
FeCoW multimetal oxide-coated W:BiVO4 photoanode for efficient oxygen evolution
Xiaorong
Liang†
ac,
Jiale
Xie†
 b,
Jinyun
Xiong
ac,
Liangping
Gong
ac and
Chang Ming
Li
*abc
b,
Jinyun
Xiong
ac,
Liangping
Gong
ac and
Chang Ming
Li
*abc
aInstitute for Clean Energy & Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing 400715, P. R. China. E-mail: ecmli@swu.edu.cn
bInstitute for Materials Science and Devices, Suzhou University of Science and Technology, Suzhou 215009, P. R. China
cChongqing Key Laboratory for Advanced Materials & Technologies of Clean Electrical Power Sources, Chongqing 400715, P. R. China
First published on 13th June 2018
Abstract
BiVO4 is one of the most promising photoanode materials due to its relatively small band gap (∼2.4 eV) for efficient light absorption, and its suitable valence band edge for the oxygen evolution reaction. However, it achieves a photocurrent which is much lower than the theoretical value of 7.5 mA cm−2. Herein we prepare a multimetal oxide-based heterojunction W:BiVO4–FeCoW by a sol–gel approach with post-annealing. The heterojunction photoanode exhibits a photocurrent which is enhanced to ∼3.8 times that of a W:BiVO4 photoanode at 1.23 V vs. the reversible hydrogen electrode (RHE), and a significant negative shift of the flat-band potential of 280 mV. The experimental results demonstrate that the performance enhancement mechanism can be attributed to the significantly enhanced charge separation/transport and light absorption due to the multimetal oxide-modified heterojunction.
Introduction
Photoelectrochemical (PEC) water splitting is one of the most promising ways to convert solar energy into hydrogen, a clean chemical energy.1–3 However, the PEC process involves the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), and the latter is very sluggish which creates a bottleneck for water splitting.4–6 Hence, much attention has been paid to the development of photoanodes for improving the water oxidation reaction. Bismuth vanadate (BiVO4) possesses a suitable valence band edge of 2.4 V (vs. the reversible hydrogen electrode; RHE) for OER along with a narrow band gap of 2.4 eV.7 However, the actual water-splitting efficiency for BiVO4 is much lower than its theoretical value owing to its poor carrier mobility, low charge separation, and slow OER kinetics.8,9 Therefore, many approaches have been proposed to improve its OER kinetics, such as doping, morphology tailoring, the use of a heterojunction, co-catalyst modification and so on.10–14 Doping is a desirable approach for improving the overall OER performance of BiVO4 because it can improve electron mobility, and increase electrical conductivity. It has been experimentally demonstrated that W-doping can decrease the trapping states and suppress the charge recombination on BiVO4 photoanodes,15 but the photocurrent density achieved by W:BiVO4 photoanodes is still far from the theoretical value.To further improve the performance of BiVO4 photoanodes, BiVO4 has been coupled with other single metal oxides to make heterojunctions, such as WO3/BiVO4,16,17 Bi2S3/BiVO4,18 TiO2/BiVO4,19 Co3O4/BiVO4,20 and ZnO/BiVO4.21 Fabrication of a heterojunction with multimetal oxides rather than a single metal oxide is considered to be more effective for the catalysis of OER by a photoanode.22–24 The OER property of a metal oxide is mainly related to the features of the metal valence states (d states) and oxygen valence states (p states), which determine the absorption energies of the metal oxides and the catalytic activity. The incorporation of some foreign metal elements could significantly influence the electronic structure due to the ligand effect and the strain effect.25,26 In addition, the heterojunction could improve the efficient separation of charge carriers, broaden the range of absorbed light, and suppress electron–hole recombination.27 Therefore, it is advisable to combine multimetal oxides with BiVO4 to form a heterojunction structure, such as ZnFe2O4/BiVO4 and FeCe-oxide/BiVO4, in order to promote PEC water splitting.28,29 It has been confirmed that the BiVO4 photoanodes with multimetal oxide coatings of either ZnFe2O4 or FeCe-oxide give greatly improved photocurrent density and stability. Furthermore, multimetal based compounds with good catalytic activity such as FeCoW oxy–hydroxide have been demonstrated in PEC water splitting and OER.30,31 Nevertheless, a heterojunction structure of W:BiVO4 modified by a ternary FeCoW multimetal oxide has not been investigated.
In this work, we synthesized a W:BiVO4–FeCoW photoanode by a sol–gel approach. The W:BiVO4–FeCoW heterojunction enhances the photocurrent density to 3.8 times that of W:BiVO4 and causes a significant negative shift of the flat-band potential of 280 mV in comparison to the pristine W:BiVO4 photoanode. The performance enhancement mechanism of this heterojunction toward water oxidation was subsequently explored. Besides, this work is very helpful in the study of the photocatalytic properties of a multiple-junction W:BiVO4–FeCoW photoanode, thus providing a new scientific insight into photoelectrochemistry.
Experimental
Synthesis of W:BiVO4 thin films
W-doped BiVO4 thin films were prepared according to a reported method with modifications. Briefly, an F-doped SnO2 (FTO) substrate was dipped into an aqueous nitric acid solution of Bi(NO3)3, NH4VO3 and H2WO4, and subsequently calcined.32 Typically, Bi(NO3)3 (2 mmol), NH4VO3 (2 mmol) and H2WO4 (7.5%) were dissolved in HNO3 solution (10 ml, 2 M). After drying at room temperature, the orange films on FTO were calcined at 723 K in air for 2 h. After cooling to room temperature, a W:BiVO4 nanoworm-like thin film was obtained.Fabrication of the W:BiVO4–FeCoW photoanode
FeCl3·H2O/CoCl2·6H2O/WCl6 (2.56 mmol) was dissolved in 2 ml of ethanol. In a separate vial, 0.23 ml of deionized water was mixed with 2 ml of ethanol. Both vials were then chilled for 2 h. The two solutions were then mixed together while 1 ml of propylene oxide was slowly added, forming a transparent gel. The films were fabricated by dip-coating of the W:BiVO4/FTO electrodes. The W:BiVO4–FeCoW films were completely crystallized after annealing at 673 K for 0.5 h in air.Fabrication of the FeCoW photoanode
The FeCoW photoanode was fabricated using the same procedure as that used for the W:BiVO4–FeCoW photoanode but without the W:BiVO4 coating step.Material characterizations
The morphologies of the synthesized photoanodes were characterized by scanning electron microscopy (SEM) together with X-ray energy dispersive spectroscopy (EDS) using a JSM-7800F scanning electron microscope equipped with an X-ray energy dispersive spectrometer. The transmission electron microscopy (TEM) analysis was carried out on a JSM-2100 electron microscope instrument operated at an accelerating voltage of 200 kV. The crystallographic analysis was conducted using X-ray diffraction (XRD) in the scan range from 15° to 80° at a scanning speed of 2° min−1 with a Shimadzu diffractometer 7000. UV-vis absorption spectra were measured using a UV-2550 spectrophotometer in the wavelength range from 300–800 nm (with BaSO4 as a reference sample). X-ray photoelectron spectroscopy (XPS) data were obtained using a Thermo Scientific instrument (ESCALAB 250Xi).Photoelectrochemical measurements
The PEC measurements were carried out using a potentiostat (CHI 660, CH Instruments) in a three-electrode configuration with Pt foil as the counter electrode (0.5 cm2), Ag/AgCl as the reference electrode and the as-prepared sample as the working electrode. The electrolyte was NaOH aqueous solution (1.0 M). The light source was a Xe lamp with a light power density of 100 mW cm−2 providing AM 1.5 G simulated sunlight, and illuminated from the back-side of the photoanodes. It is noteworthy that PEC efficiency is limited by the sluggish OER, which involves a four-hole nucleophilic reaction process. An alkaline solution was selected here since it is better than other pH electrolytes for nucleophilic reactions.The PEC performances were evaluated by measuring the current density–voltage (J–V) curves with a potentiostat (CHI660D, CH Instruments) at a scan rate of 20 mV s−1 with chopped light illumination. Stabilities were obtained by potentiostatic (I–t) measurements under intermittent illumination at a bias of 0.2 V vs. the saturated calomel electrode (SCE). Electrochemical impedance spectroscopy (EIS) was conducted in the frequency range of 0.1–105 Hz at an amplitude of 10 mV under AM 1.5 G illumination at open circuit potential using a CHI 660D workstation. Mott–Schottky plots were measured at the three frequencies of 1 kHz, 5 kHz, and 10 kHz in the dark. Intensity modulated photocurrent spectroscopy (IMPS) and impedance vs. potential measurements were carried out using a CIMPS photoelectrochemical workstation (Zahner) under 100 mW cm−2 visible light.
Results and discussion
The morphologies of the W:BiVO4 and W:BiVO4–FeCoW photoanodes were examined by SEM. Fig. 1a shows that the W:BiVO4 has an irregular worm-like nanostructure. The characteristic size is around 100 nm. Furthermore, TEM was employed to characterize the detailed structure. As shown in Fig. 1b, the TEM image of W:BiVO4 clearly exhibits the worm-like structure, which is consistent with the observation in Fig. 1a. Fig. 1c shows a number of smaller nanoparticles on the W:BiVO4 nanostructures for the W:BiVO4–FeCoW photoanode. The diameter of the nanoparticles is in the range of 10–30 nm. In addition, Fig. 1d shows the section-view of the W:BiVO4–FeCoW photoanode with 650 nm thickness. TEM images of the W:BiVO4–FeCoW photoanode are displayed in Fig. 1e and f. A distinct FeCoW coating layer with the thickness of 30–50 nm can be identified. The high magnification TEM image of the FeCoW multimetal oxide region shows that the FeCoW-oxide nanoparticles consist of some small sized particles with the size of several nanometres. Fig. 1g shows a distinct heterojunction structure in the composite photoanode. The distinct lattice fringes with measured interplanar spacings of 0.29 nm and 0.27 nm can be observed, which are consistent with the (121) plane of W:BiVO4 and a crystal plane of the FeCoW multimetal oxide, respectively. Due to the incorporation of foreign metal elements, the spacing value is not the same as that of the single Fe-, Co-, and W-based metal oxides. However, these results clearly confirm the combination of W:BiVO4 and the FeCoW multimetal oxide in a heterojunction structure. During W-doping, W6+ replaces V5+ to modify the energy level structure of BiVO4 for enhanced light absorption, thus improving the PEC performance.33 The W-doping concentration was optimized and the results are shown in Fig. 1h, in which I represents the photocurrent produced at 1.6 V vs. RHE by different W-doping concentrations, and I0 is the optimized photocurrent of 7.5 (at.)% W-doped BiVO4. Further experiments were conducted under the optimal conditions. In Fig. 2, EDS mapping of the W:BiVO4–FeCoW photoanode shows that the elements of Bi, V, Fe, Co and W are distributed uniformly, thus proving the successful formation of the W:BiVO4–FeCoW heterojunction. | ||
| Fig. 1 (a) SEM image of the as-prepared W:BiVO4 photoanode. (b) TEM image of W:BiVO4. (c and d) SEM images of the W:BiVO4–FeCoW photoanode. (e and f) TEM images of W:BiVO4–FeCoW. (g) HRTEM image of W:BiVO4–FeCoW. (h) Optimization of W-doping concentration. | ||
 | ||
| Fig. 2 EDS mappings of the W:BiVO4–FeCoW photoanode: (a0) SEM image, (a) Bi, (b) V, (c) Fe, (d) Co, (e) W. | ||
The crystal structures of the W:BiVO4 and W:BiVO4–FeCoW photoanodes were detected by XRD to confirm their compositions. The XRD patterns of the W:BiVO4 and W:BiVO4–FeCoW photoanodes are shown in Fig. 3a. The diffraction peaks from the FTO glass are marked with stars (*). The patterns of both W:BiVO4 and W:BiVO4–FeCoW photoanodes exhibit the peaks from the monoclinic structure of BiVO4 (PDF card no. 14-0688).34 This suggests that the W-doping does not change the crystalline structure of BiVO4. In the pattern of W:BiVO4, the peak marked with a cross (+) at 2θ = 28° corresponds to the (201) plane of Bi2O3 (PDF card no. 43-0452). This may be attributed to phase separation during the high temperature annealing. The XRD pattern of the W:BiVO4–FeCoW photoanode shows not only several characteristic peaks of W:BiVO4, but also three characteristic peaks (#) of the FeCoW multimetal oxide. However, these three peaks cannot be attributed to one of the known Fe-, Co- or W-based oxides. All these results suggest that the W:BiVO4–FeCoW photoanode exhibits a coexistence of both the W:BiVO4 and FeCoW multimetal oxide phases.
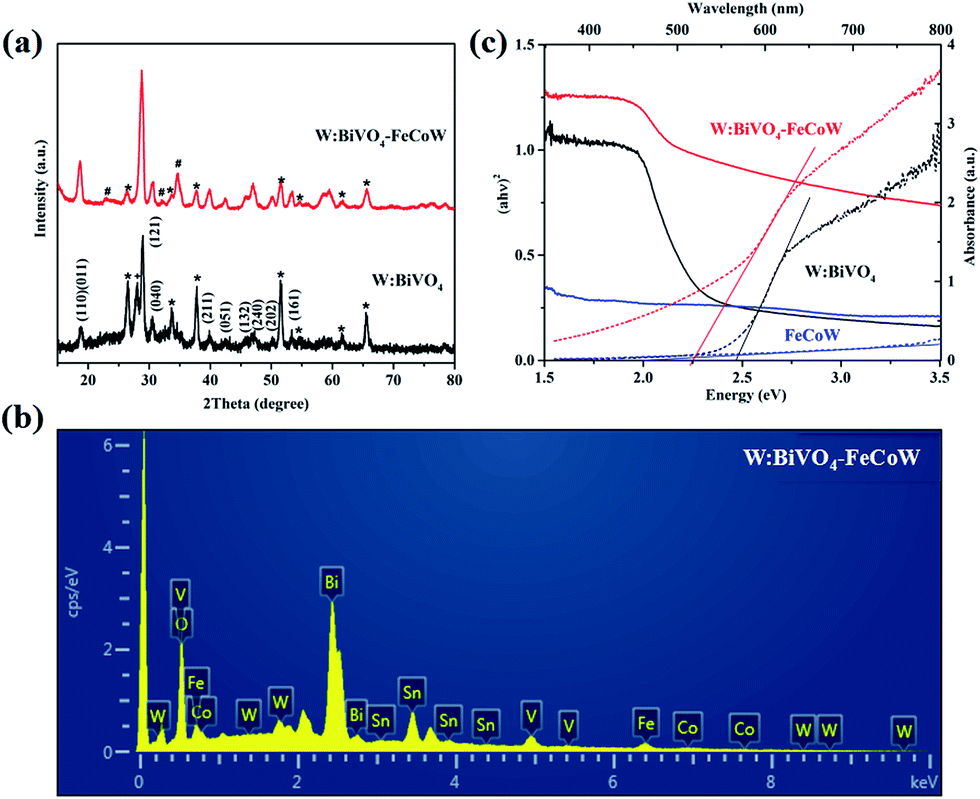 | ||
| Fig. 3 (a) XRD patterns of W:BiVO4 and W:BiVO4–FeCoW films. (b) EDS pattern of W:BiVO4–FeCoW films. (c) UV-vis absorption spectra and Tauc plots of W:BiVO4, FeCoW and W:BiVO4–FeCoW films. | ||
The EDS pattern in Fig. 3b shows the existence of Fe, Co, W, Bi, V and O elements in the prepared W:BiVO4–FeCoW films. The optical absorption properties of the pristine W:BiVO4 and W:BiVO4–FeCoW photoanodes were investigated by UV-vis absorption spectra which are shown in Fig. 3c. The W:BiVO4–FeCoW photoanode has a wider wavelength absorption range than the pristine W:BiVO4 and FeCoW photoanodes. In contrast to the W:BiVO4 photoanode, the absorption edge of W:BiVO4–FeCoW shifts from 504 nm to 551 nm. In other words, a smaller band gap of ∼2.25 eV is achieved after FeCoW multimetal oxide modification and the band gap of the FeCoW photoanode is ∼2.30 eV. The optical band gap energy is calculated according to the equation,
| λg = 1239.8/Eg | (1) |
The surface chemical states of the W:BiVO4–FeCoW photoanode were investigated by XPS and the results are shown in Fig. 4. According to the XPS spectra in Fig. 4a–f, elements including Bi, V, O, Fe, Co and W were detected. The Bi 4f peaks located at 164.0 and 158.7 eV are assigned to Bi 4f5/2 and Bi 4f7/2; the spin orbit splitting energy between the two peaks is 5.3 eV, confirming Bi3+ cations in W:BiVO4–FeCoW. The V 2p peaks located at 523.9 and 516.5 eV are assigned to V 2p1/2 and V 2p3/2; the spin orbit splitting energy between the two peaks is 7.4 eV, confirming V5+ cations in W:BiVO4–FeCoW.35–37 Moreover, the binding energy for O 1s at 529.5 eV may be attributed to the lattice oxygen in W:BiVO4–FeCoW, while binding energies between 530 eV and 532 eV are from the surface hydroxyl groups and/or oxygen deficient region.36,38
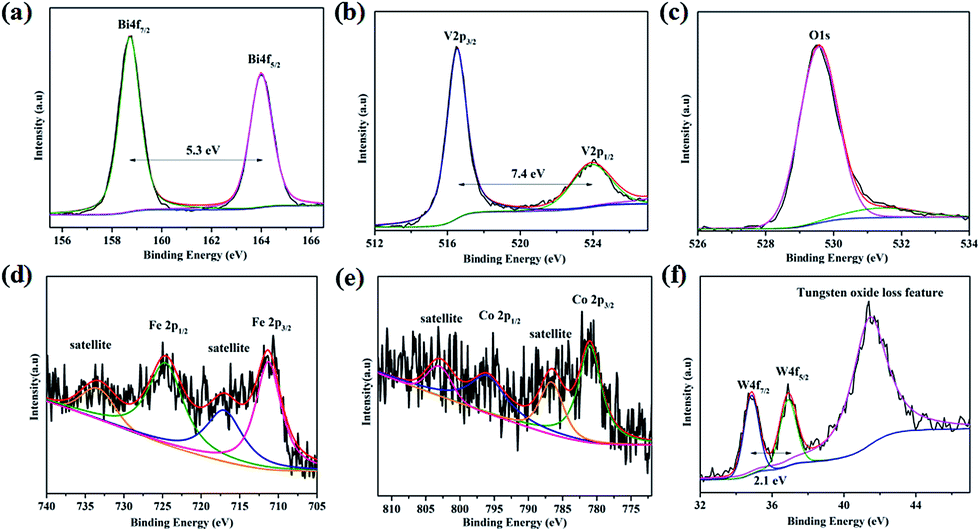 | ||
| Fig. 4 XPS spectra of (a) Bi 4f, (b) V 2p, (c) O 1s, (d) Fe 2p, (e) Co 2p and (f) W 4f of the W:BiVO4–FeCoW photoanode. | ||
Fig. 4d shows the XPS spectrum of Fe 2p, which is characterized by two peak maxima located at 724.9 eV and 711.3 eV corresponding to Fe 2p1/2 and Fe 2p3/2 respectively, which are typical values of Fe3+. The other two marked peaks are attributed to two characteristic satellites of Fe3+.39 No peaks correspond to Fe (2p3/2 at 707 eV), Fe2+ (2p3/2 at 708 eV) or Fe4+ (2p3/2 at 713 eV),40 confirming the presence of the Fe(III) metal oxide in W:BiVO4–FeCoW. The XPS spectrum of Co 2p is analysed as displayed in Fig. 3e. The two peaks at binding energies of 781.0 eV and 796.3 eV, correspond to the Co 2p3/2 and Co 2p1/2 peaks, respectively. Besides, the peaks at 803.2 eV and 786.6 eV correspond to Co 2p1/2 and Co 2p3/2 satellite peaks. These are all consistent with the presence of Co2+, as reported before.41,42 Furthermore, this result indicates the presence of the Co(II) metal oxide in the FeCoW multimetal oxide. In Fig. 4f, the spin orbit splitting energy of W 4f between the W 4f7/2 and W 4f5/2 peaks is 2.1 eV. In addition, a broad peak at 41.5 eV corresponds to the tungsten oxide loss feature, confirming that V5+ is replaced by W6+ at VO43− tetrahedral sites; a similar result has been reported previously for W:BiVO4 photoanodes.36
Chopped linear sweep voltammetry (LSV) curves of W:BiVO4–FeCoW and W:BiVO4 photoanodes were produced by measuring the chopped polarization curve at a scan rate of 20 mV s−1 under standard AM 1.5 G irradiation in 1.0 M NaOH (Fig. 5a). They show that with an increase in applied potential, the photocurrent steeply increases and does not reach saturation. The photocurrent density and the onset potential of W:BiVO4–FeCoW are significantly improved in comparison to those of the pristine W:BiVO4 and FeCoW photoanodes. At 1.23 V (vs. RHE), the photocurrent density of W:BiVO4–FeCoW reaches 0.49 mA cm−2, while only 0.13 and 0.07 mA cm−2 are achieved for W:BiVO4 and FeCoW, respectively. Thus, the photocurrent density is increased by 280% due to the multimetal oxide coating. Similar improvement effects have also been observed on a hematite photoanode.43 It is noted that the PEC performance of FeCoW is the worst among those of the three photoanodes.
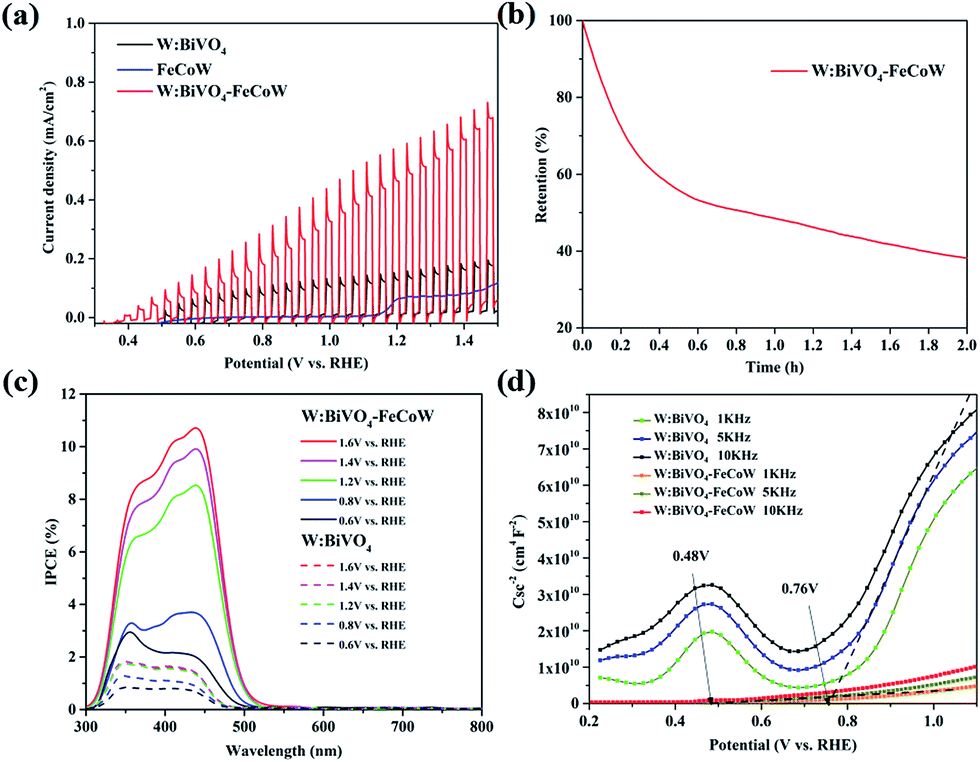 | ||
| Fig. 5 (a) LSV curves of W:BiVO4, FeCoW and W:BiVO4–FeCoW photoanodes with chopped light in 1.0 M NaOH under AM 1.5 G illumination. Scan rate: 20 mV s−1. (b) Stability testing of W:BiVO4–FeCoW at 1.23 V vs. RHE in 1.0 M NaOH under the same irradiation conditions. (c) IPCE curves of W:BiVO4 and W:BiVO4–FeCoW photoanodes measured in the potential range of 0.6–1.6 V. (d) Mott–Schottky plots of W:BiVO4 and W:BiVO4–FeCoW photoanodes at different frequencies in the dark in 1.0 M NaOH. | ||
To further examine the stability of the W:BiVO4–FeCoW photoanode, I–t curves were measured at 1.23 V (vs. RHE) under illumination as shown in Fig. 5b. It can be observed that the photocurrent density of W:BiVO4–FeCoW decreases to 38% of its initial value after 2 h, which may be due to the corrosion of the photoanodes in the PEC reactions.44
The incident-photon-to-current-conversion efficiencies (IPCE) of the W:BiVO4–FeCoW and W:BiVO4 photoanodes at various bias potentials show a functional relationship between the photoelectric conversion efficiency and the incident light wavelength (Fig. 5c). With an increase of the voltage from 0.6 V (vs. RHE) to 1.6 V (vs. RHE), the corresponding IPCE increases at a given wavelength for a specific electrode, demonstrating the enhanced hole–electron separation of W:BiVO4–FeCoW for water oxidation. In addition, after applying a specific bias, the IPCE of the W:BiVO4–FeCoW photoanode is higher than that of the W:BiVO4 electrode at the same wavelength. The maximum IPCE of W:BiVO4–FeCoW occurs at ∼440 nm, while it occurs at ∼350 nm for W:BiVO4. This red shift is consistent with the UV-vis spectra (Fig. 3c), which show the reduced band gap of W:BiVO4–FeCoW; therefore a narrow band gap can effectively harvest solar energy, enlarge the range of the absorption wavelength and lead to the shifting of the maximum absorption peak to longer wavelength. The maximum IPCE value of W:BiVO4–FeCoW (10.71%) is 6.02 times higher than that of W:BiVO4 (1.78%) at 1.2 V (vs. RHE). Moreover, the visible light response range of the W:BiVO4–FeCoW photoelectrode is extended, which indicates that the FeCoW modification increases the visible light absorption obviously and leads to more efficient charge carriers.
Fig. 5d shows Mott–Schottky plots of W:BiVO4–FeCoW and W:BiVO4 photoanodes at 1 kHz, 5 kHz and 10 kHz, respectively. The slopes of these curves are all positive, indicating n-type semiconductor characteristics with electrons as the majority carriers. The slopes of these photoanodes become smaller with increasing frequency and the flat potential shows a frequency dependency due to the non-ideality of the electrode surface.45 W:BiVO4–FeCoW exhibits lower slopes than W:BiVO4, confirming its higher donor density in comparison to W:BiVO4. This result suggests that the W:BiVO4–FeCoW photoanode could improve charge transport by introducing huge numbers of electrons. At 5 kHz, the flat band potential of the W:BiVO4–FeCoW photoanode is ∼0.48 V vs. RHE, which is markedly lower than that of W:BiVO4 (∼0.76 V vs. RHE). The significant shift should be mainly due to the passivation of the surface states of W:BiVO4. It should be mentioned that the onset potential is not always equal to the flat band potential because the photocurrent in the vicinity of the flat band potential is suppressed by the recombination process.46
Charge recombination behaviors at the semiconductor–electrolyte interface (SEI) of the photoanodes were investigated by PEC impedance spectroscopy. The Nyquist and Bode plots of the W:BiVO4–FeCoW and W:BiVO4 photoanodes measured in 1.0 M NaOH under AM 1.5 G illumination at open circuit potential are given in Fig. 6a and b. Moreover, the equivalent circuits of W:BiVO4–FeCoW and W:BiVO4 used to fit the Nyquist plots are shown in the inset of Fig. 6a. Nyquist plots of both W:BiVO4–FeCoW and W:BiVO4 are composed of two semicircles, of which the semicircle at low frequency is related to the charge transfer process in the semiconductor depletion layer, and the one at high frequency is ascribed to the electron transfer in the Helmholtz layer. R1 (R′1) is the series resistance of the cell; R2 (R′2) and CPE1 (CPE′1) represent the semiconductor depletion layer resistance and the chemical capacitance, respectively. R3 represents the charge transfer resistance in the Helmholtz layer, while the CPE2 element corresponds to the recharged Helmholtz layer. According to the fitting results in Table 1, the W:BiVO4–FeCoW photoelectrode shows the lowest R2 of 300.8 Ω, suggesting its superior charge transfer process at the SEI, which can be attributed to the modification with FeCoW multimetal oxide. In contrast, the W:BiVO4 results in an R′2 value which is considerably higher (4612 Ω). This reveals that the improved performance is due to the more efficient charge separation and transport. The high value of CPE1 for W:BiVO4–FeCoW contrasts to the low value of CPE′1 for W:BiVO4 and also indicates better electrochemical activity after FeCoW multimetal oxide modification.
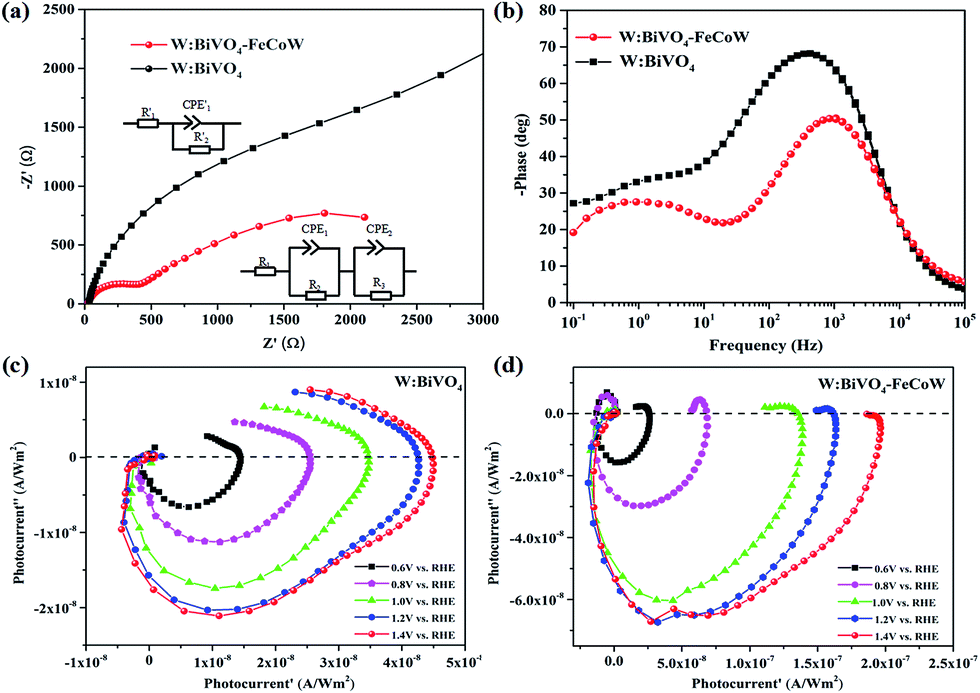 | ||
| Fig. 6 (a) Nyquist plots of W:BiVO4 and W:BiVO4–FeCoW photoanodes in 1.0 M NaOH under AM 1.5 G illumination. The insets show the equivalent circuits used to fit the impedance data. (b) Bode plots of W:BiVO4 and W:BiVO4–FeCoW photoanodes. (c) IMPS spectra of the W:BiVO4 photoanode at various potentials in 1.0 M NaOH. (d) IMPS spectra of the W:BiVO4–FeCoW photoanode at various potentials in 1.0 M NaOH. | ||
| Photoanode | R 1/R1′ (Ω) | R 2/R2′ (Ω) | CPE1/CPE1′ (F) | R 3 (Ω) | CPE2 (F) |
|---|---|---|---|---|---|
| W:BiVO4–FeCoW | 25.71 | 300.8 | 9.38 × 10−6 | 3412 | 3.65 × 10−4 |
| W:BiVO4 | 24.62 | 4612 | 5 × 10−6 | — | — |
Intensity-modulated photocurrent spectroscopy (IMPS) can efficiently minimize the photoinduced changes in band bending and was further used to identify the role of the FeCoW layer of the W:BiVO4–FeCoW photoanode. Two important parameters, namely the rate constants for charge transfer (ktr) and surface recombination (krec), were calculated using IMPS.47 An IMPS spectrum consists of two semicircles in the lower and upper quadrants, which correspond to the resistor–capacitor (RC) attenuation and the competition between charge transfer and recombination, respectively. The ratio of krec/ktr is positively proportional to the upper semicircle and a small value could indicate a charge transfer that is faster than the charge recombination.38 The W:BiVO4–FeCoW photoanode shows a smaller upper semicircle than the W:BiVO4 photoanode (Fig. 6c and d) at a specific voltage, suggesting that the charge recombination of W:BiVO4–FeCoW is much smaller than that of W:BiVO4. With an increase in the applied potential from 0.6 to 1.4 V vs. RHE, the upper semicircle of the W:BiVO4–FeCoW photoanode becomes progressively smaller. This is consistent with the theoretical relationship between water oxidation and oxidation potential, in which the reaction rate becomes faster with the increased applied potential. In contrast, this is not obvious in the upper semicircle of the W:BiVO4 photoanode with increasing applied potential, demonstrating that a higher level of charge recombination still occurs in the W:BiVO4 photoanode across a wide potential range.
The energy level diagram of the W:BiVO4–FeCoW photoanode is shown in Fig. 7. Both the FeCoW multimetal oxide and W:BiVO4 have a narrow band gap which favours the absorption of photons, consistent with the result from Fig. 3c. Under AM 1.5 G illumination, electrons are stimulated from the valence band to the conduction band in both the FeCoW multimetal oxide and the W:BiVO4 layers. More importantly, the heterojunction between the FeCoW multimetal oxide and W:BiVO4 can boost the charge separation and transport due to the triggered energy level structure. Moreover, the photocurrent of W:BiVO4–FeCoW is much larger than that of W:BiVO4 and FeCoW, mainly due to the charge transport and recombination rates (Fig. 5a). W:BiVO4–FeCoW can show improved charge transport due to the introduction of huge numbers of electrons and the electrochemical activity toward oxygen production. The lower charge transfer resistance of W:BiVO4–FeCoW can directly support this (Fig. 6a). Thus, the FeCoW multimetal oxide modification can not only enhance the charge separation and interfacial transfer, but also greatly suppress the charge recombination at the photoanode/electrolyte interface and remove the surface states of W:BiVO4 to favour the water oxidation reaction. In this stepwise band-edge energy structure, the FeCoW multimetal oxide acts as a charge transporter/sensitizer which can efficiently decrease the charge-transfer resistance, increase the electron density and consequently improve the PEC performance of the W:BiVO4–FeCoW photoanode.
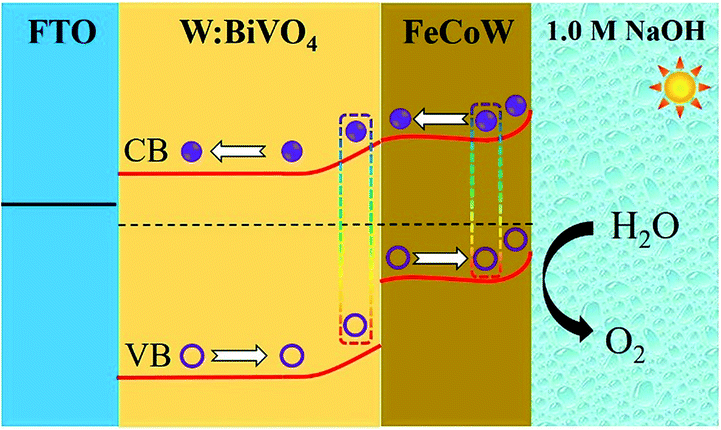 | ||
| Fig. 7 Energy level diagram of the W:BiVO4–FeCoW electrode and the charge transfer processes. | ||
Conclusions
In summary, a W:BiVO4–FeCoW photoanode prepared by a sol–gel procedure exhibits a much more efficient water oxidation performance in a PEC process than a W:BiVO4 photoanode due to its better charge separation, faster charge transport and higher charge recombination resistance. The EIS data show that the W:BiVO4–FeCoW and W:BiVO4 photoanodes have different charge transfer resistances, which reveals that the improved performance is mainly due to the more efficient charge separation and transport of W:BiVO4–FeCoW. After FeCoW multimetal oxide modification, the heterojunction photoanode shows better light absorption properties. The flat-band potential of the heterojunction W:BiVO4–FeCoW is ∼280 mV lower than that of the W:BiVO4 photoanode and the photocurrent density is increased by 280%. This work demonstrates that the FeCoW modification of a heterojunction structure not only holds great promise as a facile approach to greatly improve the PEC performance, but also sheds light on the photoelectron-catalytic process for water splitting systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (grant no. 21703150), the China Postdoctoral Science Foundation (grant no. 2015M582495), a start-up grant from Southwest University (grant no. SWU111071), Suzhou University of Science and Technology (grant no. 331812148) and the Chongqing Science and Technology Commission (grant no. cstc2012gjhz90002).References
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- R. Bhosale, S. Kelkar, G. Parte, R. Fernandes, D. Kothari and S. Ogale, ACS Appl. Mater. Interfaces, 2015, 7, 20053–20060 CrossRef PubMed.
- Z. S. Li, W. J. Luo, M. L. Zhang, J. Y. Feng and Z. G. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- J. W. Sun, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2010, 3, 1252–1261 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed.
- W. Yuan, P. K. Shen and S. P. Jiang, J. Mater. Chem. A, 2014, 2, 123–129 RSC.
- Z. F. Huang, L. Pan, J. J. Zou, X. W. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- P. M. Rao, L. L. Cai, C. Liu, I. S. Cho, C. H. Lee, J. M. Weisse, P. D. Yang and X. L. Zheng, Nano Lett., 2014, 14, 1099–1105 CrossRef PubMed.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef.
- J. H. Zhang, M. S. Deng, F. Z. Ren, Y. Wu and Y. X. Wang, RSC Adv., 2016, 6, 12290–12297 RSC.
- M. Zhou, H. B. Wu, J. Bao, L. Liang, X. W. Lou and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 8579–8583 CrossRef PubMed.
- S. K. Pilli, T. G. Deutsch, T. E. Furtak, L. D. Brown, J. A. Turner and A. M. Herring, Phys. Chem. Chem. Phys., 2013, 15, 3273–3278 RSC.
- F. Lin, D. G. Wang, Z. X. Jiang, Y. Ma, J. Li, R. G. Li and C. Li, Energy Environ. Sci., 2012, 5, 6400–6406 RSC.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef PubMed.
- B. Pattengale, J. Ludwig and J. Huang, J. Phys. Chem. C, 2016, 120, 1421–1427 CrossRef.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928–1933 CrossRef PubMed.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 RSC.
- X. Gao, H. B. Wu, L. Zheng, Y. Zhong, Y. Hu and X. W. Lou, Angew. Chem., 2014, 53, 5917–5921 CrossRef PubMed.
- B.-Y. Cheng, J.-S. Yang, H.-W. Cho and J.-J. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20032–20039 CrossRef PubMed.
- X. Chang, T. Wang, P. Zhang, J. Zhang, A. Li and J. Gong, J. Am. Chem. Soc., 2015, 137, 8356–8359 CrossRef PubMed.
- E. T. D. Kumar, K. Thirumalai, S. Balachandran, R. Aravindhan, M. Swaminathan and J. R. Rao, Surf. Interfaces, 2017, 8, 147–153 CrossRef.
- J. S. Jang, S. M. Ji, S. W. Bae, H. C. Son and J. S. Lee, J. Photochem. Photobiol., A, 2007, 188, 112–119 CrossRef.
- Y. Tak, S. J. Hong, J. S. Lee and K. Yong, J. Mater. Chem., 2009, 19, 5945–5951 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- P. Zhang, B. Y. Guan, L. Yu and X. W. Lou, Chem, 2018, 4, 162–173 Search PubMed.
- J. T. Li and N. Q. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- T. W. Kim and K.-S. Choi, J. Phys. Chem. Lett., 2016, 7, 447–451 CrossRef PubMed.
- A. Shinde, D. Guevarra, G. Liu, I. D. Sharp, F. M. Toma, J. M. Gregoire and J. A. Haber, ACS Appl. Mater. Interfaces, 2016, 8, 23696–23705 CrossRef PubMed.
- B. Zhang, X. L. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. L. Han, J. X. Xu, M. Liu, L. R. Zheng, F. P. G. de Arquer, C. T. Dinh, F. J. Fan, M. J. Yuan, E. Yassitepe, N. Chen, T. Regier, P. F. Liu, Y. H. Li, P. De Luna, A. Janmohamed, H. L. L. Xin, H. G. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef PubMed.
- J. Xiao, H. Huang, Q. Huang, X. Li, X. Hou, L. Zhao, R. Ma, H. Chen and Y. Li, Appl. Catal., B, 2017, 212, 89–96 CrossRef.
- J. Xie, C. Guo, P. Yang, X. Wang, D. Liu and C. M. Li, Nano Energy, 2017, 31, 28–36 CrossRef.
- B. Pattengale, J. Ludwig and J. Huang, J. Phys. Chem. C, 2016, 120, 1421–1427 CrossRef.
- A. J. E. Rettie, H. C. Lee, L. G. Marshall, J. F. Lin, C. Capan, J. Lindemuth, J. S. McCloy, J. S. Zhou, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 11389–11396 CrossRef PubMed.
- Z. Q. He, Y. Q. Shi, C. Gao, L. N. Wen, J. M. Chen and S. Song, J. Phys. Chem. C, 2014, 118, 389–398 CrossRef.
- J. Choi, P. Sudhagar, J. H. Kim, J. Kwon, J. Kim, C. Terashima, A. Fujishima, T. Song and U. Paik, Phys. Chem. Chem. Phys., 2017, 19, 4648–4655 RSC.
- M. Yan, Y. L. Wu, Y. An, X. Yan, F. F. Zhu, Y. Q. Hua and W. D. Shi, ACS Sustainable Chem. Eng., 2016, 4, 757–766 CrossRef.
- J. L. Xie, C. X. Guo, P. P. Yang, X. D. Wang, D. Y. Liu and C. M. Li, Nano Energy, 2017, 31, 28–36 CrossRef.
- V. Srivastava and S. Kumar, CrystEngComm, 2014, 16, 11122–11126 RSC.
- Y. C. Ling, G. M. Wang, J. Reddy, C. C. Wang, J. Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 4074–4079 CrossRef PubMed.
- J. Yan, S. Y. Yang, Z. K. Xie, X. Li, W. Y. Zhou, X. C. Zhang, Y. P. Fang, S. S. Zhang and F. Peng, J. Solid State Electrochem., 2017, 21, 455–461 CrossRef.
- X. M. He, X. Y. Song, W. Qiao, Z. W. Li, X. Zhang, S. M. Yan, W. Zhong and Y. W. Du, J. Phys. Chem. C, 2015, 119, 9550–9559 CrossRef.
- J. Xiao, H. Huang, Q. Huang, X. Li, X. Hou, L. Zhao, R. Ma, H. Chen and Y. Li, Appl. Catal., B, 2017, 212, 89–96 CrossRef.
- M.-W. Kim, K. Kim, T. Y. Ohm, B. Joshi, E. Samuel, M. T. Swihart, H. Yoon, H. Park and S. S. Yoon, J. Alloys Compd., 2017, 726, 1138–1146 CrossRef.
- Q. Shi, X. Song, H. Wang and Z. Bian, J. Electrochem. Soc., 2017, 165, H3018–H3027 CrossRef.
- R. Beranek, Adv. Phys. Chem., 2011, 786759 Search PubMed.
- H. K. Dunn, J. M. Feckl, A. Muller, D. Fattakhova-Rohlfing, S. G. Morehead, J. Roos, L. M. Peter, C. Scheu and T. Bein, Phys. Chem. Chem. Phys., 2014, 16, 24610–24620 RSC.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |