
ZIF-derived CoP as a cocatalyst for enhanced photocatalytic H2 production activity of g-C3N4†
Xiao-Jun
Sun
*a,
Dou-Dou
Yang
a,
Hong
Dong
 ab,
Xiang-Bin
Meng
a,
Jing-Li
Sheng
a,
Xin
Zhang
a,
Jin-Zhi
Wei
a and
Feng-Ming
Zhang
*a
ab,
Xiang-Bin
Meng
a,
Jing-Li
Sheng
a,
Xin
Zhang
a,
Jin-Zhi
Wei
a and
Feng-Ming
Zhang
*a
aKey Laboratory of Green Chemical Engineering and Technology of College of Heilongjiang Province, College of Chemical and Environmental Engineering, Harbin University of Science and Technology, Harbin 150040, P. R. China. E-mail: zhangfm80@163.com; Fax: +86 451 8639 2713
bMinistry of Education, Key Laboratory of Engineer Dielectrics and Its Application, College of Material Science and Engineering, Harbin University of Science and Technology, Harbin 150040, P. R. China
First published on 24th April 2018
Abstract
The graphitic carbon nitride (g-C3N4) photocatalyst has drawn widespread attention owing to its excellent photocatalytic hydrogen production activity. For the first time, an efficient g-C3N4-based non-precious-metal photocatalyst, CoP/g-C3N4, with ZIF-67-derived CoP nanoparticles as cocatalysts was prepared by a two-step calcination under different atmospheres. It demonstrates that CoP nanoparticles derived from ZIF-67 can extensively improve the photocatalytic activity of g-C3N4. When the amount of CoP in the photocatalyst was calculated to be 1.42 wt%, a high photocatalytic hydrogen production rate of 201.5 μmol g−1 h−1 was obtained, which was almost 23 times higher than that of bulk g-C3N4 and also surpassed that of bulk g-C3N4 with Pt as the cocatalyst. In addition, electrochemical experiments and photoluminescence spectra indicate that the CoP/g-C3N4 photocatalysts exhibit the advantage of a low recombination rate of photogenerated electrons and holes, resulting in a high rate of electron transport.
Introduction
With the increasing need for energy, developing renewable energy sources has been given the most urgent attention. Hydrogen is considered to be a substitute for fossil energy due to the highest energy content per unit mass of chemical fuel, and photocatalytic water splitting is the best route to utilize solar energy.1–7 The application of photocatalysts in photocatalytic reactions can improve the efficiency of hydrogen production visibly.8,9 During the past 20 years, g-C3N4, as a metal-free and visible-light-responsive photocatalyst, has stood out from various photocatalysts.10–12 It shows great potential in the area of photocatalytic hydrogen generation.13–16 Particularly, it exhibits high thermal stability, high chemical stability, and non-toxicity, and has abundant resources and a simple preparation process.17–20 Despite the advantages mentioned above, g-C3N4 is limited by a fatal defect, which is the high recombination rate of excited charges.21 The design and modification of g-C3N4 to facilitate hydrogen production remains to be fully explored.To further improve its properties, combining g-C3N4 with other semiconductors or compounds as cocatalysts is an efficient strategy.22,23 The main function of cocatalysts is to promote the transfer of photo-generated electrons. Appropriate cocatalysts guarantee a decreased recombination rate of excited charges and an improved photocatalytic hydrogen generation.24 In the process of photocatalysis, precious metal species such as Ag, Pt and Au are usually used in most cases as cocatalysts to promote the transfer of photoinduced electrons although precious metals are rare and expensive.25,26 Thus, it is necessary to develop non-precious metal cocatalysts. Additionally, Co2+ can be an efficient cocatalyst for photocatalytic hydrogen production. Developed cobalt-based systems, such as Co–Pi, CoP, and CoOOH, are reported to be suitable catalysts.27–31
Metal–organic frameworks (MOFs), as a class of porous and crystalline materials assembled from metal ions/clusters as nodes and organic linkers as struts, have attracted widespread research interest because of their versatile functionalities and attractive properties.32–36 The advantages mentioned above endow MOFs with the ability to be excellent precursors and templates of porous carbons37 and metal composites.38,39 These MOF-derived nanostructures exhibit controlled porosity which may facilitate the access of water molecules to catalysts, easy tuning of the chemical composition and remarkable shortening of charge diffusion lengths.40 Notably, CoP derived from ZIF-67, which is one kind of MOF, can supply catalytically active species. Besides, the interaction of g-C3N4 and CoP will contribute to faster electron transfer in photocatalysis. However, there is no report on photocatalytic hydrogen evolution with ZIF-67-derived CoP particles to increase the activity of g-C3N4.
In order to improve the photocatalytic hydrogen generation efficiency of g-C3N4 in the absence of precious-metal species, for the first time, we designed a modified g-C3N4-based photocatalyst, CoP/g-C3N4, with a hierarchical morphology and significantly enhanced light-driven H2 production from H2O through a calcination method using ZIF-67, melamine and sodium hypophosphite as the precursor. Details of the preparation of CoP/g-C3N4 are illustrated in Scheme 1. It is worth noting the following advantages of this strategy. First, using ZIF-67 as the precursor endowed the photocatalysts with small quantities of catalytically active species, which are significant for electron transfer. Furthermore, the solar spectral region can be broadened. Besides, an exceptionally long-lived excited state can be generated due to the formation of a charge-separated state.41 Therefore, we believe that g-C3N4-based non-precious-metal photocatalysts with ZIF-67-derived CoP particles as cocatalysts are fascinating and worth exploring.
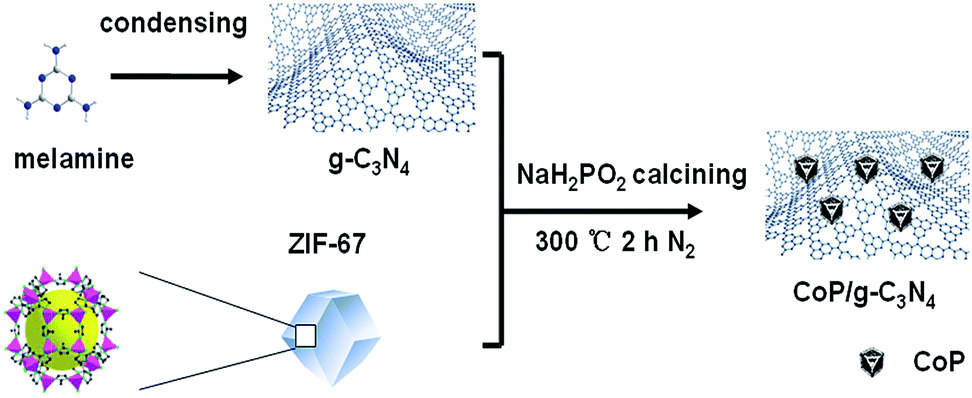 | ||
| Scheme 1 Schematic representation of the synthesis processes of CoP/g-C3N4. | ||
Experimental section
To synthesize the ZIF-67 material, cobalt nitrate hexahydrate, 2-methylimidazole (2-Hmim), and methanol were mixed in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:1100 (Co/2-Hmim/methanol). The mixture was subjected to vigorous agitation with ultrasonic dispersion for 1 h at room temperature. Then, ZIF-67 was obtained from the purple colloidal solution by centrifugation. The dispersions were centrifuged at 8000 rpm for 5 min to obtain ZIF-67, washed with methanol at least 5 times to remove excess soluble ions, and dried in a drying cabinet at 60 °C overnight.
:40:1100 (Co/2-Hmim/methanol). The mixture was subjected to vigorous agitation with ultrasonic dispersion for 1 h at room temperature. Then, ZIF-67 was obtained from the purple colloidal solution by centrifugation. The dispersions were centrifuged at 8000 rpm for 5 min to obtain ZIF-67, washed with methanol at least 5 times to remove excess soluble ions, and dried in a drying cabinet at 60 °C overnight.
Bulk g-C3N4 can be simply prepared by the thermal condensation of melamine. In a typical fabrication, melamine in a crucible was heated at 550 °C for 4 h with a heating ramp rate of 5 °C min−1 and cooling to room temperature. Thus, the light-yellow g-C3N4 was obtained. To prepare the CoP/g-C3N4, ZIF-67 and g-C3N4 were milled in a mortar for 15 min (the mass percentages of ZIF-67 in the precursors were 3 wt%, 5 wt%, and 7 wt%). Then, the mixture of ZIF-67 and g-C3N4 was placed in a quartz boat in a pipe furnace with the solid phase of NaH2PO2 put in a different quartz boat upstream. The precursor was directly heated to 300 °C and kept for 2 h in a 60 mL min−1 flow of N2. The product was put into a mortar and ground into fine powder for further use. The composites with 3 wt%, 5 wt%, and 7 wt% of ZIF-67 mixed in the precursors were synthesized and named CoP/g-C3N4-3, CoP/g-C3N4-5, and CoP/g-C3N4-7.
To evaluate the effect of the photocatalytic activity of Co2+, a Ni2+ doped ZIF-67 material was prepared and heated to obtain Ni2+ doped CoP/g-C3N4. The samples without g-C3N4, NaH2PO2 and ZIF-67 in the precursors were obtained as the contrast group for the control experiments as well.
Results and discussion
Characterization of the catalysts
From the SEM images, XRD patterns and FTIR spectra (Fig. S1–S3†), it is evident that ZIF-67 has been synthesized successfully. The XRD patterns (Fig. S4†) of g-C3N4 feature two pronounced diffraction peaks at 27.4° (002) and 13.0° (100).42 The first one can be attributed to interlayer stacking of aromatic systems, and the second one can be assigned to the peak of the interplanar separation, indicating the formation of g-C3N4.42 This is consistent with the observation from FTIR spectra (Fig. S5†). The characteristic stretching modes of CN heterocycles in g-C3N4 are between 1200 and 1700 cm−1. Besides, the absorption band at 890 cm−1 corresponds to the deformation mode of N–H, and the bands in the range of 3000–3500 cm−1 correspond to the residual N–H components and the O–H bands.43 Furthermore, the TG curve shows an initial decomposition temperature of approximately 550 °C (Fig. S6†), indicating the excellent thermal stability of g-C3N4, which is beneficial to preserving the basic structural unit of g-C3N4 during calcination.In Fig. 1a, the XRD patterns of the CoP/g-C3N4 photocatalysts exhibit characteristics essentially similar to those of the g-C3N4 sample. On the one hand, the g-C3N4 and CoP/g-C3N4 photocatalysts exhibit similar sharp diffraction peaks in their XRD patterns. But the clear decrease of the main peak located at 27.4° can be attributed to the low quantity of CoP introduced into the photocatalysts.44,45 These results demonstrate that the basic structural unit of g-C3N4 did not change during the calcination. On the other hand, the magnified XRD pattern in Fig. 1b shows that CoP particles exist, corresponding to the XRD pattern of the photocatalyst without g-C3N4 in the precursors in Fig. 1c. Peaks at 31.6°, 36.3°, 48.1° and 56.7°, attributed to CoP from PDF cards, can be observed clearly. In the FTIR spectra in Fig. 1d, the characteristic peaks of g-C3N4 can also be observed in CoP/g-C3N4. The presence of small amounts of Co and P in the CoP/g-C3N4 photocatalysts is further indicated by the XPS and EDS results described below.
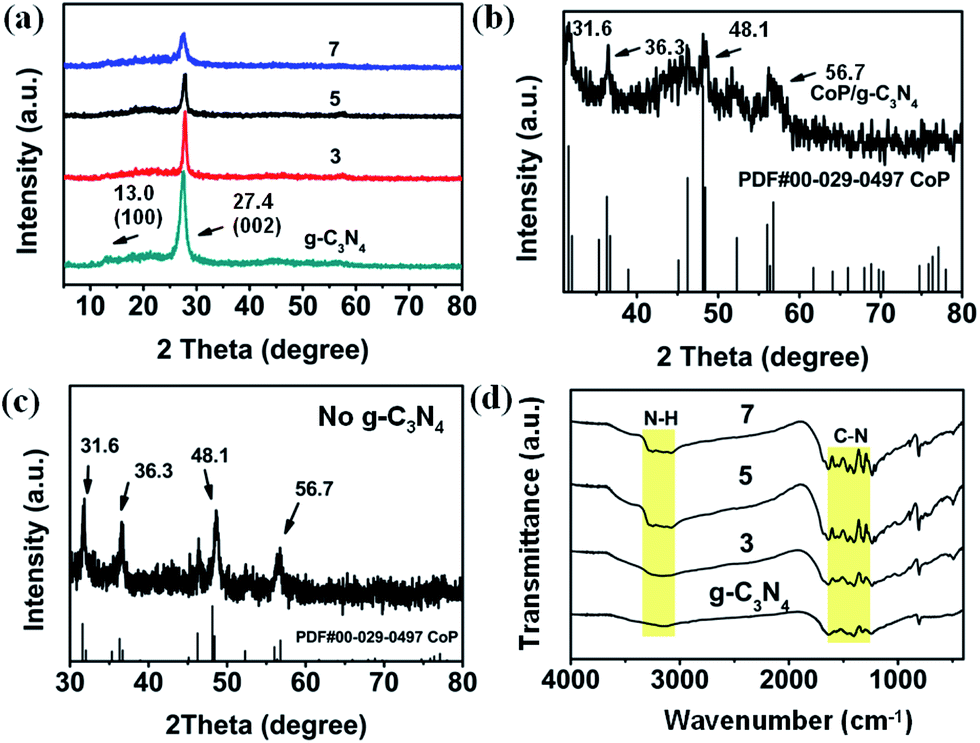 | ||
| Fig. 1 (a) XRD patterns of g-C3N4, CoP/g-C3N4-3, CoP/g-C3N4-5 and CoP/g-C3N4-7 (CoP/g-C3N4-3, CoP/g-C3N4-5 and CoP/g-C3N4-7 correspond to 3 wt%, 5 wt%, and 7 wt% of ZIF-67 mixed in the precursors of the resulting photocatalysts). (b) The magnified XRD pattern of CoP/g-C3N4. (c) XRD pattern of the CoP photocatalyst without g-C3N4 in the precursors. (d) FTIR spectra of g-C3N4, CoP/g-C3N4-3, CoP/g-C3N4-5 and CoP/g-C3N4-7. | ||
A thorough inquiry was made into the micromorphology of CoP/g-C3N4. As shown in Fig. 2a, the as-prepared g-C3N4 formed thin layers structure. The TEM image in Fig. 2b confirms that the CoP/g-C3N4 sample contains particles 100 nm in diameter embedded in channels and between layers which can be attributed to CoP. Additionally, the lattice spacing in Fig. 2c is 0.188 nm, corresponding to the (211) lattice plane of CoP. Furthermore, the corresponding selected area electron diffraction pattern exhibits a series of concentric circles, attributed to the (211), (202), (103) and (020) lattice planes of CoP (Fig. 2d). In addition, the SEM image and the corresponding EDS elemental mapping images confirm the successful introduction of Co and P into the resulting CoP/g-C3N4 (Fig. 2e). Simultaneously, the existence of CoP indicates that ZIF-67 reacted with NaH2PO2 as expected. The as-synthesized CoP/g-C3N4 exhibits an aggregated, lamellar morphology with clusters due to random doping after calcination. It can also be concluded that CoP particles exist on the g-C3N4 substrate with an average size of 100 nm. Additionally, when the amount of ZIF-67 in the precursor was 5 wt%, the amount of CoP in the as-prepared photocatalyst was calculated to be 1.42 wt% (Fig. S7†).
 | ||
| Fig. 2 (a) TEM image of g-C3N4. (b and c) TEM image of CoP/g-C3N4. (d) The corresponding selected area electron diffraction pattern of CoP/g-C3N4. (e) SEM image of CoP/g-C3N4 and the corresponding EDS elemental mapping images. | ||
To shed more light on the chemical composition and bonding states of CoP/g-C3N4, an X-ray photoelectron spectroscopy (XPS) experiment was conducted. The C 1s spectrum exhibits three peaks at 284.8, 285.96 and 288.32 eV, corresponding to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and N–CN, respectively (Fig. 3a).46 The N 1s core level at 398.8 eV corresponds to bicoordinated nitrogen atoms in CN–C. The peak centered at 400.53 eV corresponds to tricoordinated nitrogen atoms (Fig. 3b). The peak at 401.3 eV is attributed to amino functional groups in C-NHx.47 The Co 2p window and the P 2p window demonstrate that Co and P exist, respectively (Fig. 3c and d). The Co 2p spectrum further validates the Co2+ electronic state. The peaks at 780.5 and 796.7 eV in the Co 2p window are identified as the major binding energies of Co2+.48 The P 2p binding energy peaks at 133.6 and 134.4 eV are typical for P–N and P–O bonds. The P–N coordination indicates that a portion of P is doped into the skeleton of g-C3N4.49 Due to the low content of CoP, the peaks of P–Co at 129.5 eV are not clear enough. Considering all the results of XRD, FTIR, TEM, SEM, EDX and XPS, we suggest that the original graphitic structure of polymeric g-C3N4 is essentially retained.50 Meanwhile, CoP particles as cocatalysts were successfully synthesized.
N and N–CN, respectively (Fig. 3a).46 The N 1s core level at 398.8 eV corresponds to bicoordinated nitrogen atoms in CN–C. The peak centered at 400.53 eV corresponds to tricoordinated nitrogen atoms (Fig. 3b). The peak at 401.3 eV is attributed to amino functional groups in C-NHx.47 The Co 2p window and the P 2p window demonstrate that Co and P exist, respectively (Fig. 3c and d). The Co 2p spectrum further validates the Co2+ electronic state. The peaks at 780.5 and 796.7 eV in the Co 2p window are identified as the major binding energies of Co2+.48 The P 2p binding energy peaks at 133.6 and 134.4 eV are typical for P–N and P–O bonds. The P–N coordination indicates that a portion of P is doped into the skeleton of g-C3N4.49 Due to the low content of CoP, the peaks of P–Co at 129.5 eV are not clear enough. Considering all the results of XRD, FTIR, TEM, SEM, EDX and XPS, we suggest that the original graphitic structure of polymeric g-C3N4 is essentially retained.50 Meanwhile, CoP particles as cocatalysts were successfully synthesized.
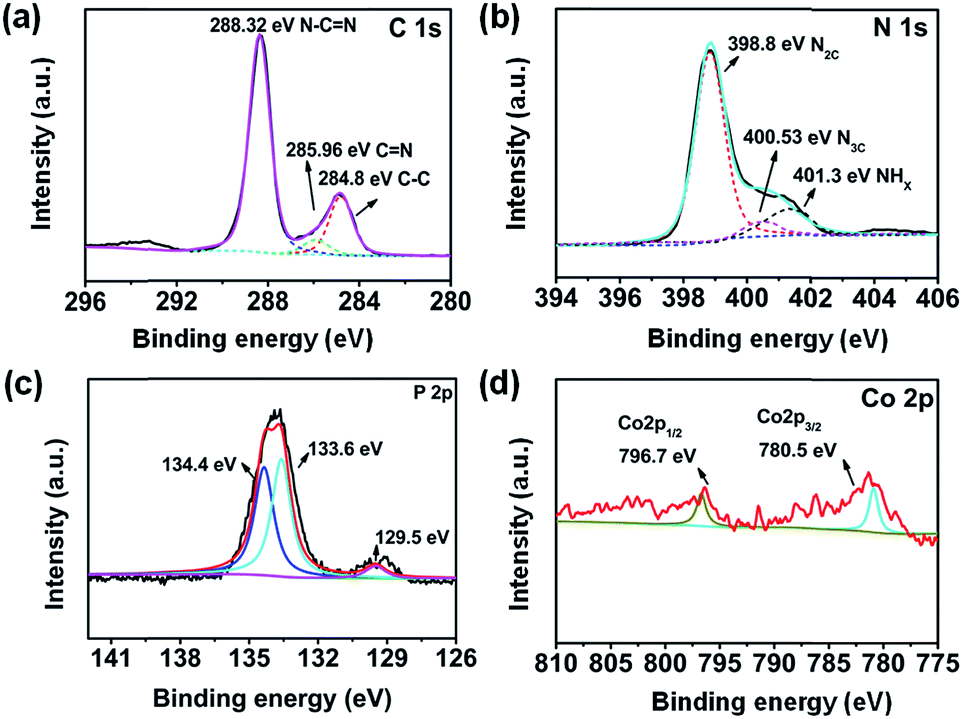 | ||
| Fig. 3 XPS spectra of (a) C 1s, (b) N 1s, (c) P 2p, and (d) Co 2p for CoP/g-C3N4-5. | ||
The optical absorption of pure g-C3N4 and CoP/g-C3N4 was determined by UV-vis diffuse reflectance spectroscopy. CoP/g-C3N4 exhibited an absorption edge at 435 nm and a corresponding band gap of 2.85 eV similar to g-C3N4 (Fig. S8a†). It also shows that visible illumination can be absorbed by CoP/g-C3N4. Compared with pure g-C3N4, the absorption of light is enhanced after introducing CoP. The photoluminescence spectra of g-C3N4 and CoP/g-C3N4 both show peaks at 469 nm due to the direct electron–hole recombination. The peaks result from the n–π* electronic transitions involving the lone pairs of nitrogen atoms in g-C3N4 (Fig. S8b†).51,52 Another weak emission at 446 nm is observed, which is attributed to π–π* transitions, trap-related electron–hole recombination and surface state emission.53 Notably, the emission intensity of g-C3N4 is almost 6.5 times the emission intensity of CoP/g-C3N4, which indicates that there is a greater resistance to the process of recombination of electrons and holes in CoP/g-C3N4. As a result, a higher electron transfer rate and more efficient hydrogen production are achieved.
Hydrogen generation
Hydrogen production experiments were carried out over the g-C3N4 and CoP/g-C3N4 samples and the results are presented in Fig. 4a and b. Control experiments were also carried out over materials without g-C3N4, without ZIF-67, without NaH2PO2, or with nickel partially substituting for cobalt, respectively (Fig. S9 and S10a†). But only a small amount of hydrogen from the contrast group can be detected. The properties of g-C3N4 and the CoP/g-C3N4 catalysts prepared at various mixing ratios were also studied (Fig. S10b and c†). With the content of the ZIF-67 precursor increasing, the hydrogen evolution rate improved sharply until the mass percentage of ZIF-67 was 5 wt% (1.42 wt% CoP) (Fig. S10b and c†). Upon further increasing the content of the ZIF-67 precursor, the hydrogen evolution rate of the resulting composite showed a decreasing trend. It can be inferred that excessive amounts of CoP hindered the absorption of light. Thus, a lower number of active sites are exposed on the photochemical accessible surface of CoP/g-C3N4. Additionally, the CoP/g-C3N4 photocatalysts exhibit excellent hydrogen production activities, which surpass that of g-C3N4, indicating that photocatalysts with high efficiency have been obtained and CoP is indispensable. The hydrogen production rates of the CoP/g-C3N4 photocatalysts also exceed that of bulk g-C3N4 with Pt photodeposited as the cocatalyst (Fig. S10d†). Besides, recycling experiments were also performed to evaluate the stability of CoP/g-C3N4. The hydrogen evolution rate of CoP/g-C3N4 was almost constant after several cycles, suggesting the excellent stability of CoP/g-C3N4 (Fig. S11 and S12†). The control hydrogen production experiments also prove that the good performance of CoP/g-C3N4 is a result of the combined action of the components and individual components can not produce a high amount of hydrogen. It should also be noted that among all the samples, CoP/g-C3N4-5 demonstrates extremely high activity: the hydrogen production occurred at a maximum rate of 67.2 μmol g−1 h−1 under visible-light irradiation (λ > 420 nm) and 201.5 μmol g−1 h−1 under light irradiation (λ > 320 nm), while the benchmark g-C3N4 was not active enough. An induction process of CoP/g-C3N4-5 in Fig. 4a and b was observed, which has also been reported in other H2 generation systems based on Co molecular catalysts.54,55 Under the same conditions, the measured apparent quantum yield (AQY) of CoP/g-C3N4-5 calculated after four hours is 0.12% at 420 nm. Moreover, the AQY was consistent with that of UV-vis absorbance, revealing that light absorption played an important role in the reaction (Fig. S13†). In conclusion, it can be speculated that the outstanding activity of CoP/g-C3N4 is attributable to the synergistic effects and faster electron transfer resulting from combining the appropriate amount of CoP and g-C3N4 in the CoP/g-C3N4 photocatalysts.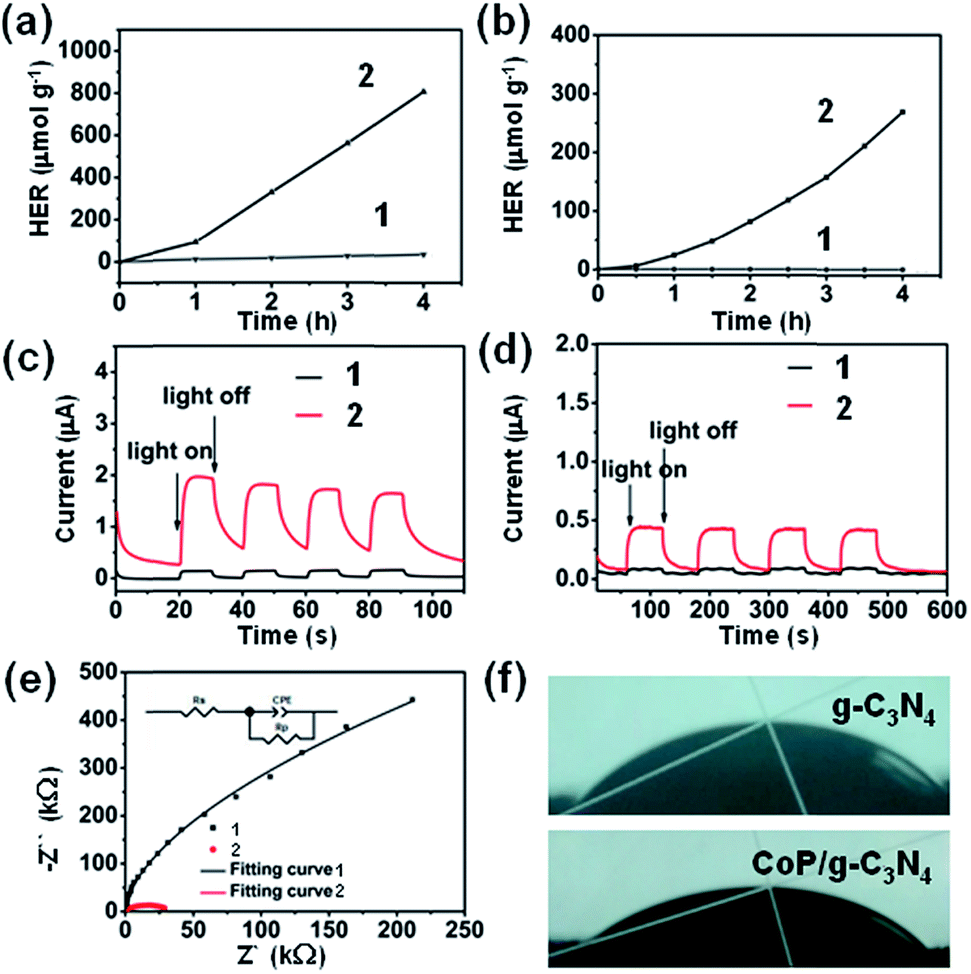 | ||
| Fig. 4 Hydrogen evolution rate (HER) at (a) λ > 320 nm and (b) λ > 420 nm. Photocurrent–time (I–T) profiles at (c) λ > 320 nm and (d) λ > 420 nm. The photocurrent was observed for each switch-on/-off event by using a 300 W xenon lamp. (e) EIS Nyquist plots of the prepared electrodes (Z′′ indicates the imaginary impedance and Z′ is the real impedance) (three electrodes; scan rate 300 mV s−1; 0.1 M Na2SO4 in water as the electrolyte; init E: −0.13 V (vs. Ag/AgCl)). The number 1 denotes g-C3N4 while the number 2 denotes CoP/g-C3N4. (f) Differences in hydrophilicity between CoP/g-C3N4 and g-C3N4 measured by contact angle measurements. | ||
The Mott–Schottky plots of CoP/g-C3N4 were measured at frequencies of 1000, 2000, 5000 and 10000 Hz. The obtained C−2vs. potential plot exhibits a positive slope typical of n-type semiconductors (Fig. S14†).56 The intersection does not change with the frequency. The corresponding flat band position (Vfb) determined from the intersection is approximately −1.22 V vs. Ag/AgCl (i.e. −1.02 V vs. NHE) for g-C3N4. In general, the bottom of the conduction band is more negative than the flat band potential in n-type semiconductors,57,58 and the conduction band (LUMO) of g-C3N4 can be estimated to be −1.12 V vs. NHE. Similarly, the conduction band (LUMO) of CoP/g-C3N4 can be calculated to be −0.97 V vs. NHE, below the redox potential of H+/H2 (−0.41 V vs. NHE, pH = 7).56,59 It can also be inferred that electron transfer from CoP/g-C3N4 to H+ is thermodynamically favorable. As a result of that, the ability of CoP/g-C3N4 for photocatalytic H2 production can be further proved.
Photocurrent experiments were conducted to investigate the charge carrier separation of the as-prepared CoP/g-C3N4 (Fig. 4c and d).59 Notably, the corresponding photocurrent responses of g-C3N4 and CoP/g-C3N4 are fast and reversible. Besides, CoP/g-C3N4 has a high photocurrent, which is about 5 times and 19 times as high as the photocurrent of g-C3N4 under visible-light irradiation (λ > 420 nm) and light irradiation (λ > 320 nm), respectively, indicating an improvement in the transport and separation of photoinduced charge carriers in CoP/g-C3N4.
In Fig. 4e, the electrochemical impedance spectra (EIS) display the charge transport efficiency of CoP/g-C3N4 indirectly. The semicircular diameter of the Nyquist curves is much smaller in comparison with that of pure g-C3N4, which indicates a decreased charge transfer resistance in the solid state interface and a faster charge-transfer process after the introduction of CoP. Surface hydrophilicity was tested by performing contact angle measurements (Fig. 4f). The CoP/g-C3N4 photocatalyst shows a contact angle of 33° compared with 45.5° measured on g-C3N4, indicating a more hydrophilic surface. Thus, CoP/g-C3N4 exhibits improved water adsorption and subsequent proton reduction.60
Photocatalytic mechanism
We further conducted DFT calculations to determine the band structures and partial density of states (PDOS) of CoP and g-C3N4. In Fig. 5a, the calculated band structure reveals that the conduction band minimum (CBM) of CoP crosses the Fermi energy, showing the properties of a conductor. The PDOS of each element revealed that CoP is mainly derived from the Co 3d state. The 3p electronic state of P atoms makes only a small PDOS contribution to the valence band maximum (VBM) and CBM, which suggests that the electrical conductance of CoP is mainly through Co atoms. As shown in Fig. 5b, the VBM and CBM of g-C3N4 show a moderate direct band gap (Eg) of about 2.66 eV at the Γ points, slightly smaller than that of g-C3N4 in the experiment, indicating semiconductor properties. What has been mentioned above proved that CoP can be an excellent electronic conductor. Thus, photogenerated electrons from g-C3N4 can be transferred to CoP rapidly.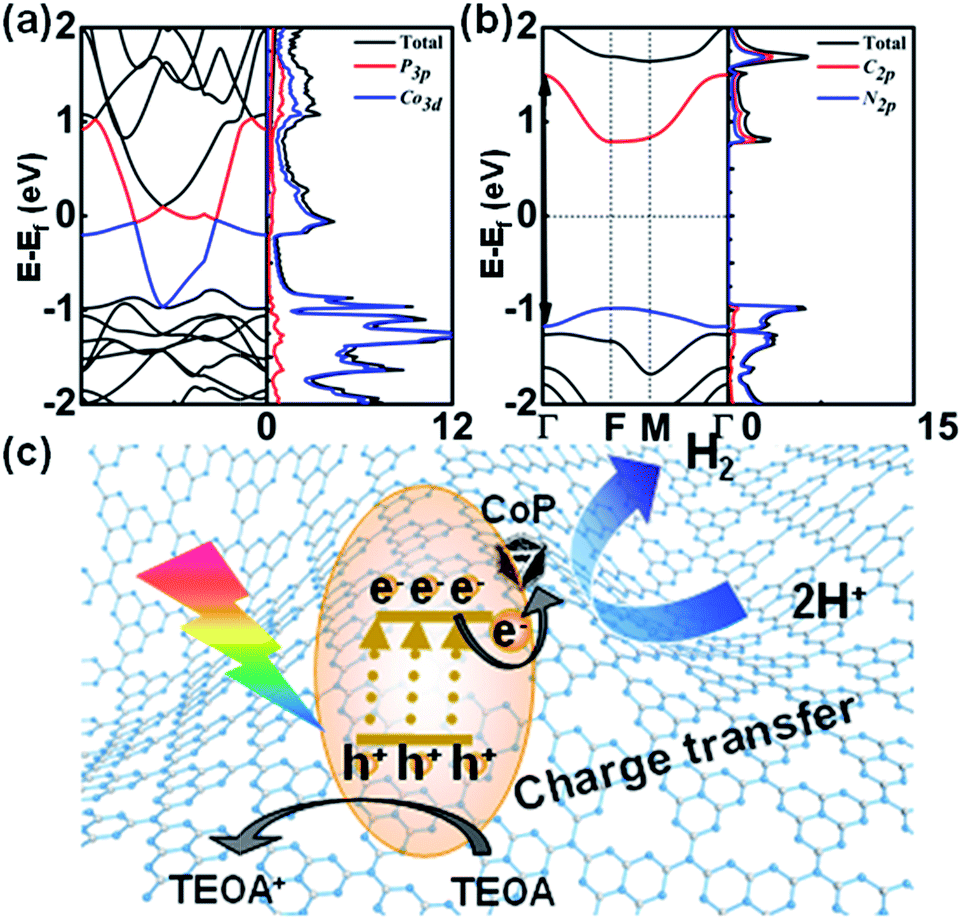 | ||
| Fig. 5 (a) Band structures and density of states for CoP. (b) Band structures and density of states for g-C3N4. (c) Proposed mechanism of CoP/g-C3N4 in the photocatalytic hydrogen production reaction. | ||
From these results, the photocatalytic mechanism of CoP/g-C3N4 in the photocatalytic hydrogen production reaction has been proposed (Fig. 5). The photoinduced electrons will be trapped and transferred from the modified g-C3N4 to CoP for hydrogen production from water, while the recombination rate of photocurrent carriers is reduced. That leads to high electron utilization efficiency during the photocatalysis reaction. Besides, introducing CoP enhances both the exposure of active sites and mass transport in hydrogen production. As a consequence, the photocatalytic activities of the CoP/g-C3N4 photocatalysts surpass that of g-C3N4.
Conclusions
In summary, to enhance the photoinduced charge separation and photocatalytic efficiency in the absence of precious metals, for the first time, we have explored an efficient g-C3N4-based non-precious-metal photocatalyst, CoP/g-C3N4, with varying mass percentages of ZIF-67 and g-C3N4 as precursors. The CoP/g-C3N4 photocatalysts were obtained after calcination under a N2 atmosphere, with NaH2PO2 as the P source upstream. The coupling of g-C3N4 and CoP derived from ZIF-67 allows enhanced light absorption ability, a faster charge-transfer process and an improvement in the separation of photoinduced charge carriers. The photocatalytic hydrogen production rate of the as-prepared CoP/g-C3N4 can reach 201.5 μmol g−1 h−1, almost 23 times higher than that of bulk g-C3N4. The new design presented in this study constitutes an important step towards exploring g-C3N4-based photocatalysts, calling for a re-thinking of ZIF materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21676066 and 21501036).Notes and references
- W. Gu, F. Lu, W. Chao, S. Kuga, L. Wu, H. Yong and W. Min, ACS Appl. Mater. Interfaces, 2017, 34, 28674–28684 Search PubMed.
- I. Majeed, M. A. Nadeem, A. Badshah, F. K. Kanodarwala, H. Ali, M. A. Khan, J. A. Stride and M. A. Nadeem, Catal. Sci. Technol., 2017, 7, 677–686 CAS.
- D. P. Kumar, M. I. Song, E. H. K. Sangyeob Hong, M. Gopannagari, D. A. Reddy and T. K. Kim, ACS Sustainable Chem. Eng., 2017, 5, 7651–7658 CrossRef CAS.
- G. Yang and J. Wang, Energy Fuels, 2017, 31, 9633–9641 CrossRef CAS.
- Y. Ge, Z. H. Shah, X.-J. Lin, R. Lu, Z. Liao and S. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 1675–1684 CrossRef CAS.
- L. Kong, Y. Dong, P. Jiang, G. Wang, H. Zhang and N. Zhao, J. Mater. Chem. A, 2016, 25, 9998–10007 Search PubMed.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 14–34 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- L. Jing, J. Zhou, J. R. Durrant, J. Tang, D. Liu and H. Fu, Energy Environ. Sci., 2012, 5, 6552–6558 CAS.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- M. Zhou, Z. Hou, L. Zhang, Y. Liu, Q. Gao and X. Chen, Sustainable Energy Fuels, 2017, 1, 317–323 CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- Y. Gong, M. Li and Y. Wang, ChemSusChem, 2015, 8, 931–946 CrossRef CAS PubMed.
- S. Yin, J. Han, T. Zhou and R. Xu, Catal. Sci. Technol., 2015, 5, 5048–5061 CAS.
- S. Ye, R. Wang, M.-Z. Wu and Y.-P. Yuan, Appl. Surf. Sci., 2015, 358, 15–27 CrossRef CAS.
- F. Guo, J. Chen, M. Zhang, B. Gao, B. Lina and Y. Chena, J. Mater. Chem. A, 2016, 4, 10806–10809 CAS.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. Patnaik, S. Martha, S. Acharya and K. M. Parida, Inorg. Chem. Front., 2016, 3, 336–347 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- W. Yu, J. Chen, T. Shang, L. Chen, L. Gu and T. Peng, Appl. Catal., B, 2017, 219, 693–704 CrossRef CAS.
- L. Zhang, L. Li, X. Sun, P. Liu, D. Yang and X. Zhao, Materials, 2016, 9, 16 CrossRef PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- F. Chen, H. Yang, W. Luo, P. Wang and H. Yu, Chin. J. Catal., 2017, 38, 1990–1998 CrossRef.
- P.-W. Chen, K. Li, Y.-X. Yu and W.-D. Zhang, Appl. Surf. Sci., 2017, 392, 608–615 CrossRef CAS.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H.-M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- J.-H. Huang, J.-T. Chen, T. Yao, J.-F. He, S. Jiang, Z.-H. Sun, Q.-H. Liu, W.-R. Cheng, F.-C. Hu, Y. Jiang, Z.-Y. Pan and S.-Q. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- W. Liu, L.-L. Cao, W.-R. Cheng, Y.-J. Cao, X.-K. Liu, W. Zhang, X.-L. Mou and L.-L. Jin, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef CAS PubMed.
- S. S. Yi, J. M. Yan, B. R. Wulan, S. J. Li, K. H. Liu and Q. Jiang, Appl. Catal., B, 2017, 200, 477–483 CrossRef CAS.
- S. Yang, B. Pattengale, E. L. Kovrigin and J. Huang, ACS Energy Lett., 2017, 2, 75–80 CrossRef CAS.
- Y.-Q. Lan, H.-L. Jiang, S.-L. Li and Q. Xu, Adv. Mater., 2011, 23, 5015–5020 CrossRef CAS PubMed.
- F.-M. Zhang, L.-Z. Dong, J.-S. Qin, W. Guan, J. Liu, S.-L. Li, M. Lu, Y.-Q. Lan, Z.-M. Su and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 6183–6189 CrossRef CAS PubMed.
- X. Li, Z. Guo, C. Xiao, W. G. Tian, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 6512–6520 Search PubMed.
- J. R. M. Hu, S. Furukawa, L. Radhakrishnan, Y. Zhang, P. Srinivasu, H. Iwai, H. Wang, Y. Nemoto, N. Suzuki, S. Kitagawa and Y. Yamauchi, Chem. Commun., 2011, 47, 8124–8126 RSC.
- W. Xia, R. Q. Zou, L. An, D. G. Xia and S. J. Guo, Energy Environ. Sci., 2015, 8, 568–576 CAS.
- Y. Hou, J. Li, Z. Wen, S. Cui, C. Yuan and J. Chen, Nano Energy, 2015, 12, 1–8 CrossRef CAS.
- Z. Xie, X. Cui, W. Xu and Y. Wang, Electrochim. Acta, 2017, 229, 361–370 CrossRef CAS.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Angew. Chem., Int. Ed. Engl., 2016, 55, 4016–4020 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed.
- C. Y. Cao, A. Xie and X. G. Yu, Nanoscale, 2015, 7, 2471–2479 RSC.
- J. B. James and Y. S. Lin, J. Phys. Chem. C, 2016, 120, 14015–14026 CAS.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 16850–16856 CAS.
- S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- X. Wang, R. Tong, Y. Wang, H. Tao, Z. Zhang and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 34270–34279 CAS.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., 2016, 55, 1830 CrossRef CAS PubMed.
- Y. Zhang and M. Antonietti, Chem.–Asian J., 2010, 5, 1307–1311 CAS.
- Q.-J. Xiang, J.-G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- R. Jin, S. Hu, J. Gui and D. Liu, Bull. Korean Chem. Soc., 2015, 36, 17–23 CrossRef CAS.
- S. Cao, Y. Chen, C.-J. Wang, P. He and W.-F. Fu, Chem. Commun., 2014, 50, 10427–10429 RSC.
- P. W. Du, J. Schneider, G. G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS PubMed.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063–7066 RSC.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, Dalton Trans., 2013, 42, 13649–13657 RSC.
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC.
- Y. Lu, D. Chu, M. Zhu, Y. Du and P. Yang, Phys. Chem. Chem. Phys., 2015, 17, 17355–17361 RSC.
- Y. Wang, M. K. Bayazit, S. J. A. Moniz, Q. Ruan, C. Lau, N. Martsinovich and J. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00063h |
| This journal is © The Royal Society of Chemistry 2018 |