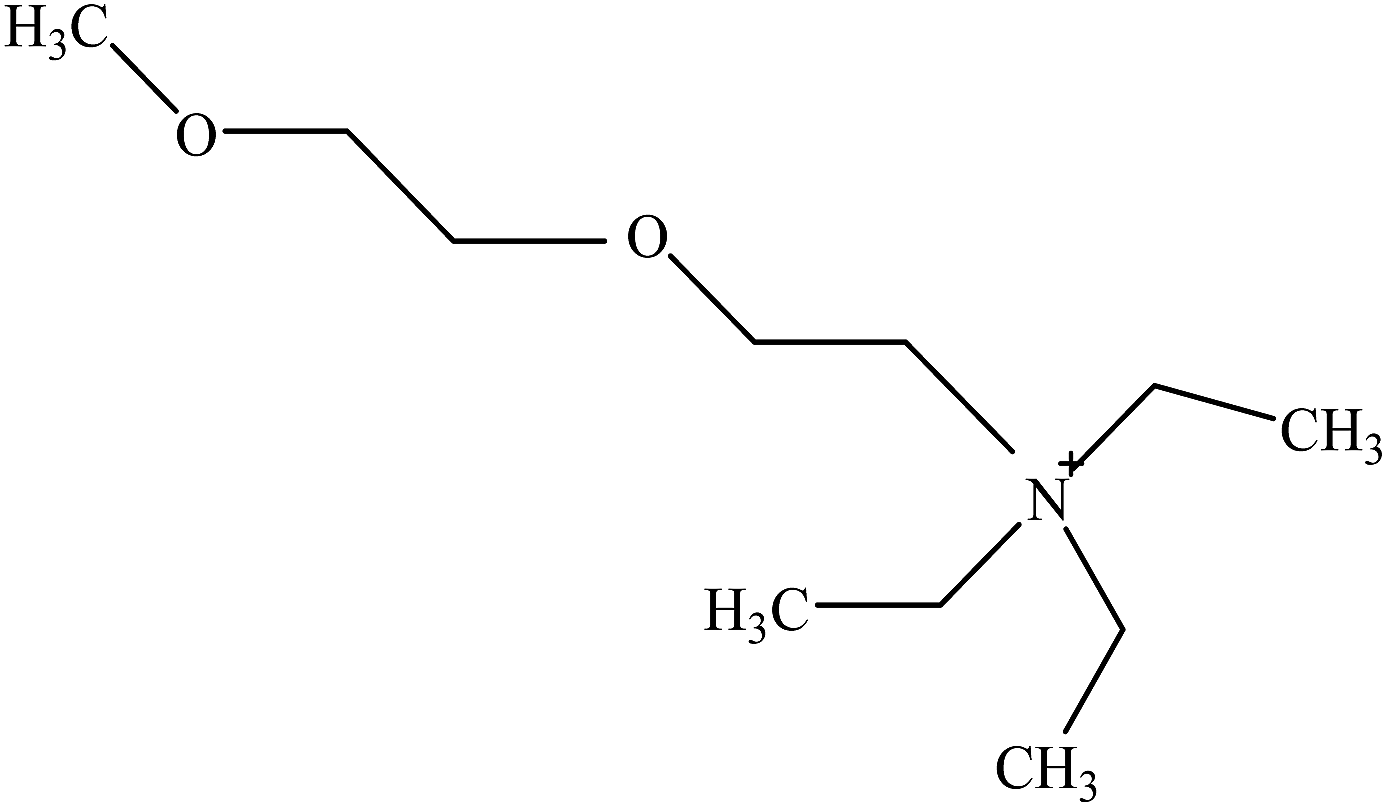
|
N,N,N-Triethyl-3,6,9-trioxadecylammonium |
Sets of TSILs, or so-called tailor-made ILs, were also designed and characterized for the dissolution and depolymerization of cellulose under mild conditions.459 Thermal heating especially by microwave or sonication, degree of polymerization of cellulose, and the IL viscosity are among the non-IL-intrinsic effective factors in cellulose dissolution.108,460–463
8.2.2.1 Role of solvent in the regeneration of cellulose from IL solutions. Hauru et al.472 characterized the Kamlet–Taft (KT) values of [EMIM][OAc], [TMGH][EtCO2], and [TMGH][OAc], and NMMO at several water contents and temperatures to investigate the role of the solvent in cellulose regeneration from the IL solution. The regeneration of cellulose was reported to start at threshold values of approximately β < 0.8 (β − α < 0.35). Shi et al.473 investigated the pretreatment of switchgrass with different [EMIM][OAc] and water concentrations (50–80%) at 160 °C and concluded a strong dependency of the chemical composition and crystallinity of the pretreated biomass as well as the corresponding lignin dissolution and depolymerization on the IL concentration. They found the hydrogen-bond basicity of the [EMIM][OAc]–water mixture to be a suitable indicator of predicting the cellulose dissolution, lignin depolymerization, and sugar yields. Besides, their molecular simulation indicated that water acts as a co- and anti-solvent in cellulose dissolution at below and above 50% [EMIM][OAc] concentration, respectively. The role of anti-solvent, e.g., ethanol, water, and acetone, in cellulose regeneration from a cellulose/[BMIM][OAc] mixture was studied by molecular simulation.474 Structural analysis based on radial distribution function revealed that among the three studied solvents, water was the most effective solvent at breaking the cellulose–[Ac]− H-bonds, leading to the subsequent formation of cellulose–cellulose H-bonds, and was demonstrated to be the best solvent for cellulose regeneration. Another molecular dynamics study was conducted to investigate the interaction of [EMIM][OAc], a cellulose oligomer, and water as an antisolvent.475Fig. 12 shows the proposed intermediate formed during the regeneration of a cellulose oligomer from the IL solution by using water, based on the simulation.
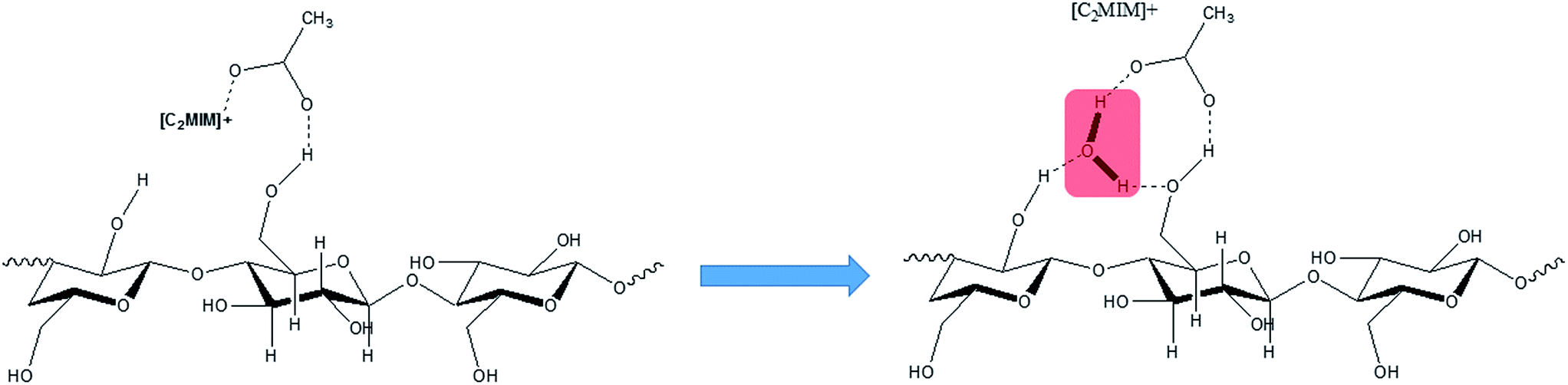 | ||
| Fig. 12 Intermediate structure of celluloses regenerated from an IL in the presence of water as an anti-solvent proposed by Liu et al.475 using MD simulation, picture adapted from Liu et al.475 | ||
8.2.2.2 Biomass loading. Some studies on IL pretreatment have focused on high biomass loading in the pretreatment, instead of typical approximate 5.0 wt%, since it is a crucial factor for process economy. Besides, a minimum amount of consumed IL and waste generation happen at high biomass loading.476 Cruz et al.477 investigated the effects of switchgrass loading on [EMIM][OAc] pretreatment in terms of viscosity, cellulose crystallinity, chemical composition, saccharification kinetics, and sugar yield. The IL pretreatment caused reduction in biomass recalcitrance for 3, 10, 20, 30, 40, and 50 wt% biomass loading and a “solid” like behavior was observed when the biomass loading increased. Moreover, the IL pretreatment caused the transformation of the cellulose crystalline structure from I to II for 3, 10, 20 and 30 wt% samples, while a mostly amorphous structure was found for 40 and 50 wt% samples. Likewise, Wu et al.478 reported the feasibility of [EMIM][OAc] pretreatment of corn stover at 125 °C for 1 h at 50 wt% biomass loadings in dramatically reducing the recalcitrance of the biomass. In another study, da Silva et al.479 used a twin-screw extruder with high shear force to pretreat sugarcane bagasse at high solid loadings in [AMIM][Cl]. They obtained the maximum glucan digestibility of 90% after 24 h of enzymatic saccharification of the pretreated substrate at a loading as high as 25 wt% at 140 °C for 8 min. The pretreatment decreased the crystallinity significantly and increased the specific surface area (SSA) by more than 100-fold. At a higher biomass loading of 50 wt%, still 76.4% glucose yield was obtained. Li et al.480 obtained 99.8% fermentable sugars from switchgrass by [EMIM][OAc] pretreatment at 15% (w/w) biomass loading during a 600- and 60-fold process scale-up for the pretreatment and enzymatic hydrolysis, respectively. Ninomiya et al.481 investigated the cholinum IL pretreatment as a function of IL/biomass weight ratio of bamboo. They obtained a critical IL/biomass ratio of 3 g g−1 to obtain a cellulose saccharification of 80%, in a solid-state pretreatment.
The capability of ILs in dissolving lignin can be employed in the delignification of lignocelluloses. Fu et al.485 chose [EMIM][OAc] amongst six ILs as the best candidate for the selective extraction of lignin to improve the enzymatic hydrolysis of triticale and wheat straw at various temperatures (70–150 °C) and time intervals (0.5–24 h). Lee et al.486 also reported the enhancement in cellulose digestibility caused by partial delignification of wood flour by [EMIM][CH3COO]. They reported the maximum digestibility of cellulose to be 95% for triticale straw pretreated at 150 °C for 90 min. Wen et al.487 used [EMIM][OAc] under varying IL pretreatment conditions (i.e., 110–170 °C and 1–16 h) to isolate poplar alkaline lignin. Chemical transformation monitoring of the isolated lignin via elemental analysis, 2D-HSQC spectra, quantitative 13C-NMR spectra, 31P NMR, and GPC analyses revealed a decrease of aliphatic OH, mainly as a result of cleavage of the β-O-4′ linkage happened at high temperatures, and an increase in phenolic hydroxyl groups in lignin, attributed to the dehydration reaction during the pretreatment. The same study confirmed the β-O-4′ linkage broken with the dehydration and demethoxylation reactions during kraft lignin dissolution.488 2D NMR bond abundance data and size exclusion chromatography (SEC) results also revealed that lignin was depolymerized during [EMIM][OAc] pretreatment at 120 and 160 °C of wheat straw, miscanthus, and Loblolly pine,489 and lignin with different molecular mass was released in different stages of the pretreatment. Brandt et al.490 obtained the same result in lignin characteristics isolated from miscanthus after extraction with the protic ionic liquid 1-butylimidazolium hydrogen sulfate ([HC4im][HSO4]). Their 13C-NMR, 1H–13C HSQC NMR, 31P-NMR, Py-GC-MS, GPC, and elemental analyses showed that the lignin-hemicellulose linkages break and more than 80% depolymerization of lignin through the cleavage of glycosidic, ester, and β-O-4 ether bonds occurs during the early stage of the pretreatment. As the pretreatment proceeded, repolymerization of lignin happened, which was evidenced by the increased lignin molecular weight determined by GPC, and the increased phenolic hydroxyl group content and C/H ratio in the lignin prepared at the later stage. In another study, Varanasi et al.491 pretreated Panicum virgatum and Eucalyptus globulus with [EMIM][OAc] at different temperatures and studied compositional changes in lignin. Preferential breakdown of S-lignin in both eucalyptus and switchgrass at high pretreatment temperature (160 °C) and breakdown of G-lignin for eucalyptus and no preferential breakdown of either S- or G-lignin in switchgrass were observed at lower pretreatment temperatures (120 °C), which may be linked to its hydrogen-bond accepting capacities at these temperatures. Accordingly, they suggested the mechanism similarity of the IL pretreatment to alkali pretreatment at lower temperatures and to acid pretreatment at higher temperatures. S-G-H type lignin was obtained from bamboo by [AMIM][Cl] treatment, where partial degradation of lignin and hemicellulose was observed.492
Thermochemical analysis is also used for the characterization of lignin extracted with ILs. The depolymerization and breakdown of lignin pretreated with [EMIM][OAc] at 120 °C and 160 °C for 1, 3, 6, and 12 h in model biomass compounds and bioenergy feedstocks, by thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC) was reported.493 Lignin dissolution in cholinium ILs resulted in a higher maximal decomposition temperature (Tm) and a higher glass transition temperature (Tg) of kraft lignin.488 Moghaddam et al.494 compared the physicochemical properties of lignin isolated from sugarcane bagasse pretreated with acidified aqueous ethylene glycol (EG) and ILs, and soda lignin from NaOH pretreatment of bagasse. Accordingly, depolymerization of thermally stable IL and EG lignins occurred at higher temperatures compared to soda lignin. Moreover, unlike soda lignin, IL and EG lignins contained less/no carbohydrates, with slightly lower hydrogen and higher oxygen contents.494
George et al.495 investigated the effects of imidazolium-based IL cation and anion combinations on the macromolecular structure of three lignins, i.e., organosolv, alkali, and alkali low sulphonate. The results showed a significant reduction in molecular mass and a remarkable structural change of the lignins, primarily influenced by the anion, with anion influence in the reduction in the order sulfates > lactate > acetate > chlorides > phosphates, meanwhile cleavage of different linkages within the lignins was caused by different anions. However, at least 40% of the original large-lignin molecules, from each of the lignins studied, were observed to remain intact. On the other hand, extraction of lignin, with relatively uniform molecular weight without significant structural changes, from bagasse using an ionic liquid mixture [EMIM][ABS] at atmospheric pressure and elevated temperatures (170–190 °C) with a maximum yield of 93% was reported by Tan et al.496 They also concluded that ILs with better phase separation properties would be desirable for higher lignin extraction.
8.3 Effects of IL pretreatment on the cellulose structure
A majority of studies on the characterization of IL-treated lignocelluloses have focused on the transition of the cellulose crystalline structure and the surface morphology of biomass (e.g., Fig. 13 for macroscopic morphological changes). Zhang et al.497 studied the changes in the cellulose crystalline structure of three different feedstocks, switchgrass, corn stover, and rice husk, pretreated with [BMIM][OAc] at temperatures of 50–130 °C for 6 h by XRD. Increasing the treatment time led to a drop in biomass CrI, which was due to the swelling of crystalline cellulose and transition of cellulose I to cellulose II. Cheng et al.498,499 pretreated Avicel cellulose, switchgrass, pine, and eucalyptus with [EMIM][OAc] at 120 °C and 160 °C for 1, 3, 6, and 12 h, and investigated the structural transformation and crystalline structure of cellulose. Although for Avicel the transformation to cellulose II occurred under all processing conditions, higher temperatures and times were required for the same transformation process for the other feedstocks, and only an expanded cellulose I lattice was observed under the mild conditions applied. Comparable with these results, XRD analysis showed a decrease in CrI from 39.2% to ∼0.09% and 28.6% to ∼0.03% for switchgrass and agave bagasse, respectively, after [EMIM][OAc] pretreatment at 120 °C for 3 h.500 The regenerated cellulose from rice husk resulted from [EMIM][DEP] pretreatment at 100 °C for 10 h (1.5% (w/v) loading) showed the highest decrease in the crystallinity index from 46.0 to 32.0, amongst the different ILs used.501 | ||
| Fig. 13 The macroscopic effects of [EMIM][OAC] pretreatment on spruce softwood (picture taken from Shafiei et al.507) | ||
The morphological characterization of the wood cell wall treated with 1-ethylpyridinium bromide ([EtPy][Br]) and [EMIM][Cl] was studied by Kanbayashi and Miyafuji.502,503 The analyses of three hardwood by light microscopy and SEM revealed that treatment with [EMIM][Cl] at 120 °C for 72 h caused significant swelling of all the woods. However, depending on the wood species, various behavior and different morphological changes in pits have occurred mainly due to their chemical component and the microfibril angle.504 Similarly, treatment of Japanese cedar with [EtPy][Br] caused the cell wall swelling and elimination of warts, while it did not change pit membranes and the cellulose crystalline structure.503 Additionally, Raman microscopic analysis showed that chemical changes in the cell walls were different for different cell wall layers in that lignin in the compound middle lamella and the cell corner was resistant to interaction with [EtPy][Br]. Singh et al.504 used auto-fluorescent mapping to visualize cellulose and lignin in switchgrass stems for determining the mechanism of biomass dissolution during 1-n-ethyl-3-methylimidazolium acetate pretreatment. Swelling of the secondary cell wall followed by complete dissolution of biomass within 3 h at 120 °C, and subsequent lignin removal by adding an anti-solvent was observed. The surface roughness of switchgrass, pine, and eucalyptus samples pretreated with [EMIM][OAc] at 120 °C for 1, 3, 6, and 12 h showed that switchgrass possessed much rougher internal surfaces than eucalyptus and pine.499 Zhang et al.505 monitored the swelling and dissolution behavior of poplar during [EMIM][OAc] pretreatment by employing confocal Raman microscopy. They concluded that the dissolution of the biomass was divided into two parts: slow penetration of the IL, which determined the process reaction rate, and rapid dissolution of lignin and carbohydrates. Therefore, enhancement of the penetration capacity of the IL, which was suggested to depend upon the properties of the IL, was crucial for improving the pretreatment efficiency. Confocal Raman microscopy and confocal fluorescence microscopy were also used to analyze the changes in different cell types including tracheids, sclerenchyma cells, and parenchyma cells of corn stover during [EMIM][OAc] pretreatment.506 A direct correlation was then observed between changes in the morphologies and chemical composition and swelling occurred mainly in the secondary plant cell walls.
8.4 Acid-catalyzed hydrolysis in ionic liquids
Different acids, e.g., mineral acids, Brönsted acids,508,509 solid protic-acid resin,510 and even amino acids,511 have been functionalized or co-utilized to enhance the effect of the IL pretreatment on enzymatic hydrolysis. The research conducted on using acidic ionic liquid solutions for the pretreatment of lignocelluloses can be divided into three parts. The use of acids for direct depolymerization of polymeric carbohydrates in the presence of ILs,512–514 the use of an acid in ILs as a boosting pretreatment agent which can enhance the effectiveness of pretreatment in enzymatic hydrolysis,510,515,516 and using acid-functionalized ILs for either direct hydrolysis or enhanced enzymatic hydrolysis of lignocelluloses.508,517–521 Although the use of homogeneous acid catalysts has its drawbacks, acid catalysts are currently used in IL pretreatment. One of the main reasons is the economic viability of the IL used for pretreatment. For example, even though [BMIM][Cl] costs ca. 1/60th of [EMIM][OAc], it is not a highly efficient solvent for pretreatment; however, the addition of an acid catalyst can boost the performance of cheaper ILs for pretreatment.510 Another reason is that acid hydrolysis of carbohydrates in such systems occurs at lower temperatures than in the aqueous phase.513 Besides, because of the presence of lignin in the solid phase, sugar-lignin fractionation is easily achieved in such systems compared with aqueous phase reactions.513Development in the acidic IL pretreatment was first focused on the direct conversion of lignocelluloses into monomeric sugars. Li et al.512 developed a method for direct hydrolysis of cellulose in [BMIM][Cl] at 100 °C under atmospheric pressure catalyzed by mineral acids. The maximum glucose and total reducing sugar (TRS) yield of 43% and 77%, respectively, was obtained at 0.11 sulfuric acid/cellulose mass ratio for 540 min reaction time. Likewise, a maximum TRS yield of 65% was obtained from corn stover pretreated with [AMIM][Cl] at 100 °C for 90 min in the presence of 2.0 mmol HCl per gram lignocellulosic substrate.514 Pretreatment of three wood species including eucalyptus, pine, and spruce thermomechanical pulp was performed at 120 °C for 3 h in [AMIM][Cl] followed by dilute hydrochloric acid hydrolysis for 5 h.513 This IL-based acid pretreatment resulted in near-complete conversion of the woods' cellulose and hemicellulose at acid concentrations of 1.4–1.5 mole of HCl per g wood. However, at higher acid concentrations, the presence of several degradation compounds, such as 5-hydroxymethylfurfural (HMF), furan-2-carboxylic acid, catechol, methylcatechol, methylguaiacol, acetoguaiacone, and acetol, were detected in recycled ILs.
Although the dissolution step was conducted at low-water content, the hydrolysis step required much more water. Consequently, the high processing cost for sugar separation is a major barrier for industrialization of this process. da Costa Lopes and Bogel-Łukasik522 comprehensively reviewed the challenges and possibilities of direct IL acid-catalyzed conversion of cellulose and lignocellulosic biomass.
A majority of studies on acidic IL pretreatment, however, have recently focused on acid co-solvent IL pretreatment for enhanced enzymatic hydrolysis. Partial saccharification of carbohydrates is inevitable in such systems; however, a 2- to 12-fold higher glucose conversion rate was reported from combined acid-IL pretreatment of pine than the single pretreatment of acid or IL.523 Besides, the sole use of ILs in the pretreatment usually requires a high temperature and longer reaction time.515 Moreover, using an IL solution containing significant amounts of water and acid solution can reduce the expensive IL usage, in spite of significantly reducing the solubility of lignocelluloses in most ILs at above 1% water concentration. The action of acids in ILs is a catalytic role in the hydrolysis of ether linkages between adjacent glucose in the cellulose chain, consequently reducing the length of the cellulose chain510 (Fig. 14). Zhang et al.515 developed an optimized sugarcane bagasse pretreatment process using aqueous [BMIM][Cl] containing 1.2% HCl in the presence of 10–30% water at 130 °C for 30 min. Accordingly, a glucan digestibility of 94–100% was obtained after 72 h of enzymatic hydrolysis using HCl, in the pretreatment medium, as a more effective catalyst than H2SO4 and FeCl3. Hydrochloric acid was also reported to be the most effective catalyst, amongst seven other inorganic acids studied, with [MMIM][DMP] in the pretreatment of corn stover at 110 °C for 2 h.524 Under these conditions, a maximum TRS yield of 92.7% was obtained after 96 h enzymatic hydrolysis.
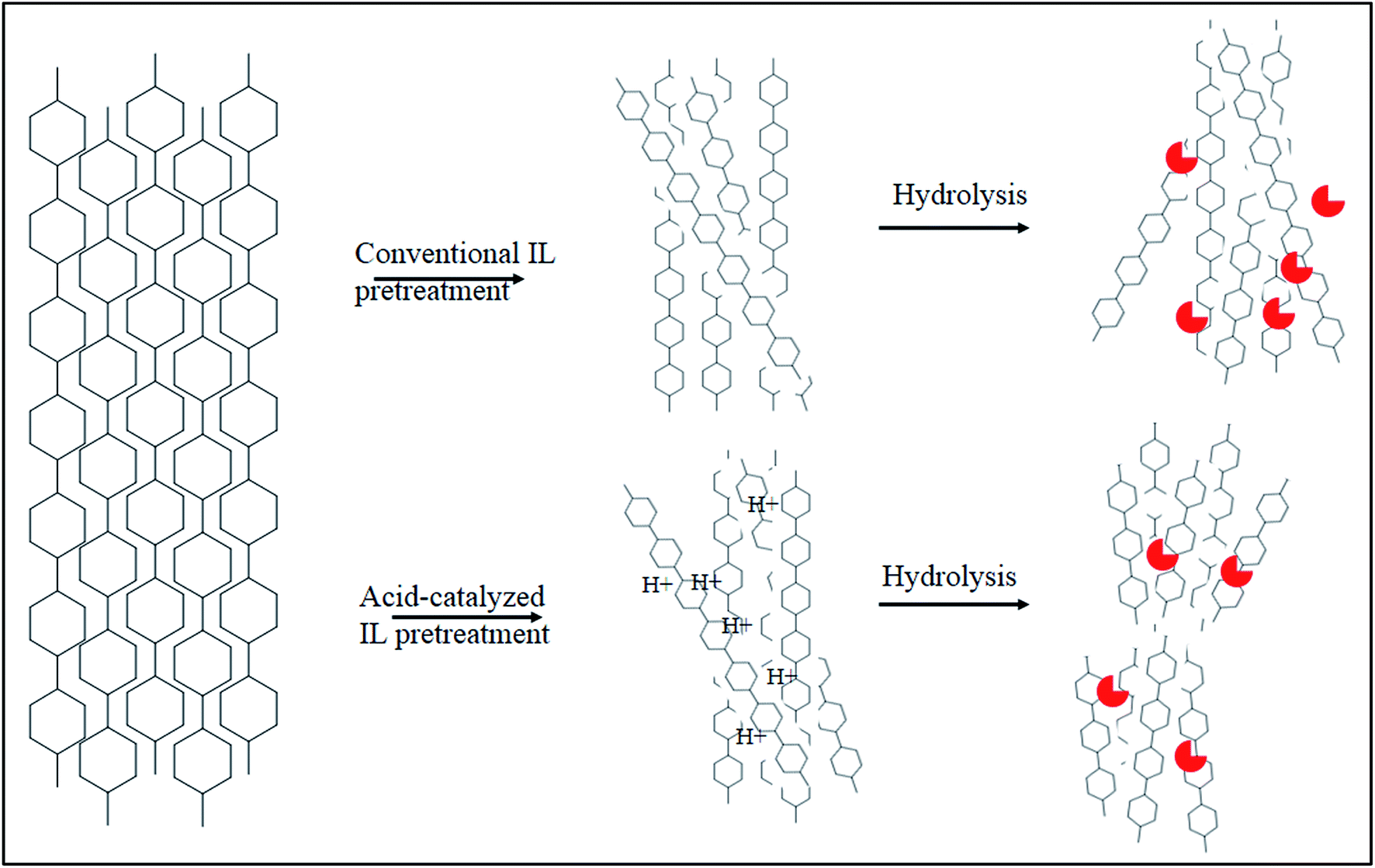 | ||
| Fig. 14 A comparison between lignocellulosic biomass pretreatment with conventional ILs and acid-based ILs (modified from ref. 510 with permission). | ||
The first attempt for the dissolution and hydrolysis of cellulose (DP ≈ 450) in Brönsted acidic ionic liquids 1-(1-propylsulfonic)-3-methylimidazolium chloride and 1-(1-butylsulfonic)-3-methylimidazolium chloride at moderate reaction temperatures was reported by Amarasekara and Owereh525 in 2009. A maximum TRS yield of 62% was obtained after 1 h of preheating at 70 °C followed by 30 min heating after adding 2.0 moles equivalent of water per glucose unit. Then, they discovered a more effective dilute aqueous solution of 1-(1-propylsulfonic)-3-methylimidazolium chloride and p-toluenesulfonic acid to be a better catalyst than aqueous sulfuric acid with the same H+ ion concentration for the degradation of cellulose at moderate temperatures and pressures.526 Amarasekara and Shanbhag527 dissolved switchgrass biomass in 1-(alkylsulfonic)-3-methylimidazolium Brönsted acidic ILs by heating at 70 °C for 2 h (0.22 g water per g switchgrass) and obtained maximum 58.1% and 15.3% TRS and glucose yields, respectively. Li et al.517 used six kinds of SO3H-functionalized ILs based on 1-methylimidazole, 1-vinylimidazole, and triethylamine to promote the hydrolysis of MCC in [BMIM][Cl]. The acidic ILs resulted in over 83% TRS yield at 100 °C with the maximum yield of 99% for triethyl-(3-sulfo-propyl)-ammonium hydrogen sulfate. Zhuo et al.519 synthesized and used six acidic ILs based on 2-phenyl-2-imidazoline for the hydrolysis of cellulose in [BMIM][Cl]. A maximum TRS yield of 85.1% was obtained by using 1-propyl sulfonic acid-2-phenyl imidazoline hydrogen sulfate, functionalized with HSO4− and Cl− instead of H2PO4−, at 100 °C for 60 min and a dosage of 0.2 g water per g cellulose. The hydrolysis activity was reported to be directly related to the activity of the catalyst and also the possibility of further degradation of the resulting carbohydrates in acidic IL to HMF.528 Tao et al.529 investigated the effect of acidity and structure of fifteen SO3H-functionalized ILs on the MCC hydrolysis and selectivity for HMF, furfural, levulinic acid (LA), and TRS yields. A maximum MCC conversion of 91.2% and selectivities for HMF, furfural, and LA of 45.7%, 26.2%, and 10.5%, respectively, were achieved in MnCl2-containing ILs. The efficiency of Brönsted acidic ILs for the conversion of hardwood hemicellulose to pentose sugars at 160 °C was reported to be related to acid strength in the following order: [C3SO3HMIM][HSO4] > [C3SO3HMIM][PTS] > [C3SO3HMIM][Cl] > [BMIM][Cl].509 Besides the activity of catalysts, solution pH is also another factor, which was investigated by Zhang et al.520 using different acid-catalyzed imidazolium IL solutions (80% in water) at 130 °C for 30 min for sugarcane bagasse. The pretreatment effectiveness was reported to be similar by using [BMIM][CH3SO3], [BMIM][CH3SO4], and [EMIM][Cl], at the same solution pH. Besides, by decreasing solution pH from 6.0 to 0.4, an increase in bagasse delignification, xylan removal, and consequently glucan digestibility was reported. Apart from Brönsted acidic ionic liquids, Muhammad et al.518 synthesized and used an amino acid-based ionic liquid, namely 1-ethyl-3-methylimidazolium glycinate, which was capable of effectively dissolving bamboo and changed its cellulose from type I to type II.
8.5 Enhancement in enzymatic digestibility of IL-pretreated lignocelluloses
The key and widely studied role of the IL pretreatment is to enhance the enzymatic digestibility of lignocelluloses.530–534Table 8 reviews some reports on improving the enzymatic hydrolysis of different lignocelluloses due to IL pretreatment. ILs with [BMIM] and [EMIM] cations and [Cl], [OAc], and [CH3COO] anions have been vastly used for the pretreatment of different lignocellulosic substrates. The pretreatment conditions applied were temperature 90–160 °C, reaction times ranging from several minutes to a few hours, and a biomass loading of ca. 2–15% depending on the biomass and IL types. As reviewed in the table, enzymatic hydrolysis of different (ligno)celluloses improved significantly as a result of IL pretreatment. From the reported data, maximum 100% digestibility was reported for MCC by using [BMIM][Cl] at 90 °C for 20 min. For lignocelluloses, the TRS yield was sometimes reported and in these cases the xylose yield was also significantly improved due to IL pretreatment. For Typha capensis, for instance, a maximum reducing sugar yield of 82.4 g/100 g biomass was obtained by [BMIM][OAc] pretreatment. A high xylose yield of 87% was also obtained from switchgrass pretreated with [EMIM][Lys] at 140 °C for 1 h.| Ionic liquid | Substrate | Pretreatment conditions | Enzymatic digestibility | Ref. |
|---|---|---|---|---|
| [BMIM][Cl] | Avicel | 130, 140, or 150 °C for 10, 30, 60, 120, or 180 min, 5% biomass (w/w) loading | Maximum ∼80% cellulose conversion to glucose after 24 h of enzymatic hydrolysis | 552 |
| [EMIM][CH3COO] | α-Cellulose | 110 °C for 40 min, 2% (w/v) cellulose loading | 61% yield of glucose at 76 h | 553 |
| Medium fibers of cellulose | 69% yield of glucose at 76 h | |||
| Long fibers of cellulose | 75% yield of glucose at 76 h | |||
| Microcrystalline cellulose | 71% yield of glucose at 76 h | |||
| [EMIM][MeO(H)PO2] | α-Cellulose | 67% yield of glucose at 76 h | ||
| Medium fibers of cellulose | 86% yield of glucose at 76 h | |||
| Long fibers of cellulose | 88% yield of glucose at 76 h | |||
| Microcrystalline cellulose | 75% yield of glucose at 76 h | |||
| [BMIM][OAc] | Typha capensis (TC) | 110 °C for 6 h, 5.0 g IL per 0.26 g TC | Maximum reducing sugar yield of 82.4 g/100 g | 554 |
| [EMIM][OAc] | Cellulose isolated from sugarcane bagasse | 90 °C for 6 h, 33.3 g IL per g cellulose | 95.2% glucose yield | 555 |
| Cholinium amino acids ILs | Rice straw | 90 °C for 5 h | Maximum glucose and xylose yields of 84.0% and 42.1%, respectively | 556 |
| Cholinium lysine IL ([Ch][Lys] IL)-water mixtures | Rice straw | 20% [Ch][Lys]-water mixture at 90 °C for 1 h | Maximum sugar yields of 81% for glucose and 48% for xylose | 557 |
| [EMIM][OAc] | Sugarcane bagasse | 150 °C, 90 min and 5% bagasse in IL | 83% and 21% glucan and xylan digestibility, respectively | 558 |
| [EMIM][OAc] | Energy cane bagasse | 120 °C for 30 min, 5% (w/w) biomass loading | 87.0% and 64.3% glucan and xylan digestibility, respectively | 125 |
| [EMIM][OAc] | Pinus radiate compression wood | 120 °C for 3 h | 93% glucan digestibility, 65% xylan digestibility, and 39% mannan digestibility after 24 h | 559 |
| 1-Hexylpyridinium chloride | Avicel and bagasse | 80 °C or 100 °C, 5% (w/w) loading | Over 95% conversion to glucose after 24 h for Avicel, and 1–3-fold higher conversion than untreated biomass for bagasse | 560 |
| 1-Butylimidazolium hydrogen sulfate | Miscanthus giganteus | 120 °C for 15 min up to 24 h, 10% (w/v) biomass loading | Recovery of up to 90% of the glucan as fermentable glucose and up to 39% saccharification yield for hemicellulose | 561 |
| [EMIM][OAc]–DMSO solutions | Eucalyptus | Ratios of 4:1, 3:2, 2:3 and 1:4 (v/v) [EMIM][OAc]-to-DMSO, 15% (w/v) biomass loading, at temperatures ranging from 80 to 140 °C |
95% of glucose theoretical maximum yield and up to 65% xylose yield | 562 |
| Chloride, acetate, and formate based IL | MCC | 90 °C for 20 min | 100% digestibility by using [BMIM][Cl] | 563 |
| [EMIM][OAc] | Rice husk | 100 °C for 10 h, 1.5% (w/v) biomass loading | 42.1% reducing sugar yield | 501 |
| [EMIM][OAc] | Agave bagasse (AGB) and switchgrass (SWG) | 120 and 160 °C for 3 h and 15% biomass loading | Increase in TRS by 100% for SWG and by 183% for AGB | 500 |
| [EMIM][OAc] | Bagasse | Optimum conditions: 145 °C, 15 min and 14 wt% solid loading | 69.7% of RS yield | 564 |
| [EMIM][OAc] | Sugarcane bagasse | 8 min at 140 °C, 25 wt% biomass loading | Glucose yields of more than 90% after 24 h of enzymatic saccharification and maximum xylose yield of ca. 85% | 479 |
| [EMIM][OAc] | Switchgrass | 160 °C and 3 h, 15% biomass loading | Glucose and xylose yields of 94.8% and 62.2%, respectively | 480 |
| 1-Butyl-3-methylimidazolium methyl sulfate and 1-butyl-3-methylimidazolium hydrogen sulfate | Miscanthus giganteus, pine (Pinus sylvestris), and willow (Salix viminalis) | 120 °C and 2 h, 10% (w/v) biomass loading | Up to 90% of the glucose and 25% of the hemicellulose by the combined ionic liquid pretreatment and the enzymatic hydrolysis | 565 |
| [EMIM][OAc] | Eucalyptus globulus | 120 °C for 3 h, 9.7 g IL and 0.3 g biomass | 37 and 30% glucose and xylose yields, respectively, after 4 h enzymatic hydrolysis | 566 |
| 1-Ethyl-3-methylimidazolium acetate | Wheat straw | Temperature (130–170 °C), time (0.5–5.5 h) and ionic liquid concentration (0–100%), biomass loading 5% (w/w) | 71.4% sugars recovery under optimum conditions of 158 °C, IL concentration, 49.5% (w/w), and 3.6 h | 567 |
| [EMIM][OAc] | Triticale straw | 150 °C for 90 min, 1.5 g straw to 48.5 g of water-IL mixture | 81% fermentable sugar yield | 568 |
| [EMIM][OAc] and [BMIM][Cl] | Cotton cellulose | Microwave irradiation or 110 °C for 30 min, 2% (w/v) biomass loading | At least 12-fold and by 50-fold enhancement in enzymatic hydrolysis at 110 °C and microwave irradiation, respectively | 569 |
| [BMIM][Cl] | Sugarcane bagasse | Temperatures (110–160 °C) and times (30–180 min); 0.25 g bagasse in 5 g IL (≤5% impurities and 2% moisture) | Optimum conditions: 150 °C for 90 min complete (100%) and rapid (3 h) glucan saccharification up to 70% xylan solubilization | 570 |
| [BMIM][Cl] | Cotton | 130 °C for 20 min, 5% w/w | At least 4-fold enhancement on cellulose saccharification conversion | 571 |
| [EMIM][CH3COO] | Wood flour | Various temperatures, 5% w/w biomass loading | >90% conversion of cellulose | 486 |
| Choline acetate (ChOAc) | Bagasse | IL/ultrasound-assisted pretreatment (60 min at 24 kHz and a power of 35 W), 0.25 g bagasse in 5 g IL | Cellulose and hemicellulose saccharification percentages 80% and 72%, respectively, in situ saccharification for 48 h | 572 |
| Choline formate (ChFor), choline acetate (ChOAc), and choline propionate (ChPro) | Kenaf powders | Microwave heating or 110 °C for 20 min, 5% w/w biomass loading | 20% cellulose conversion for regular heating and 60–90% for microwave heating | 573 |
| [BMIM][Cl] | Sweet sorghum bagasse | 110 °C for 1 h, 10% w/w biomass loading | Approximately 40% conversion of cellulose after 60 h enzymatic hydrolysis | 574 |
| [BMIM][Cl] | Populus tomentosa Carr. | 130 °C, 0.5 g of the substrate and 9.5 g of the IL | 92% glucose yield after 72 h enzymatic hydrolysis | 349 |
| [EMIM][Cl] and [EMIM][OAc] | Pine wood | 80, 100, or 120 °C for 3 h with stirring, 0.35 g wood and 7.0 g IL | Glucan conversions ranging from 23% to 84% with [EMIM][OAc] being more effective than [EMIM][Cl] | 575 |
| [BMIM][Cl] | Oil palm frond (OPF) | Temperatures less than 100 °C and times less than 1 h, and maximum loadings of 10% | 100% glucose recovery with pretreatment conditions: 80 °C, 15 min, and 10% solid loading | 576 |
| [EMIM][OAc] | Panicum virgatum (switchgrass) | 90 °C for 5 h, 10% (w/w) biomass loading | Glucose yield: 31%; xylose yield: 29% | 577 |
| [EMIM][Lys] | Glucose yield: 70%; xylose yield: 68% | |||
| [Ch][Lys] | Glucose yield: 42%; xylose yield: 58% | |||
| [Ch][OAc] | Glucose yield: 27%; xylose yield: 23% | |||
| [EMIM][OAc] | 140 °C for 1 h, 10% (w/w) biomass loading | Glucose yield: 65%; xylose yield: 86% | ||
| [EMIM][Lys] | Glucose yield:59%;xylose yield: 87% | |||
| [Ch][Lys] | Glucose yield: 61%; xylose yield: 82% | |||
| [Ch][OAc] | Glucose yield: 55%; xylose yield: 79% | |||
| [EMIM][OAc] | Poplar and switchgrass | 120 °C for 30 min, 5% (w/w) biomass loading | ∼70% and 46% glucan conversion after 24 h enzymatic hydrolysis for poplar and switchgrass, respectively | 578 |
| [EMIM][MeO(H)PO2] and [EMIM][CH3COO] | Cotton cellulose | 45 and 25 °C for 20 min, 2%, w/v biomass loading | Glucose yield after 24 h of enzymatic hydrolysis: 58.5% and 45.4% | 579 |
| [EMIM][OAc] | Spruce and oak sawdust | 110 °C for 40 min, 2% w/v biomass loading | Up to 7 times increase in enzymatic saccharification compared with the untreated substrate | 580 |
| [AMIM][Cl] | Cotton-based waste textiles were | 90, 110, and 130 °C until 2% (w/w) biomass was dissolved | 7 times higher yield of fermentable sugars than untreated fabrics | 581 |
| [EMIM][OAc] | Oil palm empty fruit bunch (OPEFB) | 130 °C, 2 h, 5% (w/w) biomass loading | 95.5% enzymatic digestibility of glucan | 582 |
| [BMIM][Cl] | 54.8% enzymatic digestibility of glucan | |||
| MTBS | 22.0% enzymatic digestibility of glucan | |||
| [EMIM][DEP] | 48.9% enzymatic digestibility of glucan | |||
| [EMIM][DEP] | Wheat straw | 130 °C for 30 min, 4% (w/w) biomass loading | 54.8% reducing sugar yield after being enzymatically hydrolyzed for 12 h | 583 |
| ChOAc | Bamboo powder | 110 °C for 60 min, and ultrasonic pretreatment in the same IL at 25 °C for 60 min, 0.5 g bamboo in 5 g IL | 55% and 92% cellulose saccharification for regular heating and ultrasonic pretreatment, respectively | 584 |
| [EMIM][OAc] | Beechwood chips | 115 °C for 1.5 h, 500 mg or 1 g of the wood in IL to obtain a mass of 10 g | Cellulose conversion of 90.2 wt% for a hydrolysis time of 72 h | 585 |
8.6 Challenges with in situ enzymatic hydrolysis of lignocelluloses in aqueous-IL media
Unlike water and buffers, which are capable of dissolving enzymes without unfolding their active structure, the biocatalysis activity in organic solvents may be hampered by a variety of factors. Most cellulases and other hydrolytic enzymes are deactivated in the presence of ILs, even at low concentrations.535,536 A comprehensive review on the enzymatic hydrolysis of lignocelluloses in the presence of ionic liquids, or the so-called in situ or one-pot pretreatment and hydrolysis, has been recently published by Wahlström and Suurnäkki.537 This review was mainly focused on the ways to keep the cellulase enzyme active for enzymatic hydrolysis in the presence of ILs. The hydrolytic enzyme stabilization techniques include enzyme immobilization, e.g., by encapsulation538 or thermostabilization.539 Besides, the discovery and development of IL-tolerant enzymes,540,541e.g., enzymes isolated from thermophilic and halophilic microbes,542,543 are of great importance in this regard. A review on various cellulase stabilization techniques for the single-step process and the design of enzyme compatible biomass-dissolving ILs was recently published by Elgharbawy et al.544 The recent trends in IL-tolerant enzymes and microorganisms were also critically reviewed by Portillo and Saadeddin545 and Xu et al.546It is notable here to mention that the residual ILs in the enzymatic hydrolysates inhibit the growth and productivity of microorganisms in downstream and fermentation processes.547,548 The residual [EMIM][OAc] in the hydrolysates (higher than 0.1%) was reported to inhibit the growth and ethanol production by S. cerevisiae, suggested due to a potential synergistic effect between this particular combination of anions and cations.549 The water-wash step results in a significant sugar loss and generation of large amounts of wastewater. To address this issue, Xu et al.550 recently developed a one-pot conversion process via using dilute bio-based ILs to produce high-titer cellulosic ethanol. Moreover, a novel CBP process was developed for ethanol production using IL pretreatment by cellulase-displaying yeast and approximately 90% ethanol yield was reported.551
Another challenge in enzymatic in situ saccharification of lignocelluloses is the recovery of sugars produced during the hydrolysis in IL media. Chromatographic techniques586 and membrane-based methods587 have been suggested for the separation and recovery of sugars and ILs from biomass hydrolysates. This challenge also applies to the case of acid-catalyzed hydrolysis in ILs,588 and it is considered a major challenge and a drawback in using ILs as pretreatment agents. However, in most cases, the recovery of sugars and the recycling of ILs occur simultaneously in a single process.
8.7 Recovery and reuse of ionic liquids
Due to the current high price of ILs for an economically viable pretreatment process, efficient recovery and recycling of ILs is vital.589–592 Besides, the wastage of ILs can cause environmental issues associated with slow degradation and toxicity to downstream processes589,590 Mai et al.593 reviewed different methods for the recovery of ILs in detail. Here, we discuss briefly the methods of IL recovery with application in the IL pretreatment of lignocelluloses.The most widely used method for the recovery and recycling of ILs from IL-anti-solvent–lignocellulose systems is distillation.580,590,594–596 The method consists of evaporating anti-solvent (e.g., water and alcohol) after removing precipitated lignocellulose from the pretreatment media. Since a large quantity of precipitating solvent is required to prevent gel phase formation, the evaporation step needs a lot of energy and often presents environmental problems. When water is used for the precipitation, the situation is even worse, because of its high specific heat capacity and the high solubility of produced biomass compounds, e.g., monomeric and small oligomeric carbohydrates.590,596 Approximately, 85–90% recovery of [EMIM][OAc] was reported in an IL–water system via distillation by Qui et al.594
The ability of ILs to form aqueous biphasic systems with a kosmotropic anion, e.g., phosphate, carbonate, or sulfate, was first reported by Gutowski et al.,597 which can be utilized to recycle hydrophilic ILs from aqueous solutions. The upper IL-rich phase can be easily recovered by simple decantation or a magnetic field. However, further separation is required in order to extract water and remaining monosaccharide hydrolysates from the IL solution.598 The recovery of ILs in these systems depends on the salt type and concentration as well as the IL cation and anion type.599 A recovery of over 95.0% for [BMIM][OAc] in K3PO4-containing systems (pH 12–13) was reported.599
Apart from these traditional separation methods, recently, the so-called green processes were developed based on chromatography (resin and alumina column chromatography)598,600 and electrodialysis601–603 for the IL recovery.
Unfortunately, the recovered ILs do not sometimes show the same performance in the pretreatment as their virgin forms.594,604 Qiu et al.594 reported a decrease in recycled [EMIM][OAc], by evaporation, performance in pretreatment of energy cane bagasse. This phenomenon could be attributed to the accumulation of IL degradation products in the pretreatment process which affect the recycling efficiency and properties of ILs. Besides, the recycled ILs may contain carbohydrate monomers and oligomers and biomass decomposition products. It is most likely for recycled ILs to have both of the mentioned impurities that negatively affect their performance in pretreatment.175 On the other hand, Auxenfans et al.580 reported the same ability and similar efficiency of recycled [EMIM][OAc], via distillation by rotary evaporation, to the fresh IL in enzymatic saccharification performance for pretreatment of industrial wood sawdust. In another study, they again reported the similar performance of two recycled imidazolium-based ILs in maintaining their efficiency to pretreat cellulose.579
8.8 IL pretreatment of lignocelluloses for enhanced biogas and renewable chemical production
Different pretreatment methods for enhanced biogas production from lignocelluloses have been comprehensively reviewed by Zheng et al.69 Mancini et al.605 also reviewed the solvent pretreatments of lignocellulosic materials to enhance biogas production. Nonetheless, there are few studies in the literature on the enhancement of biogas production from lignocelluloses by using IL pretreatment. Gao et al.606 pretreated water hyacinth, rice straw, mango leaves, and spruce with [CnMIM][Cl] (n = 2, 4, and 6) under different conditions and evaluated the effect of the pretreatment on biogas production. The maximum enhancement of biogas production was obtained by pretreatment with [BMIM][Cl] at 120 °C for 2 h for water hyacinth followed by spruce, while maximum methane production from rice straw and mango leaves, i.e., 233 and 125 mL g−1 carbohydrates, was obtained for pretreatment at 140 °C for 2 h and 140 °C for 8 h, respectively. They also obtained an increase in biogas yield and methane concentration by 16.3–97.6% and 13.2–28.3%, respectively, from water hyacinth by [BMIM][Cl] and DMSO co-solvent pretreatment at 120 °C for 120 min.607 Li and Xu608 reported severe toxicity of imidazolium-based ILs in anaerobic digestion of grass (1:10 ratio). However, they reported a low toxicity and high recyclability potential for [BMIM][OAc] in the methane production of 221 mL g−1-VS from grass.
Most of the studies on using IL pretreatment for the production of renewable products from lignocelluloses have focused on the hydrolysis of pretreated biomass into sugar-rich hydrolysates (cf. Section 8.5), which are then used by microorganisms for carbon and energy sources, e.g., for microbial lipid production. For example, Gong et al.609 prepared corn stover solids by [EMIM][OAc]–N-methylpyrrolidone (NMP) pretreatment at 140 °C and converted the pretreated solids to microbial lipids using Cryptococcus curvatus via a simultaneous saccharification and enhanced lipid production process. They obtained maximum 112 mg g−1 pretreated biomass lipid yield with efficient co-utilization of cellulose and hemicellulose. Xie et al.610 also used 20% (mole fraction) [EMIM][OAc] in NMP at 140 °C for 60 min to pretreat corn stover followed by enzymatic hydrolysis for the cultivation of Rhodosporidium toruloides Y4 for lipid production. The oleaginous microorganism utilized both C6 and C5 sugars in the hydrolyzates and produced a moderate 15.2 g L−1 biomass yield and 36.4% lipid yield. Bokinsky et al.611 reported the synthesis of biofuels, i.e., fatty acid ethyl esters, butanol, and pinene, from [EMIM][OAc] pretreated switchgrass using engineered Escherichia coli which can express cellulase, xylanase, β-glucosidase, and xylobiosidase enzymes. They reported that the IL pretreatment made the biomass completely susceptible to hydrolysis.
On the use of ILs for renewable chemical production from lignocelluloses, Huang et al.612 evaluated the effects of residual [EMIM][Cl], [EMIM][DEP], and [EMIM][OAc] on the lipid production by oleaginous yeast Rhodosporidium toruloides AS 2.1389. By adjusting pH to 6.0 in the presence of 30 mM ILs, minor inhibition effects were reported, while the presence of 60 mM ILs caused a significant inhibition on the yeast. Liu et al.613 also reported that the residual ILs in the hydrolysate of rice straw inhibited the growth and lipid accumulation by Geotrichum fermentans. The inhibition was induced by both the anion and cation of the ILs used, and the side chain of the cation showed a clear inhibition.
Varanasi et al.614 focused on the production of lignin-based renewable chemicals from different lignocelluloses by selective breakdown of lignin using [EMIIM][OAc] pretreatment at 120 and 160 °C for 6 h. The generated chemicals, (i.e., phenols, guaiacols, syringols, eugenol, and catechols), their oxidized products (i.e., vanillin, vanillic acid, and syringaldehyde), and their easily derivatized hydrocarbons (i.e., benzene, toluene, xylene, styrene, biphenyls, and cyclohexane) were produced from lignin by tuning the process conditions. The production of levulinic acid directly during the IL pretreatment was reported in some studies.615,616 Muranaka et al.616 successfully converted 60.7% (72.9 mol%) of cellulose into levulinic acid by using [EMIM][Br] and [EMIM][P] at 80–120 °C for 1–6 h under stirring. Sun et al.615 used a series of heteropolyacid (HPA) ILs to catalyze one-pot depolymerization of cellulose into glucose and subsequent levulinic acid (up to a 60% yield) in a water–methyl isobutyl ketone (MIBK) biphasic system. Xiao et al.617 optimized the catalytic conversion of cellulose into HMF by AlCl3 in DMSO–[BMIM][Cl] mixtures. They obtained a maximum HMF yield of 54.9% from cellulose at 150 °C after 9 h in a mixed solvent of DMSO–[BMIM][Cl] (10 wt%).
8.9 Techno-economic analysis of ionic liquid pretreatment
For economic evaluation of IL pretreatment of lignocelluloses, Sen et al.618 identified the IL cost as the major cost driver in IL pretreatment. They suggested lower IL consumption and/or effective separation strategies to improve the economy of IL pretreatment. Konda et al.619 compared the cost drivers and economic potential of two variants of IL pretreatment for ethanol production: first based on complete removal of the IL prior to hydrolysis and second based on a one-pot process. At a high biomass loading of 50%, both routes were reported to be economically viable with a minimum ethanol selling price of $6.3 per gal. With more reduced water and acid/base consumption in the first and second routes, respectively, improved pretreatment efficiency, and by lignin valorization, the minimum ethanol selling price could be reduced to $3.2 for the former route and $2.8 for the latter route. Baral and Shah620 conducted a techno-economic comparison study on [EMIM][OAc] and sulfuric acid pretreatment of corn stover, poplar, and switchgrass, and estimated a sugar production cost of 2.7, 3.2, and 3.0 $ per kg, respectively. They further reported that optimistic considerations of at least 97% IL recovery, less than $1 per kg IL cost, and >90% heat recovery are required to have an economically competitive IL pretreatment. To improve the process economy, George et al.621 attempted to lower the cost of IL, which is one of the main impediments to IL utilization in the pretreatment step, by designing a number of low-cost and stable protic ILs based upon the [HSO4] anion with promising potential in the pretreatment. The most effective solvent, triethylammonium hydrogen sulfate IL, demonstrated approximately 75% as effective as [C2C1IM][OAc] for switchgrass. A set of new low-cost RTILs based on butylammonium prepared by reacting carboxylic acids with aliphatic amine cations under ambient conditions was also synthesized and characterized by de Andrade Neto et al.622 for lignocellulose hydrolysis applications. Among them, n-butylammonium acetate favored the subsequent acid hydrolysis of corn fiber. Oleskowicz-Popiel et al.623 also reported the acidolysis of IL pretreated lignocelluloses as a more economically viable route than using costly enzymes for saccharification. They further calculated that the minimum ethanol selling price could be reduced to $4.00 per gal, when the performance of the hydrolysis, extraction, and sugar recovery is improved. Socha et al.624 synthesized some tertiary amine-based ILs from aromatic aldehydes derived from vanillin, p-anisaldehyde, and furfural, and confirmed their effectiveness in switchgrass pretreatment. Their approach of producing renewable ILs from lignocelluloses in a so-called “closed-loop” can be a solution for the drawback of expensive ILs in the pretreatment.8.10 Present status and future prospects of IL pretreatment
Taking all these points into consideration, ILs, as powerful non-derivatizing cellulose solvents, have been recently subjected to vast studies for lignocellulose dissolution and regeneration. The promising features of using ILs for biomass pretreatment are negligible vapor pressure, thermal stability, non-flammability, and high polarity, and being “green” solvents in many cases.625–627 They are capable of fractionating a variety of lignocelluloses, reordering or restructuring the hydrogen bonds in the cellulose network, decreasing cellulose crystallinity, and increasing cellulose accessibility to cellulases. The pretreatment requires low equipment costs with low energy consumption. The regenerated materials are more susceptible to enzymatic hydrolysis than the untreated form, with comparable or even superior yields of fermentable sugars, than the conventional pretreatments.628,629 By adjusting the ILs' anion and cation, different ILs are synthesized to tune their properties. Mora-Pale et al.630 stated that lignin released during RTIL pretreatment of lignocelluloses is likely to be far more “pristine” than Kraft lignin, and has general applications in phenol-formaldehyde replacements, conversion into liquid fuels following hydrogenative depolymerization, or possibly into specific low molecular weight chemicals.A process diagram for bioconversion of lignocelluloses to ethanol and biogas using IL pretreatment is proposed in Fig. 15. The process can be applied for the production of other fermentative chemicals via a sugar platform as well. Besides, this process is advantageous in the production of other byproducts, which can improve the overall process economy.
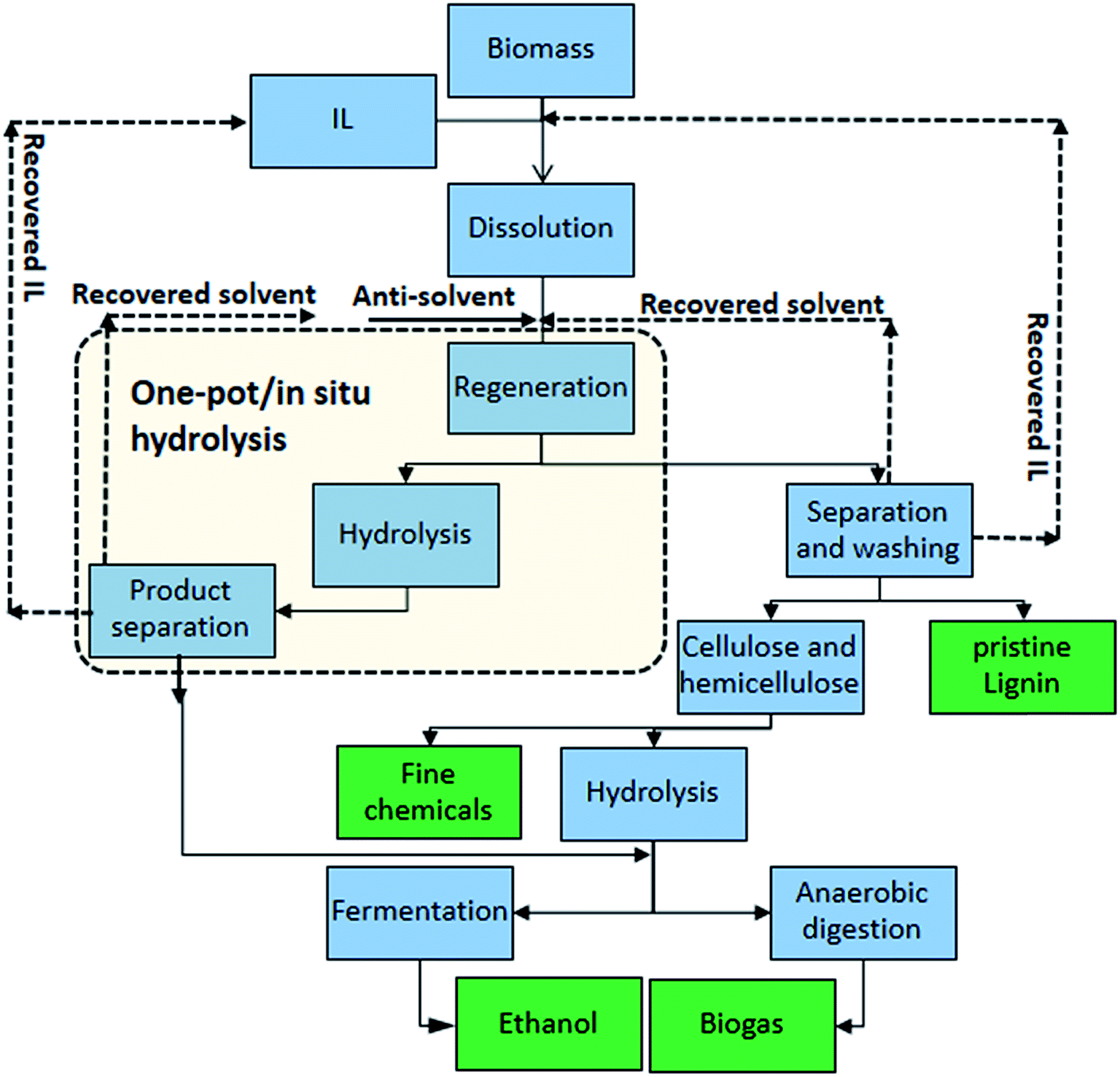 | ||
| Fig. 15 A process diagram for IL pretreatment of lignocelluloses for ethanol or biogas production applying two routes: (1) one-pot/in situ hydrolysis and (2) separated enzymatic hydrolysis and fermentation, with IL and solvent recycling. | ||
However, IL pretreatment of lignocelluloses is facing several technological and economic challenges. Although some efforts have been made to design low-cost ILs621,631 (as discussed in Section 8.9), still a major obstacle in implication of many ILs on a large scale is their high price. Efficient recovery and recycling of ILs are crucial in order to reduce the inhibitory effects of ILs on subsequent enzymatic hydrolysis and fermentation. Besides, the residual ILs in waste streams can cause environmental problems depending on their degradability.632 Although research is under way using amino acids to synthesize biodegradable ILs, most ILs for biomass processing are not easily biodegradable. However, a cost-effective technology, despite the ease of recycling via distillation, is needed to make the process competitive to conventional pretreatment strategies. This problem is not only for IL recovery, but also for the anti-solvent used in the regeneration process. Although many ILs, e.g., [EMIM][OAc] and [AMIM][Cl], were reported to be excellent solvents for cellulose dissolution,632 the selection of ILs for biomass pretreatment should compromise between solubilizing power and compatibility with enzymes and/or organisms. In the case of acidic ILs, the rate of formation of degradation products should be manipulated by the side chains of the cation. Not all, but some ILs are corrosive, toxic, and hygroscopic, which should be utilized with care.
9 Comparison of IL, CPA, and NMMO pretreatments
A few studies compared the effectiveness of the IL, CPA, and NMMO pretreatments in improving enzymatic hydrolysis yield and fuel/chemical production.165,386,571,633–637 Wheat straw was pretreated with CPA (85%, 50 °C for 1.5 h), NMMO (130 °C for 2 h), and IL ([AMIM][Cl], 110 °C for 1 h) and the results showed that the most prominent difference in the chemical composition of wheat straw was >90% solubilization of xylan due to CPA pretreatment, while for the other two treatments, xylan was solubilized <10%.633 Phosphoric acid acts as a Brønsted acid catalyst and generally promotes the hydrolysis of glycosidic bonds in cellulose/hemicellulose, which leads to considerably higher solubilization of xylan. Moreover, cellulose hydrolysis followed the order of CPA > NMMO > [AMIM][Cl]. Similarly, CPA was more successful in pretreatment of corn stover than IL, whereas, compared to 96% glucan digestibility of corn stover pretreated with CPA, a 55% glucan digestibility was obtained after pretreatment with [BMIM][Cl]. A tradeoff between cellulose disruption and the inhibitory effects of the presence of residual lignin and residual cellulose solvent on the pretreated biomass was reported as the main reason for incomplete hydrolysis for IL pretreatment. However, cellulose digestibilities of 100% and 92% were obtained with CPA and [BMIM][Cl], respectively, for Avicel cellulose as a substrate.634In a study, high glucose yields were obtained for rice straw following pretreatment with NMMO and [BMIM][OAc], and the obtained yields were comparable for both pretreatments.381 The crystallinity index and total crystallinity of the substrate were quite similar for both pretreatments. Similarly, the glucan conversion (after 72 h) for Populus tomentosa pretreated with IL ([BMIM][Cl] at 130 °C), NMMO (130 °C for 30 min), and CPA (85% at room temperature) was reported to be 80%, 82%, and 92%, respectively.635 The results also showed that the hydrolysis rate for the IL pretreated sample was higher than that of the CPA pretreated sample. These results were possibly due to the transformation of cellulose I to amorphous cellulose and cellulose II in IL and CPA pretreatments, respectively. CPA was reported as a better cellulose solvent than NMMO and [BMIM][Cl] in improving the saccharification rate and yield of cotton cellulose due to the high specific surface area and low DP for CPA pretreated cellulose.636
On the other hand, IL pretreatment ([EMIM][Br] and [EMIM][P]) was reported to be more effective than CPA pretreatment in converting cellulosic substrates to levulinic acid.637 Decrease in cellulose crystallinity, solubilization of cellulose, and IL interaction with cellulose were the determinant factors for higher yields.
In summary, the effectiveness of different cellulose solvents in the pretreatment of lignocelluloses is strongly dependent on biomass type and final chemicals produced. In contrast to phosphoric acid, residual ILs and NMMO cause inhibitory effects on biotechnological downstream processes.
10 Concluding remarks
Regarding the high worldwide fuel demand and the significant potential for biomass conversion to offset fossil fuel usage, a high number of studies and efforts have been made in the past several decades in the cellulosic fuel area. The cellulosic fuel production process involves four major steps of biomass preparation, pretreatment, hydrolysis, and fermentation, with the pretreatment step being one of the most cost contributing and the rate and yield limiting step. Given their significant ability to fractionate the lignocellulosic structure, cellulose solvents are excellent starting points for industrial biorefinery applications. Although this category of pretreatment has several advantages over other conventional pretreatments, which was the focus of this review, several issues should be addressed to make the process economically and environmentally viable. One of the promising features of cellulose solvents for the pretreatment of lignocelluloses is associated with their properties as “green” solvents. They should have negligible vapor pressure to help their recyclability, and should be easily biodegradable. Using cellulose solvents for the pretreatment of lignocelluloses is advantageous due to their application at high solid loadings and relatively low pressure/temperatures with no/less chemical modification and inhibitory byproduct formation. The solvents can be oriented towards different purposes for the pretreatment of lignocelluloses. They can be selected for (1) the separation of mainly cellulose, (2) the separation of mainly hemicellulose, (3) the separation of mainly lignin, (4) opening the compact structure, (5) regeneration and structural modification, and (6) cellulose crystallinity reduction, although more than one of these actions typically take place. Organic cellulose solvents, e.g., concentrated phosphoric acid and concentrated NaOH, are capable of removing and/or reorganizing the hydrogen bond network structure of cellulose, decreasing cellulose crystallinity, and enhancing cellulose accessibility to cellulases, and consequently enhancing glucan digestibility even at low cellulase loadings that is vital to reduce the overall process cost. There are some cellulose solvents, e.g., NMMO, that modify the structure of lignocelluloses by dissolution and regeneration of the whole biomass, without significant lignin/hemicellulose removal. Meanwhile, ILs are target-oriented solvents that can be designed to separate specific parts of lignocelluloses. The high revenues from co-products (acetic acid, lignin, and hemicelluloses) of the pretreatment can drastically improve the economy of cellulosic fuels. However, the cellulose solvent-based pretreatment of lignocelluloses is still in its early stage of development, mainly in the laboratory scale, and facing several technological and economic challenges. The efficient recovery of the solvents, which is usually a high energy intensive process, is necessary because of not only the high price of the solvents, but also because of the inhibitory effects of the solvents on subsequent processes. Furthermore, substantial reduction in the use of chemicals (both the cellulose solvents and organic solvents) is required in order to have an economically competitive process.Conflicts of interest
There are no conflicts to declare.Abbreviation
| AD | Anaerobic digestion |
| [BMIM][Cl] | 1-Butyl-3-methylimidazolium chloride |
| CBM | Cellulose binding module |
| CPA | Concentrated phosphoric acid |
| CBP | Consolidated bioprocessing |
| CAC | Cellulose accessibility to cellulase |
| COSLIF | Cellulose solvent- and organic solvent-based lignocellulosic fractionation |
| CrI | Crystallinity index |
| [EMIM][OAc | 1-Ethyl-3-methylimidazolium acetate |
| GHG | Greenhouse gas |
| IL | Ionic liquid |
| LOI | Lateral order index |
| NREL | National renewable energy laboratory |
| NMMO or NMO | N-Methyl-morpholine-N-oxide |
| RTIL | Room-temperature ionic liquid |
| SCO | Single cell oil |
| SAA | Soaking in aqueous ammonia |
| SS | Simons' staining |
| SSA | Specific surface area |
| TCI | Total crystallinity index |
| TRS | Total reducing sugar |
| VFA | Volatile fatty acid |
| WRV | Water retention value |
Acknowledgements
The corresponding author would like to acknowledge support from the Office of Biological and Environmental Research in the US Department of Energy (DOE) Office of Science through the BioEnergy Science Center (BESC) and the Center for Bioenergy Innovation (CBI), both at Oak Ridge National Laboratory.References
- F. H. Reboredo, F. Lidon, F. Pessoa and J. C. Ramalho, Trends Biotechnol., 2016, 34, 3–6 CrossRef CAS PubMed.
- N. Gaurav, S. Sivasankari, G. S. Kiran, A. Ninawe and J. Selvin, Renewable Sustainable Energy Rev., 2017, 73, 205–214 CrossRef CAS.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS.
- W. J. Orts and C. M. McMahan, BioEnergy Res., 2016, 9, 430–446 CrossRef CAS.
- C. Dellomonaco, F. Fava and R. Gonzalez, Microb. Cell Fact., 2010, 9, 1–15 CrossRef PubMed.
- S. Zinoviev, F. Müller-Langer, P. Das, N. Bertero, P. Fornasiero, M. Kaltschmitt, G. Centi and S. Miertus, ChemSusChem, 2010, 3, 1106–1133 CrossRef CAS PubMed.
- B. Antizar-Ladislao and J. L. Turrion-Gomez, Biofuels, Bioprod. Biorefin., 2008, 2, 455–469 CrossRef CAS.
- L. R. Lynd, J. H. Cushman, R. J. Nichols and C. E. Wyman, Science, 1991, 251, 1318–1323 CrossRef CAS PubMed.
- L. M. Fulton, L. R. Lynd, A. Körner, N. Greene and L. R. Tonachel, Biofuels, Bioprod. Biorefin., 2015, 9, 476–483 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS PubMed.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- S. Behera, R. Arora, N. Nandhagopal and S. Kumar, Renewable Sustainable Energy Rev., 2014, 36, 91–106 CrossRef CAS.
- A. Alaswad, M. Dassisti, T. Prescott and A. G. Olabi, Renewable Sustainable Energy Rev., 2015, 51, 1446–1460 CrossRef CAS.
- K. Kumar, S. Ghosh, I. Angelidaki, S. L. Holdt, D. B. Karakashev, M. A. Morales and D. Das, Renewable Sustainable Energy Rev., 2016, 65, 235–249 CrossRef CAS.
- E. Molina Grima, E. H. Belarbi, F. G. Acién Fernández, A. Robles Medina and Y. Chisti, Biotechnol. Adv., 2003, 20, 491–515 CrossRef CAS PubMed.
- C. Carere, R. Sparling, N. Cicek and D. Levin, Int. J. Mol. Sci., 2008, 9, 1342 CrossRef CAS PubMed.
- S. V. Vassilev and C. G. Vassileva, Fuel, 2016, 181, 1–33 CrossRef CAS.
- J. Lu, C. Sheahan and P. Fu, Energy Environ. Sci., 2011, 4, 2451–2466 RSC.
- J. Hill, E. Nelson, D. Tilman, S. Polasky and D. Tiffany, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11206–11210 CrossRef CAS PubMed.
- W. H. Liew, M. H. Hassim and D. K. S. Ng, J. Cleaner Prod., 2014, 71, 11–29 CrossRef CAS.
- K. Dutta, A. Daverey and J.-G. Lin, Renewable Energy, 2014, 69, 114–122 CrossRef CAS.
- D. C. Holzman, Environ. Health Perspect., 2008, 116, A246–A252 Search PubMed.
- S. Spatari, Y. Zhang and H. L. MacLean, Environ. Sci. Technol., 2005, 39, 9750–9758 CrossRef CAS PubMed.
- R. Kumar, M. Tabatabaei, K. Karimi and I. Sárvári Horváth, Biofuel Res. J., 2016, 3, 347–356 CrossRef CAS.
- M. A. Carriquiry, X. Du and G. R. Timilsina, Energy Policy, 2011, 39, 4222–4234 CrossRef.
- H. Chen and W. Qiu, Biotechnol. Adv., 2010, 28, 556–562 CrossRef CAS PubMed.
- P. R. Lennartsson, P. Erlandsson and M. J. Taherzadeh, Bioresour. Technol., 2014, 165, 3–8 CrossRef CAS PubMed.
- H. Zabed, J. N. Sahu, A. Suely, A. N. Boyce and G. Faruq, Renewable Sustainable Energy Rev., 2017, 71, 475–501 CrossRef CAS.
- B. Satari and K. Karimi, Appl. Microbiol. Biotechnol., 2018, 102, 1097–1117 CrossRef CAS PubMed.
- P. V. Harris, F. Xu, N. E. Kreel, C. Kang and S. Fukuyama, Curr. Opin. Chem. Biol., 2014, 19, 162–170 CrossRef CAS PubMed.
- M. Balat, Energy Convers. Manage., 2011, 52, 858–875 CrossRef CAS.
- K. Karimi, M. Tabatabaei, I. Sárvári Horváth and R. Kumar, Biofuel Res. J., 2015, 2, 301–308 CrossRef CAS.
- M. Mascal, Biofuels, Bioprod. Biorefin., 2012, 6, 483–493 CrossRef CAS.
- P. Dürre, Ann. N. Y. Acad. Sci., 2008, 1125, 353–362 CrossRef PubMed.
- P. Dürre, Biotechnol. J., 2007, 2, 1525–1534 CrossRef PubMed.
- P. Dürre, Curr. Opin. Biotechnol., 2011, 22, 331–336 CrossRef PubMed.
- K. Karimi, M. Tabatabaei, I. Sárvári Horváth and R. Kumar, Biofuel Res. J., 2015, 2, 301–308 CrossRef CAS.
- M. Kumar and K. Gayen, Appl. Energy, 2011, 88, 1999–2012 CrossRef CAS.
- Y.-N. Zheng, L.-Z. Li, M. Xian, Y.-J. Ma, J.-M. Yang, X. Xu and D.-Z. He, J. Ind. Microbiol. Biotechnol., 2009, 36, 1127–1138 CrossRef CAS PubMed.
- V. García, J. Päkkilä, H. Ojamo, E. Muurinen and R. L. Keiski, Renewable Sustainable Energy Rev., 2011, 15, 964–980 CrossRef.
- J. Cousin Saint Remi, T. Rémy, V. Van Hunskerken, S. van de Perre, T. Duerinck, M. Maes, D. De Vos, E. Gobechiya, C. E. A. Kirschhock, G. V. Baron and J. F. M. Denayer, ChemSusChem, 2011, 4, 1074–1077 CrossRef CAS PubMed.
- S. H. Ha, N. L. Mai and Y.-M. Koo, Process Biochem., 2010, 45, 1899–1903 CrossRef CAS.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef CAS PubMed.
- S. Sreekumar, Z. C. Baer, A. Pazhamalai, G. Gunbas, A. Grippo, H. W. Blanch, D. S. Clark and F. D. Toste, Nat. Protoc., 2015, 10, 528–537 CrossRef CAS PubMed.
- G. Liu, W. Wei and W. Jin, ACS Sustainable Chem. Eng., 2014, 2, 546–560 CrossRef CAS.
- N. Qureshi and H. P. Blaschek, Renewable Energy, 2001, 22, 557–564 CrossRef CAS.
- A. Morone and R. A. Pandey, Renewable Sustainable Energy Rev., 2014, 37, 21–35 CrossRef CAS.
- T. C. Ezeji, N. Qureshi and H. P. Blaschek, Curr. Opin. Biotechnol., 2007, 18, 220–227 CrossRef CAS PubMed.
- J. Bertsch and V. Müller, Biotechnol. Biofuels, 2015, 8, 210 CrossRef PubMed.
- M. Tabatabaei, K. Karimi, I. Sárvári Horváth and R. Kumar, Biofuel Res. J., 2015, 2, 258–267 CrossRef CAS.
- Q. Hu, M. Sommerfeld, E. Jarvis, M. Ghirardi, M. Posewitz, M. Seibert and A. Darzins, Plant J., 2008, 54, 621–639 CrossRef CAS PubMed.
- Q. Li, W. Du and D. Liu, Appl. Microbiol. Biotechnol., 2008, 80, 749–756 CrossRef CAS PubMed.
- X. Meng, J. Yang, X. Xu, L. Zhang, Q. Nie and M. Xian, Renewable Energy, 2009, 34, 1–5 CrossRef CAS.
- L. A. Garay, I. R. Sitepu, T. Cajka, I. Chandra, S. Shi, T. Lin, J. B. German, O. Fiehn and K. L. Boundy-Mills, J. Ind. Microbiol. Biotechnol., 2016, 43, 887–900 CrossRef CAS PubMed.
- Y. Zheng, X. Yu, J. Zeng and S. Chen, Biotechnol. Biofuels, 2012, 5, 1–10 CrossRef PubMed.
- M.-H. Liang and J.-G. Jiang, Prog. Lipid Res., 2013, 52, 395–408 CrossRef CAS PubMed.
- I. R. Sitepu, M. Jin, J. E. Fernandez, L. da Costa Sousa, V. Balan and K. L. Boundy-Mills, Appl. Microbiol. Biotechnol., 2014, 98, 7645–7657 CrossRef CAS PubMed.
- C. Huang, X.-f. Chen, L. Xiong, X.-d. Chen, L.-l. Ma and Y. Chen, Biotechnol. Adv., 2013, 31, 129–139 CrossRef CAS PubMed.
- S. Bellou, A. Makri, D. Sarris, K. Michos, P. Rentoumi, A. Celik, S. Papanikolaou and G. Aggelis, J. Biotechnol., 2014, 170, 50–59 CrossRef CAS PubMed.
- K. Kurosawa, S. J. Wewetzer and A. J. Sinskey, J. Microb. Biochem. Technol., 2014, 6, 254–259 CAS.
- B. Satari, K. Karimi, M. Taherzadeh and A. Zamani, Int. J. Mol. Sci., 2016, 17, 302 CrossRef PubMed.
- B. Satari, PhD thesis, Isfahan University of Technology, 2016.
- M. Enshaeieh, A. Abdoli, M. Madani and M. Bayat, Int. J. Environ. Sci. Technol., 2015, 12, 837–846 CrossRef CAS.
- F. B. Ahmad, Z. Zhang, W. O. S. Doherty and I. M. O'Hara, Biofuels, Bioprod. Biorefin., 2016, 10, 378–392 CrossRef CAS.
- A. Yousuf, Waste Manage., 2012, 32, 2061–2067 CrossRef CAS PubMed.
- M. Jin, P. J. Slininger, B. S. Dien, S. Waghmode, B. R. Moser, A. Orjuela, L. d. C. Sousa and V. Balan, Trends Biotechnol., 2015, 33, 43–54 CrossRef CAS PubMed.
- J.-C. Frigon and S. R. Guiot, Biofuels, Bioprod. Biorefin., 2010, 4, 447–458 CrossRef CAS.
- S. Kashi, B. Satari, M. Lundin, I. S. Horváth and M. Othman, J. Environ. Chem. Eng., 2017, 5, 6156–6164 CrossRef CAS.
- Y. Zheng, J. Zhao, F. Xu and Y. Li, Prog. Energy Combust. Sci., 2014, 42, 35–53 CrossRef.
- C. Sawatdeenarunat, K. C. Surendra, D. Takara, H. Oechsner and S. K. Khanal, Bioresour. Technol., 2015, 178, 178–186 CrossRef CAS PubMed.
- S. Campanaro, L. Treu, P. G. Kougias, D. De Francisci, G. Valle and I. Angelidaki, Biotechnol. Biofuels, 2016, 9, 26 CrossRef PubMed.
- C.-L. Cheng, Y.-C. Lo, K.-S. Lee, D.-J. Lee, C.-Y. Lin and J.-S. Chang, Bioresour. Technol., 2011, 102, 8514–8523 CrossRef CAS PubMed.
- A. Ghimire, L. Frunzo, F. Pirozzi, E. Trably, R. Escudie, P. N. L. Lens and G. Esposito, Appl. Energy, 2015, 144, 73–95 CrossRef CAS.
- H. Zilouei and M. Taherdanak, in Lignocellulose-based Bioproducts, ed. K. Karimi, Springer International Publishing, Cham, 2015, pp. 253–288 Search PubMed.
- J. A. Gimpel, V. Henríquez and S. P. Mayfield, Front. Microbiol., 2015, 6 Search PubMed.
- G. Davila-Vazquez, S. Arriaga, F. Alatriste-Mondragón, A. de León-Rodríguez, L. M. Rosales-Colunga and E. Razo-Flores, Rev. Environ. Sci. Biotechnol., 2008, 7, 27–45 CrossRef CAS.
- M. Taherdanak, H. Zilouei and K. Karimi, Int. J. Hydrogen Energy, 2016, 41, 167–173 CrossRef CAS.
- P. Sinha and A. Pandey, Int. J. Hydrogen Energy, 2011, 36, 7460–7478 CrossRef CAS.
- J. Wang and W. Wan, Int. J. Hydrogen Energy, 2009, 34, 799–811 CrossRef CAS.
- S. Rittmann and C. Herwig, Microb. Cell Fact., 2012, 11, 1–18 CrossRef PubMed.
- J. X. W. Hay, T. Y. Wu, J. C. Juan and J. Md. Jahim, Biofuels, Bioprod. Biorefin., 2013, 7, 334–352 CrossRef CAS.
- D.-J. Lee, K.-Y. Show and A. Su, Bioresour. Technol., 2011, 102, 8393–8402 CrossRef CAS PubMed.
- K. Y. Show, D. J. Lee, J. H. Tay, C. Y. Lin and J. S. Chang, Int. J. Hydrogen Energy, 2012, 37, 15616–15631 CrossRef CAS.
- X. Liu, N. Ren, F. Song, C. Yang and A. Wang, Prog. Nat. Sci., 2008, 18, 253–258 CrossRef CAS.
- Y.-H. P. Zhang, Biotechnol. Adv., 2015, 33, 1467–1483 CrossRef CAS PubMed.
- I. Z. Boboescu, M. Ilie, V. D. Gherman, I. Mirel, B. Pap, A. Negrea, É. Kondorosi, T. Bíró and G. Maróti, Biotechnol. Biofuels, 2014, 7, 1–15 CrossRef PubMed.
- N. H. M. Yasin, T. Mumtaz, M. A. Hassan and N. A. Abd Rahman, J. Environ. Manage., 2013, 130, 375–385 CrossRef CAS PubMed.
- N.-Q. Ren, L. Zhao, C. Chen, W.-Q. Guo and G.-L. Cao, Bioresour. Technol., 2016, 215, 92–99 CrossRef CAS PubMed.
- I. A. Panagiotopoulos, R. R. Bakker, T. de Vrije, E. G. Koukios and P. A. M. Claassen, Int. J. Hydrogen Energy, 2010, 35, 7738–7747 CrossRef CAS.
- T. de Vrije, G. G. de Haas, G. B. Tan, E. R. P. Keijsers and P. A. M. Claassen, Int. J. Hydrogen Energy, 2002, 27, 1381–1390 CrossRef CAS.
- Y.-C. Lo, W.-C. Lu, C.-Y. Chen and J.-S. Chang, Bioresour. Technol., 2010, 101, 5885–5891 CrossRef CAS PubMed.
- L. Zhao, G.-L. Cao, A.-J. Wang, H.-Y. Ren, D. Dong, Z.-N. Liu, X.-Y. Guan, C.-J. Xu and N.-Q. Ren, Bioresour. Technol., 2012, 114, 365–369 CrossRef CAS PubMed.
- R. Datar, J. Huang, P.-C. Maness, A. Mohagheghi, S. Czernik and E. Chornet, Int. J. Hydrogen Energy, 2007, 32, 932–939 CrossRef CAS.
- M. M. Arimi, J. Knodel, A. Kiprop, S. S. Namango, Y. Zhang and S.-U. Geißen, Biomass Bioenergy, 2015, 75, 101–118 CrossRef CAS.
- P. C. Hallenbeck and D. Ghosh, Trends Biotechnol., 2009, 27, 287–297 CrossRef CAS PubMed.
- R. Kleerebezem and M. C. M. van Loosdrecht, Curr. Opin. Biotechnol., 2007, 18, 207–212 CrossRef CAS PubMed.
- C. Sambusiti, M. Bellucci, A. Zabaniotou, L. Beneduce and F. Monlau, Renewable Sustainable Energy Rev., 2015, 44, 20–36 CrossRef CAS.
- J. Woodward, S. M. Mattingly, M. Danson, D. Hough, N. Ward and M. Adams, Nat. Biotechnol., 1996, 14, 872–874 CrossRef CAS PubMed.
- J. Woodward and M. Orr, Biotechnol. Prog., 1998, 14, 897–902 CrossRef CAS PubMed.
- X. Ye, Y. Wang, R. C. Hopkins, M. W. W. Adams, B. R. Evans, J. R. Mielenz and Y. H. P. Zhang, ChemSusChem, 2009, 2, 149–152 CrossRef CAS PubMed.
- Y. H. P. Zhang, B. R. Evans, J. R. Mielenz, R. C. Hopkins and M. W. W. Adams, PLoS One, 2007, 2, e456 CrossRef PubMed.
- J. S. Van Dyk and B. I. Pletschke, Biotechnol. Adv., 2012, 30, 1458–1480 CrossRef CAS PubMed.
- L. B. Davin, A. M. Patten, M. Jourdes and N. G. Lewis, in Biomass recalcitrance: deconstructing the plant cell wall for bioenergy, ed. M. Himmel, 2009, pp. 213–305 Search PubMed.
- S. Sun, S. Sun, X. Cao and R. Sun, Bioresour. Technol., 2016, 199, 49–58 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. da Costa Sousa, D. Gao, A. M. Cheh, U. P. Agarwal, C. M. Bianchetti, G. N. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS PubMed.
- R. H. Atalla and D. L. Vanderhart, Science, 1984, 223, 283–285 CrossRef CAS PubMed.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- R. H. Atalla, J. W. Brady, J. F. Matthews, S.-Y. Ding and M. E. Himmel, in Biomass Recalcitrance, Blackwell Publishing Ltd., 2009, pp. 188–212 Search PubMed.
- B. H. Davison, J. Parks, M. F. Davis and B. S. Donohoe, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 23–38 Search PubMed.
- S.-Y. Ding and M. E. Himmel, J. Agric. Food Chem., 2006, 54, 597–606 CrossRef CAS PubMed.
- M. Jarvis, Nature, 2003, 426, 611–612 CrossRef CAS PubMed.
- S. Y. Ding, Y. S. Liu, Y. Zeng, M. E. Himmel, J. O. Baker and E. A. Bayer, Science, 2012, 338, 1055–1060 CrossRef CAS PubMed.
- C. Li, L. Sun, B. A. Simmons and S. Singh, BioEnergy Res., 2013, 6, 14–23 CrossRef CAS.
- M. Shafiei, K. Karimi and M. J. Taherzadeh, Bioresour. Technol., 2010, 101, 4914–4918 CrossRef CAS PubMed.
- C. K. Nitsos, T. Choli-Papadopoulou, K. A. Matis and K. S. Triantafyllidis, ACS Sustainable Chem. Eng., 2016, 4, 4529–4544 CrossRef CAS.
- A. Kallioinen, M. Hakola, T. Riekkola, T. Repo, M. Leskelä, N. von Weymarn and M. Siika-aho, Bioresour. Technol., 2013, 140, 414–420 CrossRef CAS PubMed.
- A. Goshadrou, K. Karimi and M. Lefsrud, Carbohydr. Polym., 2013, 96, 440–449 CrossRef CAS PubMed.
- M. S. Noori and K. Karimi, Biochem. Eng. J., 2016, 105(part A), 197–204 CrossRef CAS.
- L. Kumar, V. Arantes, R. Chandra and J. Saddler, Bioresour. Technol., 2012, 103, 201–208 CrossRef CAS PubMed.
- P. Salehian, K. Karimi, H. Zilouei and A. Jeihanipour, Fuel, 2013, 106, 484–489 CrossRef CAS.
- J. Xu, J. J. Cheng, R. R. Sharma-Shivappa and J. C. Burns, Bioresour. Technol., 2010, 101, 2900–2903 CrossRef CAS PubMed.
- H. Beheshti and K. Karimi, Eng. Life Sci., 2016, 16, 750–761 CrossRef CAS.
- B. Erdei, M. Galbe and G. Zacchi, Biomass Bioenergy, 2013, 56, 506–514 CrossRef CAS.
- Z. Qiu, G. M. Aita and M. S. Walker, Bioresour. Technol., 2012, 117, 251–256 CrossRef CAS PubMed.
- C. G. Yoo, H. Kim, F. Lu, A. Azarpira, X. Pan, K. K. Oh, J. S. Kim, J. Ralph and T. H. Kim, BioEnergy Res., 2016, 9, 67–76 CrossRef CAS.
- A. Goshadrou, K. Karimi and M. J. Taherzadeh, Ind. Crops Prod., 2011, 34, 1219–1225 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, National Renewable Energy Laboratory, 2008 Search PubMed.
- A. Faik, in Green Biomass Pretreatment for Biofuels Production, ed. T. Gu, Springer Netherlands, 2013, pp. 1–30 Search PubMed.
- C. Wyman, S. R. Decker, M. Himmel, J. W. Brady, C. E. Skopec and L. Viikari, in Polysaccharides, CRC Press, 2004 Search PubMed.
- M. Tenkanen, in Hemicelluloses: Science and Technology, American Chemical Society, 2003, pp. 292–311 Search PubMed.
- T. Zhang, R. Kumar, Y.-D. Tsai, R. T. Elander and C. E. Wyman, Green Chem., 2015, 17, 394–403 RSC.
- R. Kumar and C. E. Wyman, Carbohydr. Res., 2008, 343, 290–300 CrossRef CAS PubMed.
- R. D. Fagan, H. E. Grethlein, A. O. Converse and A. Porteous, Environ. Sci. Technol., 1971, 5, 545–547 CrossRef CAS.
- J. F. Saeman, Ind. Eng. Chem., 1945, 37, 43–52 CrossRef CAS.
- M. Taherzadeh and K. Karimi, Int. J. Mol. Sci., 2008, 9, 1621–1651 CrossRef CAS PubMed.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315 CrossRef CAS PubMed.
- U. Bornscheuer, K. Buchholz and J. Seibel, Angew. Chem., Int. Ed., 2014, 53, 10876–10893 CrossRef CAS PubMed.
- R. Ciriminna, N. Chavarría-Hernández, A. Inés Rodríguez Hernández and M. Pagliaro, Biofuels, Bioprod. Biorefin., 2015, 9, 368–377 CrossRef CAS.
- D. Mohnen, Curr. Opin. Plant Biol., 2008, 11, 266–277 CrossRef CAS PubMed.
- A. L. Galant, G. A. Luzio, W. W. Widmer and R. G. Cameron, Food Hydrocolloids, 2014, 35, 661–669 CrossRef CAS.
- M. B. Sticklen, Nat. Rev. Genet., 2008, 9, 433–443 CrossRef CAS PubMed.
- C. Xiao and C. T. Anderson, Front. Plant Sci., 2013, 4, 67 Search PubMed.
- B. Satari, J. Palhed, K. Karimi, M. Lundin, M. J. Taherzadeh and A. Zamani, BioResources, 2017, 12, 1706–1722 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS PubMed.
- M. J. Taherzadeh and A. Jeihanipour, in Biogas Production, John Wiley & Sons, Inc., 2012, pp. 27–54 Search PubMed.
- X. Zhao, L. Zhang and D. Liu, Biofuels, Bioprod. Biorefin., 2012, 6, 465–482 CrossRef CAS.
- B. Yang and C. E. Wyman, Biofuels, Bioprod. Biorefin., 2008, 2, 26–40 CrossRef CAS.
- P. Alvira, E. Tomás-Pejó, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed.
- R. P. Chandra, R. Bura, W. E. Mabee, A. Berlin, X. Pan and J. N. Saddler, Adv. Biochem. Eng./Biotechnol., 2007, 108, 67–93 CrossRef CAS PubMed.
- R. Kumar, G. Mago, V. Balan and C. E. Wyman, Bioresour. Technol., 2009, 100, 3948–3962 CrossRef CAS PubMed.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- L. da Costa Sousa, S. P. S. Chundawat, V. Balan and B. E. Dale, Curr. Opin. Biotechnol., 2009, 20, 339–347 CrossRef PubMed.
- S. S. Hassan, G. A. Williams and A. K. Jaiswal, Bioresour. Technol., 2018, 262, 310–318 CrossRef CAS PubMed.
- E. C. Bensah and M. Mensah, Int. J. Chem. Eng., 2013, 2013, 21 Search PubMed.
- G. Brodeur, E. Yau, K. Badal, J. Collier, K. B. Ramachandran and S. Ramakrishnan, Enzyme Res., 2011, 2011, 17 Search PubMed.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- Y.-L. Loow, T. Y. Wu, J. Md. Jahim, A. W. Mohammad and W. H. Teoh, Cellulose, 2016, 23, 1491–1520 CrossRef CAS.
- B. Yang, L. Tao and C. E. Wyman, Biofuels, Bioprod. Biorefin., 2017, 12, 125–138 CrossRef.
- M. Kumar, A. O. Oyedun and A. Kumar, Renewable Sustainable Energy Rev., 2018, 81, 1742–1770 CrossRef.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- S. Wang, A. Lu and L. Zhang, Prog. Polym. Sci., 2016, 53, 169–206 CrossRef CAS.
- Y. Cao, R. Zhang, T. Cheng, J. Guo, M. Xian and H. Liu, Appl. Microbiol. Biotechnol., 2017, 101, 521–532 CrossRef CAS PubMed.
- T. Li, Q. Fang, H. Chen, F. Qi, X. Ou and X. Zhao, et al. , RSC Adv., 2017, 7, 10609–10617 RSC.
- B. Medronho and B. Lindman, Curr. Opin. Colloid Interface Sci., 2014, 19, 32–40 CrossRef CAS.
- T. Kanda, K. Wakabayashi and K. Nisizawa, J. Biochem., 1980, 87, 1635–1639 CrossRef CAS PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- M. Wada, M. Ike and K. Tokuyasu, Polym. Degrad. Stab., 2010, 95, 543–548 CrossRef CAS.
- E. Husson, T. Auxenfans, M. Herbaut, M. Baralle, V. Lambertyn and H. Rakotoarivonina, et al. , Bioresour. Technol., 2018, 251, 280–287 CrossRef CAS PubMed.
- G. Papa, T. Feldman, K. L. Sale, F. Adani, S. Singh and B. A. Simmons, Bioresour. Technol., 2017, 241, 627–637 CrossRef CAS PubMed.
- N. Sathitsuksanoh, A. George and Y. H. P. Zhang, J. Chem. Technol. Biotechnol., 2013, 88, 169–180 CrossRef CAS.
- T. Dutta, N. G. Isern, J. Sun, E. Wang, S. Hull and J. R. Cort, et al. , ACS Sustainable Chem. Eng., 2017, 5, 10116–10127 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- S. Singh and B. A. Simmons, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 223–238 Search PubMed.
- K. Karimi, M. Shafiei and R. Kumar, in Biofuel Technologies: Recent Developments, ed. K. V. Gupta and G. M. Tuohy, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 53–96 Search PubMed.
- P. Domínguez de María, J. Chem. Technol. Biotechnol., 2014, 89, 11–18 CrossRef.
- H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913–3929 RSC.
- X. Zhao, L. Zhang and D. Liu, Biofuels, Bioprod. Biorefin., 2012, 6, 561–579 CrossRef CAS.
- J. Luo, M. Cai and T. Gu, in Green Biomass Pretreatment for Biofuels Production, ed. T. Gu, Springer Netherlands, Dordrecht, 2013, pp. 127–153 Search PubMed.
- T. Vancov, A.-S. Alston, T. Brown and S. McIntosh, Renewable Energy, 2012, 45, 1–6 CrossRef CAS.
- B. Satari and K. Karimi, Resour., Conserv. Recycl., 2018, 129, 153–167 CrossRef.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC.
- Q. Hou, M. Ju, W. Li, L. Liu, Y. Chen and Q. Yang, Molecules, 2017, 22, 490 CrossRef PubMed.
- M. Ghasemi, M. Tsianou and P. Alexandridis, Bioresour. Technol., 2017, 228, 330–338 CrossRef CAS PubMed.
- R. Katzen and D. F. Othmer, Ind. Eng. Chem., 1942, 34, 314–322 CrossRef CAS.
- N. Gilbert, I. A. Hobbs and J. D. Levine, Ind. Eng. Chem., 1952, 44, 1712–1720 CrossRef CAS.
- M. J. Taherzadeh and K. Karimi, BioResources, 2007, 2, 472–499 CAS.
- M. J. Taherzadeh and K. Karimi, BioResources, 2007, 2, 707–738 CAS.
- B. Satari Baboukani, M. Vossoughi and I. Alemzadeh, Biosyst. Eng., 2012, 111, 166–174 CrossRef.
- C. E. Wyman, Trends Biotechnol., 2007, 25, 153–157 CrossRef CAS PubMed.
- D. Klein-Marcuschamer, P. Oleskowicz-Popiel, B. A. Simmons and H. W. Blanch, Biotechnol. Bioeng., 2012, 109, 1083–1087 CrossRef CAS PubMed.
- H. Jørgensen, J. B. Kristensen and C. Felby, Biofuels, Bioprod. Biorefin., 2007, 1, 119–134 CrossRef.
- Z. Zhang, A. A. Donaldson and X. Ma, Biotechnol. Adv., 2012, 30, 913–919 CrossRef CAS PubMed.
- E. Ximenes, Y. Kim and M. R. Ladisch, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 39–60 Search PubMed.
- M. Kostylev and D. Wilson, Biofuels, 2012, 3, 61–70 CrossRef CAS.
- R. C. Kuhad, D. Deswal, S. Sharma, A. Bhattacharya, K. K. Jain, A. Kaur, B. I. Pletschke, A. Singh and M. Karp, Renewable Sustainable Energy Rev., 2016, 55, 249–272 CrossRef CAS.
- Y. H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88, 797–824 CrossRef CAS PubMed.
- Y. H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2006, 94, 888–898 CrossRef CAS PubMed.
- T. Jeoh, M. J. Cardona, N. Karuna, A. R. Mudinoor and J. Nill, Biotechnol. Bioeng., 2017, 114, 1369–1385 CrossRef CAS PubMed.
- P. Bansal, M. Hall, M. J. Realff, J. H. Lee and A. S. Bommarius, Biotechnol. Adv., 2009, 27, 833–848 CrossRef CAS PubMed.
- D. B. Wilson, Curr. Opin. Biotechnol., 2009, 20, 295–299 CrossRef CAS PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed.
- R. Biswas, A. Persad and V. S. Bisaria, in Bioprocessing of Renewable Resources to Commodity Bioproducts, John Wiley & Sons, Inc., 2014, pp. 105–132 Search PubMed.
- H. J. Gilbert, J. P. Knox and A. B. Boraston, Curr. Opin. Struct. Biol., 2013, 23, 669–677 CrossRef CAS PubMed.
- M. Saloheimo, M. Paloheimo, S. Hakola, J. Pere, B. Swanson, E. Nyyssönen, A. Bhatia, M. Ward and M. Penttilä, Eur. J. Biochem., 2002, 269, 4202–4211 CrossRef CAS PubMed.
- S. Jung, Y. Song, H. M. Kim and H. J. Bae, Enzyme Microb. Technol., 2015, 77, 38–45 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sorlie and V. G. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- I. J. Kim, H. J. Lee, I.-G. Choi and K. H. Kim, Appl. Microbiol. Biotechnol., 2014, 98, 8469–8480 CrossRef CAS PubMed.
- S. R. Decker, M. Siika-Aho and L. Viikari, in Biomass Recalcitrance, Blackwell Publishing Ltd., 2009, pp. 352–373 Search PubMed.
- D. B. Wilson, Appl. Microbiol. Biotechnol., 2012, 93, 497–502 CrossRef CAS PubMed.
- T. Lloyd and C. Liu, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 451–469 Search PubMed.
- D. Shallom and Y. Shoham, Curr. Opin. Microbiol., 2003, 6, 219–228 CrossRef CAS PubMed.
- A. Várnai, L. Huikko, J. Pere, M. Siika-Aho and L. Viikari, Bioresour. Technol., 2011, 102, 9096–9104 CrossRef PubMed.
- R. Kumar and C. E. Wyman, Biotechnol. Prog., 2009, 25, 302–314 CrossRef CAS PubMed.
- S. P. S. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed.
- M. M. Kabir, G. Forgács and I. Sárvári Horváth, in Lignocellulose-based Bioproducts, ed. K. Karimi, Springer International Publishing, Cham, 2015, pp. 207–251 Search PubMed.
- L. R. Lynd, P. J. Weimer, W. H. van Zyl and I. S. Pretorius, Microbiol. Mol. Biol. Rev., 2002, 66, 506–577 CrossRef CAS PubMed.
- E. A. Bayer, J. P. Belaich, Y. Shoham and R. Lamed, Annu. Rev. Microbiol., 2004, 58, 521–554 CrossRef CAS PubMed.
- G. Salehi Jouzani and M. J. Taherzadeh, Biofuel Res. J., 2015, 2, 152–195 CrossRef.
- D. Cannella and H. Jørgensen, Biotechnol. Bioeng., 2014, 111, 59–68 CrossRef CAS PubMed.
- W. F. Gauss, S. Suzuki and M. Takagi, US Pat., US3990944A, 1976.
- T. B. Vinzant, W. S. Adney, S. R. Decker, J. O. Baker, M. T. Kinter, N. E. Sherman, J. W. Fox and M. E. Himmel, in Twenty-Second Symposium on Biotechnology for Fuels and Chemicals, ed. B. H. Davison, J. McMillan and M. Finkelstein, Humana Press, Totowa, NJ, 2001, pp. 99–107 Search PubMed.
- D. B. Wilson, in Biomass Recalcitrance, Blackwell Publishing Ltd., 2009, pp. 374–392 Search PubMed.
- T. K. Ghose, Pure Appl. Chem., 1987, 59, 257–268 CAS.
- B. Adney and J. Baker, Technical Report, Natural Renewable Energy Laboratory, NREL/TP-510-42628, 2008 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS PubMed.
- S. P. Chundawat, M. S. Lipton, S. O. Purvine, N. Uppugundla, D. Gao, V. Balan and B. E. Dale, J. Proteome Res., 2011, 10, 4365–4372 CrossRef CAS PubMed.
- M. Selig and N. Y. J. Weiss, in Technique report, NREL/TP-510-42629, 2008 Search PubMed.
- R. Kumar and C. E. Wyman, Woodhead Publ. Ser. Energy, 2010, 3, 73–121 CAS.
- R. Kumar and C. E. Wyman, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 281–310 Search PubMed.
- S. B. A. Hamid, M. M. Islam and R. Das, Cellulose, 2015, 22, 2157–2182 CrossRef.
- A. P. Sinitsyn, A. V. Gusakov and E. Y. Vlasenko, Appl. Biochem. Biotechnol., 1991, 30, 43–59 CrossRef CAS.
- J. Wallace, M. Brienzo, M. P. García-Aparicio and J. F. Görgens, New Biotechnol., 2016, 33, 361–371 CrossRef CAS PubMed.
- L. Zhu, J. P. O'Dwyer, V. S. Chang, C. B. Granda and M. T. Holtzapple, Bioresour. Technol., 2008, 99, 3817–3828 CrossRef CAS PubMed.
- Z. Ling, S. Chen, X. Zhang, K. Takabe and F. Xu, Sci. Rep., 2017, 7, 10230 CrossRef PubMed.
- V. Pihlajaniemi, M. H. Sipponen, H. Liimatainen, J. A. Sirvio, A. Nyyssola and S. Laakso, Green Chem., 2016, 18, 1295–1305 RSC.
- J. A. Rollin, Z. Zhu, N. Sathitsuksanoh and Y. H. P. Zhang, Biotechnol. Bioeng., 2011, 108, 22–30 CrossRef CAS PubMed.
- B. Yang, Z. Dai, S.-Y. Ding and C. E. Wyman, Biofuels, 2011, 2, 421–450 CrossRef CAS.
- K. Karimi and M. J. Taherzadeh, Bioresour. Technol., 2016, 200, 1008–1018 CrossRef CAS PubMed.
- S. Park, J. Baker, M. Himmel, P. Parilla and D. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- A. Peciulyte, K. Karlström, P. T. Larsson and L. Olsson, Biotechnol. Biofuels, 2015, 8, 1–13 CrossRef CAS PubMed.
- B. Yang, D. M. Willies and C. E. Wyman, Biotechnol. Bioeng., 2006, 94, 1122–1128 CrossRef CAS PubMed.
- M. Hall, P. Bansal, J. H. Lee, M. J. Realff and A. S. Bommarius, FEBS J., 2010, 277, 1571–1582 CrossRef CAS PubMed.
- A. Mittal, R. Katahira, M. E. Himmel and D. K. Johnson, Biotechnol. Biofuels, 2011, 4, 1–16 CrossRef PubMed.
- T. Cui, J. Li, Z. Yan, M. Yu and S. Li, Biotechnol. Biofuels, 2014, 7, 134 CrossRef PubMed.
- M. Ioelovich and E. Morag, BioResources, 2011, 6, 2818–2835 CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- A. D. French and M. Santiago Cintrón, Cellulose, 2013, 20, 583–588 CrossRef CAS.
- P. J. Harris and B. A. Stone, in Biomass Recalcitrance, Blackwell Publishing Ltd., 2009, pp. 61–93 Search PubMed.
- K. Karimi and M. J. Taherzadeh, Bioresour. Technol., 2016, 203, 348–356 CrossRef CAS PubMed.
- Y. H. P. Zhang and L. R. Lynd, Biomacromolecules, 2005, 6, 1510–1515 CrossRef CAS PubMed.
- V. P. Puri, Biotechnol. Bioeng., 1984, 26, 1219–1222 CrossRef CAS PubMed.
- V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2010, 3, 1–11 CrossRef PubMed.
- V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2011, 4, 1–17 CrossRef PubMed.
- X. Meng and A. J. Ragauskas, Curr. Opin. Biotechnol., 2014, 27, 150–158 CrossRef CAS PubMed.
- M. Wiman, D. Dienes, M. A. T. Hansen, T. van der Meulen, G. Zacchi and G. Lidén, Bioresour. Technol., 2012, 126, 208–215 CrossRef CAS PubMed.
- R. Kumar and C. E. Wyman, Biotechnol. Bioeng., 2009, 103, 252–267 CrossRef CAS PubMed.
- J. Hu, K. Gourlay, V. Arantes, J. S. Van Dyk, A. Pribowo and J. N. Saddler, ChemSusChem, 2015, 8, 901–907 CrossRef CAS PubMed.
- R. Kumar and C. E. Wyman, Enzyme Microb. Technol., 2008, 42, 426–433 CrossRef CAS.
- S. A. Maurer, C. N. Bedbrook and C. J. Radke, Ind. Eng. Chem. Res., 2012, 51, 11389–11400 CrossRef CAS.
- C. Tai and D. R. Keshwani, Energy Fuels, 2014, 28, 1956–1961 CrossRef CAS.
- D. Gao, S. P. S. Chundawat, N. Uppugundla, V. Balan and B. E. Dale, Biotechnol. Bioeng., 2011, 108, 1788–1800 CrossRef CAS PubMed.
- Z. Zhu, N. Sathitsuksanoh and Y. H. Percival Zhang, Analyst, 2009, 134, 2267–2272 RSC.
- Y. K. Mok, V. Arantes and J. N. Saddler, Appl. Biochem. Biotechnol., 2015, 176, 1564–1580 CrossRef CAS PubMed.
- A. Várnai, L. Viikari, K. Marjamaa and M. Siika-aho, Bioresour. Technol., 2011, 102, 1220–1227 CrossRef PubMed.
- D. L. Machado, J. Moreira Neto, J. G. da Cruz Pradella, A. Bonomi, S. C. Rabelo and A. C. da Costa, Biotechnol. Appl. Biochem., 2015, 62, 681–689 CrossRef CAS PubMed.
- J. Zhang, M. Tang and L. Viikari, Bioresour. Technol., 2012, 121, 8–12 CrossRef CAS PubMed.
- T. Jeoh, D. K. Johnson, W. S. Adney and M. E. Himmel, in ACS Division of Fuel Chemistry, Preprints, 2005, pp. 673–674 Search PubMed.
- A. Várnai, M. Siika-aho and L. Viikari, Enzyme Microb. Technol., 2010, 46, 185–193 CrossRef.
- M. J. Selig, T. B. Vinzant, M. E. Himmel and S. R. Decker, Appl. Biochem. Biotechnol., 2009, 155, 94–103 CrossRef PubMed.
- R. Kumar, F. Hu, P. Sannigrahi, S. Jung, A. J. Ragauskas and C. E. Wyman, Biotechnol. Bioeng., 2013, 110, 737–753 CrossRef CAS PubMed.
- X. Wang, K. Li, M. Yang and J. Zhang, Carbohydr. Polym., 2016, 148, 362–370 CrossRef CAS PubMed.
- R. Kumar and C. E. Wyman, Bioresour. Technol., 2009, 100, 4203–4213 CrossRef CAS PubMed.
- J. Zhang and L. Viikari, Appl. Biochem. Biotechnol., 2014, 174, 1393–1402 CrossRef CAS PubMed.
- K. Öhgren, R. Bura, J. Saddler and G. Zacchi, Bioresour. Technol., 2007, 98, 2503–2510 CrossRef PubMed.
- R. Kumar and C. E. Wyman, Biotechnol. Bioeng., 2014, 111, 1341–1353 CrossRef CAS PubMed.
- Q. Qing, B. Yang and C. E. Wyman, Bioresour. Technol., 2010, 101 Search PubMed.
- J. Shi, M. A. Ebrik, B. Yang, R. J. Garlock, V. Balan, B. E. Dale, V. Ramesh Pallapolu, Y. Y. Lee, Y. Kim, N. S. Mosier, M. R. Ladisch, M. T. Holtzapple, M. Falls, R. Sierra-Ramirez, B. S. Donohoe, T. B. Vinzant, R. T. Elander, B. Hames, S. Thomas, R. E. Warner and C. E. Wyman, Bioresour. Technol., 2011, 102, 11080–11088 CrossRef CAS PubMed.
- S. Xue, N. Uppugundla, M. J. Bowman, D. Cavalier, L. Da Costa Sousa, B. E Dale and V. Balan, Biotechnol. Biofuels, 2015, 8, 195 CrossRef PubMed.
- D. Xin, M. Yang, X. Chen and J. Zhang, RSC Adv., 2016, 6, 73859–73868 RSC.
- R. Kumar and C. E. Wyman, Biotechnol. Bioeng., 2009, 102, 457–467 CrossRef CAS PubMed.
- H. Yang, J. Chen, Q. Chen, K. Wang and R.-C. Sun, BioEnergy Res., 2015, 8, 1847–1855 CrossRef CAS.
- Q. Sun, M. Foston, X. Meng, D. Sawada, S. V. Pingali, H. M. O'Neill, H. Li, C. E. Wyman, P. Langan, A. J. Ragauskas and R. Kumar, Biotechnol. Biofuels, 2014, 7, 150 CrossRef PubMed.
- S. Nakagame, R. P. Chandra, J. F. Kadla and J. N. Saddler, Bioresour. Technol., 2011, 102, 4507–4517 CrossRef CAS PubMed.
- Y. X. An, M. H. Zong, S. Q. Hu and N. Li, Chem. Eng. Sci., 2017, 161, 48–56 CrossRef CAS.
- Y. Zeng, S. Zhao, S. Yang and S.-Y. Ding, Curr. Opin. Biotechnol., 2014, 27, 38–45 CrossRef CAS PubMed.
- S. Nakagame, R. P. Chandra, J. F. Kadla and J. N. Saddler, Biotechnol. Bioeng., 2011, 108, 538–548 CrossRef CAS PubMed.
- W. Wang, Y. Zhu, J. Du, Y. Yang and Y. Jin, Bioresour. Technol., 2015, 181, 7–12 CrossRef CAS PubMed.
- C. Lai, M. Tu, Z. Shi, K. Zheng, L. G. Olmos and S. Yu, Bioresour. Technol., 2014, 163, 320–327 CrossRef CAS PubMed.
- Z. Yu, K.-S. Gwak, T. Treasure, H. Jameel, H.-m. Chang and S. Park, ChemSusChem, 2014, 7, 1942–1950 CrossRef CAS PubMed.
- Z. Yan, J. Li, S. Chang, T. Cui, Y. Jiang, M. Yu, L. Zhang, G. Zhao, P. Qi and S. Li, Fuel, 2015, 158, 152–158 CrossRef CAS.
- K. C. Nlewem and M. E. Thrash Jr, Bioresour. Technol., 2010, 101, 5426–5430 CrossRef CAS PubMed.
- F. Ghorbani, M. Karimi, D. Biria, H. R. Kariminia and A. Jeihanipour, Biochem. Eng. J., 2015, 101, 77–84 CrossRef CAS.
- R. Kumar and C. E. Wyman, Biotechnol. Prog., 2009, 25, 807–819 CrossRef CAS PubMed.
- B. Yang and C. E. Wyman, Biotechnol. Bioeng., 2006, 94, 611–617 CrossRef CAS PubMed.
- B. Yang and C. Wyman, US Pat., US20110076725A1, 2004.
- J. Rahikainen, S. Mikander, K. Marjamaa, T. Tamminen, A. Lappas, L. Viikari and K. Kruus, Biotechnol. Bioeng., 2011, 108, 2823–2834 CrossRef CAS PubMed.
- F. Guo, W. Shi, W. Sun, X. Li, F. Wang, J. Zhao and Y. Qu, Biotechnol. Biofuels, 2014, 7, 1–10 CrossRef PubMed.
- J. K. Ko, E. Ximenes, Y. Kim and M. R. Ladisch, Biotechnol. Bioeng., 2015, 112, 447–456 CrossRef CAS PubMed.
- J. L. Rahikainen, R. Martin-Sampedro, H. Heikkinen, S. Rovio, K. Marjamaa, T. Tamminen, O. J. Rojas and K. Kruus, Bioresour. Technol., 2013, 133, 270–278 CrossRef CAS PubMed.
- L. Qin, W.-C. Li, L. Liu, J.-Q. Zhu, X. Li, B.-Z. Li and Y.-J. Yuan, Biotechnol. Biofuels, 2016, 9, 1–10 CrossRef PubMed.
- E. Ximenes, Y. Kim, N. Mosier, B. Dien and M. Ladisch, Enzyme Microb. Technol., 2010, 46, 170–176 CrossRef CAS.
- E. Walch, A. Zemann, F. Schinner, G. Bonn and O. Bobleter, Bioresour. Technol., 1992, 39, 173–177 CrossRef CAS.
- L. J. Jönsson, B. Alriksson and N.-O. Nilvebrant, Biotechnol. Biofuels, 2013, 6, 1–10 CrossRef PubMed.
- Y. Li, B. Qi and Y. Wan, Bioresour. Technol., 2014, 167, 324–330 CrossRef CAS PubMed.
- A. Oliva-Taravilla, E. Tomás-Pejó, M. Demuez, C. González-Fernández and M. Ballesteros, J. Biotechnol., 2016, 218, 94–101 CrossRef CAS PubMed.
- J. K. Ko, Y. Um, Y. C. Park, J. H. Seo and K. H. Kim, Appl. Microbiol. Biotechnol., 2015, 99, 4201–4212 CrossRef CAS PubMed.
- L. J. Jönsson and C. Martín, Bioresour. Technol., 2016, 199, 103–112 CrossRef PubMed.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, J. Chem. Technol. Biotechnol., 2014, 89, 2–10 CrossRef CAS.
- C. M. Cai, N. Nagane, R. Kumar and C. E. Wyman, Green Chem., 2014, 16, 3819–3829 RSC.
- T. Zhang, R. Kumar and C. E. Wyman, RSC Adv., 2013, 3, 9809–9819 RSC.
- J. Shen and C. E. Wyman, AIChE J., 2012, 58, 236–246 CrossRef CAS.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 RSC.
- V. Reginatto and R. V. Antônio, Braz. J. Microbiol., 2015, 46, 323–335 CrossRef PubMed.
- X. Jing, X. Zhang and J. Bao, Appl. Biochem. Biotechnol., 2009, 159, 696–707 CrossRef CAS PubMed.
- A. Arora, E. M. Martin, M. H. Pelkki and D. J. Carrier, ACS Sustainable Chem. Eng., 2013, 1, 23–28 CrossRef CAS.
- Z. Xiao, X. Zhang, D. J. Gregg and J. N. Saddler, Appl. Biochem. Biotechnol., 2004, 113–116, 1115–1126 CrossRef CAS PubMed.
- Q. Qing and C. E. Wyman, Biotechnol. Biofuels, 2011, 4, 1–12 CrossRef PubMed.
- X. Wang, K. Li, M. Yang, J. Wang and J. Zhang, Cellulose, 2017, 24, 35–47 CrossRef CAS.
- R. Zhai, J. Hu and J. N. Saddler, ACS Sustainable Chem. Eng., 2016, 4, 3429–3436 CrossRef CAS.
- L. Zhang, X. Li, Q. Yong, S.-T. Yang, J. Ouyang and S. Yu, Bioresour. Technol., 2016, 203, 173–180 CrossRef CAS PubMed.
- Y. Lu, R. Warner, M. Sedlak, N. Ho and N. S. Mosier, Biotechnol. Prog., 2009, 25, 349–356 CrossRef CAS PubMed.
- C. S. Walseth, Tappi, 1952, 35, 228–233 CAS.
- K. R. Sharrock, J. Biochem. Biophys. Methods, 1988, 17, 81–105 CrossRef CAS PubMed.
- T. M. Wood, in Methods Enzymol., ed. S. T. K. Willis and A. Wood, Academic Press, 1988, pp. 19–25 Search PubMed.
- T. M. Wood and K. M. Bhat, in Methods Enzymol., ed. S. T. K. Willis and A. Wood,, Academic Press, 1988, pp. 87–112 Search PubMed.
- W. D. Bellamy and F. F. Holub, US Pat., US7447713B1, 1977.
- Y. H. P. Zhang, J. Cui, L. R. Lynd and L. R. Kuang, Biomacromolecules, 2006, 7, 644–648 CrossRef CAS PubMed.
- D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, in Comprehensive Cellulose Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2004, pp. 9–29 Search PubMed.
- J. Zhang, J. Zhang, L. Lin, T. Chen, J. Zhang, S. Liu, Z. Li and P. Ouyang, Molecules, 2009, 14, 5027–5041 CrossRef CAS PubMed.
- Y.-H. P. Zhang, S.-Y. Ding, J. R. Mielenz, J.-B. Cui, R. T. Elander, M. Laser, M. E. Himmel, J. R. McMillan and L. R. Lynd, Biotechnol. Bioeng., 2007, 97, 214–223 CrossRef CAS PubMed.
- P. Conte, A. Maccotta, C. De Pasquale, S. Bubici and G. Alonzo, J. Agric. Food Chem., 2009, 57, 8748–8752 CrossRef CAS PubMed.
- J. Zhang, B. Zhang, J. Zhang, L. Lin, S. Liu and P. Ouyang, Biotechnol. Adv., 2010, 28, 613–619 CrossRef CAS PubMed.
- X. Jia, Y. Chen, C. Shi, Y. Ye, P. Wang and X. Zeng, et al. , J. Agric. Food Chem., 2013, 61, 12405–12414 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu, T.-J. Ho, M.-D. Bai and Y.-H. P. Zhang, Bioresour. Technol., 2010, 101, 4926–4929 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu, N. Templeton, J. A. Rollin, S. P. Harvey and Y. H. P. Zhang, Ind. Eng. Chem. Res., 2009, 48, 6441–6447 CrossRef CAS.
- N. Sathitsuksanoh, Z. Zhu and Y. H. P. Zhang, Bioresour. Technol., 2012, 117, 228–233 CrossRef CAS PubMed.
- Z. Zhu, N. Sathitsuksanoh, T. Vinzant, D. J. Schell, J. D. McMillan and Y. H. P. Zhang, Biotechnol. Bioeng., 2009, 103, 715–724 CrossRef CAS PubMed.
- B. Satari, K. Karimi and A. Zamani, J. Chem. Technol. Biotechnol., 2016, 91, 1835–1843 CrossRef CAS.
- K. Karimi and A. Zamani, Biotechnol. Adv., 2013, 31, 466–481 CrossRef CAS PubMed.
- D. C. Nieves, K. Karimi and I. S. Horváth, Ind. Crops Prod., 2011, 34, 1097–1101 CrossRef CAS.
- S. Mirmohamadsadeghi, K. Karimi and I. Sárvári Horváth, BioResources, 2016, 11, 2 CrossRef.
- N. K. Mund, D. Dash, C. R. Barik, V. V. Goud, L. Sahoo, P. Mishra and N. R. Nayak, Bioresour. Technol., 2017, 236, 97–105 CrossRef CAS PubMed.
- P. Siripong, P. Duangporn, E. Takata and Y. Tsutsumi, Bioresour. Technol., 2016, 203, 303–308 CrossRef CAS PubMed.
- V. Jafari, S. R. Labafzadeh, A. Jeihanipour, K. Karimi and M. J. Taherzadeh, Renewable Energy, 2011, 36, 2771–2775 CrossRef CAS.
- K. Wang, H. Y. Yang, F. Xu and R. C. Sun, Bioresour. Technol., 2011, 102, 4524–4529 CrossRef CAS PubMed.
- G. Moxley, Z. Zhu and Y. H. P. Zhang, J. Agric. Food Chem., 2008, 56, 7885–7890 CrossRef CAS PubMed.
- H. Li, N.-J. Kim, M. Jiang, J. W. Kang and H. N. Chang, Bioresour. Technol., 2009, 100, 3245–3251 CrossRef CAS PubMed.
- X. Ge, D. M. Burner, J. Xu, G. C. Phillips and G. Sivakumar, Biotechnol. J., 2011, 6, 66–73 CrossRef CAS PubMed.
- X. Ge, V. S. Green, N. Zhang, G. Sivakumar and J. Xu, Process Biochem., 2012, 47, 335–339 CrossRef CAS.
- N. Sathitsuksanoh, Z. Zhu and Y. H. P. Zhang, Cellulose, 2012, 19, 1161–1172 CrossRef CAS.
- M. M. Ishola, Isroi and M. J. Taherzadeh, Bioresour. Technol., 2014, 165, 9–12 CrossRef CAS PubMed.
- A. Goshadrou, K. Karimi and M. Lefsrud, CSBE/SCGAB 2013 Annual Conference, University of Saskatchewan, Saskatoon, SK, 7–10 July, 2013 Search PubMed.
- K. Gourlay, V. Arantes and J. Saddler, Biotechnol. Biofuels, 2012, 5, 51 CrossRef CAS PubMed.
- Y. H. P. Zhang, Z. Zhiguang, R. Joe and S. Noppadon, in Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society, 2010, pp. 365–379 Search PubMed.
- A. R. Esteghalian, V. Srivastava, N. Gilkes, D. J. Gregg and J. N. Saddler, in Glycosyl Hydrolases for Biomass Conversion, ed. M. E. Himmel, J. O. Baker and J. N. Saddler, American Chemical Society, Washington DC, 2000 Search PubMed.
- T. Jeoh, C. I. Ishizawa, M. F. Davis, M. E. Himmel, W. S. Adney and D. K. Johnson, Biotechnol. Bioeng., 2007, 98 CrossRef CAS PubMed.
- Y. H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2006, 94, 888–898 CrossRef CAS PubMed.
- Y. Liu, Q. Zhong, S. Wang and Z. Cai, Biomacromolecules, 2011, 12, 2626–2632 CrossRef CAS PubMed.
- J. Hong, X. Ye and Y. H. P. Zhang, Langmuir, 2007, 23, 12535–12540 CrossRef CAS PubMed.
- J. A. Rollin, Z. Zhu, N. Sathitsuksanoh and Y. H. P. Zhang, Biotechnol. Bioeng., 2011, 108, 22–30 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu, S. Wi and Y. H. Percival Zhang, Biotechnol. Bioeng., 2011, 108, 521–529 CrossRef CAS PubMed.
- B. John, J. K. Francis and H. G. Kenncorwin, in Cellul. Chem. Technol., American Chemical Society, 1977, pp. 42–55 Search PubMed.
- G. T. Beckham, Y. J. Bomble, E. A. Bayer, M. E. Himmel and M. F. Crowley, Curr. Opin. Biotechnol., 2011, 22, 231–238 CrossRef CAS PubMed.
- G. T. Beckham, J. F. Matthews, B. Peters, Y. J. Bomble, M. E. Himmel and M. F. Crowley, J. Phys. Chem. B, 2011, 115, 4118–4127 CrossRef CAS PubMed.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- M. Bergenstråhle, J. Wohlert, M. E. Himmel and J. W. Brady, Carbohydr. Res., 2010, 345, 2060–2066 CrossRef PubMed.
- L. Charlier and K. Mazeau, J. Phys. Chem. B, 2012, 116, 4163–4174 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu and Y. H. P. Zhang, Bioresour. Technol., 2012, 117, 228–233 CrossRef CAS PubMed.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- K. Wang, F. Xu and R.-C. Sun, in Biological Conversion of Biomass for Fuels and Chemicals: Explorations from Natural Utilization Systems, The Royal Society of Chemistry, 2014, pp. 318–343 Search PubMed.
- C. Graenacher and R. Sallmann, US Pat., US2179181A, 1939.
- D. L. Johnson, US Pat., US3447939A, 1969.
- C. C. McCorsley and J. K. Varga, US Pat., US4142913A, 1979.
- H. P. Fink, P. Weigel, H. J. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CrossRef CAS.
- D. L. Johnson, US Pat., US3508941A, 1970.
- C. Woodings, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000 Search PubMed.
- R. Asachi and K. Karimi, Process Biochem., 2013, 48, 1524–1531 CrossRef CAS.
- A. Goshadrou, K. Karimi and M. J. Taherzadeh, Biomass Bioenergy, 2013, 49, 95–101 CrossRef CAS.
- A. Jeihanipour, K. Karimi and M. J. Taherzadeh, Biotechnol. Bioeng., 2010, 105, 469–476 CrossRef CAS PubMed.
- C.-H. Kuo and C.-K. Lee, Bioresour. Technol., 2009, 100, 866–871 CrossRef CAS PubMed.
- P. R. Lennartsson, C. Niklasson and M. J. Taherzadeh, Bioresour. Technol., 2011, 102, 4425–4432 CrossRef CAS PubMed.
- N. Poornejad, K. Karimi and T. Behzad, Ind. Crops Prod., 2013, 41, 408–413 CrossRef CAS.
- S. Aslanzadeh, A. Berg, M. Taherzadeh and I. Sárvári Horváth, Appl. Biochem. Biotechnol., 2014, 172, 2998–3008 CrossRef CAS PubMed.
- S. Aslanzadeh, M. J. Taherzadeh and I. S. Horváth, BioResources, 2011, 6, 5193–5205 CAS.
- A. Jeihanipour, S. Aslanzadeh, K. Rajendran, G. Balasubramanian and M. J. Taherzadeh, Renewable Energy, 2013, 52, 128–135 CrossRef CAS.
- A. Jeihanipour, K. Karimi, C. Niklasson and M. J. Taherzadeh, Waste Manage., 2010, 30, 2504–2509 CrossRef CAS PubMed.
- A. Jeihanipour, C. Niklasson and M. J. Taherzadeh, Process Biochem., 2011, 46, 1509–1514 CrossRef CAS.
- M. M. Kabir, M. d. P. Castillo, M. J. Taherzadeh and I. S. Horváth, BioResources, 2013, 8, 5409–5423 CrossRef.
- M. M. Kabir, C. Niklasson, M. J. Taherzadeh and I. S. Horváth, Bioresour. Technol., 2014, 161, 446–450 CrossRef CAS PubMed.
- R. Millati, F. A. Purwandari, A. P. Sanjaya, M. N. Cahyanto, I. S. Horváth, C. Niklasson and M. J. Taherzadeh, Use of NMMO Pretreatment for Biogas Production from Oil Palm Empty Fruit Bunch, World Academy of Science, Engineering and Technology, 2012, vol. 6, pp. 1010–1014 Search PubMed.
- F. A. Purwandari, A. P. Sanjaya, R. Millati, M. N. Cahyanto, I. S. Horváth, C. Niklasson and M. J. Taherzadeh, Bioresour. Technol., 2013, 128, 461–466 CrossRef CAS PubMed.
- A. Teghammar, K. Karimi, I. Sárvári Horváth and M. J. Taherzadeh, Biomass Bioenergy, 2012, 36, 116–120 CrossRef CAS.
- F. Wendler, A. Konkin and T. Heinze, Macromol. Symp., 2008, 262, 72–84 CrossRef CAS.
- O. Biganska and P. Navard, Polymer, 2003, 44, 1035–1039 CrossRef CAS.
- C. Cuissinat and P. Navard, Macromol. Symp., 2006, 244, 1–18 CrossRef CAS.
- H. Zhao, J. H. Kwak, Y. Wang, J. A. Franz, J. M. White and J. E. Holladay, Carbohydr. Polym., 2007, 67, 97–103 CrossRef CAS.
- L. Zhang, H. Lu, J. Yu, Z. Wang, Y. Fan and X. J. Zhou, J. Agric. Food Chem., 2017, 65, 9587–9594 CrossRef CAS PubMed.
- G. Brodeur, Electronic Theses, Treatises and Dissertations, Paper 7312, Florida State University, 2013.
- N. Le Moigne and P. Navard, in Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society, 2010, pp. 137–148 Search PubMed.
- M. L. Nelson and R. T. O'Connor, J. Appl. Polym. Sci., 1964, 8, 1325–1341 CrossRef CAS.
- S. Y. Oh, D. I. Yoo, Y. Shin, H. C. Kim, H. Y. Kim, Y. S. Chung, W. H. Park and J. H. Youk, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef CAS PubMed.
- M. L. Nelson and R. T. O'Connor, J. Appl. Polym. Sci., 1964, 8, 1311–1324 CrossRef CAS.
- M. Khodaverdi, A. Jeihanipour, K. Karimi and M. Taherzadeh, J. Ind. Microbiol. Biotechnol., 2012, 39, 429–438 CrossRef CAS PubMed.
- S. Sasmal, V. V. Goud and K. Mohanty, Biomass Bioenergy, 2012, 45, 212–220 CrossRef CAS.
- T. Virtanen and S. Maunu, Cellulose, 2014, 21, 153–165 CrossRef CAS.
- B. Satari, K. Karimi and M. Molaverdi, Bioprocess Biosyst. Eng., 2018, 41, 249–264 CrossRef CAS PubMed.
- R. Chandra, S. Ewanick, P. Chung, K. Au-Yeung, L. Rio, W. Mabee and J. Saddler, Biotechnol. Lett., 2009, 31, 1217–1222 CrossRef CAS PubMed.
- R. Chandra, S. Ewanick, C. Hsieh and J. N. Saddler, Biotechnol. Prog., 2008, 24, 1178–1185 CrossRef CAS PubMed.
- F. Simons, Tappi J., 1950, 33, 312–314 CAS.
- X. Meng, M. Foston, J. Leisen, J. DeMartini, C. E. Wyman and A. J. Ragauskas, Bioresour. Technol., 2013, 144, 467–476 CrossRef CAS PubMed.
- A. Teghammar, R. P. Chandra, J. N. Saddler, M. J. Taherzadeh and I. S. Horváth, BioResources, 2012, 7, 3921–3934 Search PubMed.
- Y. Ogiwara and K. Arai, Text. Res. J., 1968, 38, 885–891 CrossRef CAS.
- A. Kongdee, T. Bechtold, E. Burtscher and M. Scheinecker, Carbohydr. Polym., 2004, 57, 39–44 CrossRef CAS.
- O. Biganska and P. Navard, Biomacromolecules, 2005, 6, 1948–1953 CrossRef CAS PubMed.
- K. E. Perepelkin, Fibre Chem., 2007, 39, 163–172 CrossRef CAS.
- M. Shafiei, K. Karimi and M. J. Taherzadeh, Bioresour. Technol., 2011, 102, 7879–7886 CrossRef CAS PubMed.
- A. Teghammar, G. Forgács, I. Sárvári Horváth and M. J. Taherzadeh, Appl. Energy, 2014, 116, 125–133 CrossRef CAS.
- A. Konkin, F. Wendler, F. Meister, H. K. Roth, A. Aganov and O. Ambacher, Spectrochim. Acta, Part A, 2008, 69, 1053–1055 CrossRef CAS PubMed.
- T. Rosenau, A. Potthast, I. Adorjan, A. Hofinger, H. Sixta, H. Firgo and P. Kosma, Cellulose, 2002, 9, 283–291 CrossRef CAS.
- Y.-C. He, D.-Q. Xia, C.-L. Ma, L. Gong, T. Gong, M.-X. Wu, Y. Zhang, Y.-J. Tang, J.-H. Xu and Y.-Y. Liu, Bioresour. Technol., 2013, 135, 18–22 CrossRef CAS PubMed.
- G. Meister and M. Wechsler, Biodegradation, 1998, 9, 91–102 CrossRef CAS PubMed.
- H. Stockinger, O. M. Kut and E. Heinzle, Water Res., 1996, 30, 1745–1748 CrossRef CAS.
- M. A. P. Martins, C. P. Frizzo, A. Z. Tier, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2014, 114, PR1–PR70 CrossRef PubMed.
- P. Walden, Bull. Acad. Imp. Sci. St.-Petersbourg, 1914, 8, 405–422 Search PubMed.
- F. H. Hurley and T. P. WIer, J. Electrochem. Soc., 1951, 98, 207–212 CrossRef CAS.
- J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263–1264 CrossRef CAS.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- J. H. Davis, Chem. Lett., 2004, 33, 1072–1077 CrossRef CAS.
- J. L. Anderson, D. W. Armstrong and G.-T. Wei, Anal. Chem., 2006, 78, 2892–2902 CrossRef PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- M. Freemantle, Chem. Eng. News, 1998, 76, 32–37 CrossRef.
- G. Laus, G. Bentivoglio, H. Schottenberger, V. Kahlenberg, H. Kopacka, T. Röder and H. Sixta, Lenzinger Ber., 2005, 84, 71–85 CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- Q.-P. Liu, X.-D. Hou, N. Li and M.-H. Zong, Green Chem., 2012, 14, 304–307 RSC.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- C. Hardacre, J. D. Holbrey, M. Nieuwenhuyzen and T. G. A. Youngs, Acc. Chem. Res., 2007, 40, 1146–1155 CrossRef CAS PubMed.
- O. A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629–2647 CrossRef CAS PubMed.
- T. C. Schutt, G. A. Hegde, V. S. Bharadwaj, A. J. Johns and C. M. Maupin, J. Phys. Chem. B, 2017, 121, 843–853 CrossRef CAS PubMed.
- T. G. A. Youngs, C. Hardacre and J. D. Holbrey, J. Phys. Chem. B, 2007, 111, 13765–13774 CrossRef CAS PubMed.
- T. G. A. Youngs, J. D. Holbrey, M. Deetlefs, M. Nieuwenhuyzen, M. F. Costa Gomes and C. Hardacre, ChemPhysChem, 2006, 7, 2279–2281 CrossRef CAS PubMed.
- Q. Liu, M. H. A. Janssen, F. van Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39–42 RSC.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- F. Hermanutz, F. Gähr, E. Uerdingen, F. Meister and B. Kosan, Macromol. Symp., 2008, 262, 23–27 CrossRef CAS.
- C. Graenacher, US Pat., US1943176A, 1934.
- N. Sun, H. Rodriguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405–1421 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557–1559 RSC.
- Y. Li, X. Liu, Y. Zhang, K. Jiang, J. Wang and S. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 3417–3428 CrossRef CAS.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- T. R. Dawsey and C. L. McCormick, J. Macromol. Sci., Part C: Polym. Rev., 1990, 30, 405–440 CrossRef.
- N. J. Cao, Q. Xu and L. F. Chen, Appl. Biochem. Biotechnol., 1995, 51, 21–28 CrossRef.
- J. Vitz, T. Erdmenger, C. Haensch and U. S. Schubert, Green Chem., 2009, 11, 417–424 RSC.
- H. Ohno and Y. Fukaya, Chem. Lett., 2009, 38, 2–7 CrossRef CAS.
- L. Feng and Z.-l. Chen, J. Mol. Liq., 2008, 142, 1–5 CrossRef.
- V. Rigual, T. M. Santos, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Bioresour. Technol., 2018, 251, 197–203 CrossRef CAS PubMed.
- X. Yu, X. Bao, C. Zhou, L. Zhang, A. E.-G. A. Yagoub and H. Yang, et al. , Ultrason. Sonochem., 2018, 41, 410–418 CrossRef CAS PubMed.
- S. K. Paul and S. Chakraborty, Bioresour. Technol., 2018, 253, 85–93 CrossRef CAS PubMed.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- H. Xie, S. Li and S. Zhang, Green Chem., 2005, 7, 606–608 RSC.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63–69 RSC.
- B. Li, J. Asikkala, I. Filpponen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2010, 49, 2477–2484 CrossRef CAS.
- K. C. Badgujar and B. M. Bhanage, Bioresour. Technol., 2015, 178, 2–18 CrossRef CAS PubMed.
- M. G. Freire, A. R. R. Teles, M. A. A. Rocha, B. Schröder, C. M. S. S. Neves, P. J. Carvalho, D. V. Evtuguin, L. M. N. B. F. Santos and J. A. P. Coutinho, J. Chem. Eng. Data, 2011, 56, 4813–4822 CrossRef CAS.
- T. V. Doherty, M. Mora-Pale, S. E. Foley, R. J. Linhardt and J. S. Dordick, Green Chem., 2010, 12, 1967–1975 RSC.
- W. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038–2047 RSC.
- L. K. J. Hauru, M. Hummel, A. W. T. King, I. Kilpeläinen and H. Sixta, Biomacromolecules, 2012, 13, 2896–2905 CrossRef CAS PubMed.
- J. Shi, K. Balamurugan, R. Parthasarathi, N. Sathitsuksanoh, S. Zhang, V. Stavila, V. Subramanian, B. A. Simmons and S. Singh, Green Chem., 2014, 16, 3830–3840 RSC.
- K. M. Gupta, Z. Hu and J. Jiang, RSC Adv., 2013, 3, 12794–12801 RSC.
- H. Liu, K. L. Sale, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2011, 115, 10251–10258 CrossRef CAS PubMed.
- K. Ninomiya, A. R. I. Utami, Y. Tsuge, K. Kuroda, C. Ogino and T. Taima, et al. , Chem. Eng. J., 2018, 334, 657–663 CrossRef CAS.
- A. G. Cruz, C. Scullin, C. Mu, G. Cheng, V. Stavila, P. Varanasi, D. Xu, J. Mentel, Y.-D. Chuang, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 1–10 CrossRef PubMed.
- H. Wu, M. Mora-Pale, J. Miao, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 2865–2875 CrossRef CAS PubMed.
- A. S. A. da Silva, R. S. S. Teixeira, T. Endo, E. P. S. Bon and S.-H. Lee, Green Chem., 2013, 15, 1991–2001 RSC.
- C. Li, D. Tanjore, W. He, J. Wong, J. L. Gardner, K. L. Sale, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 1–13 CrossRef PubMed.
- K. Ninomiya, H. Soda, C. Ogino, K. Takahashi and N. Shimizu, Bioresour. Technol., 2013, 128, 188–192 CrossRef CAS PubMed.
- W. Ji, Z. Ding, J. Liu, Q. Song, X. Xia, H. Gao, H. Wang and W. Gu, Energy Fuels, 2012, 26, 6393–6403 CrossRef CAS.
- Y. Wang, L. Wei, K. Li, Y. Ma, N. Ma, S. Ding, L. Wang, D. Zhao, B. Yan, W. Wan, Q. Zhang, X. Wang, J. Wang and H. Li, Bioresour. Technol., 2014, 170, 499–505 CrossRef CAS PubMed.
- A. Diop, A. H. Bouazza, C. Daneault and D. Montplaisir, BioResources, 2013, 8, 4270–4282 CrossRef.
- D. Fu, G. Mazza and Y. Tamaki, J. Agric. Food Chem., 2010, 58, 2915–2922 CrossRef CAS PubMed.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- J. L. Wen, T. Q. Yuan, S. L. Sun, F. Xu and R. C. Sun, Green Chem., 2014, 16, 181–190 RSC.
- Y.-X. An, N. Li, H. Wu, W.-Y. Lou and M.-H. Zong, ACS Sustainable Chem. Eng., 2015, 3, 2951–2958 CrossRef CAS.
- N. Sathitsuksanoh, K. M. Holtman, D. J. Yelle, T. Morgan, V. Stavila, J. Pelton, H. Blanch, B. A. Simmons and A. George, Green Chem., 2014, 16, 1236–1247 RSC.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- P. Varanasi, P. Singh, R. Arora, P. D. Adams, M. Auer, B. A. Simmons and S. Singh, Bioresour. Technol., 2012, 126, 156–161 CrossRef CAS PubMed.
- D. Yang, L.-X. Zhong, T.-Q. Yuan, X.-W. Peng and R.-C. Sun, Ind. Crops Prod., 2013, 43, 141–149 CrossRef CAS.
- S. Singh, P. Varanasi, P. Singh, P. D. Adams, M. Auer and B. A. Simmons, Biomass Bioenergy, 2013, 54, 276–283 CrossRef CAS.
- L. Moghaddam, Z. Zhang, R. M. Wellard, J. P. Bartley, I. M. O'Hara and W. O. S. Doherty, Biomass Bioenergy, 2014, 70, 498–512 CrossRef CAS.
- A. George, K. Tran, T. J. Morgan, P. I. Benke, C. Berrueco, E. Lorente, B. C. Wu, J. D. Keasling, B. A. Simmons and B. M. Holmes, Green Chem., 2011, 13, 3375–3385 RSC.
- S. S. Y. Tan, D. R. MacFarlane, J. Upfal, L. A. Edye, W. O. S. Doherty, A. F. Patti, J. M. Pringle and J. L. Scott, Green Chem., 2009, 11, 339–345 RSC.
- J. Zhang, Y. Wang, L. Zhang, R. Zhang, G. Liu and G. Cheng, Bioresour. Technol., 2014, 151, 402–405 CrossRef CAS PubMed.
- G. Cheng, P. Varanasi, C. Li, H. Liu, Y. B. Melnichenko, B. A. Simmons, M. S. Kent and S. Singh, Biomacromolecules, 2011, 12, 933–941 CrossRef CAS PubMed.
- G. Cheng, P. Varanasi, R. Arora, V. Stavila, B. A. Simmons, M. S. Kent and S. Singh, J. Phys. Chem. B, 2012, 116, 10049–10054 CrossRef CAS PubMed.
- J. A. Perez-Pimienta, M. G. Lopez-Ortega, P. Varanasi, V. Stavila, G. Cheng, S. Singh and B. A. Simmons, Bioresour. Technol., 2013, 127, 18–24 CrossRef CAS PubMed.
- T. N. Ang, G. C. Ngoh, A. S. M. Chua and M. G. Lee, Biotechnol. Biofuels, 2012, 5, 1–10 CrossRef PubMed.
- T. Kanbayashi and H. Miyafuji, J. Wood Sci., 2014, 60, 152–159 CrossRef CAS.
- T. Kanbayashi and H. Miyafuji, Planta, 2015, 242, 509–518 CrossRef CAS PubMed.
- S. Singh, B. A. Simmons and K. P. Vogel, Biotechnol. Bioeng., 2009, 104, 68–75 CrossRef CAS PubMed.
- X. Zhang, J. Ma, Z. Ji, G. H. Yang, X. Zhou and F. Xu, Microsc. Res. Tech., 2014, 77, 609–618 CrossRef CAS PubMed.
- L. Sun, C. Li, Z. Xue, B. A. Simmons and S. Singh, RSC Adv., 2013, 3, 2017–2027 RSC.
- M. Shafiei, Ph.D. Thesis, Isfahan University of Technology, 2014.
- A. S. Amarasekara and P. Shanbhag, BioEnergy Res., 2012, 6, 719–724 CrossRef.
- B. M. Matsagar and P. L. Dhepe, Catal. Sci. Technol., 2015, 5, 531–539 RSC.
- D. Groff, A. George, N. Sun, N. Sathitsuksanoh, G. Bokinsky, B. A. Simmons, B. M. Holmes and J. D. Keasling, Green Chem., 2013, 15, 1264–1267 RSC.
- J. Tao, T. Kishimoto, M. Hamada and N. Nakajima, Holzforschung, 2017, 71, 21–26 CAS.
- C. Li and Z. K. Zhao, Adv. Synth. Catal., 2007, 349, 1847–1850 CrossRef CAS.
- B. Li, I. Filpponen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2010, 49, 3126–3136 CrossRef CAS.
- C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177–182 RSC.
- Z. Zhang, I. M. O'Hara and W. O. Doherty, Bioresour. Technol., 2012, 120, 149–156 CrossRef CAS PubMed.
- B. J. Cox and J. G. Ekerdt, Bioresour. Technol., 2013, 134, 59–65 CrossRef CAS PubMed.
- Y. Liu, W. Xiao, S. Xia and P. Ma, Carbohydr. Polym., 2013, 92, 218–222 CrossRef CAS PubMed.
- N. Muhammad, Z. Man, M. A. Bustam, M. I. A. Mutalib, C. D. Wilfred and S. Rafiq, Appl. Biochem. Biotechnol., 2011, 165, 998–1009 CrossRef CAS PubMed.
- K. Zhuo, Q. Du, G. Bai, C. Wang, Y. Chen and J. Wang, Carbohydr. Polym., 2015, 115, 49–53 CrossRef CAS PubMed.
- Z. Zhang, I. M. O'Hara and W. O. S. Doherty, Green Chem., 2013, 15, 431–438 RSC.
- H. Satria, K. Kuroda, T. Endo, K. Takada, K. Ninomiya and K. Takahashi, ACS Sustainable Chem. Eng., 2017, 5, 708–713 CrossRef CAS.
- A. M. da Costa Lopes and R. Bogel-Łukasik, ChemSusChem, 2015, 8, 947–965 CrossRef CAS PubMed.
- Uju, K. Abe, N. Uemura, T. Oshima, M. Goto and N. Kamiya, Bioresour. Technol., 2013, 138, 87–94 CrossRef CAS PubMed.
- Q. Qing, R. Hu, Y. He, Y. Zhang and L. Wang, Appl. Microbiol. Biotechnol., 2014, 98, 5275–5286 CrossRef CAS PubMed.
- A. S. Amarasekara and O. S. Owereh, Ind. Eng. Chem. Res., 2009, 48, 10152–10155 CrossRef CAS.
- A. S. Amarasekara and B. Wiredu, Ind. Eng. Chem. Res., 2011, 50, 12276–12280 CrossRef CAS.
- A. S. Amarasekara and P. Shanbhag, BioEnergy Res., 2013, 6, 719–724 CrossRef CAS.
- F. Jiang, Q. Zhu, D. Ma, X. Liu and X. Han, J. Mol. Catal. A: Chem., 2011, 334, 8–12 CrossRef CAS.
- F. Tao, H. Song and L. Chou, Bioresour. Technol., 2011, 102, 9000–9006 CrossRef CAS PubMed.
- S. Kahani, M. Shafiei, A. Abdolmaleki and K. Karimi, J. Cleaner Prod., 2017, 168, 952–962 CrossRef CAS.
- Y. Han Song, C.-W. Park, N.-H. Kim and S.-H. Lee, Holzforschung, 2017, 71, 43–50 Search PubMed.
- J. A. Perez-Pimienta, N. Sathitsuksanoh, V. S. Thompson, K. Tran, T. Ponce-Noyola and V. Stavila, et al. , Biotechnol. Biofuels, 2017, 10, 72 CrossRef PubMed.
- J. Xu, J. Xu, S. Zhang, J. Xia, X. Liu and X. Chu, et al. , Bioresour. Technol., 2018, 249, 1058–1061 CrossRef CAS PubMed.
- C. L. B. Reis, L. M. A. Silva, T. H. S. Rodrigues, A. K. N. Félix, R. S. Santiago-Aguiar and K. M. Canuto, et al. , Bioresour. Technol., 2017, 224, 694–701 CrossRef CAS PubMed.
- J. Sun, N. V. S. N. M. Konda, R. Parthasarathi, T. Dutta, M. Valiev and F. Xu, et al. , Green Chem., 2017, 19, 3152–3163 RSC.
- T. Auxenfans, E. Husson and C. Sarazin, Biochem. Eng. J., 2017, 117(part A), 77–86 CrossRef CAS.
- R. M. Wahlström and A. Suurnäkki, Green Chem., 2015, 17, 694–714 RSC.
- J. Xu, X. Liu, J. He, L. Hu, B. Dai and B. Wu, J. Chem. Technol. Biotechnol., 2015, 90, 57–63 CrossRef CAS.
- H. Li, A. Kankaanpaa, H. Xiong, M. Hummel, H. Sixta, H. Ojamo and O. Turunen, Enzyme Microb. Technol., 2013, 53, 414–419 CrossRef CAS PubMed.
- J. Xu, B. Wu, L. Hu, Z. Wu, N. Xu, B. Dai and J. He, Chem. Eng. J., 2015, 267, 163–169 CrossRef CAS.
- J. Xu, B. He, B. Wu, B. Wang, C. Wang and L. Hu, Bioresour. Technol., 2014, 157, 166–173 CrossRef CAS PubMed.
- A. A. N. Gunny, D. Arbain, R. Edwin Gumba, B. C. Jong and P. Jamal, Bioresour. Technol., 2014, 155, 177–181 CrossRef CAS PubMed.
- J. I. Park, E. J. Steen, H. Burd, S. S. Evans, A. M. Redding-Johnson, T. Batth, P. I. Benke, P. D'Haeseleer, N. Sun, K. L. Sale, J. D. Keasling, T. S. Lee, C. J. Petzold, A. Mukhopadhyay, S. W. Singer, B. A. Simmons and J. M. Gladden, PLoS One, 2012, 7, e37010 CrossRef CAS PubMed.
- A. A. Elgharbawy, M. Z. Alam, M. Moniruzzaman and M. Goto, Biochem. Eng. J., 2016, 109, 252–267 CrossRef CAS.
- C. Portillo Mdel and A. Saadeddin, Crit. Rev. Biotechnol., 2015, 35, 294–301 CrossRef PubMed.
- J. Xu, P. Xiong and B. He, Bioresour. Technol., 2016, 200, 961–970 CrossRef CAS PubMed.
- C. Yu, B. A. Simmons, S. W. Singer, M. P. Thelen and J. S. VanderGheynst, Appl. Microbiol. Biotechnol., 2016, 100, 10237–10249 CrossRef CAS PubMed.
- A. S. Dotsenko, G. S. Dotsenko, O. V. Senko, N. A. Stepanov, I. V. Lyagin and E. N. Efremenko, et al. , Bioresour. Technol., 2018, 250, 429–438 CrossRef CAS PubMed.
- M. Ouellet, S. Datta, D. C. Dibble, P. R. Tamrakar, P. I. Benke, C. Li, S. Singh, K. L. Sale, P. D. Adams, J. D. Keasling, B. A. Simmons, B. M. Holmes and A. Mukhopadhyay, Green Chem., 2011, 13, 2743–2749 RSC.
- F. Xu, J. Sun, N. V. S. N. M. Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Energy Environ. Sci., 2016, 9, 1042–1049 RSC.
- K. Nakashima, K. Yamaguchi, N. Taniguchi, S. Arai, R. Yamada, S. Katahira, N. Ishida, H. Takahashi, C. Ogino and A. Kondo, Green Chem., 2011, 13, 2948–2953 RSC.
- A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS PubMed.
- E. Husson, S. Buchoux, C. Avondo, D. Cailleu, K. Djellab, I. Gosselin, O. Wattraint and C. Sarazin, Bioresour. Technol., 2011, 102, 7335–7342 CrossRef CAS PubMed.
- I. G. Audu, N. Brosse, L. Desharnais and S. K. Rakshit, Energy Fuels, 2013, 27, 189–196 CrossRef CAS.
- J. Bian, F. Peng, X.-P. Peng, X. Xiao, P. Peng, F. Xu and R.-C. Sun, Carbohydr. Polym., 2014, 100, 211–217 CrossRef CAS PubMed.
- X. D. Hou, T. J. Smith, N. Li and M. H. Zong, Biotechnol. Bioeng., 2012, 109, 2484–2493 CrossRef CAS PubMed.
- X.-D. Hou, N. Li and M.-H. Zong, Bioresour. Technol., 2013, 136, 469–474 CrossRef CAS PubMed.
- S. K. Karatzos, L. A. Edye and W. O. S. Doherty, Biotechnol. Biofuels, 2012, 5, 1–12 CrossRef PubMed.
- K. M. Torr, K. T. Love, O. P. Cetinkol, L. A. Donaldson, A. George, B. M. Holmes and B. A. Simmons, Green Chem., 2012, 14, 778–787 RSC.
- Uju, A. Nakamoto, Y. Shoda, M. Goto, W. Tokuhara, Y. Noritake, S. Katahira, N. Ishida, C. Ogino and N. Kamiya, Bioresour. Technol., 2013, 135, 103–108 CrossRef CAS PubMed.
- P. Verdia, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617–1627 RSC.
- L. Wu, S.-H. Lee and T. Endo, Bioresour. Technol., 2013, 140, 90–96 CrossRef CAS PubMed.
- W. Xiao, W. Yin, S. Xia and P. Ma, Carbohydr. Polym., 2012, 87, 2019–2023 CrossRef CAS.
- L. W. Yoon, T. N. Ang, G. C. Ngoh and A. S. M. Chua, Biomass Bioenergy, 2012, 36, 160–169 CrossRef CAS.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489–2499 RSC.
- Ö. Çetinkol, D. Dibble, G. Cheng, M. Kent, B. Knierim, M. Auer, D. Wemmer, J. Pelton, Y. Melnichenko, J. Ralph, B. Simmons and B. Holmes, Biofuels, 2010, 1, 33–46 CrossRef.
- D. Fu and G. Mazza, Bioresour. Technol., 2011, 102, 8003–8010 CrossRef CAS PubMed.
- D. Fu and G. Mazza, Bioresour. Technol., 2011, 102, 7008–7011 CrossRef CAS PubMed.
- S. H. Ha, N. L. Mai, G. An and Y. M. Koo, Bioresour. Technol., 2011, 102, 1214–1219 CrossRef CAS PubMed.
- S. Karatzos, L. A. Edye and W. O. S. Doherty, Bioresour. Technol., 2011, 102, 9325–9329 CrossRef PubMed.
- C.-H. Kuo and C.-K. Lee, Carbohydr. Polym., 2009, 77, 41–46 CrossRef CAS.
- K. Ninomiya, A. Kohori, M. Tatsumi, K. Osawa, T. Endo, R. Kakuchi, C. Ogino, N. Shimizu and K. Takahashi, Bioresour. Technol., 2015, 176, 169–174 CrossRef CAS PubMed.
- K. Ninomiya, T. Yamauchi, C. Ogino, N. Shimizu and K. Takahashi, Biochem. Eng. J., 2014, 90, 90–95 CrossRef CAS.
- J. Zhang, X. Ma, J. Yu, X. Zhang and T. Tan, Bioresour. Technol., 2011, 102, 4585–4589 CrossRef CAS PubMed.
- K. M. Torr, K. T. Love, B. A. Simmons and S. J. Hill, Biotechnol. Bioeng., 2016, 113, 540–549 CrossRef CAS PubMed.
- H. T. Tan, K. T. Lee and A. R. Mohamed, Carbohydr. Polym., 2011, 83, 1862–1868 CrossRef CAS.
- N. Sun, R. Parthasarathi, A. M. Socha, J. Shi, S. Zhang, V. Stavila, K. L. Sale, B. A. Simmons and S. Singh, Green Chem., 2014, 16, 2546–2557 RSC.
- I. P. Samayam and C. A. Schall, Bioresour. Technol., 2010, 101, 3561–3566 CrossRef CAS PubMed.
- T. Auxenfans, S. Buchoux, K. Djellab, C. Avondo, E. Husson and C. Sarazin, Carbohydr. Polym., 2012, 90, 805–813 CrossRef CAS PubMed.
- T. Auxenfans, S. Buchoux, D. Larcher, G. Husson, E. Husson and C. Sarazin, Energy Convers. Manage., 2014, 88, 1094–1103 CrossRef CAS.
- F. Hong, X. Guo, S. Zhang, S.-f. Han, G. Yang and L. J. Jönsson, Bioresour. Technol., 2012, 104, 503–508 CrossRef CAS PubMed.
- W. Katinonkul, J.-S. Lee, S. H. Ha and J.-Y. Park, Energy, 2012, 47, 11–16 CrossRef CAS.
- Q. Li, Y. He, M. Xian, G. Jun, X. Xu and J. Yang, Bioresour. Technol., 2009, 100, 3570–3575 CrossRef CAS PubMed.
- K. Ninomiya, A. Ohta, S. Omote, C. Ogino, K. Takahashi and N. Shimizu, Chem. Eng. J., 2013, 215–216, 811–818 CrossRef CAS.
- J. Viell, H. Wulfhorst, T. Schmidt, U. Commandeur, R. Fischer, A. Spiess and W. Marquardt, Bioresour. Technol., 2013, 146, 144–151 CrossRef CAS PubMed.
- B. R. Caes, T. R. Vanoosbree, F. Lu, J. Ralph, C. T. Maravelias and R. T. Raines, ChemSusChem, 2013, 6, 2083–2089 CrossRef CAS PubMed.
- C. Abels, K. Thimm, H. Wulfhorst, A. C. Spiess and M. Wessling, Bioresour. Technol., 2013, 149, 58–64 CrossRef CAS PubMed.
- N. Sun, H. Liu, N. Sathitsuksanoh, V. Stavila, M. Sawant, A. Bonito, K. Tran, A. George, K. Sale, S. Singh, B. Simmons and B. Holmes, Biotechnol. Biofuels, 2013, 6, 1–15 CrossRef PubMed.
- K. Shill, S. Padmanabhan, Q. Xin, J. M. Prausnitz, D. S. Clark and H. W. Blanch, Biotechnol. Bioeng., 2011, 108, 511–520 CrossRef CAS PubMed.
- D. C. Dibble, C. Li, L. Sun, A. George, A. Cheng, O. P. Cetinkol, P. Benke, B. M. Holmes, S. Singh and B. A. Simmons, Green Chem., 2011, 13, 3255–3264 RSC.
- J. Xu, B. Liu, H. Hou and J. Hu, Bioresour. Technol., 2017, 234, 406–414 CrossRef CAS PubMed.
- U. Hamidah, T. Arakawa, Y. Y. H'ng, A. Nakagawa-izumi and M. Kishino, J. Wood Sci., 2018, 64, 149–156 CrossRef CAS.
- N. L. Mai, K. Ahn and Y.-M. Koo, Process Biochem., 2014, 49, 872–881 CrossRef CAS.
- Z. Qiu and G. M. Aita, Bioresour. Technol., 2013, 129, 532–537 CrossRef CAS PubMed.
- Y. X. An, M. H. Zong, H. Wu and N. Li, Bioresour. Technol., 2015, 192, 165–171 CrossRef CAS PubMed.
- N. Sathitsuksanoh, M. Sawant, Q. Truong, J. Tan, C. G. Canlas, N. Sun, W. Zhang, S. Renneckar, T. Prasomsri, J. Shi, Ö. Çetinkol, S. Singh, B. A. Simmons and A. George, BioEnergy Res., 2015, 8, 973–981 CrossRef CAS.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632–6633 CrossRef CAS PubMed.
- D. Feng, L. Li, F. Yang, W. Tan, G. Zhao, H. Zou, M. Xian and Y. Zhang, Appl. Microbiol. Biotechnol., 2011, 91, 399–405 CrossRef CAS PubMed.
- K. Shill, S. Padmanabhan, Q. Xin, J. Prausnitz, D. Clark and H. Blanch, Biotechnol. Bioeng., 2010, 108, 511–520 CrossRef PubMed.
- N. L. Mai, N. T. Nguyen, J.-I. Kim, H.-M. Park, S.-K. Lee and Y.-M. Koo, J. Chromatogr., 2012, 1227, 67–72 CrossRef CAS PubMed.
- L. T. P. Trinh, Y. J. Lee, J.-W. Lee, H.-J. Bae and H.-J. Lee, Sep. Purif. Technol., 2013, 120, 86–91 CrossRef CAS.
- X. Liang, Y. Fu and J. Chang, Bioresour. Technol., 2016, 220, 289–296 CrossRef CAS PubMed.
- X. Liang, Y. Fu and J. Chang, Bioresour. Technol., 2017, 245, 760–767 CrossRef CAS PubMed.
- V. Rigual, T. M. Santos, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, ACS Sustainable Chem. Eng., 2017, 5, 2384–2392 CrossRef CAS.
- G. Mancini, S. Papirio, P. N. L. Lens and G. Esposito, Energy Fuels, 2016, 30, 1892–1903 CrossRef CAS.
- J. Gao, L. Chen, K. Yuan, H. Huang and Z. Yan, Bioresour. Technol., 2013, 150, 352–358 CrossRef CAS PubMed.
- J. Gao, L. Chen, Z. Yan and L. Wang, Bioresour. Technol., 2013, 132, 361–364 CrossRef CAS PubMed.
- W. Li and G. Xu, Environ. Technol., 2016, 1–9 Search PubMed.
- Z. Gong, H. Shen, Q. Wang, X. Yang, H. Xie and Z. K. Zhao, Biotechnol. Biofuels, 2013, 6, 1–12 CrossRef PubMed.
- H. Xie, H. Shen, Z. Gong, Q. Wang, Z. K. Zhao and F. Bai, Green Chem., 2012, 14, 1202–1210 RSC.
- G. Bokinsky, P. P. Peralta-Yahya, A. George, B. M. Holmes, E. J. Steen, J. Dietrich, T. Soon Lee, D. Tullman-Ercek, C. A. Voigt, B. A. Simmons and J. D. Keasling, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19949–19954 CrossRef CAS PubMed.
- Q. Huang, Q. Wang, Z. Gong, G. Jin, H. Shen, S. Xiao, H. Xie, S. Ye, J. Wang and Z. K. Zhao, Bioresour. Technol., 2013, 130, 339–344 CrossRef CAS PubMed.
- L.-P. Liu, M.-H. Zong, R. J. Linhardt, W.-Y. Lou, N. Li, C. Huang and H. Wu, Biotechnol. Biofuels, 2016, 9, 266 CrossRef PubMed.
- P. Varanasi, P. Singh, M. Auer, P. D. Adams, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 1–9 CrossRef PubMed.
- Z. Sun, M. Cheng, H. Li, T. Shi, M. Yuan, X. Wang and Z. Jiang, RSC Adv., 2012, 2, 9058–9065 RSC.
- Y. Muranaka, T. Suzuki, H. Sawanishi, I. Hasegawa and K. Mae, Ind. Eng. Chem. Res., 2014, 53, 11611–11621 CrossRef CAS.
- S. Xiao, B. Liu, Y. Wang, Z. Fang and Z. Zhang, Bioresour. Technol., 2014, 151, 361–366 CrossRef CAS PubMed.
- S. M. Sen, J. B. Binder, R. T. Raines and C. T. Maravelias, Biofuels, Bioprod. Biorefin., 2012, 6, 444–452 CrossRef CAS.
- N. M. Konda, J. Shi, S. Singh, H. W. Blanch, B. A. Simmons and D. Klein-Marcuschamer, Biotechnol. Biofuels, 2014, 7, 1–11 CrossRef PubMed.
- N. R. Baral and A. Shah, Biofuels, Bioprod. Biorefin., 2016, 10, 70–88 CrossRef CAS.
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC.
- J. C. de Andrade Neto, A. de Souza Cabral, L. R. D. de Oliveira, R. B. Torres and A. d. A. Morandim-Giannetti, Carbohydr. Polym., 2016, 143, 279–287 CrossRef CAS PubMed.
- P. Oleskowicz-Popiel, D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Bioresour. Technol., 2014, 158, 294–299 CrossRef CAS PubMed.
- A. M. Socha, R. Parthasarathi, J. Shi, S. Pattathil, D. Whyte, M. Bergeron, A. George, K. Tran, V. Stavila, S. Venkatachalam, M. G. Hahn, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3587–E3595 CrossRef CAS PubMed.
- N. Muhammad, Z. Man and M. A. Bustam Khalil, Eur. J. Wood Wood Prod., 2012, 70, 125–133 CrossRef CAS.
- C.-Z. Liu, F. Wang, A. R. Stiles and C. Guo, Appl. Energy, 2012, 92, 406–414 CrossRef CAS.
- D. J. G. P. van Osch, L. J. B. M. Kollau, A. van den Bruinhorst, S. Asikainen, M. A. A. Rocha and M. C. Kroon, Phys. Chem. Chem. Phys., 2017, 19, 2636–2665 RSC.
- N. Uppugundla, L. da Costa Sousa, S. P. Chundawat, X. Yu, B. Simmons, S. Singh, X. Gao, R. Kumar, C. E. Wyman, B. E. Dale and V. Balan, Biotechnol. Biofuels, 2014, 7, 1–14 CrossRef PubMed.
- X. Gao, R. Kumar, S. Singh, B. A. Simmons, V. Balan, B. E. Dale and C. E. Wyman, Biotechnol. Biofuels, 2014, 7, 1–13 CrossRef PubMed.
- M. Mora-Pale, L. Meli, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 1229–1245 CrossRef CAS PubMed.
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan and J. Weale, et al. , Green Chem., 2017, 19, 3078–3102 RSC.
- M. Zavrel, D. Bross, M. Funke, J. Buchs and A. C. Spiess, Bioresour. Technol., 2009, 100, 2580–2587 CrossRef CAS PubMed.
- T. Li, Q. Fang, H. Chen, F. Qi, X. Ou, X. Zhao and D. Liu, RSC Adv., 2017, 7, 10609–10617 RSC.
- N. Sathitsuksanoh, Z. Zhu and Y.-H. P. Zhang, Cellulose, 2012, 19, 1161–1172 CrossRef CAS.
- K. Wang, H. Y. Yang, F. Xu and R. C. Sun, Bioresour. Technol., 2011, 102, 4524–4529 CrossRef CAS PubMed.
- C.-H. Kuo and C.-K. Lee, Carbohydr. Polym., 2009, 77, 41–46 CrossRef CAS.
- Y. Muranaka, T. Suzuki, H. Sawanishi, I. Hasegawa and K. Mae, Ind. Eng. Chem. Res., 2014, 53, 11611–11621 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- Y. Nguyen Thanh, M. Cai Charles, R. Kumar and E. Wyman Charles, ChemSusChem, 2015, 8, 1716–1725 CrossRef PubMed.
- R. Kumar, S. Bhagia, M. D. Smith, L. Petridis, R. G. Ong, C. M. Cai, A. Mittal, M. H. Himmel, V. Balan, B. E. Dale, A. J. Ragauskas, J. C. Smith and C. E. Wyman, Green Chem., 2018, 20, 921–934 RSC.
- B. J. Watson, B. Hammouda, R. M. Briber and S. W. Hutcheson, J. Appl. Polym. Sci., 2013, 127, 2620–2627 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |