
Facile synthesis of 2D nitrogen-containing porous carbon nanosheets induced by graphene oxide for high-performance supercapacitors†
Xu
Zhang
 *a,
Qiuyu
Fan
a,
Yingyuan
Zhao
c,
Man
Wang
b,
Yulan
Meng
a,
Yonghou
Xiao
a,
Xiaoting
Lei
a and
Juan
Yang
*b
*a,
Qiuyu
Fan
a,
Yingyuan
Zhao
c,
Man
Wang
b,
Yulan
Meng
a,
Yonghou
Xiao
a,
Xiaoting
Lei
a and
Juan
Yang
*b
aState Key Laboratory of Fine Chemicals, School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin 124221, China. E-mail: zhangxu@dlut.edu.cn
bSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: juanyangdlut@126.com
cCollege of Chemical Engineering and Safety, Binzhou University, Binzhou 256603, PR China
First published on 24th July 2018
Abstract
Two-dimensional (2D) nitrogen-containing porous carbon nanosheets are prepared in the presence of graphene oxide (GO) as a structure-directing agent by a simple yet facile hydrothermal method. It is found that GO can effectively tune the morphologies of the hydrothermal carbonaceous products, resulting in the unique 2D structures. The as-obtained 2D nitrogen-containing porous carbon nanosheets as electrodes for supercapacitors exhibit a high specific capacitance of 301.6 F g−1 at a current density of 0.5 A g−1 and superior rate capability with a capacitance retention of 70.5% at a high current density of 20 A g−1. The 2D nitrogen-containing porous carbon nanosheet electrodes also exhibit good cycling performance, possessing a high capacitance retention of over 92.3% after 5000 charge–discharge cycles at a current density of 5 A g−1. Such superior electrochemical performance of the 2D nitrogen-containing porous carbon nanosheets could be attributed to the high pseudocapacitive effect of the nitrogen-containing species and the peculiar structural characteristics featuring a short ion transport distance and abundant porous channels.
1. Introduction
Carbon electrode materials for supercapacitors have been studied extensively over the past few decades due to their high surface area, excellent electrochemical stability, and low cost.1,2 However, these carbon-based electrodes usual involve an electrical double-layer mechanism, and their low specific capacitances and energy densities still pose a huge challenge in meeting the requirements of high energy storage devices. One of the efficient ways to further improve the specific capacitances and energy densities of electrodes is to dope heteroatoms, such as nitrogen (N), oxygen (O) and phosphorus (P), into the carbon skeletons.3,4 It is believed that this kind of electrode offers not only electrical double-layer capacitances but also pseudocapacitances through the surface redox reactions of the doped heteroatoms with the electrolyte.5–8 Of these available dopants, N-doping has attracted much attention and shows a significant improvement in energy storage ability.9,10 In general, N-doped carbons can be prepared from various precursors including fossil fuels,11 polymers,12,13 metal–organic frameworks14 and biomass.2,15,16 In particular, biomass-based precursors, by virtue of their advantages of renewability, environmental friendliness, and low cost, are being exploited as an ideal medium for the fabrication of N-containing carbon nanomaterials.Engineering the morphology of carbon materials and their pore structures is also a vitally important strategy to further increase the electrochemical performance of carbon-based electrodes, in which the electrolyte permeability, ion transport/diffusion, and ion-adsorbing surface area can well be modulated.17–19 Recently, two-dimensional (2D) structural carbon nanosheets with large aspect ratios and porous structures have been fabricated and employed as supercapacitor electrodes, featuring a high ion-accessible surface area for charge storage, an open shortened diffusion distance for electrolyte ion-transport, and superior continuous conducting pathways for electrons transfer.20,21 Because of this, various approaches have been developed for the synthesis of 2D porous carbon nanosheets with greatly improved ion-transport properties, such as the template method, carbonization-activation method, and space-confinement strategy.15,22,23 Of these approaches, the template method has been shown to be one of the most effective strategies to fabricate 2D carbon nanosheets.22–24 Nanostructured inorganic frameworks, such as magnesium oxide, porous silica, and layered materials, have been used as templates to construct carbon nanosheets.25–28 However, the tedious etching and washing process, and the inevitable use of hazardous reagents such as hydrogen fluoride, strong sodium hydroxide solution for template removal, still limit the fabrication of 2D carbon nanosheets on a large scale.26,28,29
Graphene oxide (GO) has received great attention because of its rich surface chemistry properties and 2D flexible structure.30 GO can be used as a structure-directing agent to induce the formation of highly conductive carbon nanosheets.23,31 For instance, 2D polymerization can be directed by GO and then the porous carbon nanosheets can be converted by the pyrolysis of the 2D polymers.13,32 It is noteworthy that the graphene should not be removed after carbonization and the existing graphene can improve the transfer process of electrons and electrolyte ions because of the improved pore structures and electrical conductivity of the carbon nanosheets.7
Herein, inspired by molecules functionalizing GO, we report a simple yet facile hydrothermal method to fabricate 2D nitrogen-containing porous carbon nanosheets using GO as a structure-directing agent, fructose as the carbohydrate precursor and glycine as the nitrogen source. Glycine can react with fructose to produce nitrogen containing compounds via a Maillard type reaction. The reaction, known as the non-enzymatic browning reaction, is a complex chemical reaction between carbonyl groups of reducing sugars and free amino groups of N-containing compounds.33 The GO has a significant effect on promoting hydrothermal product morphological conversion due to its good water processability and 2D structure. In addition, the GO is also favourable for preventing nitrogen loss of the carbon nanosheets to some degree after KOH activation. The electrochemical performance of the as-obtained 2D nitrogen-containing porous carbon nanosheets as electrode materials for supercapacitors is investigated, showing a high specific capacitance and excellent rate capability.
2. Methods
2.1 Sample preparation
GO was prepared according to a modified Hummers' method, which has been described in previous reports.34–36 10 mg GO was mixed with a solution which contained 1 g fructose and 0.84 g glycine dissolved in 35 mL deionized water. Then the mixture was transferred to a Teflon autoclave and treated at 160 °C for 12 h. After cooling, the product was washed with deionized water and freeze-dried for 24 h, in which the product was marked as FGA. For comparison, a nitrogen-doped product without GO as a template was prepared by hydrothermal treatment of fructose and glycine at 160 °C for 12 h, which was marked as FA. A carbonaceous material without glycine was prepared by hydrothermal treatment of fructose and GO at 160 °C for 12 h, which was marked as FG.The FA, FG and FGA samples were activated with potassium hydroxides (KOH) at 600 °C for 2 h with a mass ratio of KOH to carbonaceous materials 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under an argon (Ar) atmosphere. The as-obtained products were treated with diluted hydrochloric acid and washed with deionized water into neutral and then freeze-dried overnight. The activated products were denoted as FA-a, FG-a, and FGA-a, respectively.
:1 under an argon (Ar) atmosphere. The as-obtained products were treated with diluted hydrochloric acid and washed with deionized water into neutral and then freeze-dried overnight. The activated products were denoted as FA-a, FG-a, and FGA-a, respectively.
FGAG-a and FGAS-a samples with glutamic acid and serine as the nitrogen source were also fabricated in a similar way to that of FGA-a.
2.2 Characterization methods
Field emission scanning electron microscopy (FE-SEM) and energy dispersive spectroscopy (EDS) were carried out on a FEI NOVA NanoSEM 450 system. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were performed on a FEI Tecnai G2 F30 system. Nitrogen sorption isotherms were measured at 77 K on a physical sorption instrument (Autosorb-iQ-C). The samples were degassed at 150 °C prior to nitrogen sorption measurement. Elemental compositions were measured using a Vario EL cube analyzer. The data of X-ray photoelectron spectroscopy (XPS) were collected on a Thermo ESCALAB 250 photoelectron spectrometer. X-ray diffraction (XRD) was used to examine the graphitization degree of the obtained samples. The samples were heated from room temperature to 800 °C at 10 °C min−1 under a nitrogen atmosphere for thermogravimetric analysis (TGA, STA 449 F3).2.3 Electrochemical measurement
The electrochemical properties of the samples were evaluated by using a CHI 760E electrochemistry workstation with a three-electrode system in 6 M KOH solution. The as-obtained samples were used as the working electrode, a Hg/HgO electrode was used as the reference electrode, and platinum foil was used as the counter electrode. The working electrode was fabricated by mixing the as-prepared samples, acetylene black and polytetrafluoroethylene at a weight ratio of 80:10:10. Then the mixture was dried at 80 °C overnight. The prepared mixture was pressed onto nickel foam to make electrodes. The specific capacitance of the samples was calculated from the discharge curves according to the following equation: for the three-electrode testand for the two electrode test
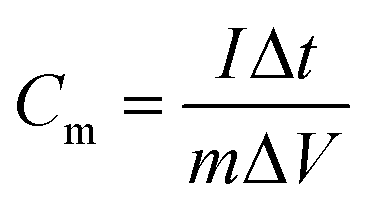
where Cm is the specific capacitance (F g−1), I is the discharging current (A), Δt is the discharge time (s), m is the mass of active material in a single electrode (g), and ΔV is the potential change during the discharge process.
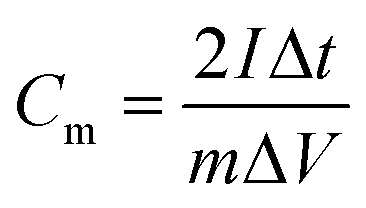
The energy density (W h kg−1) and power density (W kg−1) of the electrodes were calculated based on the following equations:
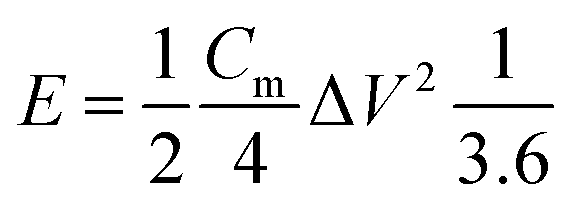
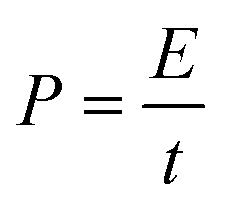
3. Results and discussion
A facile two-step approach, which included the preparation of a carbon precursor modified with nitrogen-containing groups followed by activation at an elevated temperature, was designed to fabricate the carbon materials (Fig. 1). The typical scanning electron microscopy (SEM) images of the FA, FG, and FGA samples are shown in Fig. 2 and S1.† During the hydrothermal reaction of fructose and glycine, discrete carbonaceous microspheres with a smooth surface are obtained (Fig. 2a and b). When a certain amount of GO is added to the reaction solution, the morphology of the hydrothermal reaction product is completely altered, highlighting the unique role of GO. As shown in Fig. 2c, d and S1b,† there are almost no carbonaceous spheres and the resulting materials appear to be nanosheets, indicating that GO can suppress the carbonaceous spheres formation. The SEM images of FG without glycine in Fig. 2e, f and S1a† also display the 2D sheet morphologies. This implies that GO sheets can act as the structure-directing agent for seeding the hydrothermal reaction product.23 The thermogravimetric analysis (TGA) curves of the FA and FGA products are shown in Fig. S2.† The major weight loss of both the samples occurred between 200 and 800 °C, which corresponds to the cracking of organic compounds. FGA exhibits a better thermal stability, showing around 45% total weight loss when heated to 800 °C, in comparison with the final weight loss of FA (about 53%). Glycine could be used as the N source to introduce N atoms into hydrothermal carbonaceous materials via a Maillard type reaction. The N contents of FA and FGA are calculated by X-ray photoelectron spectroscopy to be 8.6 at% and 8.5 at%, respectively, indicating that the carbonaceous materials with high nitrogen content can be achieved through the hydrothermal reaction of fructose and glycine.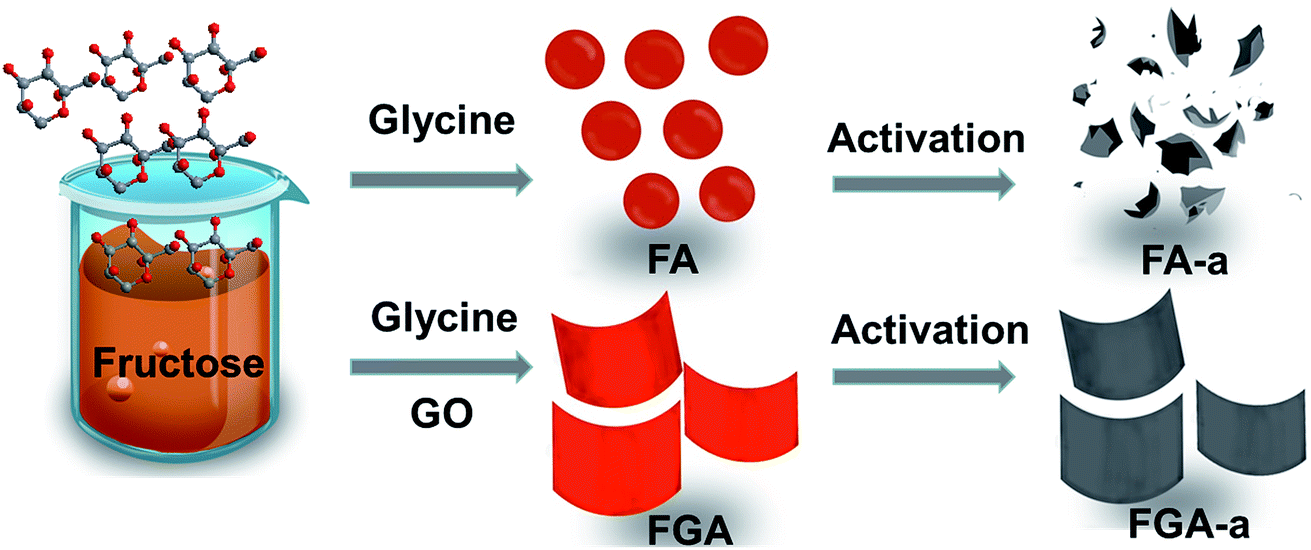 | ||
| Fig. 1 Schematic representation of the synthesis of the FA-a and FGA-a samples. | ||
 | ||
| Fig. 2 SEM images of the as-made (a and b) FA, (c and d) FGA, (e and f) FG, (g and h) FA-a, (i and j) FGA-a, and (k and l) FG-a samples. | ||
Chemical activation is a general yet efficient method to increase the specific surface area and pore volume of carbon materials, and helps to obtain electrode materials with excellent electrochemical performances.37 Therefore, the FA, FG, and FGA samples were further activated with KOH at 600 °C for 2 h to obtain the products FA-a, FG-a, and FGA-a, respectively. For the FA sample, the morphologies of carbonaceous spheres convert into typical morphologies of activated carbon composed of broken carbon bulk particles with an etched surface after activation (Fig. 2g and h). However, both FG-a and FGA-a exhibit wrinkled carbon nanosheet assemblies as shown in Fig. 2i–l and S3.† The X-ray diffraction (XRD) results of FGA and FGA-a are shown in Fig. S4.† It can be found that only broad peaks are present, indicating that the amorphous carbon nanostructures of FGA-a are retained after the activation process. In addition, the corresponding energy dispersive spectroscopy (EDS) elemental mapping images (Fig. S5†) reveal the presence and uniform distribution of C, N, and O elements throughout the FGA-a sample. The transmission electron microscopy (TEM) images (Fig. 3 and S6†) of the as-made samples further confirm the particle structures of FA-a and the 2D sheet-like structures of FG-a and FGA-a, respectively. The selected area electron diffraction (SAED) pattern of FGA-a in the inset in Fig. 3c further suggests that the carbon nanosheets have an amorphous structure, which is consistent with XRD results. The high-resolution TEM (HR-TEM) image of the FGA-a sample reveals that abundant micropores with a very small diameter are present in the carbon nanosheets as shown in Fig. 3d. This will provide rich transport channels for electrolyte ions for enhancing the capacitance.
 | ||
| Fig. 3 TEM images of (a) FA-a, (b) FG-a, and (c) FGA-a and (d) HR-TEM image of FGA-a. The inset is the SEAD pattern of FGA-a. | ||
Raman spectra of FA-a, FG-a, and FGA-a exhibit two broad peaks at around 1350 cm−1and 1587 cm−1, corresponding to the disorder band (D band) and the sp2 carbon lattice (G band), respectively.36 Broad D and G peaks can be observed in Fig. S7,† and the ID/IG ratios of all the samples are larger than 0.9, indicating amorphous carbon nanostructures. FGA-a has the highest ID/IG ratio (1.01), maybe because the high N doping level increases the defects of carbon nanosheets. The porous structures of the FA-a, FG-a, and FGA-a samples were analyzed by the nitrogen sorption technique and the corresponding nitrogen adsorption/desorption isotherms are shown in Fig. S7a.† The Brunauer–Emmett–Teller (BET) specific surface areas of FA-a, FG-a, and FGA-a are about 706, 643 and 783 m2 g−1, respectively. All the samples exhibit the combined characteristics of type I/IV isotherms, indicating that the samples contain micro- and mesopores. Pore size distributions are obtained by the density functional theory (DFT) method and are shown in Fig. S7b† and Table 1. The mesopore volumes of FG-a and FGA-a reach 0.26 and 0.10 cm3 g−1, respectively, which are higher than that of FA-a (0.074 cm3 g−1). Importantly, the mesoporous carbon will be a positive factor for the rate capability of the electrodes for supercapacitors.28 These results demonstrate that the existence of 2D graphene has a unique effect on the enhancement of the surface area and porosity of the as-obtained samples.
The Fourier-transform infrared (FTIR) spectra (Fig. S9†) of all the samples demonstrate a broad peak at 3435 cm−1, which is attributed to the stretching vibration of the O–H bond. The peaks at 1566 and 1140 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O bonds.38 The peak of the C–N band in FGA-a and FA-a is not detected, which may be covered up by the stretching vibration of oxygen-containing functional groups. XPS analysis was employed to further determine the surface chemistry of representative FG-a, FA-a, and FGA-a (Fig. 4, S10†). As shown in Fig. 4a, the peaks of C1s, and O1s at ca. 285 and 531 eV are identified in all the samples, and N1s peaks can be found in FA-a and FGA-a. The C 1s spectra of the samples in Fig. S10a–c† can be divided into four components, and the carbon species correspond to graphitic carbon (∼284.6 eV); carbon in C–O–C or R–OH (∼285.6 eV); carbon in CO (∼286.6 eV); and carbon in COOH or C(O)–O–C (∼288.3 eV).39 No obvious differences are observed in the carbon components of the samples. The oxygen contents of FG-a, FA-a and FGA-a are 13.3 at%, 12.7 at% and 12.9 at%, respectively. The oxygen functional groups in the samples can provide pseudocapacitance to increase the capacitive performance.4 The oxygen species of all the samples contains three components, corresponding to oxygen double bonded to carbon (CO), oxygen single bonded to carbon in C–O and oxygen in hydroxyl OH (Fig. S10d–f†).7 The nitrogen content of FA-a on the surface is 5.1 at%, and the N 1s peak shows three components at 398.4, 400.0 and 400.8 eV, which can be ascribed to pyridinic (N-6), pyrrolic (N-5) and quaternary (N-Q) nitrogen, respectively (Fig. 4c).38,40,41 In the case of FGA-a, the surface nitrogen content increases up to 5.7 at% and the N/C ratio increases from 0.062 of FA-a to 0.070 of FGA-a. Compared with the N contents of FA and FGA, the existence of graphene may prevent the loss of the nitrogen component in the chemical activation process to some degree. The nitrogen species mainly include N-6, N-5, and N-Q (Fig. 4d). It was believed that the N-6 could promote electron transfer to the carbon layers, while the N-Q could enhance the electronic conductivity of carbon materials, which were both important for electrode application.42,43 Furthermore, the N-6 and N-5 located at the edges of carbon layers could participate in redox reactions giving rise to pseudocapacitance and thus increase the total capacitive performance of electrodes.15,44
C and C–O bonds.38 The peak of the C–N band in FGA-a and FA-a is not detected, which may be covered up by the stretching vibration of oxygen-containing functional groups. XPS analysis was employed to further determine the surface chemistry of representative FG-a, FA-a, and FGA-a (Fig. 4, S10†). As shown in Fig. 4a, the peaks of C1s, and O1s at ca. 285 and 531 eV are identified in all the samples, and N1s peaks can be found in FA-a and FGA-a. The C 1s spectra of the samples in Fig. S10a–c† can be divided into four components, and the carbon species correspond to graphitic carbon (∼284.6 eV); carbon in C–O–C or R–OH (∼285.6 eV); carbon in CO (∼286.6 eV); and carbon in COOH or C(O)–O–C (∼288.3 eV).39 No obvious differences are observed in the carbon components of the samples. The oxygen contents of FG-a, FA-a and FGA-a are 13.3 at%, 12.7 at% and 12.9 at%, respectively. The oxygen functional groups in the samples can provide pseudocapacitance to increase the capacitive performance.4 The oxygen species of all the samples contains three components, corresponding to oxygen double bonded to carbon (CO), oxygen single bonded to carbon in C–O and oxygen in hydroxyl OH (Fig. S10d–f†).7 The nitrogen content of FA-a on the surface is 5.1 at%, and the N 1s peak shows three components at 398.4, 400.0 and 400.8 eV, which can be ascribed to pyridinic (N-6), pyrrolic (N-5) and quaternary (N-Q) nitrogen, respectively (Fig. 4c).38,40,41 In the case of FGA-a, the surface nitrogen content increases up to 5.7 at% and the N/C ratio increases from 0.062 of FA-a to 0.070 of FGA-a. Compared with the N contents of FA and FGA, the existence of graphene may prevent the loss of the nitrogen component in the chemical activation process to some degree. The nitrogen species mainly include N-6, N-5, and N-Q (Fig. 4d). It was believed that the N-6 could promote electron transfer to the carbon layers, while the N-Q could enhance the electronic conductivity of carbon materials, which were both important for electrode application.42,43 Furthermore, the N-6 and N-5 located at the edges of carbon layers could participate in redox reactions giving rise to pseudocapacitance and thus increase the total capacitive performance of electrodes.15,44
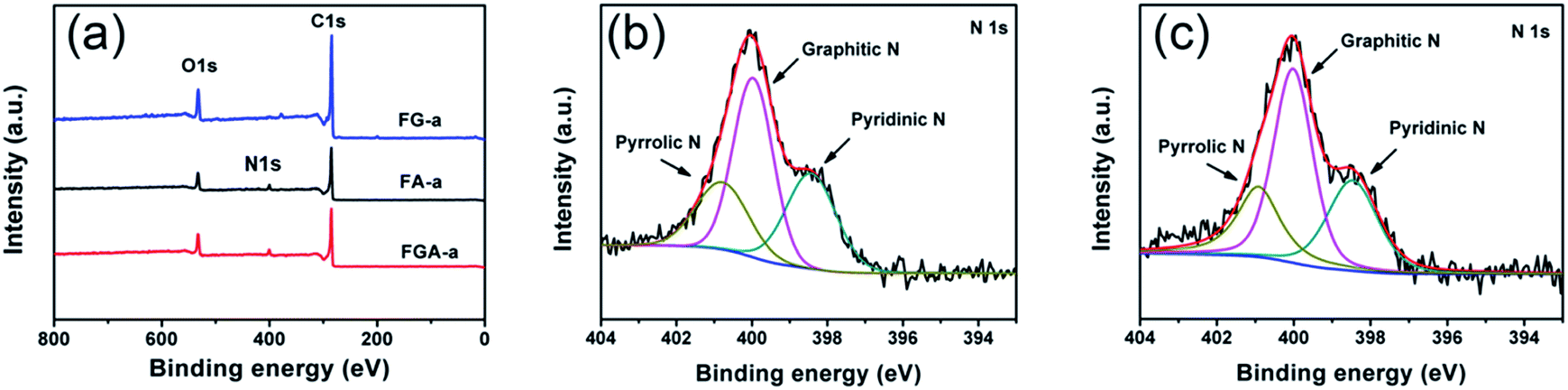 | ||
| Fig. 4 (a) XPS spectra of FG-a, FA-a and FGA-a. High-resolution N 1s spectra of the (b) FA-a and (c) FGA-a samples. | ||
The electrochemical performance of FA-a, FG-a, and FGA-a as electrodes for supercapacitors was analyzed using a three-electrode cell in a 6 M KOH aqueous solution. Fig. 5a illustrates the cyclic voltammograms (CVs) of FA-a, FG-a, and FGA-a measured at a scan rate of 5 mV s−1. FG-a exhibits a good rectangular shape corresponding to a typical double-layer capacitive behavior. Both FA-a and FGA-a exhibit a typical capacitive behavior with quasi-rectangular shaped voltammetry characteristics as well as broad humps in the CVs, indicating that the capacitance results from the combination of electrical double-layer capacitance and pseudocapacitance. The pseudocapacitance is mainly attributed to the surface redox reactions of the O and N atoms with the electrolyte.39,45 When increasing the scan rate to 100 mV s−1 (Fig. 5b), the FG-a and FGA-a electrodes become somewhat distorted but still retain the rectangular-like voltammogram profiles, suggesting an excellent high-rate capacitive behavior in contrast with poorly performing FA-a electrodes. This indicates that FG-a and FGA-a have a better rate capability at a high scan rate.
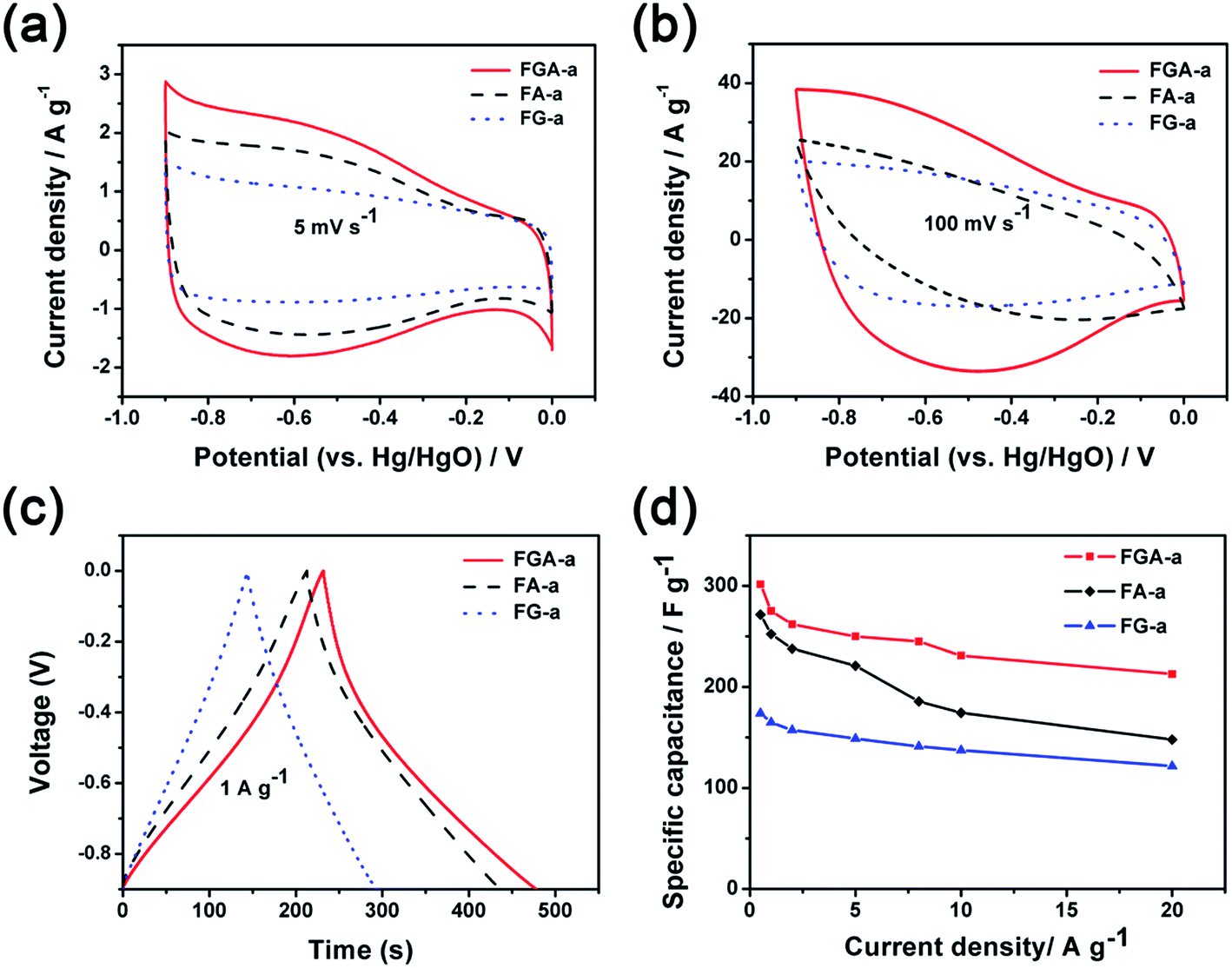 | ||
| Fig. 5 Cyclic voltammograms of FA-a, FG-a, and FGA-a at the scan rates of (a) 5 mV s−1 and (b) 100 mV s−1; (c) galvanostatic charge–discharge curves of FA-a, FG-a, and FGA-a at a current density of 1 A g−1; (d) specific capacitances of FA-a, FG-a, and FGA-a at different current densities. | ||
Fig. 5c shows the galvanostatic charge–discharge (GC) profiles of FA-a, FG-a, and FGA-a at a current density of 1 A g−1. The GC curve of FG-a displays a triangle shape, indicating an ideal capacitive behavior. However, deviations from the linear curves can be observed in the GC tests of FA-a and FGA-a, indicating the presence of redox reactions during the charge–discharge process.5 This is consistent with the results of CVs. The specific capacitances of the samples calculated from the results of GC profiles are shown in Fig. 5d. FGA-a possesses a specific capacitance of 301.6 F g−1 at 0.5 A g−1 in 6 M KOH aqueous solution, and the specific capacitance values are 271.5 F g−1 and 173.9 F g−1 for FA-a and FG-a, respectively. It should be noted that high capacitance retentions can be achieved for FG-a and FGA-a, being 70.0% and 70.5% at a high current density of 20 A g−1, respectively, compared with the capacitance retention of 54.5% for FA-a (148.0 F g−1).
In order to measure the charge transport and ion diffusion properties of the electrode materials, electrochemical impedance spectroscopy (EIS) is performed in the range from 10−2 to 105 Hz. The corresponding Nyquist plots are shown in Fig. 6a. The semicircle of the Nyquist plot in the high-frequency range is associated with the surface properties of the electrode and corresponds to the charge transfer resistance. The smaller diameter of the semicircle corresponds to lower charge transfer resistance. The diameter of the semicircle in the Nyquist plot of FGA-a is much smaller than that of FA-a and FG-a, respectively, indicating that the FGA-a electrode can guarantee faster charge transportation. The line of the Nyquist plot in the low-frequency range is related to the capacitive behavior of the electrode. Compared to FA-a, nearly vertical lines can be observed in the Nyquist plots of the FG-a and FGA-a electrodes, especially for FG-a, which reflect excellent electrochemical capacitive properties.46 This is in agreement with the results of CV and GC profiles.
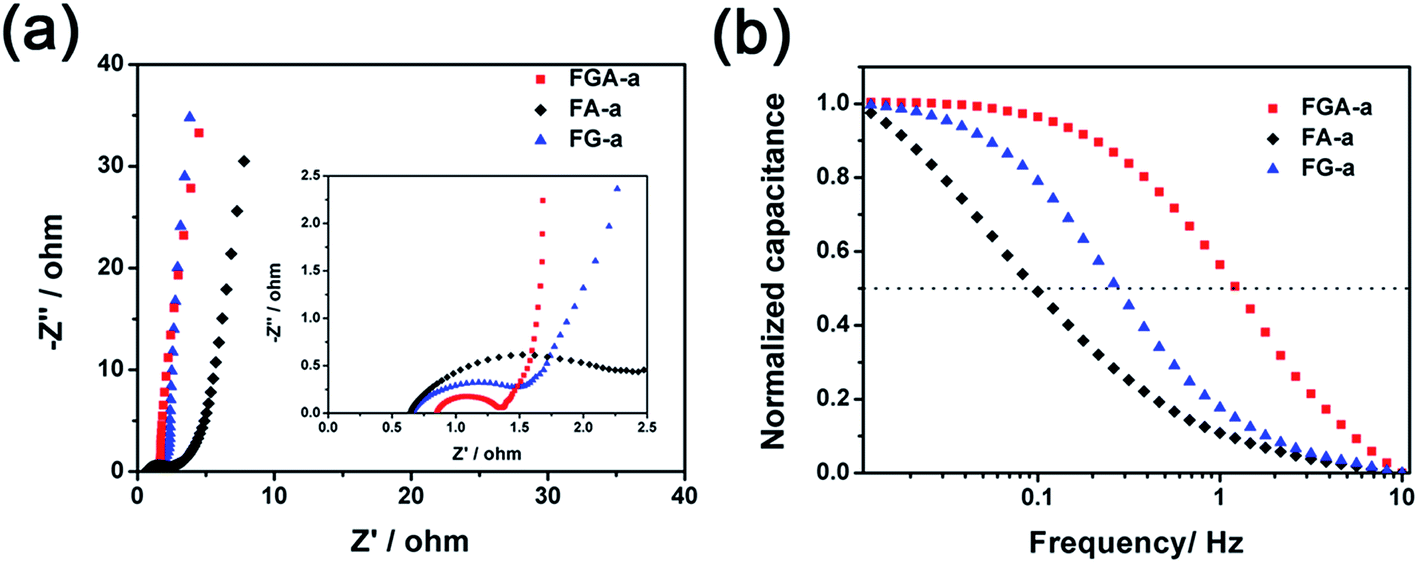 | ||
| Fig. 6 Nyquist plots (a) and frequency responses (b) of FA-a, FG-a, and FGA-a. | ||
The FG-a and FGA-a samples demonstrate a higher operating frequency, at which the capacitance equals 50% of its maximum value (Fig. 6b). The operating frequency increases from 0.10 (FA-a) to 0.26 (FG-a) and 1.21 Hz (FGA-a), demonstrating the fastest frequency response for FGA-a. The faster performance of the FG-a and FGA-a samples correlates with the graphene sheets, which can be attributed to their unique 2D structures and significantly shorten electrolyte ion transport distance, thus increasing the pore accessibility. In addition, FG-a and FGA-a electrodes with larger mesopore volumes further offer fast ion transport channels, resulting in higher capacitance retentions at a high current load. The high N content of FA-a and FGA-a also results in high pseudocapacitances. Based on the above analysis, the FGA-a electrode featuring a unique 2D structure, high N content, and large mesopore volume exhibits the best capacitive performance among all the as-made samples. Furthermore, FGA-a also exhibits good cycling performance, possessing a high capacitance retention of over 92.3% after 5000 charge–discharge cycles at 5 A g−1 (Fig. 7). In addition, Fig. S8† shows the electrochemical performance of FGA-a obtained through the two-electrode cell test which was carried out in 6 M KOH within a potential range of 0–1 V. The CVs of FGA-a (Fig. S9a†) exhibit slightly asymmetrically shaped voltammetry characteristics, indicating a high degree of reversibility, which is consistent with charge–discharge profiles (Fig. S9b†). As shown in Fig. S9c,† the calculated specific capacitance of FGA-a is 202.3 F g−1 at a current density of 0.5 A g−1 and has about 61.0% initial capacitance retention at a current density of 10 A g−1. Fig. S9d† shows the Ragone plots of FGA-a corresponding to the relationship between energy and power densities. An energy density of 7.03 W h kg−1 was achieved at a power density of 125 W kg−1 for the FGA-a based symmetric supercapacitor.
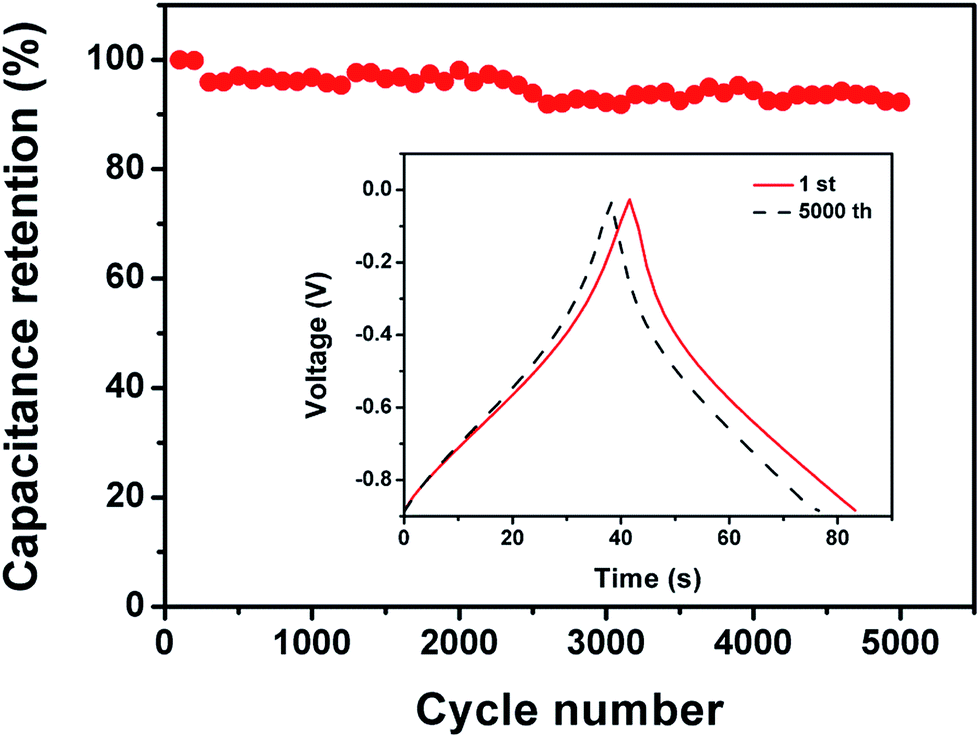 | ||
| Fig. 7 Capacitance retention of FGA-a measured at 5 A g−1. | ||
It is noted that other amino acids, such as glutamic acid and serine, can also be used as the nitrogen sources instead of glycine to obtain 2D nitrogen-containing porous carbon nanosheets, labeled FGAG-a and FGAS-a, respectively. The electrochemical performance of FGAG-a and FGAS-a electrodes is shown in Fig. S10.† The specific capacitance of FGAG-a and FGAS-a is 245.6 F g−1 and 250.6 F g−1 at a current density of 0.5 A g−1 in 6 M KOH aqueous solution, respectively. The low N content in FGAG-a (3.5 at%) and FGAS-a (3.7 at%) may cause lower specific capacitances. A high capacitance retention of 66.3% and 70.4% also can be achieved for both samples at a current load of 20 A g−1. These results indicate that amino acids are the general nitrogen sources for this strategy for the preparation of 2D nitrogen-containing carbon nanosheets for high-performance supercapacitors.
4. Conclusions
In summary, 2D N-containing porous carbon nanosheets have been successfully fabricated in the presence of GO by a simple yet facile hydrothermal method followed by a chemical activation process. The GO plays a unique role in turning the morphologies of the hydrothermal carbonaceous products. The as-obtained FGA-a electrode exhibits a high specific capacitance of 301.6 F g−1 at a current density of 0.5 A g−1 and excellent rate capability with a capacitance retention of 70.5% at a high current density of 20 A g−1, which is obviously better than that of FA-a and FG-a electrodes. It is believed that the 2D nanosheet porous structures and high N content of the FGA-a electrode can effectively improve the transport process of electrolyte ions and increase the pseudocapacitive contribution. Our current research reporting the low-cost and sustainable synthesis of 2D N-containing carbon nanosheets provides valuable insight into the development of high-performance carbon-based electrodes for supercapacitors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Science Foundation of China (21776028), Doctoral Scientific Research Foundation of Liaoning Province (20170520263), Fundamental Research Funds for the Central Universities (DUT16RC(3)008 and DUT18RC(4)059).References
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2013, 46, 1094–1103 CrossRef PubMed.
- J. Wang, P. Nie, B. Ding, S. Dong, X. Hao, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 2411–2428 RSC.
- Z. Zhang, S. Mu, B. Zhang, L. Tao, S. Huang, Y. Huang, F. Gao and Y. Zhao, J. Mater. Chem. A, 2016, 4, 2137–2146 RSC.
- Y. Zhao, S. Huang, M. Xia, S. Rehman, S. Mu, Z. Kou, Z. Zhang, Z. Chen, F. Gao and Y. Hou, Nano Energy, 2016, 28, 346–355 CrossRef.
- W. Gu, M. Sevilla, A. Magasinski, A. B. Fuertes and G. Yushin, Energy Environ. Sci., 2013, 6, 2465–2476 RSC.
- J. Zhang, J. Zhou, D. Wang, L. Hou and F. Gao, Electrochim. Acta, 2016, 191, 933–939 CrossRef.
- X. Fan, C. Yu, J. Yang, Z. Ling and J. Qiu, Carbon, 2014, 70, 130–141 CrossRef.
- S. Liu, Y. Cai, X. Zhao, Y. Liang, M. Zheng, H. Hu, H. Dong, S. Jiang, Y. Liu and Y. Xiao, J. Power Sources, 2017, 360, 373–382 CrossRef.
- S. Li, C. Yu, J. Yang, C. Zhao, X. Fan, H. Huang, X. Han, J. Wang, X. He and J. Qiu, ChemElectroChem, 2017, 4, 369–375 CrossRef.
- C. Lu, D. Wang, J. Zhao, S. Han and W. Chen, Adv. Funct. Mater., 2017, 27, 1606219 CrossRef.
- F. Sun, J. Gao, Y. Yang, Y. Zhu, L. Wang, X. Pi, X. Liu, Z. Qu, S. Wu and Y. Qin, Carbon, 2016, 109, 747–754 CrossRef.
- H. Lu, X. Sun, R. R. Gaddam, N. A. Kumar and X. S. Zhao, J. Power Sources, 2017, 360, 634–641 CrossRef.
- X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081–3086 CrossRef PubMed.
- L. Kong, Q. Chen, X. Shen, Z. Xu, C. Xu, Z. Ji and J. Zhu, Electrochim. Acta, 2018, 265, 651–661 CrossRef.
- X. Zhou, P. Wang, Y. Zhang, L. Wang, L. Zhang, L. Zhang, L. Xu and L. Liu, J. Mater. Chem. A, 2017, 5, 12958–12968 RSC.
- L. Kong, Q. Chen, X. Shen, C. Xia, Z. Ji and J. Zhu, Electrochim. Acta, 2017, 245, 241–250 CrossRef.
- Q. Wang, J. Yan, Y. Wang, T. Wei, X. Jing and Z. Fan, Carbon, 2014, 67, 119–127 CrossRef.
- B. Wang, J. Qiu, H. Feng, E. Sakai and T. Komiyama, Electrochim. Acta, 2016, 190, 229–239 CrossRef.
- S. Liu, J. Zhou and H. Song, Adv. Energy Mater., 2018, 8, 1800569 CrossRef.
- R. Ma and T. Sasaki, Acc. Chem. Res., 2015, 48, 136–143 CrossRef PubMed.
- Q. Niu, K. Gao, Q. Tang, L. Wang, L. Han, H. Fang, Y. Zhang, S. Wang and L. Wang, Carbon, 2017, 123, 290–298 CrossRef.
- J. Zhao, Y. Jiang, H. Fan, M. Liu, O. Zhuo, X. Wang, Q. Wu, L. Yang, Y. Ma and Z. Hu, Adv. Mater., 2017, 29, 1604569 CrossRef PubMed.
- D. Krishnan, K. Raidongia, J. Shao and J. Huang, ACS Nano, 2014, 8, 449–457 CrossRef PubMed.
- D. Liu, Z. Jia and D. Wang, Carbon, 2016, 100, 664–677 CrossRef.
- Z. Fan, Y. Liu, J. Yan, G. Ning, Q. Wang, T. Wei, L. Zhi and F. Wei, Adv. Energy Mater., 2012, 2, 419–424 CrossRef.
- Q. Wang, J. Yan, T. Wei, J. Feng, Y. Ren, Z. Fan, M. Zhang and X. Jing, Carbon, 2013, 60, 481–487 CrossRef.
- W. Geng, F. Ma, G. Wu, S. Song, J. Wan and D. Ma, Electrochim. Acta, 2016, 191, 854–863 CrossRef.
- X. Fan, C. Yu, J. Yang, Z. Ling, C. Hu, M. Zhang and J. Qiu, Adv. Energy Mater., 2015, 5, 201401761 Search PubMed.
- W. Li, S. Liu, N. Pan, F. Zeng, Y. Liu, M. Zheng and Y. Liang, J. Power Sources, 2017, 357, 138–143 CrossRef.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef PubMed.
- T. Liu, B. Lee, M. J. Lee, J. Park, Z. Chen, S. Noda and S. W. Lee, J. Mater. Chem. A, 2018, 6, 3367–3375 RSC.
- K. Yuan, T. Hu, Y. Xu, R. Graf, G. Brunklaus, M. Forster, Y. Chen and U. Scherf, ChemElectroChem, 2016, 3, 822–828 CrossRef.
- S. Arena, G. Renzone, C. D'Ambrosio, A. M. Salzano and A. Scaloni, Food Chem., 2017, 219, 477–489 CrossRef PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef.
- X. Zhang, Z. Wang, S. Li, C. Wang and J. Qiu, RSC Adv., 2014, 4, 59977–59980 RSC.
- L. Sun, C. Wang, Y. Zhou, X. Zhang and J. Qiu, J. Appl. Electrochem., 2014, 44, 309–316 CrossRef.
- S. Huang, Z. Li, B. Wang, J. Zhang, Z. Peng, R. Qi, J. Wang and Y. Zhao, Adv. Funct. Mater., 2018, 28, 1706294 CrossRef.
- Y. Zhao, Z. Zhang, Y. Ren, W. Ran, X. Chen, J. Wu and F. Gao, J. Power Sources, 2015, 286, 1–9 CrossRef.
- J. Zhang, G. Chen, Q. Zhang, F. Kang and B. You, ACS Appl. Mater. Interfaces, 2015, 7, 12760–12766 CrossRef PubMed.
- L. Chen, Y. Lu, L. Yu and X. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC.
- J. Xu, Z. Tan, W. Zeng, G. Chen, S. Wu, Y. Zhao, K. Ni, Z. Tao, M. Ikram, H. Ji and Y. Zhu, Adv. Mater., 2016, 28, 5222–5228 CrossRef PubMed.
- X. Lin, Y. Wang, T. Liu, H. Chen, Z. Jiang, Y. Chen, J. Liu, J. Huang and M. Liu, ChemistrySelect, 2018, 3, 4831–4837 CrossRef.
- Y. Tan, C. Xu, G. Chen, Z. Liu, M. Ma, Q. Xie, N. Zheng and S. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2241–2248 CrossRef PubMed.
- Y. Zhao, W. Ran, J. He, Y. Song, C. Zhang, D. Xiong, F. Gao, J. Wu and Y. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 1132–1139 CrossRef PubMed.
- Y. Zhao, M. Liu, X. Deng, L. Miao, P. K. Tripathi, X. Ma, D. Zhu, Z. Xu, Z. Hao and L. Gan, Electrochim. Acta, 2015, 153, 448–455 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00242h |
| This journal is © The Royal Society of Chemistry 2018 |