
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven hydrogen evolution catalyzed by a cobaloxime catalyst incorporated in a MIL-101(Cr) metal–organic framework†
Souvik
Roy‡
 a,
Asamanjoy
Bhunia‡
a,
Nils
Schuth
b,
Michael
Haumann
b and
Sascha
Ott
*a
a,
Asamanjoy
Bhunia‡
a,
Nils
Schuth
b,
Michael
Haumann
b and
Sascha
Ott
*a
aUppsala University, Department of Chemistry – Ångström Laboratory, Box 523, 751 20 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
bFreie Universität Berlin, Department of Physics, 14195 Berlin, Germany
First published on 1st May 2018
Abstract
A cobaloxime H2 evolution catalyst with a hydroxo-functionalized pyridine ligand, Co(dmgH)2(4-HEP)Cl [dmgH = dimethylglyoxime, 4-HEP = 4-(2-hydroxyethyl)pyridine] was immobilized on a chromium terephthalate metal–organic framework (MOF), MIL-101(Cr), to construct a MOF-catalyst hybrid which displays good photocatalytic H2 evolution activity. The longevity of the cobaloxime catalyst is increased by MOF incorporation, but limited by the stability of the cobalt–pyridine bond under turnover conditions.
Increasing global energy demands combined with unprecedented levels of atmospheric CO2 caused by burning fossil fuels have spurred widespread interest in renewable energies, of which solar energy plays a prominent role.1,2 Reduction of water to hydrogen has been put forward as a promising ‘solar-to-fuel’ approach to store harvested sunlight. In this context, the development of inexpensive and efficient catalysts for the hydrogen evolution reaction (HER) is crucial for achieving a sustainable carbon-neutral energy economy. While many molecular catalysts for the photocatalytic HER have been developed in recent years,3 such studies are frequently performed in homogeneous solution phase. In contrast, future solar fuels devices will most likely require heterogenized catalysts for simple practical considerations. There is thus a need to immobilize molecular catalysts that are appealing, for example, due to their high activity per metal centre, into heterogeneous supports.4,5 Moreover, scalable and technologically robust solar-H2 evolving systems require the use of inexpensive, non-noble metal based catalysts.
Metal–organic frameworks (MOFs), a class of materials comprised of metal-cluster nodes connected by organic linkers,6,7 represent an excellent scaffold for incorporating single-site molecular catalysts because of their porous and crystalline nature, high surface area, and structural versatility.8 Immobilizing molecular species within MOF-supports allows catalyst recycling, and eliminates potentially destructive collisional pathways by site isolation of the catalytic species. Encapsulation of molecular catalysts within MOF cavities was used by different groups to incorporate cobalt catalysts9,10 and a [NiFe]-hydrogenase mimic11 inside MOFs, MIL-125(Ti)-NH2 and PCN-777, respectively. While these MOF-composites were robust and recyclable, such ‘ship-in-a-bottle’ type encapsulation strategy requires precise size-matching between the catalyst and MOF-pore-windows to prevent the catalysts from leaching out, and thus, limits the choice of MOFs and catalysts. Moreover, substrate and product transport may become limiting in such systems where the MOF pores are occupied by catalysts.12
Grafting molecular catalysts into MOFs via covalent or coordination bonds offers a more versatile route towards MOF-catalysts. Surprisingly, only few such examples with HER catalysts have been reported to date, most of them based on [FeFe]-hydrogenase (H2ase) mimics.12–16 While such species offer IR spectroscopic handles for probing oxidation and protonation states of the catalysts inside the MOF and are appealing for mechanistic studies, [FeFe] H2ase models are limited as HER catalysts.17,18 For the present study, we therefore chose cobaloximes as HER catalyst, as they are among the most active molecular catalysts when considering activity, overpotential requirement, and synthesis costs.19,20
Herein, we report the first example of anchoring a cobaloxime catalyst, Co(dmgH)2(4-HEP)Cl (dmgH = dimethylglyoxime, 4-HEP = 4-(2-hydroxyethyl)pyridine) (compound 1, Scheme 1), into a MIL-101(Cr) via a coordination linkage. The resulting material, MIL-101-CH2@1, was fully characterized and was shown to be an efficient photocatalyst with a high rate for H2 evolution.
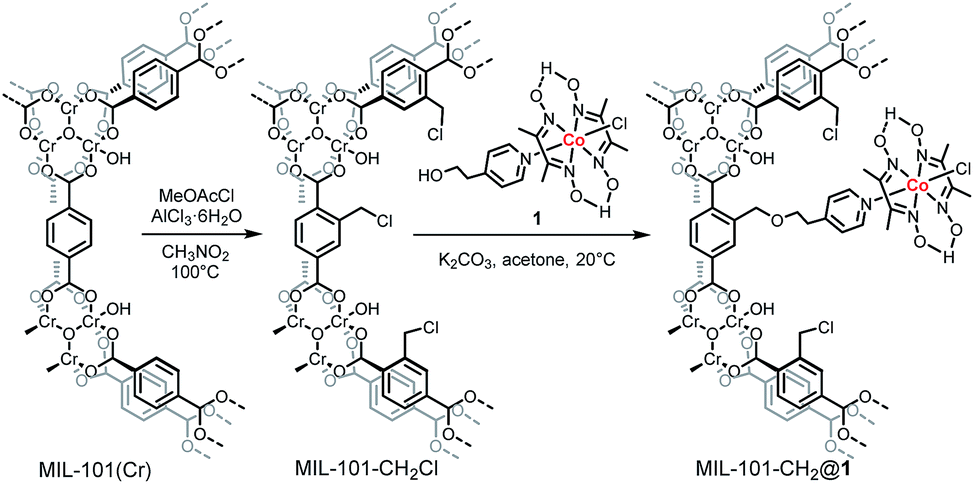 | ||
| Scheme 1 Schematic illustration of the post-synthetic chloromethylation on the aromatic rings of MIL-101(Cr) and incorporation of the molecular cobaloxime catalyst. | ||
The chromium based MIL-101 framework was selected as the scaffold due to its exceptional thermal and chemical stability.21–23 MIL-101(Cr) [Cr3(μ3-O)(OH)(BDC)3(H2O)2]·∼25H2O; (BDC = benzene-1,4-dicarboxylate), contains large inner cages of 2.9 and 3.4 nm diameters with pore aperture window diameters of 1.2 and 1.6 nm, respectively,21 and its BDC linkers can easily be functionalized via post-synthetic modifications (PSM). Thus, MIL-101(Cr) was functionalized with chloromethylene groups at the BDC linkers to provide anchoring points for catalysts incorporation. This transformation is easily done by the reaction of MIL-101(Cr) with methoxyacetyl chloride and aluminium chloride to give MIL-101-CH2Cl.24 The catalyst was grafted onto the MOF by the reaction of MIL-101-CH2Cl and 1 in the presence of K2CO3 in acetone to yield MIL-101-CH2@1 (Scheme 1). The ICP-AES of acid digested MIL-101-CH2@1 shows a Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr ratio of 1:8, which corresponds to 0.36 μmol [Co] per mg MOF (Table S1†). The cobaloxime loading within MIL-101(Cr) can easily be controlled by varying the reaction conditions, and a second MOF-catalyst with lower cobaloxime loading was also synthesized (0.07 μmol Co per mg MOF, denoted as MIL-101-CH2@1low). These catalyst loadings correspond to an average occupancy of 5 and 1 cobaloxime units in each of the large diameter pores of MIL-101-CH2@1 and MIL-101-CH2@1low, respectively, and 3 and 0.7 for the smaller pores, assuming an idealized statistical distribution throughout the MIL-101(Cr) framework (see ESI†). The powder X-ray diffraction analyses of the three materials, MIL-101(Cr), MIL-101-CH2Cl, and MIL-101-CH2@1, demonstrate that the underlying crystallinity of the MIL-101 framework is maintained during the PSM steps (Fig. 1a). Also, the scanning electron microscope (SEM) images for MIL-101(Cr), MIL-101-CH2Cl, and MIL-101-CH2@1 show that the PSM steps do not affect the MOF morphology and the cubic shape of the micro-crystals remain intact in the composites (Fig. S1†). As expected, the BET surface areas of the MOFs, measured by N2-sorption, decreased from 3160 m2 g−1 to 2272 m2 g−1 after chloromethylation, and further to 1512 m2 g−1 following immobilization of the cobaloxime units in the high [Co] loading material (Fig. S2†). The introduced cobaloximes are uniformly distributed throughout the MIL-101-CH2@1 micro-crystals as confirmed by scanning electron microscopy-energy dispersive X-ray (SEM-EDX) elemental mapping (Fig. 2b and S3†). This surface sensitive technique gave a Co:Cr ratio of 1:7 (±0.5) which is in good agreement with the ICP-AES analysis of the bulk material.
:Cr ratio of 1:8, which corresponds to 0.36 μmol [Co] per mg MOF (Table S1†). The cobaloxime loading within MIL-101(Cr) can easily be controlled by varying the reaction conditions, and a second MOF-catalyst with lower cobaloxime loading was also synthesized (0.07 μmol Co per mg MOF, denoted as MIL-101-CH2@1low). These catalyst loadings correspond to an average occupancy of 5 and 1 cobaloxime units in each of the large diameter pores of MIL-101-CH2@1 and MIL-101-CH2@1low, respectively, and 3 and 0.7 for the smaller pores, assuming an idealized statistical distribution throughout the MIL-101(Cr) framework (see ESI†). The powder X-ray diffraction analyses of the three materials, MIL-101(Cr), MIL-101-CH2Cl, and MIL-101-CH2@1, demonstrate that the underlying crystallinity of the MIL-101 framework is maintained during the PSM steps (Fig. 1a). Also, the scanning electron microscope (SEM) images for MIL-101(Cr), MIL-101-CH2Cl, and MIL-101-CH2@1 show that the PSM steps do not affect the MOF morphology and the cubic shape of the micro-crystals remain intact in the composites (Fig. S1†). As expected, the BET surface areas of the MOFs, measured by N2-sorption, decreased from 3160 m2 g−1 to 2272 m2 g−1 after chloromethylation, and further to 1512 m2 g−1 following immobilization of the cobaloxime units in the high [Co] loading material (Fig. S2†). The introduced cobaloximes are uniformly distributed throughout the MIL-101-CH2@1 micro-crystals as confirmed by scanning electron microscopy-energy dispersive X-ray (SEM-EDX) elemental mapping (Fig. 2b and S3†). This surface sensitive technique gave a Co:Cr ratio of 1:7 (±0.5) which is in good agreement with the ICP-AES analysis of the bulk material.
 | ||
| Fig. 1 (a) PXRD of MIL-101(Cr), MIL-101-CH2Cl and MIL-101(Cr)-CH2@1; (b) SEM image of MIL-101-CH2@1 and corresponding color-coded EDX elemental analysis maps (labeled images). Each scale bar represents 1 μm. | ||
X-ray absorption spectroscopy at the Co K-edge was used to assess the structural integrity and redox level of the cobalt centers in powder samples of the materials (Fig. 2). Both molecular and MOF-catalyst (1 and MIL-101-CH2@1) show a K-edge energy in the XANES (extended X-ray absorption near edge structure) which is consistent with predominantly Co(III) centers. While the overall edge shape of 1 and MIL-101-CH2@1 indicates near-octahedral Co(III) centers, the difference in XANES edge shapes indicates a change of ligation state at cobalt upon immobilization of 1 into the MOF. The primary coordination sphere around cobalt was verified using EXAFS (extended X-ray absorption fine structure) analysis (Fig. 2, Tables 1 and S2†). The cobalt–ligand bond lengths in 1 determined from EXAFS are in good agreement with the crystal structure of the analogous cobaloxime complex [Co(dmgH)2(pyr)Cl],25 and a near-quantitative presence of the axial chloride ligand was observed. Upon grafting 1 into MIL-101(Cr), however, the chloride ligand is barely detectable. Instead, a coordinated solvent molecule or hydroxo ligand could be detected with a Co–O bond length of ∼1.95 Å.26 Such a ligand exchange can be expected due to the lability of the Co(III)–Cl bond under basic conditions (K2CO3 in acetone).
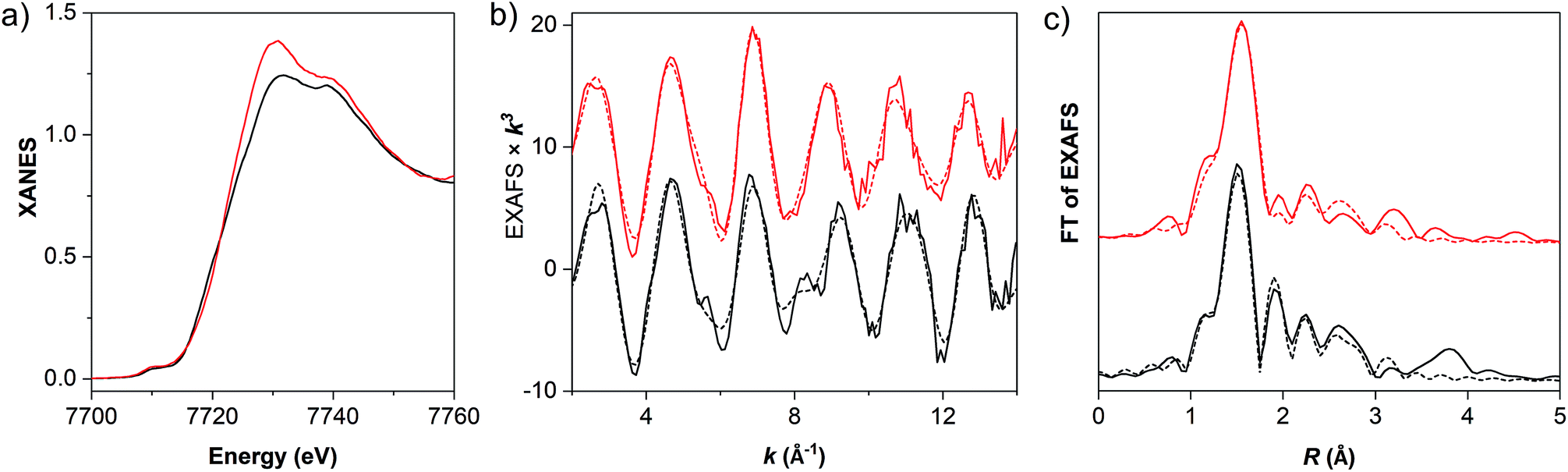 | ||
| Fig. 2 (a) Normalized Co K-edge spectra, (b) k-weighted EXAFS oscillations χ(k)k3, and (c) Fourier-transforms of EXAFS oscillations in R space for MIL-101-CH2@1 (red trace) and the molecular cobaloxime (Co(dmgH)2(4-HEP)Cl (1), black trace). In (b) and (c), solid and dashed lines show experimental and simulated data, respectively, with the parameters listed in Tables 1 and S1.† | ||
| Bond length (Å) | 1 | MIL-101-CH2@1a | Co(dmgH)2PyClb |
|---|---|---|---|
| a Bond lengths from EXAFS analysis. b Experimental bond-lengths obtained from single crystal X-ray analysis. Number of atoms around the cobalt centre is given in parenthesis. | |||
| Co–N (dmgH) | 1.90 (4) | 1.92 (4) | 1.895 (4) |
| Co–N (py) | 1.83 (1) | 1.82 (1) | 1.959 (1) |
| Co–Cl | 2.24 (0.8) | — | 2.229 (1) |
| Co–O | 2.42 (0.2) | 1.95 (1.2) | — |
The photocatalytic HER activity of MIL-101-CH2@1 was investigated using an organic photosensitizer, Eosin-Y (EY, a divalent anion in aqueous solution), that has previously been shown to be effective for photocatalytic hydrogen evolution using cobaloximes.27,28 In a typical experiment, MIL-101-CH2@1 was suspended in a 1:1 water-acetonitrile mixture containing EY (0.05 mM) and triethanolamine (5% v/v), buffered at pH 7. The suspension was irradiated with visible light (455–850 nm) and the composition of the headspace gas was monitored by gas chromatography.
The photocatalytic performances of the two materials with different Co loadings (MIL-101-CH2@1 and MIL-101-CH2@1low with 0.36 and 0.07 μmol Co per mg MOF, respectively) are shown in Fig. 3a. MIL-101-CH2@1 remains active for approximately 10–15 h with an initial H2 evolution rate of 1.5 mmol h−1 g−1 MOF (over 10 h). For MIL-101-CH2@1low, H2 evolves at a slower rate (0.7 mmol h−1 g−1 MOF) for a shorter time period (∼2 h) before gradually ceasing in ∼5 h. The total amount of evolved H2 corresponds to ∼45 (20 h) and ∼34 (10 h) turnovers vs. [Co] for MIL-101-CH2@1 and MIL-101-CH2@1low, respectively. For comparison, irradiation of a homogeneous solution of a cobaloxime reference catalyst [Co(dmgH)2(4-methylpyridine)Cl] and EY under similar conditions (2.5 μmol cobaloxime and 0.6 μmol EY) evolved ∼24.9 μmol H2 after 4 h (TON = 10) which is significantly lower than the TONs obtained for both MIL-101-CH2@1 materials after identical irradiation times (∼20 and ∼30 turnovers, Fig. 3a inset). Notably, when the rate of H2 formation is normalized to total catalyst content (Fig. 3a inset), MIL-101-CH2@1low evolves H2 at a faster rate compared to that in the high-loading material, which is consistent with the five times higher photosensitizer/catalyst ratio in MIL-101-CH2@1low experiments ([EY]:[Co] = 5:4) compared to those in MIL-101-CH2@1 and homogeneous system ([EY]:[Co] = 1:4). Control experiments show that cobaloxime catalyst, EY, TEOA, and light are all necessary components for hydrogen evolution. Pristine MIL-101 and the chloromethylated framework (MIL-101-CH2Cl) show no activity towards H2 generation under these conditions.
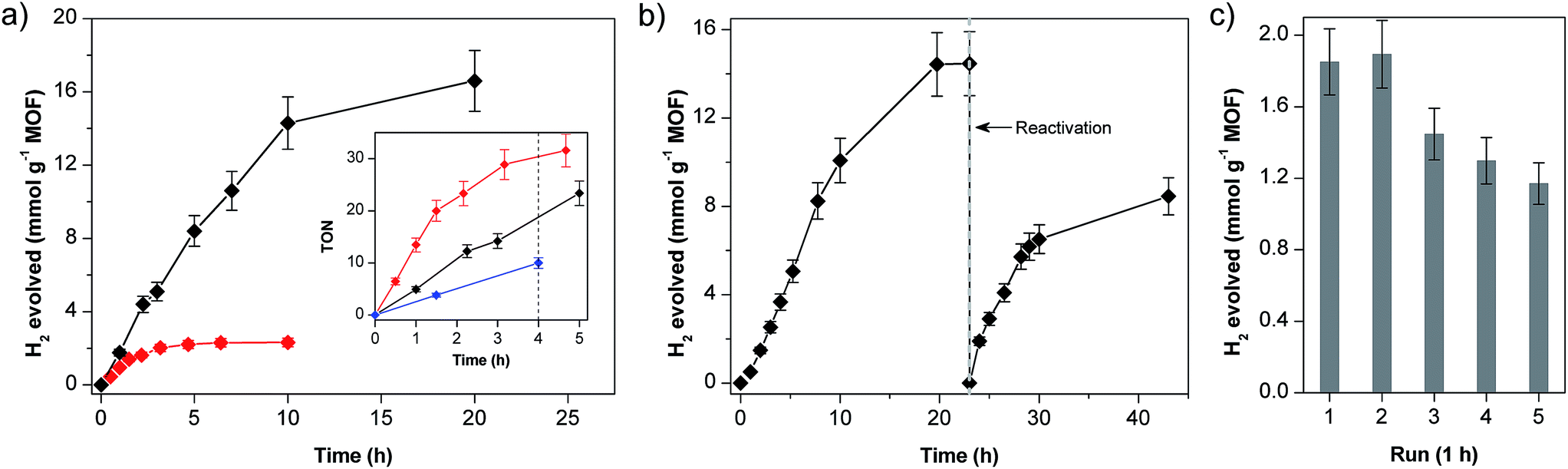 | ||
| Fig. 3 (a) Photocatalytic proton reduction using MIL-101-CH2@1 (black) and MIL-101-CH2@1low (red); inset: comparison of the corresponding catalyst turnover numbers (TON) for MIL-101-CH2@1 materials (black and red traces for high and low [Co]-loading, respectively) and that for the molecular catalyst in homogeneous solution (blue trace). (b) Reactivation of MIL-101-CH2@1 by treating the inactive MOF with Co(dmgH)2Cl2. (c) Recycling tests on MIL-101-CH2@1. Conditions for photocatalysis: 0.05 mM EY, 1:1 water/acetonitrile, 5% (v/v) TEOA, pH 7. | ||
ICP-AES analysis of the MOF-catalyst after photocatalysis shows that more than 90% of the initially present cobalt has been lost from the material. This finding is in line with earlier reports that have questioned the stability of the axial cobalt-pyridine coordination bond in the Co(I) oxidation state.29–33 Decoordination of the cobaloxime from the labile pyridine ligand during catalytic cycle and subsequent degradation of the catalyst to Co2+ ions limits the longevity of the MOF-catalyst under photocatalytic conditions. To confirm the dissociation of the Co–pyridine linkage, the inactive MOF-catalyst isolated after photocatalysis was treated with freshly prepared cobaloxime, [Co(dmgH)(dmgH2)Cl2], to reconstitute the catalyst unit [Co(dmgH)2pyCl] on MIL-101. The obtained material was washed thoroughly to remove any cobaloxime that is not coordinated to any pyridine. Gratifyingly, catalytic activity of the ‘repaired’ MOF-catalyst was restored to approximately 60% of that of the starting material (Fig. 3b).
The obtained results are consistent with the hypothesis that cobaloxime species, [CoI(dmgH)2] and [CoIII(dmgH)2(H)], that are detached from the pyridine groups during the photocatalytic cycle remain in close proximity of the pyridine anchors inside the MOF-pores. Restoration of the Co–pyridine coordination bond during catalysis is therefore far more likely to occur inside the MOF-pores compared to the situation in the homogeneous system. Consequently, the heterogeneous MOF-catalyst displays enhanced HER activity due to the presence of axial N-ligand.34 Eventual deactivation of the system likely occurs through diffusion of detached [Co(dmgH)2] units out of the MOF-cages, followed by loss of the glyoxime ligands from cobalt.29 In MIL-101-CH2@1low, the majority of the cobaloxime units may preferentially reside at the exterior of the MOF-crystals, as has been found for related PSM reactions in other MOFs.35,36 Such a localization would be consistent with the shorter lifetime of MIL-101-CH2@1low during photocatalysis (∼2 h) as the [Co(dmgH)2] intermediates would be more prone to diffuse from the MOF-crystals to the bulk solution.
To ensure that the hydrogen evolution is not catalysed by cobaloxime units (Co(dmgH)2L2; L = Cl or solvent) that leached from MIL-101 cages, photocatalytic HER of the supernatant solution isolated after 1 h of irradiation was tested and showed negligible activity. Moreover, the MIL-101-CH2@1 maintained activity during the recycling experiments up to five runs (Fig. 3c) consistent with heterogeneous catalysis. This data is consistent with our hypothesis that photocatalysis occurs inside the MOF-pores and that the axial pyridines detach and reattach to the Co centers. Powder XRD profile and SEM analyses of the MOF-material isolated after photocatalysis demonstrate that the MOF retains crystallinity and structural integrity, highlighting the stability of MIL-101 under photocatalytic conditions (Fig. S4 and S5†).
In summary, we have developed a strategy for using chloromethylated MIL-101(Cr) framework as a support for immobilizing cobaloxime catalysts. Importantly, the PSM strategy employed here does not rely on any structural resemblance between the catalyst and linker, and is thus applicable to a wide range of MOFs and catalysts. While the hybrid MOF-catalyst (MIL-101-CH2@1) catalyses photochemical hydrogen evolution at a decent rate, the limited stability of the axial cobalt–pyridine bond under turnover conditions compromises sustained catalysis. Nevertheless, the observed rate of H2 evolution ranks MIL-101-CH2@1 (1.5 mmol H2 h−1 g1 MOF over initial 10 h) among the most active MOF-based photocatalysts for HER (Table S3†). In comparison, the two previously reported hybrid MIL-125-NH2(Ti) materials with encapsulated cobalt catalysts evolved H2 at a much slower rate (0.38 and 0.55 mmol H2 h−1 g−1 MOF), but they show prolonged activity over significantly longer time period.9,10 Future developments, for example by a more robust covalent linkage of cobaloxime catalysts will most likely increase the longevity of the MOF/catalyst hybrid material even further.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Wenner-Gren Foundations (postdoctoral stipend to SR), the Swedish Research Council, the Swedish Energy Agency, and the European Research Council (ERC-CoG2015-681895_MOFcat) is gratefully acknowledged. M. H. thanks the Bundesministerium für Bildung und Forschung (BMBF) for funding within the Röntgen-Angström Cluster (grant 05K14KE1), and A. B. the DST-INSPIRE program for an INSPIRE Faculty Fellowship (DST/INSPIRE/04/2017/001072).Notes and references
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- A. Thapper, S. Styring, G. Saracco, A. W. Rutherford, B. Robert, A. Magnuson, W. Lubitz, A. Llobet, P. Kurz, A. Holzwarth, S. Fiechter, H. de Groot, S. Campagna, A. Braun, H. Bercegol and V. Artero, Green, 2013, 3, 43 CrossRef.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- G. Caserta, S. Roy, M. Atta, V. Artero and M. Fontecave, Curr. Opin. Chem. Biol., 2015, 25, 36–47 CrossRef CAS PubMed.
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 CAS.
- Z. Li, J.-D. Xiao and H.-L. Jiang, ACS Catal., 2016, 6, 5359–5365 CrossRef CAS.
- D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò and M. Gennari, Inorg. Chem., 2017, 56, 14801–14808 CrossRef CAS PubMed.
- S. Roy, V. Pascanu, S. Pullen, G. Gonzalez Miera, B. Martin-Matute and S. Ott, Chem. Commun., 2017, 53, 3257–3260 RSC.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Chem. Commun., 2014, 50, 10390–10393 RSC.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- S. Pullen, S. Roy and S. Ott, Chem. Commun., 2017, 53, 5227–5230 RSC.
- M. Watanabe, Y. Honda, H. Hagiwara and T. Ishihara, J. Photochem. Photobiol., C, 2017, 33, 1–26 CrossRef CAS.
- F. Wang, W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, ACS Catal., 2012, 2, 407–416 CrossRef CAS.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- B. Li, Y. Zhang, D. Ma, L. Li, G. Li, G. Li, Z. Shi and S. Feng, Chem. Commun., 2012, 48, 6151–6153 RSC.
- S. Bernt, V. Guillerm, C. Serre and N. Stock, Chem. Commun., 2011, 47, 2838–2840 RSC.
- M. G. Goesten, K. B. Sai Sankar Gupta, E. V. Ramos-Fernandez, H. Khajavi, J. Gascon and F. Kapteijn, CrystEngComm, 2012, 14, 4109–4111 RSC.
- S. Geremia, R. Dreos, L. Randaccio, G. Tauzher and L. Antolini, Inorg. Chim. Acta, 1994, 216, 125–129 CrossRef CAS.
- Assigning hydroxo ligand by EXAFS is tentative and it could also be nitrogen or even carbon. However, under the reaction conditions coordination of an hydroxo ligand is most likley.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632–3638 CrossRef CAS PubMed.
- T. M. McCormick, Z. Han, D. J. Weinberg, W. W. Brennessel, P. L. Holland and R. Eisenberg, Inorg. Chem., 2011, 50, 10660–10666 CrossRef CAS PubMed.
- B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917–1928 CAS.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC.
- J. Willkomm, N. M. Muresan and E. Reisner, Chem. Sci., 2015, 6, 2727–2736 RSC.
- D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS PubMed.
- C. Liu, C. Zeng, T.-Y. Luo, A. D. Merg, R. Jin and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 12045–12048 CrossRef CAS PubMed.
- W. A. Maza, R. Padilla and A. J. Morris, J. Am. Chem. Soc., 2015, 137, 8161–8168 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization of the MOF and the cobaloxime complex, and supplementary figures. See DOI: 10.1039/c8se00072g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |