
Unraveling the structural properties and reactivity trends of Cu–Ni bimetallic nanoalloy catalysts for biomass-derived levulinic acid hydrogenation†
Saikiran
Pendem
a,
Indranil
Mondal
a,
Abhijit
Shrotri
 b,
Bolla Srinivasa
Rao
a,
Nakka
Lingaiah
a and
John
Mondal
*a
b,
Bolla Srinivasa
Rao
a,
Nakka
Lingaiah
a and
John
Mondal
*a
aInorganic & Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Uppal Road, Hyderabad-500007, India. E-mail: johncuchem@gmail.com; johnmondal@iict.res.in
bInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-Ku, Sapporo, Japan 001-0021
First published on 17th April 2018
Abstract
Herein, we have developed a series of silica nanosphere supported CuNi bimetallic nanoalloy catalysts via solvothermal, impregnation and co-precipitation methods and evaluated their catalytic performance for the hydrogenation of levulinic acid, a key platform molecule in many biorefinery schemes, into γ-valerolactone. Different characterization techniques including powder X-ray diffraction (PXRD), high resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), solid state 13C CP MAS NMR, and field emission scanning electron microscopy (FE-SEM) with the corresponding elemental mapping have been employed to evaluate the compositional and structural features of the silica nanosphere encapsulated Cu–Ni bimetallic systems. The resultant catalyst asdeveloped by an impregnation method presents enhanced catalytic performance compared with other catalysts, providing 99.3% conversion of levulinic acid with 96.8% selectivity of γ-valerolactone in 13 h at 120 °C. Comparing catalytic experiments between bimetallic Cu–Ni and monometallic Cu catalysts demonstrated that the outstandingly improved catalyst activity and stability could be ascribed to the modulation of the electronic properties of the active sites and inhibition of metal particles with the introduction of another metal promoter, with good recyclability after ten recycles without notable loss in the activity. Our investigation in this study sheds light on a new insight into the structure–performance relationship of the bimetallic Cu–Ni nanoalloy phase in the low cost liquid fuel production from biomass.
Introduction
In pursuit of growing energy demand and the pollution caused by huge depletion of fossil resources, the utilization of renewable biomass to produce energy has been widely acknowledged as a promising solution to develop alternative energy sources.1,2 Levulinic acid (LA) is a lignocellulosic biomass derived promising platform molecule in many biorefinery schemes and its catalytic transformation to γ-valerolactone (GVL) has attracted much growing interest, as it can be either used directly or as a fuel additive, because of its properties such as high flash point, low toxicity and suitable octane number.3–5 However, the high water solubility of GVL restricted its practical applications in gasoline engines. In this context, many value-added fine chemicals including succinic and adipic acids, 2-methyltetrahydrofuran (MTHF), 1,4-pentanediol (PD) and valeric acid esters could be produced from biomass-derived LA involving GVL as a reaction intermediate which have enormous potential in practical applications for polymer monomers, solvents, plasticizers, and fuel components.6 Although these molecules have low water solubility with interesting fuel properties, their implementation in the gasoline industry is quite a challenging task owing to the high stability of the GVL molecule. Previous research studies demonstrated that expensive noble metals including Pd, Ru, Pt, Ir, and Rh exhibited excellent catalytic activities in the production of GVL from LA through hydrogenation reactions.7–10 Many research efforts are currently being invested in the design of efficient inexpensive non-noble metal (Cu, Ni & Co) based nanocatalysts for catalytic hydrogenation of LA.Catalytic activity enhancement with improved product selectivity and catalyst durability for bimetallic catalysts is caused by some interfacial synergistic and geometric effects, which influence their compositional and structural characteristics, features that are basically decided by the synthesis strategies. Consequently, we have recently reviewed many noble-metal free heterogeneous catalysts for catalytic application of LA hydrogenation to large-scale GVL production. Cu–Fe bimetallic catalyst,11 nickel-promoted copper–silica nanocomposite and SiO2-supported Cu–Ni bimetallic catalysts,12 Ni/SiO2,13 Cu–ZrO2,14 Cu/SiO2,15 Ni–H-ZSM-5,16 Ni/MoOx-C,17 Cu–ZrO2(Og),18 Ni/MgxAlyO(x+1.5y),19 Ni–Sn (1.4)/AlOH,20 and β-Mo2C,21 were evaluated for the hydrogenation of LA to GVL. In recent years, Cu–Ni bimetallic alloys have been largely investigated to explore the activity in the catalytic transfer hydrogenation of ethyl levulinate to GVL,22 one-pot hydrogenation of LA to 2-methyltetrahydrofuran (MTHF),23 hydrodeoxygenation of HMF, furfural hydrogenation24 and so on. Nevertheless, easy availability of the non-noble metal nanocatalysts make them economically viable with enhanced catalytic activity but some persistent restrictions including harsh reaction conditions, catalyst performance, stability, and sintering of nanoparticles become big hurdles for switching from a petrol based economy to a biomass based energy model. Moreover, a homogeneous composition with atomically uniform distribution of metal ions is a very important factor in regulating the catalytic performances of Cu–Ni alloys by promoting the synergistic interaction between Cu and Ni in the resultant CuNi alloy, which could be easily tuned by the catalyst preparation methods. It is also well documented in the literature that more arduous reaction conditions are required to develop such alloys owing to the low reduction potentials of non-noble metals compared with the noble metals. Phase separation into metallic Cu and Cu–Ni alloy is also quite famous in binary Cu–Ni systems than in bulk Cu–Ni systems. Therefore, further studies are challenging but necessary for the design and development of new active, stable catalytic systems through cost effective methods to upgrade platform molecules derived from lignocellulose. In addition, some recent review reports focused on the synthesis of γ-valerolactone from biomass and biomass-derived carbohydrates and its potential applications in biomass-derived platform chemicals to produce value-added chemicals.25 Chang and co-workers have reviewed the recent progress in the defunctionalization of functionalized feedstocks and also demonstrated nicely about the use of copper-silica nanocomposite catalysts in the efficient catalytic transformation of biomass-derived platform chemicals into cyclic compounds, such as lactones and hydrofurans, with high selectivities and yields.26
In our present investigation, we have synthesized an –NH2 group functionalized silica nanosphere encapsulated bimetallic CuNi nanoalloy following solvothermal, impregnation and coprecipitation methods and investigated the influence of the preparation methods in regulating the catalytic activity of the Cu–Ni bimetallic nanoalloy for hydrogenation of LA, a key platform molecule in many biorefinery schemes, into GVL. As shown in Scheme 1, all the silica nanosphere encapsulated Cu–Ni bimetallic materials synthesized using solvothermal, impregnation and co-precipitation methods are denoted as CuNi@SiO2-A, CuNi@SiO2-B & CuNi@SiO2-C, respectively. Different characterization techniques including powder X-ray diffraction (PXRD), high resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), solid state 13C CP MAS NMR, field emission scanning electron microscopy (FE-SEM) with the corresponding elemental mapping have been employed to evaluate the compositional and structural features of the silica nanosphere encapsulated Cu–Ni bimetallic systems and their effect on the catalytic activity trend. An enhancement in the catalytic performance of the bimetallic Cu–Ni catalyst as-prepared by the co-impregnation method is observed compared with the other bimetallic catalysts and their corresponding monometallic counterparts, in the catalytic hydrogenation of levulinic acid, providing 99.3% conversion with exclusive selectivity (96.8%) of GVL. The improved catalytic activity of the bimetallic Cu–Ni catalyst could be attributed to the larger number of surface exposed Cu0–NPs, weaker Cu–SiO2 interaction and strong Cu–Ni interfacial interaction, as experimentally evidenced by spectroscopic characterization details. An investigation on the superior catalytic activity and stability has been performed by comparing catalytic experiments between bimetallic Cu–Ni and monometallic Cu catalysts which revealed that the modulation of the electronic properties of the active sites and inhibition of metal particles with the introduction of another metal promoter play a crucial role, with good recyclability after ten recycles without notable loss in the activity. The novelty of the newly designed bimetallic Cu–Ni@SiO2 based catalytic system and our present study is associated with the following: (1) an easily scalable and cost-effective catalytic synthetic approach has been chosen; (2) the strong electronic interaction between Ni and SiO2 support is the main driving force for the development of the catalyst structure where Ni concentration reduction took place, thereby exposing more Cu sites on the catalyst surface to enhance the activity and catalyst stability without compromising the desired product selectivity; (3) the structure–activity relationships including the role of active metal sites, significance of Cu–Ni interactions and influence of the catalyst preparation method have been nicely understood; (4) selection of the SiO2 support compared with the other supports where BAS/LAS acidic site ratios are preferential and optimum for biomass-derived LA to GVL transformation rather than generation of over-hydrogenated products; (5) long-term catalyst stability, negligible leaching, no sign of deactivation, and good recyclability in LA hydrogenation to GVL compared with the other reported non-noble metal catalysts; (6) use of a suitable hydrogen donor solvent such as a secondary alcohol obtained from biomass-derived feedstock which can be used as a fuel by significant minimization of the product purification requirements thereby upgrading the process economy from a green and sustainable point of view.
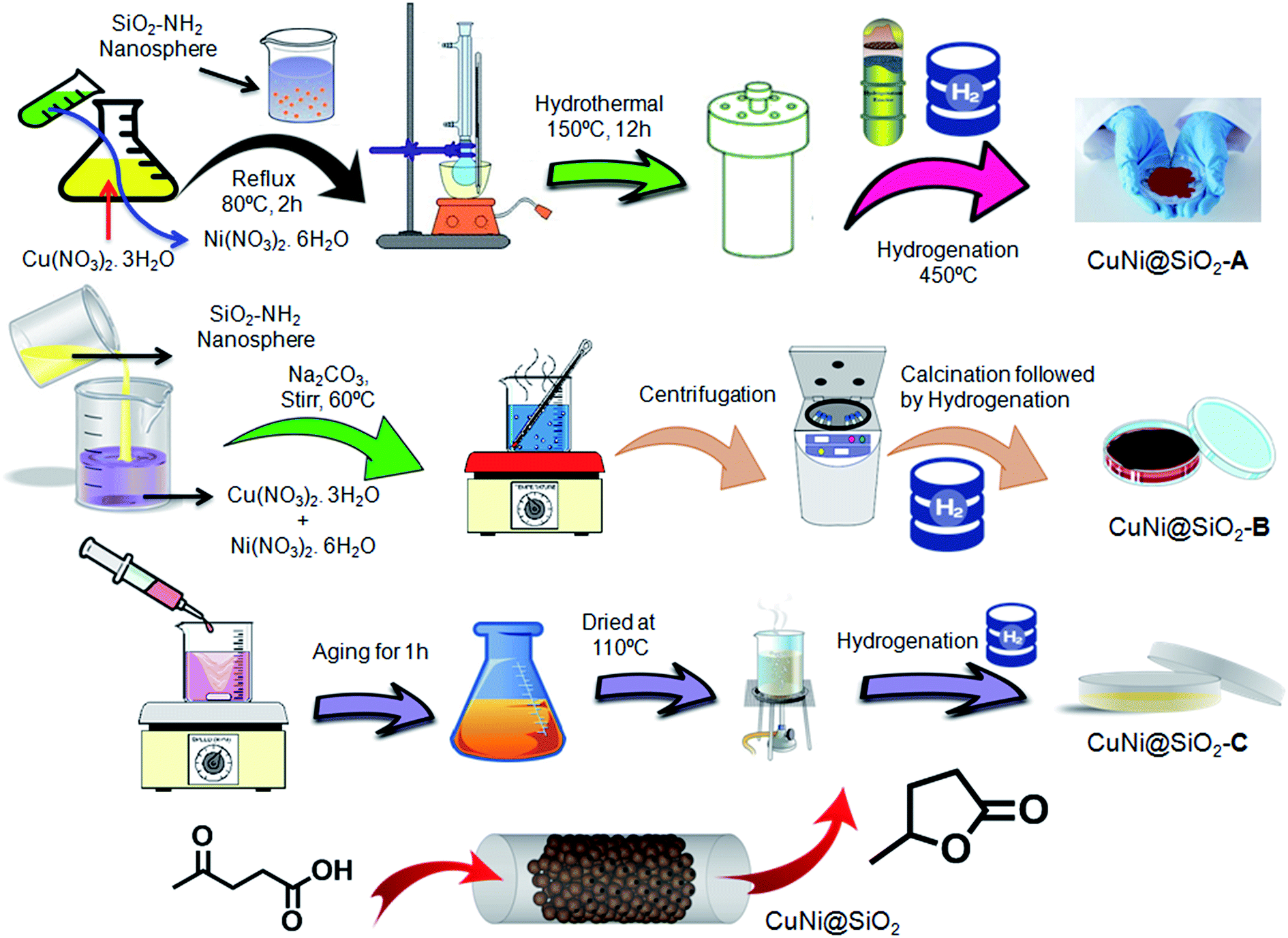 | ||
| Scheme 1 Schematic illustration showing the synthetic strategy of the Cu–Ni bimetallic nanoalloy catalyst by three different synthetic routes and its selective catalytic application in levulinic acid hydrogenation to γ-valerolactone (GVL). | ||
Experimental section
Preparation methods of CuNi@SiO2 materials
(1) Synthesis of CuNi@SiO2-A. CuNi@SiO2-A material has been synthesized by using the solvothermal method. Cu(NO3)2·3H2O (3 mmol, 0.724 mg) and Ni(NO3)2·6H2O (3 mmol, 0.872 mg) were dissolved in 20 mL distilled water to form a homogeneous solution and the solution was refluxed for 2 h in a N2 atmosphere at 80 °C. After that the amine functionalized SiO2 nanospheres were dispersed in 24 mL distilled water and 6 mL ethylene glycol (H2O
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene glycol = 4:1) to form a suspension which was slowly added to the previous solution and the mixture was refluxed for 2 h in a N2 atmosphere at 80 °C. After that the resulting reaction mixture was solvothermally treated in an autoclave at 150 °C for about 12 h. After cooling to room temperature a brown colour solid was isolated from the mixture by centrifugation followed by washing with methanol several times. The resulting solid was reduced at 450 °C in a H2 flow at the rate of 30 cm3 min−1 and designated as CuNi@SiO2-A.
:ethylene glycol = 4:1) to form a suspension which was slowly added to the previous solution and the mixture was refluxed for 2 h in a N2 atmosphere at 80 °C. After that the resulting reaction mixture was solvothermally treated in an autoclave at 150 °C for about 12 h. After cooling to room temperature a brown colour solid was isolated from the mixture by centrifugation followed by washing with methanol several times. The resulting solid was reduced at 450 °C in a H2 flow at the rate of 30 cm3 min−1 and designated as CuNi@SiO2-A.
(2) Synthesis of CuNi@SiO2-B. CuNi@SiO2-B was synthesized by the method of wet impregnation. The amine functionalized SiO2 nanospheres were dispersed in 30 mL distilled water to form a suspension and the resulting solution was stirred at 60 °C. After that Cu(NO3)2·3H2O (3 mmol, 0.724 mg) and Ni(NO3)2·6H2O (3 mmol, 0.872 mg) were dissolved in 20 mL distilled water to form a solution which was slowly added to the previous mixture through maintaining pH ±7 by adding Na2CO3 and the mixture was stirred for 1 h at 60 °C. Then the solution was aged overnight. After that the product was isolated from the mixture by centrifugation followed by washing with water several times. The resulting green colour solid was then oven-dried at 110 °C overnight. Then the solid was calcined at 300 °C for 4 h at the rate of 2 °C min−1 in a N2 atmosphere. After that the compound was reduced at 450 °C in a H2 flow at the rate of 30 cm3 min−1 and designated as CuNi@SiO2-B.
(3) Synthesis of CuNi@SiO2-C. The deposition co-precipitation method has been employed to synthesize CuNi@SiO2-C. Cu(NO3)2·3H2O (3 mmol, 0.724 mg) and Ni(NO3)2·6H2O (3 mmol, 0.872 mg) were dissolved in 20 mL distilled water and the solution was refluxed for 2 h in a N2 atmosphere at 80 °C. After that the amine functionalized SiO2 nanospheres were dispersed in 30 mL distilled water to form a suspension which was slowly added to the previous solution and stirred for 1 h at room temperature. After that the mixture was aged for 1 h then the mixture was oven-dried at 110 °C overnight and the resulting solid was reduced at 450 °C in a H2 flow at the rate of 30 cm3 min−1 and designated as CuNi@SiO2-C.
Results and discussion
Uniformly dispersed SiO2 nanospheres were synthesized by a modification of the Stöber method where tetraethyl orthosilicate (TEOS) was used as the silica source which undergoes hydrolysis in EtOH medium. The role of NH3 has been considered as a base to maintain the uniformity and smoothness of the surface of the prepared SiO2 nanospheres. Surface functionalization of the as-prepared monodisperse SiO2 nanospheres was carried out by employing (3-aminopropyl)-triethoxysilane (APTES) as the organosilane reagent. TEM images revealed the homogeneous distribution of –NH2 functionalized SiO2 with spherical morphology (Fig. S1, ESI†). A series of –NH2 functionalized SiO2 nanosphere supported Cu–Ni bimetallic nanoalloy catalysts have been synthesized via solvothermal, impregnation and co-precipitation methods. The route for stepwise structural evolution from –NH2–SiO2 to a series of Cu–Ni based bimetallic nanocatalysts is outlined in Scheme 1. The Cu and Ni contents in the respective materials as measured by ICP-OES elemental analysis are listed in Table S1, ESI.† We investigated the catalytic activity of the newly designed as-prepared Cu–Ni catalysts in the liquid-phase LA hydrogenation to GVL in a stainless-steel reactor inbuilt with a pressure gauge setup under 40 bar H2 pressure at 120 °C in isopropanol. We achieved 68.9, 99.3 and 34.6% LA conversion with GVL selectivity of 75.3, 96.8 and 27.6%, respectively, of the corresponding bimetallic Cu–Ni catalysts (entries 1, 2 & 3, Table 1). It was found that the bimetallic CuNi@SiO2-B catalyst exhibited the highest catalytic activity among all the examined bimetallic catalysts (Table 1) and its corresponding monometallic counterpart under optimized reaction conditions. Monometallic Cu@SiO2 catalyst appeared to be more efficient than the monometallic zerovalent Ni@SiO2 catalyst (entries 4 & 5, Table 1).| Entry | Catalyst | Con. (%) | GVL selectivity (%) |
|---|---|---|---|
| a Reaction conditions: 0.2 mL levulinic acid (2 mmol), temperature 120 °C, H2 pressure 40 bar, time 13 h, iPrOH 40 mL. Pd/MCM-41 and Pd/ZrMCM-41 were synthesized by following ref. 29. | |||
| 1 | CuNi@SiO2-A | 68.9 | 75.3 |
| 2 | CuNi@SiO2-B | 99.3 | 96.8 |
| 3 | CuNi@SiO2-C | 34.6 | 27.6 |
| 4 | Cu@SiO2 | 55.8 | 61.2 |
| 5 | Ni@SiO2 | 31.2 | 26.7 |
| 6 | Cu–SiO2 + Ni–SiO2 | 75.6 | 65.3 |
| 7 | CuCo@SiO2 | 42.8 | 61.3 |
| 8 | CuFe@SiO2 | 23.6 | 52.3 |
| 9 | Pd/MCM-41 | 55.9 | 62.7 |
| 10 | Pd/ZrMCM-41 | 63.1 | 70.6 |
The observed poor selectivity of the desired product GVL below 40% for CuNi@SiO2-C could be explained by the appearance of α-angelica lactone (AL) as the intermediate followed by some undesired side reaction of AL to polymeric byproducts, resulting in selectivity losses in addition to catalyst deactivation caused by carbon deposition.27 A profound rise in GVL selectivity (%) has been evidently displayed in Table 1 (entry 4), when the monometallic Ni catalyst was replaced by the monometallic Cu catalyst. These results unambiguously imply a synergetic effect between Ni and Cu, promotion effect of Ni on Cu@SiO2 to achieve high catalytic performance in the conversion of LA to GVL and selectivity improvement towards GVL due to the presence of Cu sites.28 A diminishment in catalytic activity was observed using a physical mixture of both catalysts (entry 6, Table 1), signifying the importance of an intrinsic synergistic effect between Cu and Ni generated during the process of bimetallic nanoalloy synthesis. Examination of other Cu based bimetallic catalysts (M = Co & Fe) revealed poor conversion of LA to GVL (entries 7 & 8, Table 1), implying that the presence of Cu has a special promotion effect in lowering the reduction temperature of isolated Ni species in the bimetallic Cu–Ni catalyst, thereby providing more reduced Ni sites. We have also carried out a comparison study with Pd/MCM-41 and Pd/ZrMCM-41, respectively following the previous synthesis procedure29 to get clear ideas on advancement in the activity. The catalytic activity results are provided in the entries 9 & 10 of Table 1, respectively, which suggests that the electronic interaction between two earth abundant metals (Cu & Ni) becomes more important in the advancement of catalytic activity than that of noble-metal catalysts.
In order to gain insight into the role of the support used, a series of bimetallic Cu–Ni catalysts including CuNi@ZrO2, CuNi@TiO2, CuNi@Al2O3 and CuNi@C, respectively, were developed following a similar impregnation synthesis procedure to study the catalytic performance in LA hydrogenation under optimized reaction conditions. The comparison study with various CuNi-supported catalysts, which includes the conversion (%) of LA and GVL selectivity (%), is summarized in Table 2. We have achieved 75.8%, 12.6%, 65.8% and 55.2% LA conversion for CuNi@ZrO2, CuNi@TiO2, CuNi@Al2O3 and CuNi@C, respectively. Cu–Ni catalysts supported on TiO2 and C provided much lower yields of GVL. The observed poor selectivity of GVL for TiO2 and carbon based catalysts could be explained either by the reduction of the titania support thereby causing destruction of metal support interaction or most likely owing to this intermediate not undergoing further reaction to GVL, as supported with results from the previous report by Weckhuysen and co-workers.30 Moderate GVL selectivity for CuNi@ZrO2 and CuNi@Al2O3 was observed, which could be attributed to the presence of surface acid–base sites that favours associated dehydrogenation of the alcohol by forming alkoxide on the catalyst surface. The presence of MTHF, however, indicates that either AL had undergone ring opening reactions, the rate of hydrogenation of the double bond (in AL) on the active metal site appeared to be slow over ZrO2 and Al2O3 supports, or the consequent transformation of GVL to MTHF owing to presence of a larger number of Brønsted acid sites, which could be the probable reason for the low GVL selectivity, in accordance with the previous report by Venugopal et al.31 The reason behind the observed GVL selectivity variation could be described in terms of the present Brønsted and/or Lewis acid site (BAS or LAS) ratio on the catalyst surface as measured by the respective pyridine adsorption FT-IR spectra analysis (Fig. S2, ESI†). The characteristic bands that appeared at around 1450 and 1540 cm−1 could be attributed to the presence of pyridine adsorbed on Lewis acid and Brønsted acid sites, respectively. We could find that the Cu–Ni@SiO2 catalyst exhibited a lower BAS/LAS ratio (∼0.048), which indicated the higher selectivity of GVL. In strong contrast, relatively higher BAS/LAS ratios (0.31 and 0.24) for Cu–Ni@ZrO2 and Cu–Ni@Al2O3 catalysts demonstrated the higher rate of transformation of GVL to other products, causing a decrease in GVL selectivity. Our observation is in good agreement with the previous report by Venugopal et al.31 The selective activity for LA hydrogenation to GVL of our newly designed Cu–Ni@SiO2 catalysts could be explained by the presence of both (Brønsted and/or Lewis) acid sites and their associated acidic strengths. In order to investigate in detail we systematically carried out catalytic reactions by introducing an equimolar concentration of both pyridine and 2,6-dimethylpyridine in the reaction system, which are considered as both Brønsted and Lewis acid site blocker and selective Brønsted acid site blocker, respectively. We noticed a drastic drop in LA conversion from 99% to 72.8%, with a slight decline in GVL selectivity from 96.8% to 86.6%, when the reaction was conducted with pyridine (Table 2, entry 6). From this experimental result, we can surely conclude that Lewis acid sites strongly influence LA dehydration which is not much affected by the addition of pyridine, owing to the low BAS/LAS ratio. In strong contrast, when the reaction was carried out in the presence of 2,6-dimethylpyridine, we achieved a negligible decrease in LA conversion which demonstrates that the available Lewis acid sites participate in the ring opening of GVL, which is additionally supported by the pyridine adsorption FT-IR analysis (Table 2, entry 7). Finally, we can definitely conclude that LA chemisorption on the strong Brønsted acidic catalyst surface is the driving force that leads to the ring opening of AL. The interaction between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of levulinic acid and Ni(0) could lead to the formation of monodentate and bidentate configurations. The bidentate configuration may involve either one surface Ni species which gives rise to a chelating bidentate configuration (a) or two Ni species leading to a bridging bidentate configuration (b) shown in Fig. S2, ESI.† The appearance of the peak at around ∼1565 cm−1 (Fig. S2, ESI†) could be ascribed to the presence of the bidentate configuration where both C and O atoms participate in the interaction with Ni(0) species.
O bond of levulinic acid and Ni(0) could lead to the formation of monodentate and bidentate configurations. The bidentate configuration may involve either one surface Ni species which gives rise to a chelating bidentate configuration (a) or two Ni species leading to a bridging bidentate configuration (b) shown in Fig. S2, ESI.† The appearance of the peak at around ∼1565 cm−1 (Fig. S2, ESI†) could be ascribed to the presence of the bidentate configuration where both C and O atoms participate in the interaction with Ni(0) species.
| Entry | Catalyst | Metal loading (mmol g−1) | Con (%) | GVL selectivity (%) | AL selectivity (%) | MTHF selectivity (%) | |
|---|---|---|---|---|---|---|---|
| Cu | Ni | ||||||
| a Reaction conditions: 0.2 mL levulinic acid (2 mmol), temperature 120 °C, H2 pressure 40 bar, time 13 h, iPrOH 40 mL. Reactions were carried out in the presence of pyridinea and 2,6-dimethylpyridineb. AL = α-angelica lactone, methyltetrahydrofuran (MTHF). | |||||||
| 1 | CuNi@SiO2 | 0.00347 | 0.00191 | 99.3 | 96.8 | 2.9 | 0.3 |
| 2 | CuNi@ZrO2 | 0.0041 | 0.0023 | 75.8 | 80.3 | 15.3 | 4.4 |
| 3 | CuNi@TiO2 | 0.0046 | 0.0029 | 12.6 | 2.9 | 97.1 | — |
| 4 | CuNi@Al2O3 | 0.0036 | 0.0021 | 65.8 | 86.7 | 10.9 | 2.4 |
| 5 | CuNi@C | 0.0048 | 0.0025 | 55.2 | 46.5 | 53.5 | — |
| 6a | CuNi@SiO2 | 0.00347 | 0.00191 | 72.8 | 86.6 | 13.4 | — |
| 7b | CuNi@SiO2 | 0.00347 | 0.00191 | 92.6 | 93.4 | 6.6 | — |
We have also investigated the effect of solvent in the LA to GVL conversion and iPrOH was considered as the best solvent among all the tested solvents using all the bimetallic CuNi catalysts (Fig. S3, ESI†). Polar protic solvents like H2O have an edge over polar aprotic solvents like THF in the conversion of LA to GVL. This indicated that the LA conversion with GVL selectivity was promoted in the presence of water, in good agreement with the report by Zhu et al.32 The increase of activity of the Cu–Ni catalyst in the aqueous phase is probably due to the hydrogen bond effect of liquid water. Michel et al. investigated the role of water in LA hydrogenation in a combined experimental and DFT study, which showed that a single chemisorbed water molecule and the dissociation of hydrogen-bonded water followed by transfer of the proton involved in the hydrogen bond to the alkoxy intermediate led to significant enhancement in LA conversion.33 The comparable activity in THF could be attributed to the similar solubility of H2 in THF and H2O (≈0.1203 MPa at STP).34 A comparison study of the turn over frequency (s−1) along with the GVL productivity (mmolGVL gcatalyst−1 h−1) of the three new Cu–Ni@SiO2 based catalysts is provided in Fig. 1a. Consequently, we have achieved TOFs (s−1) of 0.056 and 0.02 s−1, respectively, for the Cu–Ni@SiO2-A and Cu–NiSiO2-C catalysts, affording GVL productivities of 0.14 and 0.11 mmolGVL gcatalyst−1 h−1, respectively. In strong contrast, the bimetallic Cu–Ni@SiO2-B catalyst displayed a substantially higher catalytic activity under identical conditions, exhibiting GVL productivity of about 0.31 mmolGVL gcatalyst−1 h−1 and TOF of 0.11 s−1. In order to investigate the influence of Cu and Ni loading at different concentrations we have synthesized a variety of Cu–Ni@SiO2 based catalysts by the impregnation method and conducted the hydrogenation of LA under optimized reaction conditions and the results are presented in Fig. 1b. A prominent difference in LA conversion along with the GVL selectivity has been observed with the increase in Cu concentrations accordingly. The higher Cu concentration than Ni in the designed bimetallic CuNi@SiO2-B catalysts did not improve the GVL selectivity beyond 96.8%, though it did produce additional over-hydrogenation products, e.g. 1,4-pentane diol (1,4-PD) and 2-methyltetrahydrofuran (MTHF). In contrast, for the high Ni loading in Cu1Ni2@SiO2-B, we have achieved LA conversion of 66.8% and GVL selectivity of 55.4% along with 44.4% unreacted AL.
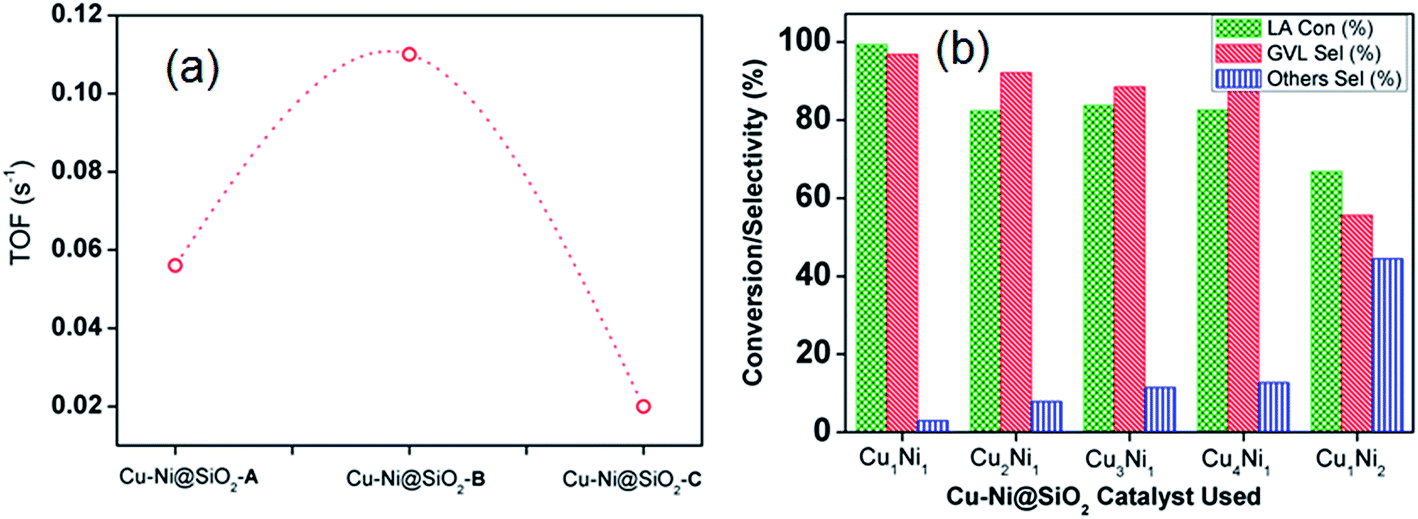 | ||
| Fig. 1 Comparison study of turn over frequency (TOF) among our three catalysts and (b) influence of Cu and Ni loading at different concentrations for the hydrogenation of LA to GVL. Reaction conditions: 0.2 mL levulinic acid (2 mmol), temperature 120 °C, H2 pressure 40 bar, time 13 h, iPrOH 40 mL. | ||
A comparison of the catalytic activities of the corresponding CuNi bimetallic catalysts tested under identical reaction conditions is presented in Fig. 2a. Complete LA consumption and excellent selectivity in the desired product formation have been noticed for the CuNi@SiO2-B catalyst. The reaction yielded 66.3% GVL after 9 h for CuNi@SiO2-B and the product yield gradually increased as the reaction proceeded. In strong contrast, the other bimetallic catalysts exhibited a comparatively lower catalytic activity, with a drop in GVL selectivity which can be attributed to the deposition of carbonaceous species generated from side products including α-angelica lactone (AL) as the intermediate thereby blocking the surface active sites on further prolonging the reaction time. We have also achieved 80.3% and 52.4% LA conversion to GVL after 15 h of continuous reaction with the corresponding CuNi@SiO2-A and CuNi@SiO2-C, respectively (Fig. 2a). Moreover, we also systematically investigated the influence of temperature in LA to GVL conversion (Fig. 2b) at a fixed H2 pressure of 40 bar. Full LA conversion to GVL was only attained after 13 h at 120 °C, while at 100 °C less than 48% GVL was obtained after the same time interval for the CuNi@SiO2-B catalyst. Instead, we have achieved 80.3 and 62.9% LA conversion with quantitative GVL yields for CuNi@SiO2-A and CuNi@SiO2-C catalysts, respectively, after a convenient 13 h of reaction at 160 °C. Fig. 2b demonstrates that the hydrogenation rate is accelerated with slight decrease in GVL selectivity due to the generation of some unknown products at higher reaction temperature accompanied by side reactions.35 Subsequently, we have also explored the profound effect of H2 pressure ranging from 10 to 50 bar on the efficiency of the CuNi bimetallic catalysts (Fig. 2c). Initially only 6.4% of GVL yield was noticed in the absence of H2 gas in the reactor. Under this condition we observed that levulinate ester was the main product which was produced through the acid site-promoted esterification of LA and isopropyl alcohol. An enhancement in LA conversion with increase in H2 pressure is noticed reaching the maximum value at a pressure of 40 bar. Enrichment of dissolved hydrogen concentration in isopropyl alcohol with the continuous increase of H2 pressure is responsible for the acceleration of catalytic hydrogenation from 10 to 40 bar H2 pressure, thereby making Ni and Cu active sites easily accessible to more hydrogen molecules. An obvious drop in GVL selectivity is noticed with further increase in hydrogen pressure from 40 to 50 bar, which could be ascribed to the formation of over-hydrogenation products of GVL including 2-methyltetrahydrofuran (MTHF) and 1,4-pentanediol (PD), analogous to what the Yuan group found in their recent work on the hydrogenolysis of HMF.36,37
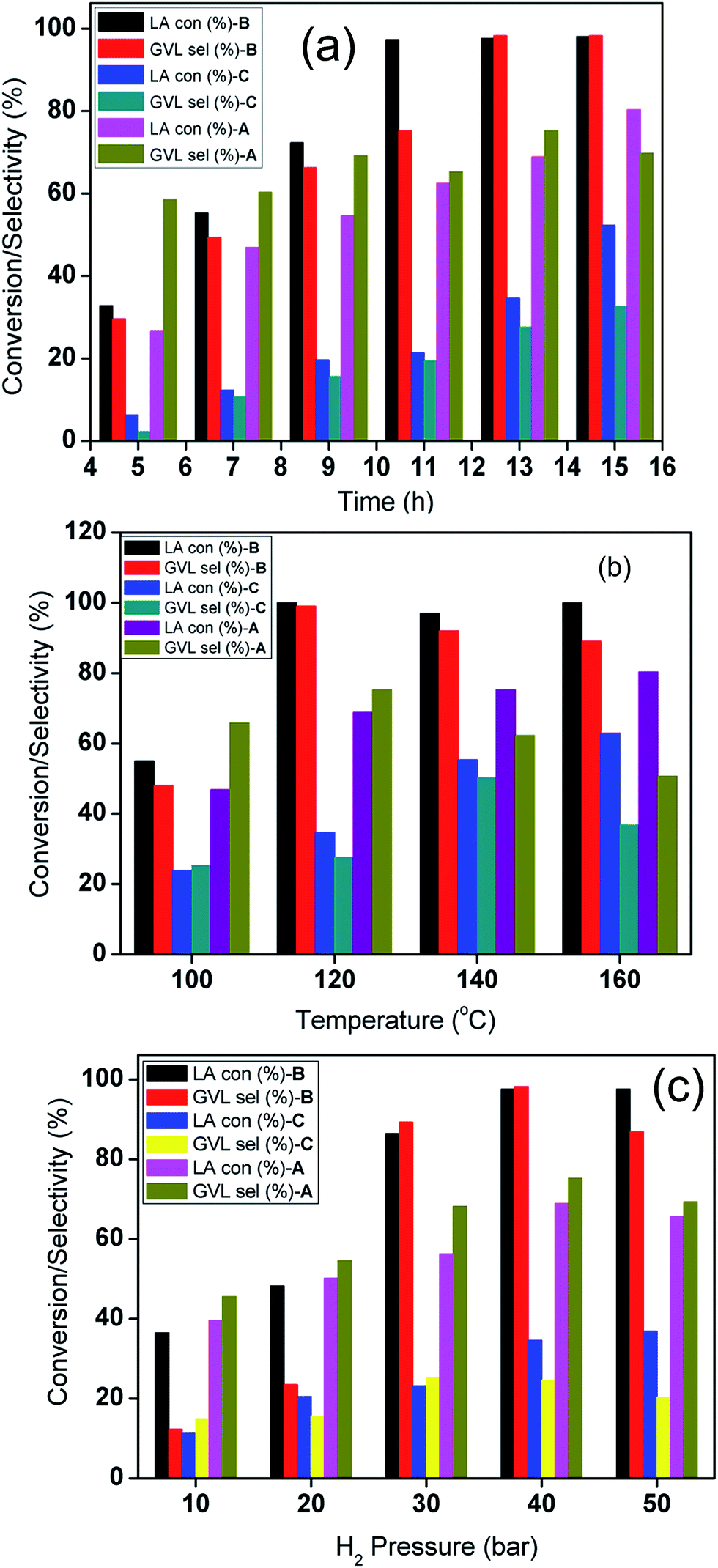 | ||
| Fig. 2 Time-on-stream profile diagram, (a) influence of reaction temperature (b) and H2 pressure (c) on the conversion of LA to GVL with CuNi@SiO2 catalysts. Here A, B & C refer to CuNi@SiO2-A, CuNi@SiO2-B & CuNi@SiO2-C catalysts, respectively. | ||
The 29Si MAS NMR spectra (Fig. 3a) exhibited four signals with chemical shifts at −101.1, −57.6 ppm, −109.9 and −67.1 ppm, respectively, which could be unambiguously ascribed to the characteristic Q3 [Si(Osi)3(OH)], T2 [C–Si(Osi)2(OH)], Q4 [Si(Osi)4] and T3 [(Osi)3Si-R] sites, respectively.38 In the 13C CP-MAS NMR spectra (Fig. 3b) three signals appeared at 10.3, 21.8 and 43.1 ppm, which clearly indicate the existence of C1, C2 and C3 carbon atoms of the aminopropyl unit attached to the silica framework.
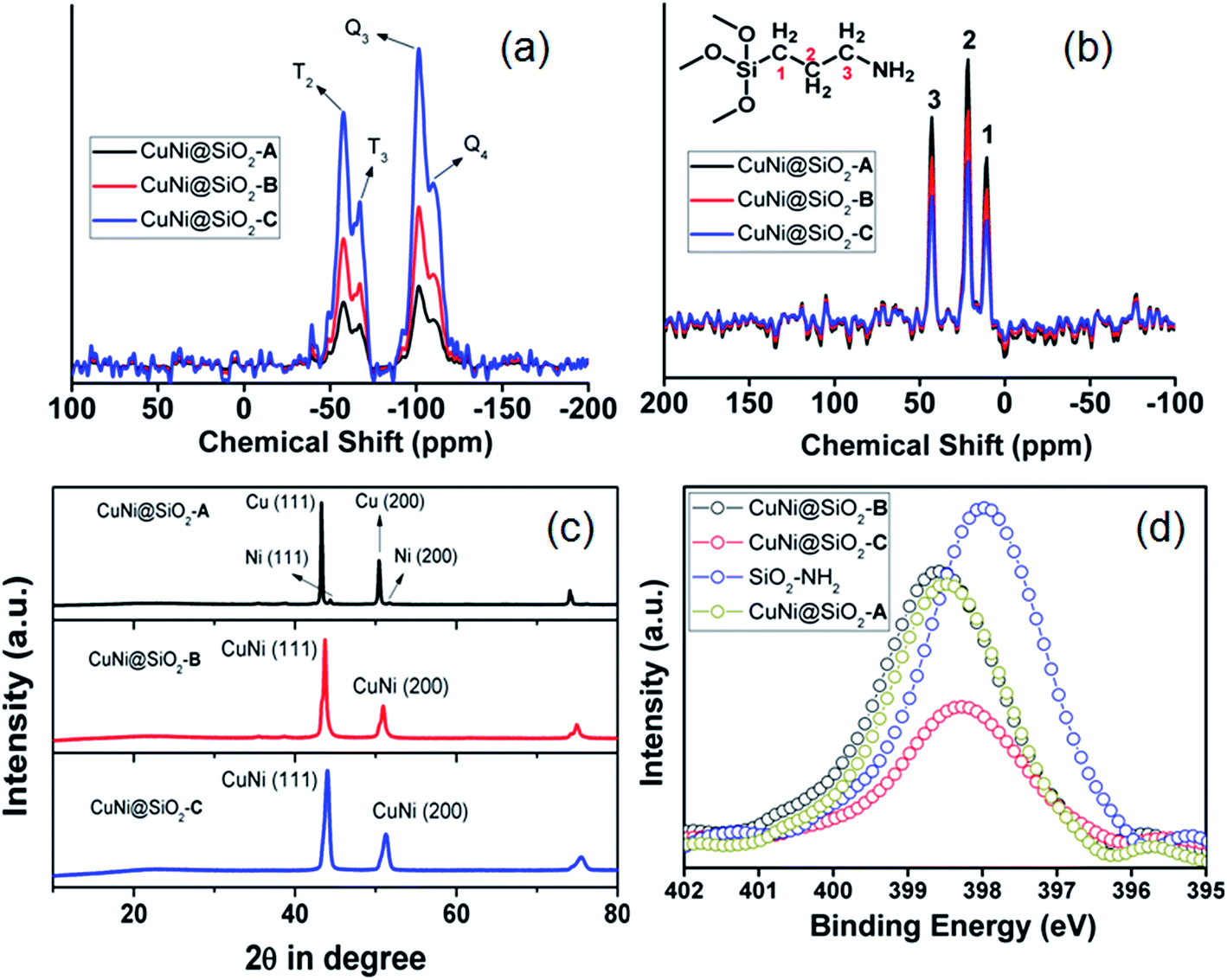 | ||
| Fig. 3 (a) 29Si and (b) 13C CP solid state MAS NMR spectra, (c) wide angle powder XRD spectra and (d) XPS spectra in the N-1S region. | ||
The molecular connectivity and structural integrity of the organic framework unit has been further confirmed by the appearance of strong absorption bands at 1145 (Si–O–Si), 2980 (–CH2 stretching) and 820 (Si–C stretching) cm−1 in the Fourier transform-infrared (FT-IR) spectral analysis (Fig. S4, ESI†). In the wide angle powder XRD pattern (Fig. 3c) CuNi@SiO2-A exhibited four distinct peaks located at 2θ ∼ 43.2°, 44.4°, 50.4° & 51.5°, respectively, which could be indexed to the (111) and (200) reflections of monometallic Cu(0) and Ni(0)-NPs, respectively. In contrast, two symmetric reflections appeared at 2θ ∼ 43.6° and 50.9° in between Cu (111) and Ni (111) reflections for CuNi@SiO2-B & CuNi@SiO2-C, respectively, clearly indicating the existence of the Cu–Ni bimetallic nanoalloy without the formation of separate metal phases.39–41 A positive displacement in the N-1S XPS binding energy for all the respective catalysts signifies strong co-ordination of the metal phases with the –NH2 groups of the organic silica ligand (Fig. 3d).
XPS technique has been employed to elucidate the oxidation state, surface composition and interaction between Cu and Ni of all synthesized catalysts. High-resolution XPS spectra (Fig. 4a) in the Cu-2p core region demonstrate significant signatures of Cu 2p3/2 binding energies (BE) at around 932.1–932.6 eV and 933.7–934.4 eV, respectively, which could be attributed to the presence of Cu0 and Cu2+ species.42–45 Cu0/Cu2+ ratios evaluated in the CuNi@SiO2-A, CuNi@SiO2-B and CuNi@SiO2-C catalysts are 1.07, 1.61 and 0.39, respectively. A down shift in the Cu0 BE (∼0.5 eV) for CuNi@SiO2-B compared with CuNi@SiO2-A could be attributed to the weaker interactions between Cu and SiO2 leading to easy partial reduction of Cu species, which is consistent with the literature report, making the Cu0 system more preferentially surface exposed.46 In the Ni-2p XPS spectra (Fig. 4b) two main peaks located in the regions at 852.4, 852.1 and 852.6 along with 854.2, 854.7 and 855.1 eV for the CuNi@SiO2-A, CuNi@SiO2-B and CuNi@SiO2-C catalysts, respectively, could to be assigned to the presence of metallic Ni0 and NiO, respectively, along with a satellite peak at higher BE.47 The third peak that appeared at 856.6 eV is indicative of a less-intense asymmetric tail for the nickel silicide.49 The slight Ni 2p3/2 BE displacement observed for the CuNi@SiO2-C catalyst from 852.2 eV demonstrates reduced Ni–Si and Cu–Ni interactions.50 It may also be noted that for the CuNi@SiO2-A catalyst the presence of Ni0 species could be readily distinguished by a BE of 852.4 eV with a positive shift around 0.3 eV compared with CuNi@SiO2-B, which is suggestive of the poor interaction between Cu and Ni driven by the lower degree of charge transfer from Cu to Ni. In the Ni-2p XPS spectra (Fig. 4a) we have clearly observed a negative displacement in the XPS binding energy by about ∼0.4 eV for CuNi@SiO2-B compared with CuNi@SiO2-C, which could be attributed to the modification of the electronic status of Cu caused by the intrinsic synergistic effect between Cu and Ni by charge transfer of electrons from Cu to Ni with the generation of more Cuδ+ charged species, in accordance with the previous report by Gorte et al.24 This result also well corroborated with the positive binding energy displacement (∼0.3 eV) for CuNi@SiO2-B compared with CuNi@SiO2-C in their respective Cu-2p XPS spectral profile (Fig. 4a). Similar XPS binding energy displacement for the bimetallic Cu–Ni/CNT catalyst has been reported by Chen et al. and they have interpreted a Cu–Ni bimetallic synergistic effect through XPS measurement.48 The peak shift to higher binding energy for CuNi@SiO2-C, assigned to NiO (Fig. 4b), was more significant than that of other catalysts, demonstrating stronger interactions and increased charge transfer at the Ni/SiO2 interface, in good agreement with the previous report by Wan et al. for the highly dispersed and stable Ni catalyst with microporous nanosilica as the support.51
 | ||
| Fig. 4 XPS spectra in Cu-2p (a) and Ni-2p (b) regions of all the Cu–Ni bimetallic catalysts, respectively. | ||
The morphology and micro-structure of the as-synthesized CuNi@SiO2 catalysts were characterized by HR-TEM and FE-SEM spectroscopy analysis (Fig. 5). HR-TEM images of CuNi@SiO2-A clearly illustrate (Fig. 5a) that black sheet-like CuNi bimetallic NPs stacked together on the surface of the SiO2 nanospheres. From HRTEM images of CuNi@SiO2-B (Fig. 5c), homogeneously distributed highly branched CuNi NPs with spherical morphology having an average particle size about 32.3 nm incorporated on the silica substrate were obviously observed.
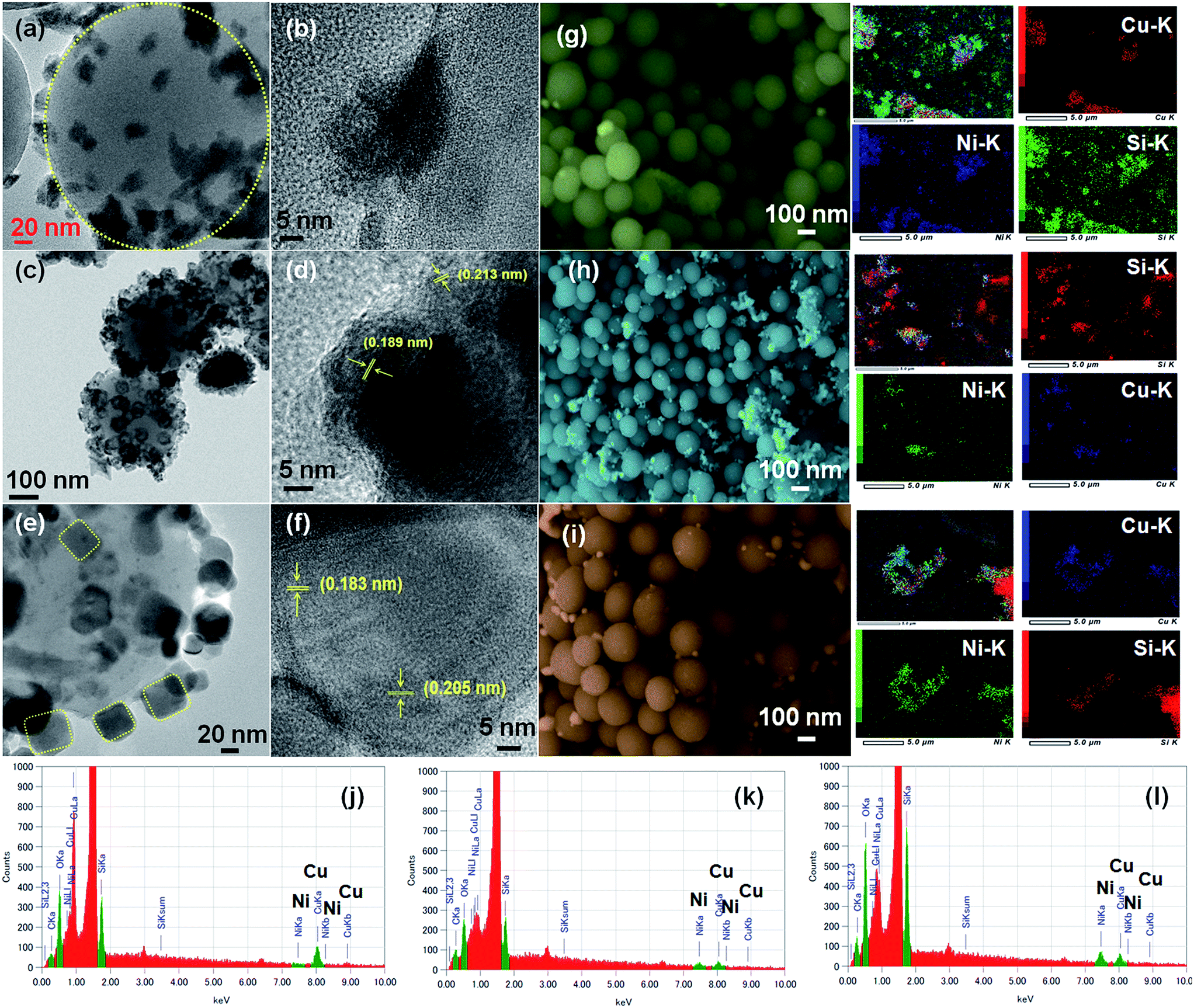 | ||
| Fig. 5 HR-TEM images of CuNi@SiO2-A (a, b), CuNi@SiO2-B (c, d) and CuNi@SiO2-C (e, f), respectively. FE-SEM images with the corresponding elemental mapping, Cu, Ni & Si (g–i), and EDX patterns (j–l) of CuNi@SiO2-A, CuNi@SiO2-B & CuNi@SiO2-C, respectively. | ||
The metallic NPs (marked in yellow) of CuNi@SiO2-C exist in irregular rounded rectangular shapes with the average diameter of 37.1–42.6 nm (Fig. 5e). Individual particles in the respective TEM images were employed to determine the average size of the metal particles. It has been observed that there is an increasing trend in the mean Cu–Ni particle size from solvothermal, impregnation to deposition co-precipitation techniques employed for the synthesis of bimetallic Cu–Ni@SiO2 based catalysts. It is well documented in the literature that the drying step of the resulting silica-supported metal nitrate composite synthesized from the silica and metal nitrate precursors is responsible for the particle size distribution.52 Severe aggregation of the metal hydroxynitrate particles took place when the drying of the composite was carried out at elevated temperature (90 °C) but drying at ambient temperatures could inhibit the formation of large crystals. In our present study, the silica-metal nitrate composite has been subjected to drying at 110 °C overnight which could help us to have an insight into the large particle size distribution as observed for both Cu–Ni@SiO2-B and Cu–Ni@SiO2-C catalysts as prepared by employing impregnation and deposition co-precipitation methods, in accordance with Jensen and co-workers.53 Furthermore, growth of Ni over the Cu NPs is expected to happen during the H2 reduction treatment at 450 °C owing to the higher standard reduction potential for Cu (0.337 eV and −0.25 eV for Cu and Ni), thereby causing formation of excessively large particles as evidenced by the previous report by Sultana et al.46 Similar HR-TEM images (Fig. 5d and f) evidently demonstrate reasonable crystallinity of the CuNi NPs, with the characteristic lattice spacing of 0.213 & 0.183 nm, corresponding to the (111) crystalline lattice plane of the face-centered cubic (fcc) CuNi alloy, well consistent with the XRD patterns.39 From the microscopy study it is quite evident that the geometry of the NPs plays a crucial role in regulating the catalytic activity. From the XPS analysis study it is quite evident that more number of Cu0 active sites are exposed on the surface of the CuNi@SiO2-B catalyst with a high Cu0/Cu2+ ratio of 1.61 and the strong Cu–Ni interfacial interaction exists which helps to modulate the electronic properties of the prepared Cu–Ni crystalline active phase. In the impregnation technique for catalyst synthesis the in situ generated Cuδ+ may play a crucial role in the activity performance where carbonyl hydrogenation is favoured. This is in good agreement with the previous observation reported by Christopher et al. with their Cu–Ni/TiO2 catalyst for co-conversion of HMF and furfural to methylated furans.42 Cuδ+ is usually generated by the gradual charge transfer from Cu to Ni which is evidently higher in CuNi@SiO2-B in comparison to CuNi@SiO2-C. This phenomenon is strongly supported by the appearance of the characteristic Ni binding energy peak displacement towards the lower region (∼0.4 eV) in the XPS profile (Fig. 4b). This greater surface exposure of Cu0-NPs, weaker Cu–SiO2 interaction and strong Cu–Ni interfacial interaction in the CuNi@SiO2-B catalyst relevant to the electronic effect of the Cu–Ni bimetallic nanoalloy is primarily attributed to the enhanced catalytic performance with a favourable carbonyl hydrogenation pathway. Also from the above TEM image analysis we can emphasize that the particle size and morphology of the Cu–Ni bimetallic alloy could also be responsible for the enhancement in catalytic activity (Fig. 5). The average size of the Cu–Ni bimetallic-NPs for CuNi@SiO2-B was estimated to be 32.3 nm, much smaller than that (42.6 nm) of the CuNi@SiO2-C catalyst. This is probably due to relatively larger particle aggregates on the inorganic organic hybrid framework facilitated by the strong interaction between the large number of available exposed crystalline sites and substrates in the branched spherical geometry of NPs. In parallel, the very large particle size of CuNi@SiO2-C reduced the active sites of the catalyst, which may be responsible for lowering the activity. The results of XPS and TEM together illustrate the superior catalytic activity of CuNi@SiO2-B to CuNi@SiO2-A and CuNi@SiO2-C catalysts, respectively. The unique structures and compositions of the prepared Cu–Ni catalysts are also visualized by FE-SEM images and the corresponding elemental mapping analysis (Fig. 5g–i, S5†) which revealed the spherical morphology of the silica nanospheres and uniform distribution of Cu, Ni & Si elements in the bimetallic catalysts, respectively, confirming the alloy structure of Cu–Ni, as experimentally evidenced with EDX analysis (Fig. 5j–l). Fig. S5† shows typical HAADF-STEM images of the as-prepared Cu–Ni based bimetallic catalysts, in good agreement with the HR-TEM results. Physicochemical surface characterization of the examined Cu–Ni@SiO2 catalysts was achieved by conducting N2-adsorption/desorption and H2-temperature programmed reduction (TPR) analysis (Fig. 6a and b). As shown in Fig. 6a, all the three examined catalysts exhibited typical type-II isotherms in the N2 adsorption–desorption analysis by continuous increase of N2 uptake at the high pressure region, suggesting the existence of interparticle void spaces. The Brunauer–Emmett–Teller (BET) surface areas of Cu–Ni@SiO2-A, Cu–Ni@SiO2-B and Cu–Ni@SiO2-C materials are measured to be 47, 83 and 68 m2 g−1, respectively. Fig. 6b shows the H2-Temperature Programmed Reduction (TPR) profiles of the catalysts. In general, Cu2+ is more easily reduced than Ni2+ under similar conditions as the standard reduction potential of Cu2+ is more than that of Ni2+. Subsequently, the reduced Cu can act as a catalyst to promote the reduction of Ni2+ and it shifted the Ni2+ reduction temperature to the Cu2+ reduction temperature region. In the meantime, the reduced Ni species could be considered as a co-active site that can accelerate the adsorption and activation of H2, thus helping the shift of reduction temperature to lower temperature regions for the bimetallic samples. The co-precipitated Cu–Ni@SiO2-C catalyst showed two reduction peaks at 280 and 333 °C corresponding to Cu2+ to Cu+ and Cu+ to Cu0, respectively. Three reduction peaks could be noticed for the Cu–Ni@SiO2-B catalyst as prepared via the impregnation method, which could be easily correlated with Cu2+ to Cu+ (320 °C), Cu+ to Cu0 (337 °C) and bulk copper species (370 °C) on the support. The shift in the peak positions to higher temperatures is attributed to the strong interaction with the support. The solvothermally synthesized Cu–Ni@SiO2-A catalyst showed a broad signal at 352 °C, which could be attributed to the reduction of Cu2+ to Cu0.12 The total H2 consumption of Cu–Ni@SiO2-A, Cu–Ni@SiO2-B and Cu–Ni@SiO2-C catalysts are 4.13, 4.79 and 4.24 mmol g−1, respectively, as calculated from the H2-TPR analysis.
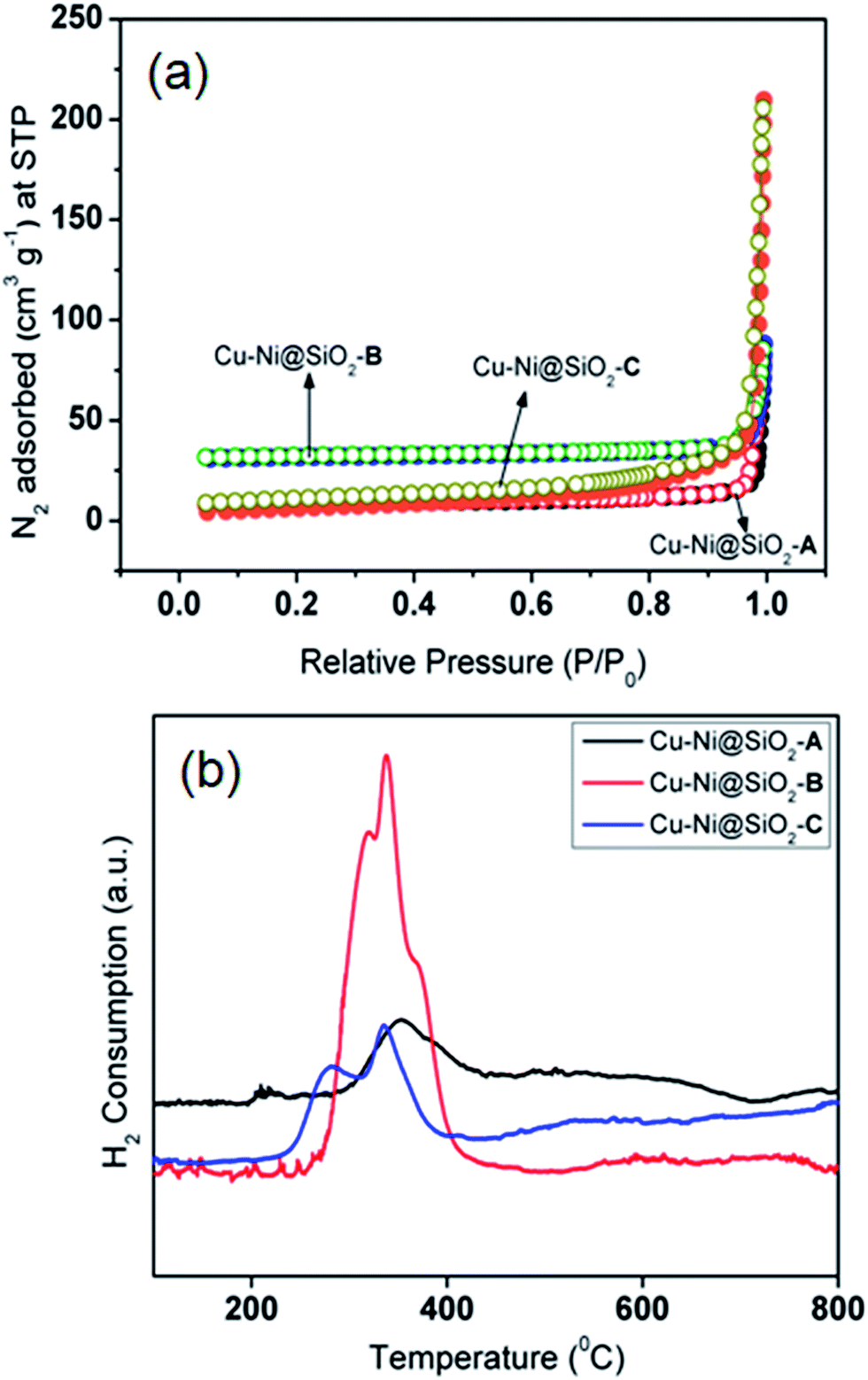 | ||
| Fig. 6 N2-adsorption/desorption isotherms (a) and H2-TPR profiles (b) of CuNi@SiO2-A, CuNi@SiO2-B & CuNi@SiO2-C, respectively. | ||
We have also examined catalyst stability for the bimetallic Cu–Ni catalyst by conducting a model reaction in comparison with the monometallic Cu catalyst after exposing in air for 1 week (Fig. 7) under optimized reaction conditions. Freshly prepared monometallic Cu@SiO2 and bimetallic CuNi@SiO2-B catalysts afforded 55.8% and 97.6% yield of GVL, respectively. After exposing in air for 1 week, we achieved 6.8% and 88.6% yield of GVL for monometallic and bimetallic catalysts, respectively. In contrast to Cu@SiO2, only a slight drop in GVL yield for CuNi@SiO2-B was observed. The observed result suggested that the stability and resistance of the bimetallic Cu–Ni catalyst toward air oxidation has been improved which could be ascribed to the modulation of the electron properties of the active sites and inhibition of metal particles with the introduction of another metal promoter, resulting in a more robust catalyst. The inherent stability of the bimetallic Cu–Ni catalyst compared with the monometallic Cu catalyst could also be rationalized to (i) the addition of Ni which will significantly contribute to the strain effect (related to the shift of the d-band density); (ii) ligand effect (related to electron density); (iii) “stabilizing effect” (the addition of a second metal often used to improve the performance of the catalytically active metal), electronic effect and stability, in accordance with the previous report by Yan et al.43 In addition, we have conducted the catalytic reactions for the next run after isolation and recycling of both catalysts from the reaction mixture; in those cases 10.3% and 95.3% GVL yields were furnished by the respective catalysts. The recyclable potential of the bimetallic Cu–Ni catalyst was investigated by performing consecutive catalytic runs under the optimized reaction conditions. After each catalytic run, the catalyst was isolated from the reaction mixture by the centrifugation technique, washed properly with acetone 3–4 times, oven-dried at 80 °C and then directly used for the next run. The recyclability results of the catalysts are presented in Fig. 8a which indicates that the catalyst could be reused for at least 10 successive catalytic runs without significant drop in LA conversion and GVL yield. To examine whether our CuNi bimetallic nanocatalyst is indeed heterogeneous in nature, a hot filtration test was performed (Fig. 8b), which clearly demonstrates the limited leaching of Cu & Ni into the reaction mixture as evidenced from Table S1, ESI.† In one test, of the hot filtration test, the catalyst was separated from the reaction mixture by centrifugation after 7 h, and we have achieved 48.6% LA conversion to GVL at that point. However, the conversion did not go beyond 48% with further continuation (Fig. 8b). This investigation evidently implies that no leaching of catalytically active species took place in the reaction mixture. In another test, after 15 h of reaction, we visualized a light blue color in the reaction mixture which indicates that a trace amount of Cu has been leached into the reaction mixture, as confirmed by the AAS analytical technique. After that we removed the catalyst, and excess levulinic acid was poured into the filtrate with the leached homogeneous Cu species, and the reaction was continued for another 22 h. But in that case we achieved only 4–5% LA conversion, which revealed that the leached copper species contributed to the hydrogenation reaction but did not catalyse the catalytic reaction up to full conversion.
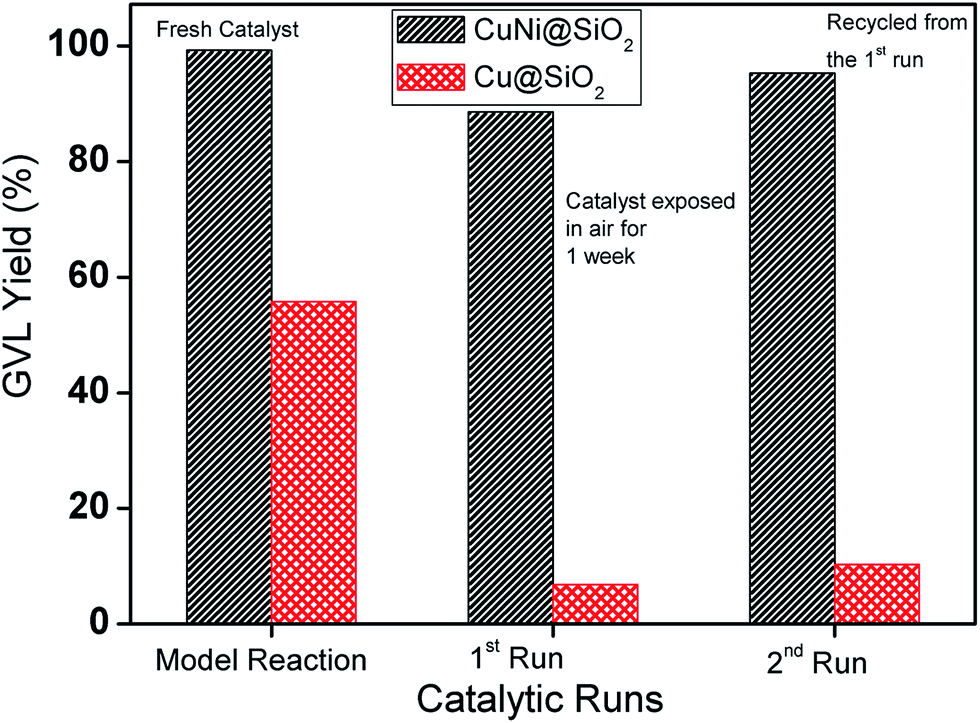 | ||
| Fig. 7 Investigation of the stability of the catalyst. Experimental conditions: 0.2 mL levulinic acid (2 mmol), temperature (120 °C), H2 pressure (40 bar), time (13 h), iPrOH (40 mL). | ||
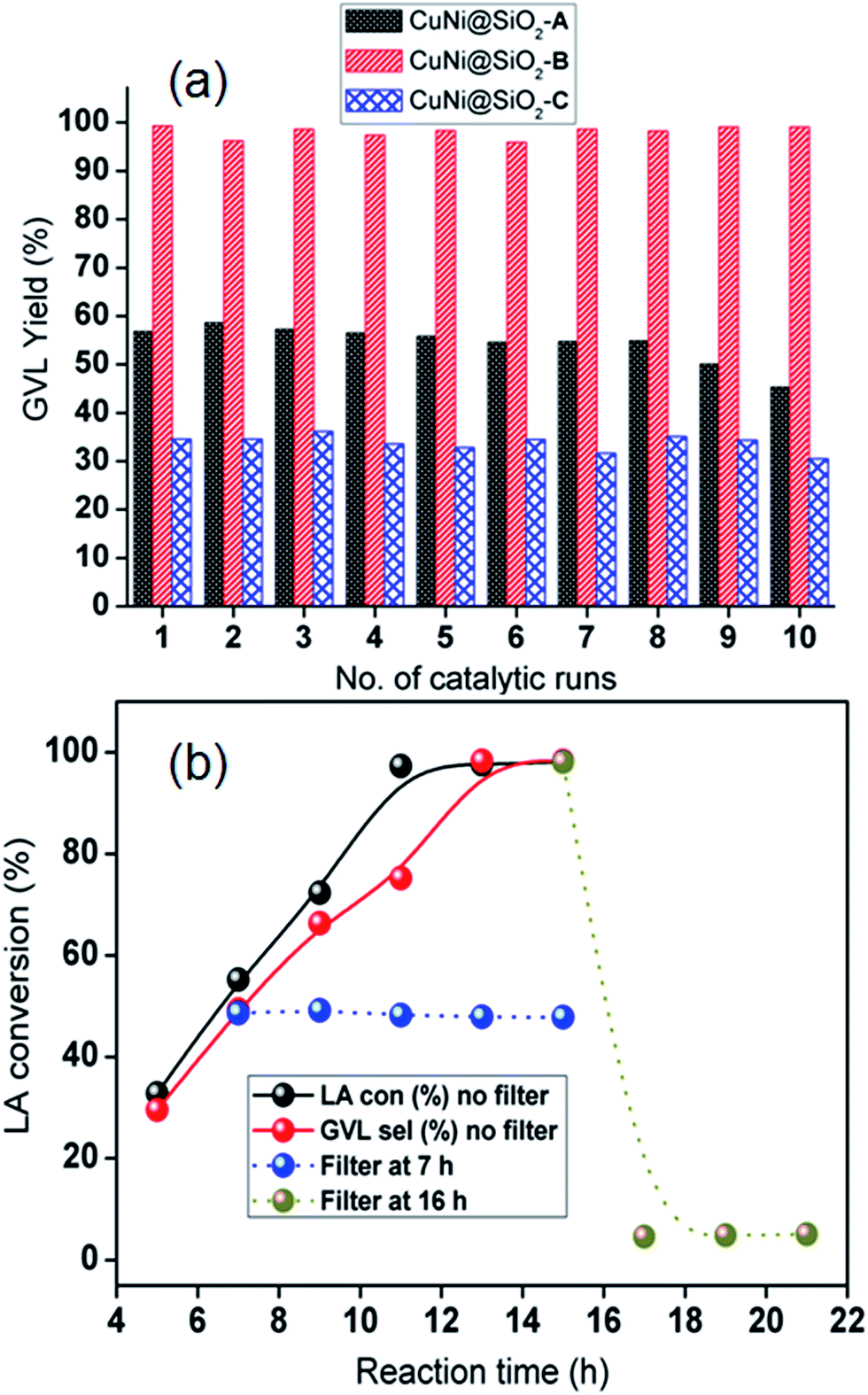 | ||
| Fig. 8 (a) Recycling test of the CuNi bimetallic catalysts. Conditions: LA 5.0 mmol, catalyst 130 mg, iPrOH 150 mL, 120 °C, 13 h; (b) hot filtration experiments in which the CuNi@SiO2-B catalyst was removed after 7 h and 16 h followed by addition of LA 2.0 mmol, catalyst 65 mg, iPrOH 40 mL, 120 °C, 13 h. | ||
For a critical comparison, the LA hydrogenation activity in terms of GVL productivity (mmolGVL gcatalyst−1 h−1) and TOF (s−1) along with GVL selectivity over various previously reported precious and non-precious metal catalysts is tabulated in Table 3. Although almost quantitative yields of GVL are obtained, in those cases either noble metals or tetrametallic catalysts and harsh conditions have been employed. We have also carried out characterization of the used CuNi@SiO2-B catalyst by TEM, HAADF-STEM, wide angle powder XRD and XPS spectrum analysis (Fig. 9) in order to evaluate whether any additional change took place during the course of the catalytic reaction. TEM and the respective HAADF-STEM analysis of the used catalyst confirmed that the nanostructure and spherical surface morphology with no obvious aggregation has been well retained after the reaction (Fig. 9a–c). Wide angle powder XRD analysis of the used catalyst (Fig. 9d) showed analogous crystalline diffraction patterns of the bimetallic Cu–Ni phase, which signifies that the catalyst surface remains unaltered with the active metal sites. The deconvoluted XPS spectrum at the Cu-2p core region (Fig. 9e) for the used CuNi@SiO2-B catalyst revealed the appearance of two peaks located at binding energies of 932.7 eV and 934.4 eV, which could be ascribed to the presence of Cu0 and Cu2+ crystallite species, respectively. The Cu0/Cu2+ ratio for the used CuNi@SiO2-B catalyst has been evaluated to be 1.49, which is very close to that of the fresh one. Comparing with the fresh catalyst, the Cu0 peak intensity remains almost constant in the reused catalyst, demonstrating further the improved stability of the catalyst. An insignificant deviation in the Cu0/Cu2+ ratio indicates that high-valent Cu2+ crystallite species were also partially reduced during the reaction to generate reduced Cu0 sites which could be the possible reason for maintaining the excellent activity and selectivity of the bimetallic Cu–Ni catalyst during the recycling experiments. Our observation is consistent with the previous report by Huang et al.22 In order to gain deep insight we have compared the XPS analysis data at the Cu-2p region with the single Cu/SiO2 catalyst (Fig. 9f), which indicated that no notable reduction from Cu2+ crystallite species to reduced Cu0 took place after the reaction. It is reported in the literature that LA hydrogenation to GVL proceeds via two pathways, either via the hydrogenation of 4-hydroxypentanoic acid followed by esterification to GVL, or via esterification of the enol form of levulinic acid to angelica lactone (α-AL) followed by hydrogenation over metal sites to give the target molecules (Fig. S7, ESI†). In order to elucidate the involved reaction pathway we monitored the reaction by GC-MS analysis. With the progress of the reaction, we found LA, α-AL, GVL and γ-hydroxyvaleric acid (very little amount) in the GC-MS spectrum (Fig. S8, ESI†). Hence from this experimental evidence we can understand and believe that our catalytic reaction proceeds via the angelica lactone formation pathway followed by further hydrogenation of angelica lactone to furnish the desired product. Also, the angelica lactone formation pathway is favoured in the presence of an acid based catalyst which is confirmed by pyridine adsorption studies.16
| Entry | Catalyst used | LA conversion (%) | GVL selectivity (%) | GVL productivity (mmolGVL gcatalyst−1 h−1) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Au–Pd/TiO2 | 99 | >99 | 1.97 | 0.1 | 54 |
| 2 | Ni/Cu/Al/Fe hydrotalcite | 100 | 98.1 | 1.2 | 0.003 | 36 |
| 3 | Ru0.7Ni0.3–OMC | 96 | 94 | 1.5 | 0.606 | 55 |
| 4 | Ni–Cu/Al2O3 | 100 | 13.3 | 0.0012 | 0.00017 | 56 |
| 5 | Pt–Mo/HAP | 99 | 67 | 0.003 | 0.001 | 57 |
| 6 | Cu–Fe catalyst | 99.4 | 51.1 | 0.16 | 0.0046 | 11 |
| 7 | 30 wt% Ni/SiO2 | 99 | 80 | 0.8506 | 0.4 | 13 |
| 8 | Cu–ZrO2 | 100 | 100 | 0.006 | 0.001 | 14 |
| 9 | Cu/SiO2 | 100 | 99.9 | 0.513 | 0.2166 | 15 |
| 10 | Cu catalyst (Cu–Cr) | 99 | 91 | 0.16 | 0.0046 | 58 |
| 11 | Ni/H-ZSM-5 | 100 | 92.2 | 0.909 | 0.05 | 16 |
| 12 | Ru/C | 97.1 | 99 | 0.190 | 0.0008 | 59 |
| 13 | Ni–MoOx/C | 100 | 97 | 0.3 | 0.05 | 17 |
| 14 | Cu–ZrO2 (Og) | 100 | 99 | 0.001 | 0.0004 | 18 |
| 15 | Ni/MgxAlyO(x+1.5y) | 100 | 99.7 | 0.02 | 0.003 | 19 |
| 16 | Ni–Sn(1.4)/AlOH | >99 | >99 | 0.01 | 0.018 | 20 |
| 17 | 20 wt% Ni/SiO2 | 100 | 90.6 | 5.1 | 1.82 | 31 |
| 18 | β-Mo2C | 60 | >85 | 7.1 | 1.7 | 21 |
| 19 | Cu–Ni@SiO 2 | 99 | 96.8 | 0.31 | 0.11 | Present study |
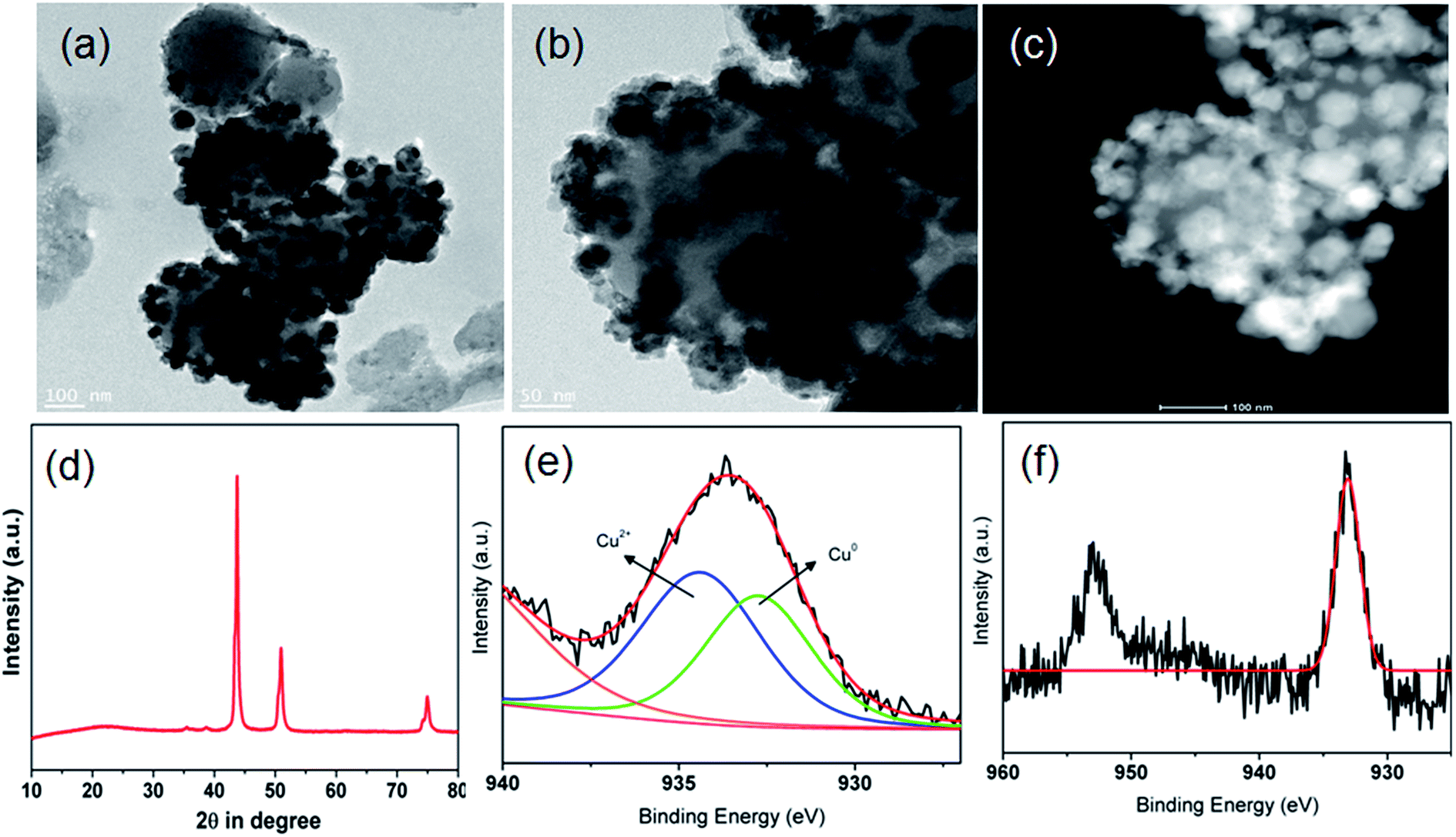 | ||
| Fig. 9 (a), (b) TEM images, (c) HAADF-STEM image, (d) wide angle powder XRD pattern and (e) XPS spectrum in the Cu-2p region of the used CuNi@SiO2-B catalyst, (f) XPS spectrum in the Cu-2p region for the single Cu catalyst. | ||
Conclusion
In summary, we have successfully developed organosilica nanospheres encapsulated bimetallic Cu–Ni catalysts via solvothermal, impregnation and co-precipitation methods, comprehensively characterized by employing several spectroscopic tools and investigated their structure–performance relationship in the liquid-phase LA hydrogenation to GVL, a key platform molecule in many biorefinery schemes. The resulting catalyst as synthesized by the impregnation method exhibited superior catalytic activity compared to other catalysts, providing 99.3% conversion of levulinic acid with 96.8% selectivity of γ-valerolactone in 13 h at 120 °C. Consequently, enhanced activity was achieved by using the impregnation preparation method, which could be ascribed to the surface composition, Cu–Ni interfacial interaction and chemical state of the active catalytic phase. From the several characterization studies of the fresh and used catalyst, the intrinsic catalyst synergy between Cu and Ni particles is thought to be responsible for the high stability and reactivity of the bimetallic catalyst, with good recyclability after ten successive cycles without considerable loss in the activity, which represents a significant step forward in promoting biomass refining.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for the financial support by the Department of Science and Technology, India, for the DST-INSPIRE Faculty Research project grant (GAP-0522) at CSIR-IICT, Hyderabad. P. S. wishes to thankfully acknowledge CSIR, New Delhi, for his CSIR-UGC fellowship.References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107(6), 2411–2502 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114(3), 1827–1870 CrossRef CAS PubMed.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10(2), 238–242 RSC.
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, ACS Catal., 2015, 5(3), 1882–1894 CrossRef CAS.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed.
- M. Li, G. Li, N. Li, A. Wang, W. Dong, X. Wang and Y. Cong, Chem. Commun., 2014, 50, 1414–1416 RSC.
- L. E. Manzer, Appl. Catal., A, 2004, 272(1–2), 249–256 CrossRef CAS.
- M. Chalid, A. A. Broekhuis and H. J. Heeres, J. Mol. Catal. A: Chem., 2011, 341(1–2), 14–21 CrossRef CAS.
- H. A. Schuette and R. W. Thomas, J. Am. Chem. Soc., 1930, 52(7), 3010–3012 CrossRef CAS.
- P. P. Upare, J.-M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J.-S. J. Chang, J. Ind. Eng. Chem., 2011, 17(2), 287–292 CrossRef CAS.
- K. Yan and A. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS.
- R. Yoshida, D. Sun, Y. Yamada, S. Sato and G. J. Hutchings, Catal. Commun., 2017, 97, 79–82 CrossRef CAS.
- M. Varkolu, V. Velpula, V. P. Chodimella, D. R. Burri and S. R. R. Kamaraju, Catal. Sci. Technol., 2014, 4, 1253–1259 Search PubMed.
- A. M. Hengnea and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- P. P. Upare, J. M. Lee, Y. K. Hwang, D. W. Hwang, J. H. Lee, S. B. Halligudi, J. S. Hwang and J. S. Chang, ChemSusChem, 2011, 4, 1749–1752 CrossRef CAS PubMed.
- M. Varkolu, R. Chakali, V. P. Chodimella, D. R. Burri and S. R. R. Kamaraju, RSC Adv., 2014, 4, 9660–9668 RSC.
- S. Ken-ichi, K. Shota and K. Kenichi, Green Chem., 2014, 16, 3899–3903 RSC.
- S. Ishikawa, D. R. Jones, S. Iqbal, C. Reece, D. J. Morgan, D. J. Willock, P. J. Miedziak, J. K. Bartley, J. K. Edwards, T. Murayama, W. Uedaa and G. J. Hutchings, Green Chem., 2017, 19, 225–236 RSC.
- J. Kun, S. Dong, Z. Zihao, F. Jie, H. Zhaoyin and L. Xiuyang, Catal. Today, 2016, 274, 55–59 CrossRef.
- Rodiansono, M. D. Astuti, T. Hara, N. Ichikuni and S. Shimazu, Catal. Sci. Technol., 2016, 6, 2955–2961 CAS.
- J. Quiroz, E. F. Mai and V. T. Silva, Top. Catal., 2016, 59, 148–158 CrossRef CAS.
- B. Cai, X.-C. Zhou, Y.-C. Miao, J.-Y. Luo and Y. B. P. H. Huang, ACS Sustainable Chem. Eng., 2017, 5(2), 1322–1331 CrossRef CAS.
- I. Obregón, I. Gandarias, A. Ocio, I. García-García, N. A. de Eulate and P. L. Arias, Appl. Catal., B, 2017, 210, 328–341 CrossRef.
- J. Luo, M. Monai, C. Wang, J. D. Lee, T. Duchoň, F. Dvořak, V. Matolin, C. B. Murray, P. Fornasiero and R. J. Gorte, Catal. Sci. Technol., 2017, 7, 1735–1743 CAS.
- Z. Zhang, ChemSusChem, 2016, 9, 156–171 CrossRef CAS PubMed.
- P. P. Upare, Y. K. Hwang, J.-M. Lee, D. W. Hwang and J. S. Chang, ChemSusChem, 2015, 8, 2345–2357 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- P. P. Upare, M.-G. Jeong, Y. K. Hwang, D. H. Kim, Y. D. Kima, D. W. Hwang, U.-H. Lee and J.-S. Chang, Appl. Catal., A, 2015, 49, 127–135 CrossRef.
- A. Wang, Y. Lu, Z. Yi, A. Ejaz, K. Hu, L. Zhang and K. Yan, ChemistrySelect, 2018, 3, 1097–1101 CrossRef CAS.
- J. Ftouni, A. Muñoz-Murillo, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, L. Lu, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Catal., 2016, 6, 5462–5472 CrossRef CAS.
- V. K. Velisoju, N. Gutta, S. Medak, A. Chatla, S. K. Bhargava, T. James, K. R. Vanga, H. P. Aytam and A. Venugopal, RSC Adv., 2016, 6, 9872–9879 RSC.
- T. Jingjing, C. Jinglei, D. Tiansheng, C. Xiaojing, D. Guoqiang, Y. Zhu and L. Yongwang, ChemCatChem, 2015, 7, 508–512 CrossRef.
- C. Michel, Z. Jérémie, M. R. Agnieszka, M. M. Joanna, J. Marcin, G. Jacek and S. Philippe, Chem. Commun., 2014, 50, 12450–12453 RSC.
- B. Erwin, J. Chem. Eng. Data, 1985, 30, 269–273 CrossRef.
- J. Wu, G. Gao, J. Li, P. Sun, X. Long and F. Li, Appl. Catal., B, 2017, 203, 227–236 CrossRef CAS.
- J. Zhang, J. Chen, Y. Guo and L. Chen, ACS Sustainable Chem. Eng., 2015, 3(8), 1708–1714 CrossRef CAS.
- B. Chen, F. Li, Z. Huang and G. Yuan, Appl. Catal., B, 2017, 200, 192–199 CrossRef CAS.
- B. Banerjee, R. Singuru, S. K. Kundu, K. Dhanalaxmi, L. Bai, Y. Zhao, B. M. Reddy, A. Bhaumik and J. Mondal, Catal. Sci. Technol., 2016, 6, 5102–5115 CAS.
- M. Wang, L. Wang, H. Li, W. Du, M. U. Khan, S. Zhao, C. Ma, Z. Li and J. Zeng, J. Am. Chem. Soc., 2015, 137, 14027–14030 CrossRef CAS PubMed.
- B. Zhao, G. Shao, B. Fan, W. Zhao and R. Zhang, RSC Adv., 2015, 5, 42587–42590 RSC.
- A. R. Naghash, T. H. Etsell and S. Xu, Chem. Mater., 2006, 18, 2480–2488 CrossRef CAS.
- B. Seemala, C. M. Cai, C. E. Wyman and P. Christopher, ACS Catal., 2017, 7, 4070–4082 CrossRef CAS.
- K. Yan, Y. Liu, Y. Lu, J. Chai and L. Sun, Catal. Sci. Technol., 2017, 7, 1622–1645 CAS.
- P. O. Larsson and A. Andersson, J. Catal., 1998, 179, 72–89 CrossRef CAS.
- A. Wolfbeisser, G. Kovács, S. M. Kozlov, K. Föttinger, J. Bernardi, B. Klötzer, K. M. Neyman and G. Rupprechter, Catal. Today, 2017, 283, 134–143 CrossRef CAS.
- S. Lomate, A. Sultana and T. Fujitani, Catal. Sci. Technol., 2017, 7, 3073–3083 CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- L. Liu, H. Lou and M. Chen, Int. J. Hydrogen Energy, 2016, 41, 14721–14731 CrossRef CAS.
- M. García-Méndez, F. F. Castillón, G. A. Hirata, M. H. Farías and G. Beamson, Appl. Surf. Sci., 2000, 161, 61–73 CrossRef.
- X. Chen, X. Wang, J. Xiu, C. T. Williams and C. Liang, J. Phys. Chem. C, 2012, 116, 24968–24976 CAS.
- M. Yang, H. Wu, H. Wu, C. Huang, W. Weng, M. Chena and H. Wan, RSC Adv., 2016, 6, 81237–81244 RSC.
- S. Catillon-Mucherie, F. Ammari, J. M. Krafft, H. Lauron-Pernot, T. Raymonde and L. Catherine, J. Phys. Chem. C, 2007, 111, 11619–11626 CAS.
- Q. Wu, W. L. Eriksen, L. D. L. Duchstein, J. M. Christensen, C. D. Damsgaard, J. B. Wagner, B. Temel, J. D. Grunwaldtae and A. D. Jensen, Catal. Sci. Technol., 2014, 4, 378–386 CAS.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- Y. Yang, G. Gao, X. Zhang and F. Li, ACS Catal., 2014, 4, 1419–1425 CrossRef CAS.
- I. Obregón, I. Gandarias, N. Miletić, A. Ocio and P. L. Arias, ChemSusChem, 2015, 8, 3483–3488 CrossRef PubMed.
- T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC.
- K. Yan, J. Liao, X. Wua and X. Xie, RSC Adv., 2013, 3, 3853–3856 RSC.
- J. M. Tukacs, R. V. Jones, F. Darvas, G. Dibó, G. Lezsáka and L. T. Mika, RSC Adv., 2013, 3, 16283–16287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization techniques, TEM images of the –NH2 functionalized SiO2 nanospheres, pyridine adsorption drift spectra, ICP-OES results, GVL yields as a function of various solvents, FT-IR, HAADF-STEM, FE-SEM images and the proposed mechanistic pathway for catalytic hydrogenation of LA to GVL. See DOI: 10.1039/c8se00138c |
| This journal is © The Royal Society of Chemistry 2018 |