
Amorphous-cathode-route towards low temperature SOFC†
Andrea
Cavallaro
 *a,
Stevin S.
Pramana‡
b,
Enrique
Ruiz-Trejo
c,
Peter C.
Sherrell
a,
Ecaterina
Ware
a,
John A.
Kilner
ad and
Stephen J.
Skinner
a
*a,
Stevin S.
Pramana‡
b,
Enrique
Ruiz-Trejo
c,
Peter C.
Sherrell
a,
Ecaterina
Ware
a,
John A.
Kilner
ad and
Stephen J.
Skinner
a
aDepartment of Materials, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: a.cavallaro@imperial.ac.uk
bSchool of Engineering, University of Newcastle, Merz Court, Newcastle, NE1 7RU, UK
cDepartment of Earth Science and Engineering, Imperial College London, Exhibition Road, London, SW7 2AZ, UK
dInternational Institute for Carbon-Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 16th February 2018
Abstract
Lowering the operating temperature of solid oxide fuel cell (SOFC) devices is one of the major challenges limiting the industrial breakthrough of this technology. In this study we explore a novel approach to electrode preparation employing amorphous cathode materials. La0.8Sr0.2CoO3−δ dense films have been deposited at different temperatures using pulsed laser deposition on silicon substrates. Depending on the deposition temperature, textured polycrystalline or amorphous films have been obtained. Isotope exchange depth profiling experiments reveal that the oxygen diffusion coefficient of the amorphous film increased more than four times with respect to the crystalline materials and was accompanied by an increase of the surface exchange coefficient. No differences in the surface chemical composition between amorphous and crystalline samples were observed. Remarkably, even if the electronic conductivities measured by the Van Der Pauw method indicate that the conductivity of the amorphous material was reduced, the overall catalytic properties of the cathode itself were not affected. This finding suggests that the rate limiting step is the oxygen mobility and that the local electronic conductivity in the amorphous cathode surface is enough to preserve its catalytic properties. Different cathode materials have also been tested to prove the more general applicability of the amorphous-cathode route.
Introduction
Research into renewable energy sources is currently focussed on finding a breakthrough solution to the increasing demand for clean energies. Solid oxide electrochemical cell studies are increasingly focusing on two different directions: reducing the operating temperature of the solid oxide fuel cell (SOFC) and studying new materials for high temperature electrolyser devices. Generally, the electrolyte and electrode materials for SOFC applications are crystalline metal oxides because the technologically important temperature range has traditionally been between 700–1000 °C. In other research fields, such as electronics, the use of amorphous metal oxide materials has been explored for many decades with remarkable results.1–8 For example, to fulfil the commercial demand for transparent electronic devices on flexible substrates, Nomura et al. firstly found in amorphous semiconductor oxides a good alternative to hydrogenated amorphous silicon (A-Si:H) and organic semiconductors.9 A-HfO2, A-ZrO2, A-TiO2, A-In2O3, A-Ga2O3, A-Al2O3 and A-Cu2O amorphous metal oxides (AMOs) have been studied as possible candidates for a wide range of applications such as high-k dielectrics,1,2 non-volatile memories,3–5 flexible-transparent electronics,6,7 and solar cells.8 Until now very little work has been done regarding fully-amorphous metal oxide materials in the SOFC field since the device operational conditions would have been incompatible with a stable amorphous metal oxide state and a detrimental large reduction of the mixed ionic electronic conductor (MIEC) electronic conductivity would also have been expected. In a future prospective, the “next generation” Low Temperature Solid Oxide Fuel Cells (LT-SOFC) will operate in the range from 400 °C to 650 °C.10 As pointed out by Gao and co-workers, lowering the operational temperature of SOFC devices as well as reducing the technological cost of the cell itself, would also benefit its use in reversible mode for energy storage promoting, for example, the H2O–CO2 co-electrolysis towards CH4 production. In our study, we explored the possibility of employing amorphous cathode materials in LT-SOFC applications. It is important to highlight two interesting works: the first by Januschewsky et al. that observed how the decrease of the La0.6Sr0.4CoO3−δ (LSC6040) film crystallinity, when employing a low PLD-deposition temperature, was linked with a strong increase of the electrochemical oxygen exchange rate;11 the second by Evans et al. who have recently reported how a partially amorphous La0.6Sr0.4CoO3−δ cathode can be successfully employed in μ-SOFC achieving significant power density at temperatures as low as 400 °C.12 In the present work we have analysed in depth the effect of the amorphisation of the La0.8Sr0.2CoO3−δ (LSC8020) cathode material on the surface oxygen reduction and diffusion properties using the Isotope Exchange Depth Profiling (IEDP) technique.13,14 A first hypothesis to explain the unexpected behaviour of the Amorphous Mixed Ionic Electronic Conducting (A-MIEC) materials has also been proposed.Results and discussion
Films of around 300 nm thickness have been prepared at different deposition temperatures: Room Temperature (RT), 200, 400, 500, 550, 600 and 750 °C by Pulsed Laser Deposition (PLD) on Si (001) single crystals.Deposition conditions can be found in the methodology section. From the X-ray Diffraction (XRD) characterization of LSC8020, textured-polycrystalline films have been obtained for all the samples grown at different PLD deposition temperatures except for those prepared at a nominal temperature lower or equal to 400 °C. These samples do not present any crystallinity within the detection limit of our diffractometer as shown in Fig. 1(a); the reflection at 69° two-theta is attributed to the silicon single crystal (004) plane. The XRD intensities have been presented on a logarithmic scale to emphasise the background. The black pattern corresponds to the sample grown at a nominal temperature of 750 °C. The (001), (011), (111), (002), (112) and (013) reflections correspond to a cubic structure. Strong (00l) and (hkl) reflections appear in the XRD patterns suggesting an out-of-plane LSC8020 preferential growth for the samples deposited at T > 400 °C. Transmission Electron Microscopy (TEM) analysis of the sample deposited at 400 °C has been performed to confirm the amorphous nature of the LSC film at low deposition temperatures. In addition this analysis will also identify the presence of any porosity in the film.
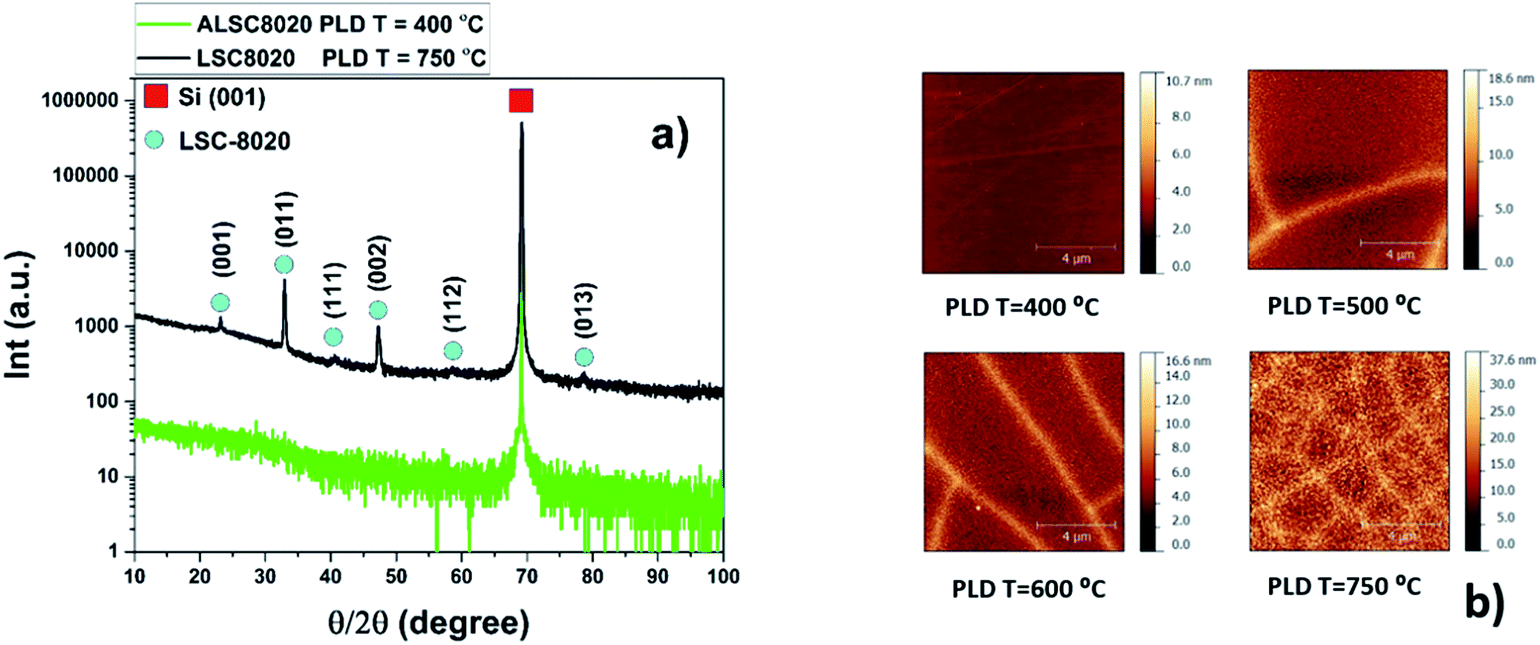 | ||
| Fig. 1 (a) XRD θ/2θ-scan of 300 nm LSC8020 PLD dense cathode films. (b) AFM 10 × 10 μm2 area analysis of 300 nm LSC films grown on silicon substrates. (a) The green pattern corresponds to the amorphous LSC8020 film grown at 400 °C. Black pattern belongs instead to the crystalline film grown at 750 °C. The XRD reflections of the latter reveal a LSC8020 preferential oriented growth. (b) The samples presented here have been deposited in the temperature range from 400 °C to 750 °C by PLD. The bright lines on the surface correspond to corrugations on the film surface. | ||
The density of the material is expected to change on passing from the amorphous to the crystalline state. Due to the small amount of deposited LSC8020 material, the use of the X-ray diffraction method to evaluate the film density has been chosen. X-ray reflectivity, normally employed to characterize the thin film thickness, could also provide information about film surface roughness and film density by fitting a simple physical model based on the Fresnel reflectivity equation.15 In the case of the mass density, ρ, calculation the model is the following:
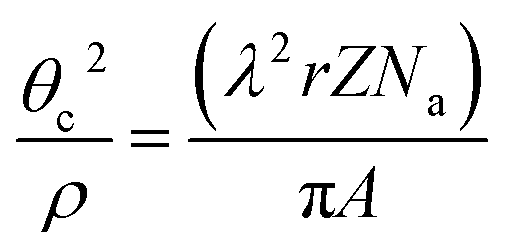
The surface topography of the LSC films has been characterized by Atomic Force Microscopy (AFM). As expected, the LSC8020 thin film surface roughness decreased with the reduction of the deposition temperature; from a root mean square of roughness (rms) of 4.8 nm for the sample grown at 750 °C to an rms of 0.3 nm for the amorphous film grown at 400 °C, Fig. 1(b). In the AFM topography images, the brighter colour relates to a higher surface; the lines on the film surfaces correspond to a film deformation occurring during the cooling step in the PLD film preparation. This phenomenon is associated with the different thermal expansion coefficients of silicon with respect to the LSC8020 perovskite material, see ESI.† The surface defect density was found to be inversely proportional to the PLD deposition temperature. From an AFM profiling analysis the defects correspond to deformed material areas that were raised from the film surface during the PLD cooling process and caused by the strong compressive forces the film is subjected to, (ESI†). As discussed later these deformed areas have a detrimental effect on the oxygen diffusion but do not affect the overall conclusion of this work.
Surface and sub-surface chemical composition thin film characterization
The LSC8020 films have been analysed by Low Energy Ion Scattering (LEIS). This unique and powerful technique can be used to determine the chemical composition of the outermost atomic layer. For more information about the LEIS technique readers are referred to the ESI.† Using the primary analysis-beam and a 0.5 keV argon sputter ion gun it is possible to chemically depth profile the first 0–10 nm of the sample surface layers. The detected back-scattered ion intensities are directly proportional to the amount of each chemical species covering the surface. Four samples prepared at 400, 500, 600 and 750 °C have been depth profiled by LEIS. No chemical cleaning of the surface was performed before the analysis to avoid altering the surface chemical composition. The normalized intensities of each cation (Co, Sr and La) have been plotted as a function of the Ar sputtering dose (normalized by the total cation intensity) (ESI†). A higher Ar dose corresponds to the chemical analysis of a deeper film layer. The intensity of each element increased slightly during the first argon sputtering cycle as the surface carbon, covering the sample, was sputtered away. The fitting error related to the first two depth profiling scans, when the surface is still partially covered by carbon, is very high and for this reason have not been considered. Each element intensity reached a plateau at a certain depth from the outermost surface. Assuming that the material 4 nm from the top surface, has the nominal stoichiometric composition, and in the absence of argon preferential sputtering, this plateau value can be used as an internal calibration to calculate the element concentration at any particular depth. In Fig. 2 each elemental composition has been plotted as a function of the total argon sputtering dose. Comparing the results obtained from samples prepared at different deposition temperatures, the elemental composition profiles are very similar. Overall it can be concluded that the chemical top surface composition of the four samples is comparable. SRIM software has been employed to simulate the sputtering rate during the analysis conditions, giving a value of 2.2 × 10−16 nm per atoms per cm2.17 Looking at the film surface (zero position of the x-axis), a change in the LSC chemical composition with respect to the bulk (around 4 nm from the atomic-surface) is observable. The surface depletion of lanthanum and the increase of strontium signals are mostly correlated: strontium segregates to the surface substituting the lanthanum in the LSC crystallographic structure. In similar perovskite materials such as La1−xSrxMnO3 and La0.6Sr0.4CoO3−δ, Sr replacing the La on the A-site of the surface has already been reported.18–20 Cobalt instead, lightly depleted at the outermost top-surface quickly reaches an over-stoichiometry of around 20% in the sub-surface. In the first nanometre from the surface a small excess of strontium not related to lanthanum depletion has also been observed. This excess may be in the form of SrO, Sr(OH)2 or SrCO3. As has been previously reported,21–27 the surfaces of strontium-doped perovskites are enriched with Sr when the films are thermally annealed for several hours. This segregation process is thermally activated and the strontium preferentially moves through the grain boundaries as observed by Kubicek et al.27 LEIS analysis of the LSC8020 thick film samples confirms the strontium enriched surface. Nevertheless, in this study the strontium top-surface segregation does not appear to be related to the PLD deposition temperature. This enrichment seems to be related to the surface chemical stability under the employed PLD deposition conditions. Most of the studies regarding Sr segregation have been performed at a constant temperature for long periods of time, and so near to the thermodynamic equilibrium. On the contrary, PLD deposition, especially when employing a high repetition rate, is mostly driven by kinetics. In addition, as shown by Niania et al.,28 even in a thermodynamically controlled segregation process, the gas atmosphere has an important role in governing the segregation rate, which is accelerated in the presence of water. Cai et al. measured by X-ray photoelectron spectroscopy (XPS) a change in the surface composition of as-grown LSC6040 films and related it to the PLD deposition temperature.23 However, two main differences with the present work should be underlined: the first is the Sr dopant content, double with respect to the LSC8020 phase and secondly the ten times higher base pressure employed in the PLD deposition that could have an effect on the strontium segregation process. In addition, in our experimental set up, the films are directly cooled down after deposition remaining at high temperature for a very short amount of time (see methodology). The short cooling step, where thermodynamics dominates the strontium segregation process is too short to produce any differences in the film surface composition. A complete study of the atomic surface composition of the LSC film is far from the objective of this publication and needs further investigation. The key factor is that the surface chemical composition does not change with respect to the PLD deposition temperature and no difference in the LSC8020 surface catalytic properties should then be expected.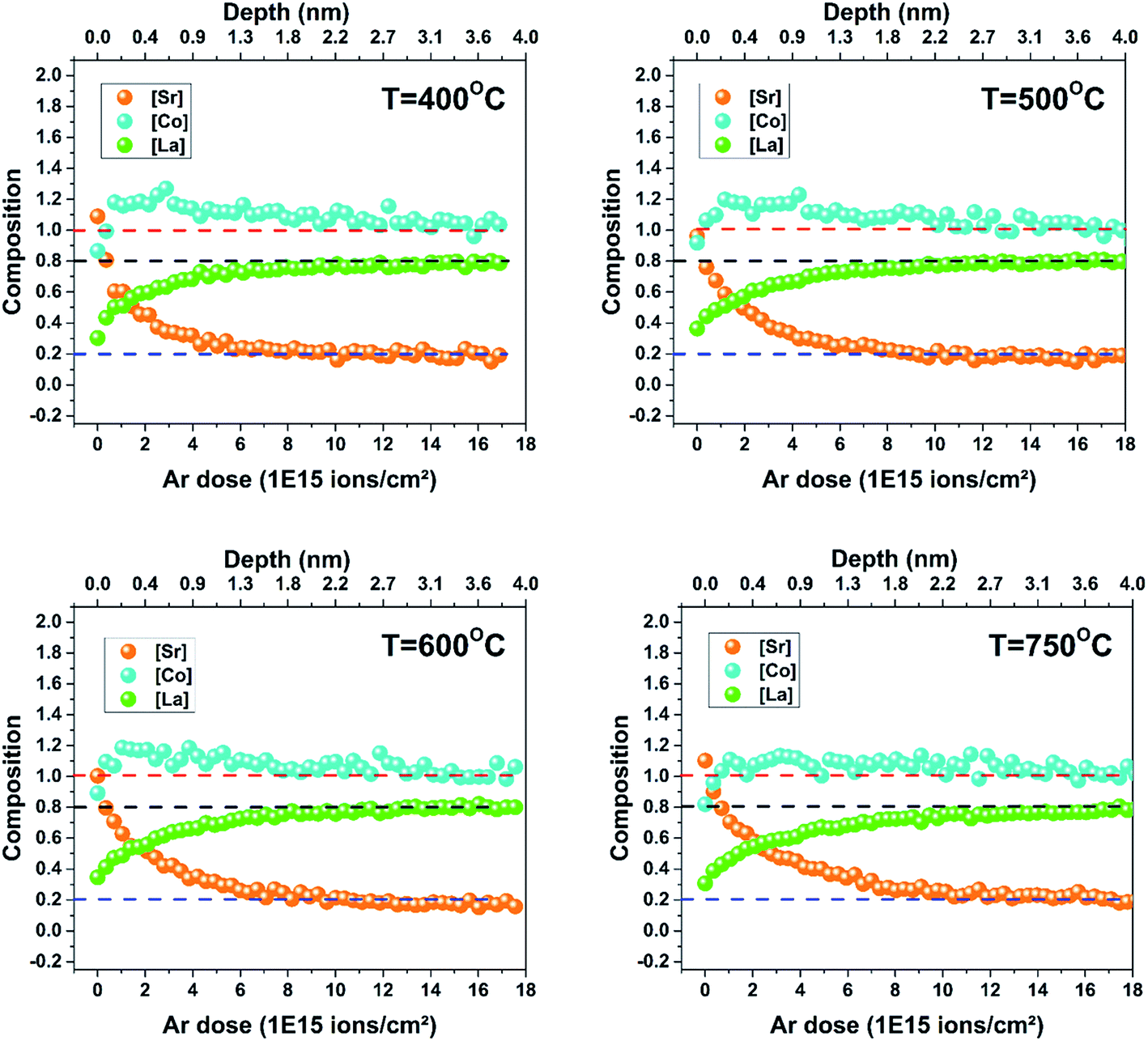 | ||
| Fig. 2 LEIS analysis of the 300 nm LSC films. Four samples prepared at 400, 500, 600 and 750 °C have been depth profiled by LEIS at the same analysis conditions: primary beam 3 keV He+ and sputtering beam 0.5 keV Ar+. Each of the different cation species intensity, cobalt, strontium and lanthanum, has been normalised and the composition calculated with respect to the ‘’bulk-surface’’ intensity. In the x-axis the total argon sputtering dose is shown: 0 ions per cm2 corresponds to the sample very top-surface. Horizontal dotted lines, blue, black and red indicate the nominal stoichiometry composition for Sr, La and Co respectively. | ||
Crystallinity study by transmission electron microscopy (TEM)
To confirm the amorphous nature of the sample prepared at 400 °C, TEM analysis has been performed. For comparison, one of the well-crystallized samples (750 °C) was also analysed by TEM. The selected area electron diffraction pattern (SAED) of the textured crystalline film (750 °C sample) together with the Si substrate is presented in Fig. 3. A SiO2 thin film is present at the interface between the silicon substrate and the LSC film. This layer forms during the heating stage in the PLD deposition chamber and its thickness is proportional to the deposition temperature as seen by comparing Fig. 3(a) and (c). The d-spacings labelled (1) and (2) correspond to 0.38 and 0.27 nm, matching the (001) and (011) reflections of the La0.8Sr0.2CoO3−δ phase respectively, Fig. 3(e) and (f). In contrast, the sample grown at 400 °C did not show any textured behaviour, but only a single polycrystalline ring with d-spacing of 0.30 nm which is not characteristic of the La0.8Sr0.2CoO3−δ phase, Fig. 3d. The plausible compound that might be formed is SrO (d111 = 0.30 nm).29 Semi-quantitative energy dispersive X-ray spectroscopy (EDS) showed an overall composition of the film close to the nominal one. The fact that polycrystalline SrO is not detectable by XRD and that the 400 °C sample composition is very close to the well crystallized sample, suggests that any SrO has a very small grain size and is homogenously distributed in the film thickness, consistent with the broad width of the polycrystalline ring pattern. Considering the general aim of this work the presence of nano-crystalline SrO inside an amorphous LSC8020 phase does not represent a limitation. The strontium oxide formation in fact is generally considered detrimental for the catalytic activity of the LSC phases.23 From the TEM images of the amorphous ALSC8020 film, Fig. 3(a) and (b), we can exclude the presence of macroscopic porosity in the sample. | ||
| Fig. 3 TEM cross section analysis. Comparison between one amorphous ALSC sample grown at 400 °C (a, b) and a crystalline one deposited by PLD at 750 °C (c). The bright layer between the silicon substrate and the LSC film is SiO2 that forms during the substrate heating in the PLD chamber. The SiO2 thickness is directly correlated to the PLD deposition temperature. (d) SAED analysis of the amorphous film; (e) and (f) corresponds to SAED analysis of the crystalline sample. | ||
LSC films μ-Raman spectroscopy
When a material crystal size shrinks to the nanometric level, Raman spectroscopy is capable of distinguishing between the amorphous and crystalline state much more precisely than the X-ray diffraction experiment commonly performed. Below a certain grain size, in fact, the broadening of the diffraction peak related to the small size merges with the background, meaning that distinguishing between truly amorphous materials and small crystallites is difficult. The Raman spectrum of a 300 nm thick LSC8020 cathode material deposited on Si provides further supporting evidence for the amorphous nature of the perovskite, Fig. 4(a) and (b). Two key features support this analysis; firstly the presence of only the crystalline silicon peak at 520.7 cm−1 and secondly the presence of a broad amorphous hump between 300 cm−1 and 700 cm−1 indicating the presence of an amorphous phase throughout the material. Mapping over a 25 × 25 μm2 area showed no significant variation in the signal indicating that the surface was highly homogenous.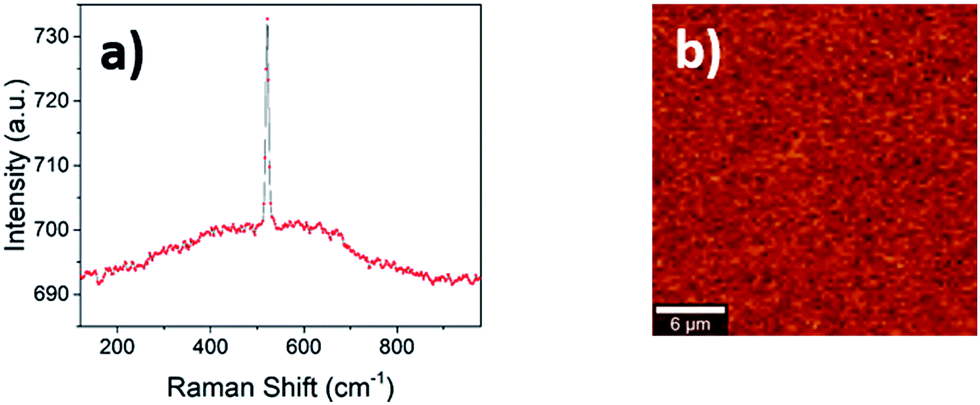 | ||
| Fig. 4 μ-Raman spectroscopy analysis. (a) Raman spectra of a sample of ALSC8020 produced on a Si/SiO2 substrate, showing the appearance of only the crystalline silicon peak at 520.7 cm−1; (b) Raman intensity map of the main perovskite peak at 670 cm−1 reported in literature, no crystallinity is observed. | ||
Oxygen-18 isotope exchange experiments
Before performing oxygen isotopic exchange, the LSC8020 films were equilibrated in dry pure oxygen (with a natural abundance of oxygen-18) for more than ten times the time required for the isotopic exchange experiment at 400 °C. This extra step is required to thermodynamically equilibrate any possible oxygen defects present in the film and to avoid oxygen chemical diffusion. The isotope exchange temperature was maintained at 400 °C for all the experiments and the exchange time in 90% 18O2 enriched atmosphere was maintained at around 15 minutes. After the exchange at 400 °C the samples were quenched to room temperature and the oxygen diffusion profiles collected by FIB-SIMS analysis. Our FIB-SIMS allows the selection of the analysis area by SEM imaging, therefore avoiding the higher density defect surfaces in the data collection. In the case of the sample treated at 750 °C this was unavoidable since the deformed patterns were too small, with features of 2 × 2 μm2. The normalized 18O diffusion signal from the surface to the silicon substrate was then fitted using the sheet-solution to the diffusion equation to extract the values of the oxygen surface exchange coefficient (k*) and the tracer diffusion coefficient (D*). For each sample, from six to eight measurements in different sample areas were collected. To analyse these large amounts of data the TraceX script developed for running in the Matlab environment was employed.30 In Fig. 5D* and k* values have been plotted as a function of the PLD deposition temperature. The calculation of errors associated with these measurements is discussed in the ESI.† With the aim of comparing our results, k* and D* values extrapolated for an exchange temperature of 400 °C from the IEDP study of polycrystalline bulk LSC8020 by De Souza et al.31 have been added to the figures (black dots). In the case of the diffusion coefficient (D*), Fig. 5(a), the values obtained for the samples PLD-grown at 500 °C and 600 °C (18O exchanged at 400 °C) were of the same order of magnitude as those extrapolated from De Souza's work. The LSC8020 film grown at 750 °C instead presents a lower value of D*. This sample possesses the highest density of surface stress-induced defects, Fig. 1(b). A lower D* value for this particular sample confirms that the surface defects are not open surface cracks (open from the surface to the substrate). A cracked surface would have allowed the isotopically enriched gas to penetrate inside the film matrix producing an overestimation of D* and k* values. Instead, the strongly compressed film area, reduces the overall oxygen-18 isotope diffusion, with a similar mechanism as the one observed in epitaxial cathode thin films under compressed strain. Surprisingly when the film crystallinity decreases into the amorphous state, the D* value increased by several times compared with the crystalline one.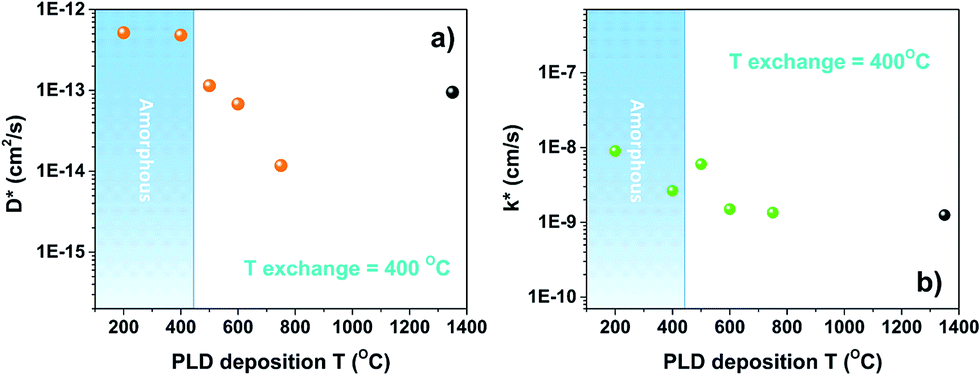 | ||
| Fig. 5 Isotope exchange depth profile experiments (IEDP). D* and k* values for LSC 300 nm thick samples that have been isotopically exchanged in an 18O2 enriched atmosphere under the same conditions (400 °C for around 15 min). The black dot represents the reference value for bulk LSC8020 extrapolated from De Souza et al.31 | ||
To evaluate the effect of an even lower deposition temperature, a 300 nm LSC8020 film was grown at 200 °C by PLD and isotopically exchanged in the same conditions. The measured D* is very close to the value observed for the amorphous LSC film deposited at 400 °C. This seems to indicate the achievement of a maximum oxygen diffusivity plateau once the amorphous state is acquired. The clear increase of oxygen diffusivity could be related to the decrease of the La0.8Sr0.2CoO3−δ material density and it will be discussed later in more detail. In Fig. 5(b) the surface exchange coefficients are presented. The k* behaviour similarly increased with the reduction of the PLD deposition temperature. In the same way, the surface exchange coefficients of the samples deposited at higher temperature are close to the k* value extrapolated from De Souza's work.31 The oxygen self-diffusion coefficient D* can be described as follows:
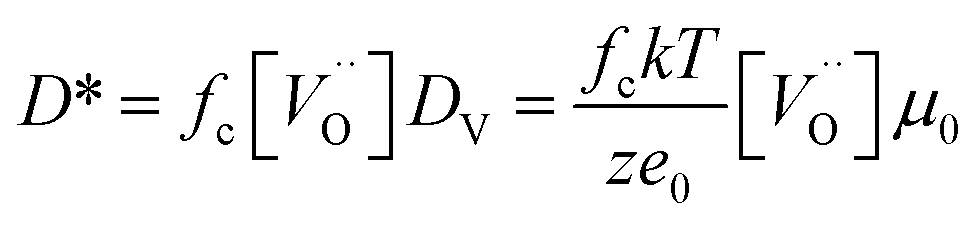
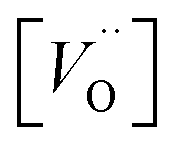 is the oxygen vacancy concentration, fc is the correlation factor for the crystal structure and typically considered to be 0.69 for perovskite type materials,32DV is the oxygen vacancy diffusion coefficient, k is the Boltzmann's constant, T the temperature, z is the charge number, e0 is the elementary charge and μ0 is the oxygen vacancy mobility. The oxygen non-stoichiometry in the LSC compound family is mainly controlled by the strontium dopant amount but it can also increase by increasing temperature or by decreasing oxygen partial pressure. Small changes of δ for LSC6040 and LSC4060 phases were measured for temperatures below 425 °C in low pO2 atmosphere (10−2 bar).33 A similar behaviour is expected also for the LSC8020. If we hypothesise a constant value of
is the oxygen vacancy concentration, fc is the correlation factor for the crystal structure and typically considered to be 0.69 for perovskite type materials,32DV is the oxygen vacancy diffusion coefficient, k is the Boltzmann's constant, T the temperature, z is the charge number, e0 is the elementary charge and μ0 is the oxygen vacancy mobility. The oxygen non-stoichiometry in the LSC compound family is mainly controlled by the strontium dopant amount but it can also increase by increasing temperature or by decreasing oxygen partial pressure. Small changes of δ for LSC6040 and LSC4060 phases were measured for temperatures below 425 °C in low pO2 atmosphere (10−2 bar).33 A similar behaviour is expected also for the LSC8020. If we hypothesise a constant value of 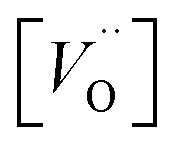 at the employed exchange temperature and pO2, the increase in D* for the amorphous LSC8020 should then be related to an increase of the oxygen vacancy mobility. Since no significant change in the chemical composition was observed between the amorphous PLD-LSC8020 and the crystalline thin films (LEIS analysis) and no change of δ should be expected, it can be argued that the difference in k* should be at least in part related to the increased mobility of the oxygen species in the LSC films.
at the employed exchange temperature and pO2, the increase in D* for the amorphous LSC8020 should then be related to an increase of the oxygen vacancy mobility. Since no significant change in the chemical composition was observed between the amorphous PLD-LSC8020 and the crystalline thin films (LEIS analysis) and no change of δ should be expected, it can be argued that the difference in k* should be at least in part related to the increased mobility of the oxygen species in the LSC films.
Description of amorphous materials
When a material becomes amorphous the long-range order satisfying the Bragg X-ray diffraction conditions no longer exists.34 Amorphous materials can further be classified into three different categories depending on their chemical bonding:In this study the amorphous LSC8020 thick films have shown enhanced oxygen diffusion and surface exchange activity. Since the PLD deposition technique is considered to transfer the stoichiometry of the target directly onto the film and the LEIS results have not shown any clear difference between amorphous and crystalline films we can speculate that the atom-position and stereochemistry should not change significantly. PLD deposited amorphous thick films could be then classified as a Type 1 amorphous material. In crystalline La0.8Sr0.2CoO3−δ perovskites, the oxygen atoms move across the oxygen vacancy created through the partial Sr2+ aliovalent substitution of the La3+ cation position. The increase in the oxygen diffusivity could be suggesting a reduction of the energetic barrier for the oxygen movement in the perovskite structure. As first suggested by Kilner35 and later by Cherry et al.,36 the oxygen instead of jumping from one site to the adjacent vacancy along the anion octahedra edge,37 goes through a ‘‘saddle point’’ in between two A-site cations and one B-site cation. The increased perovskite cell volume of the amorphous cathode material, and corresponding lower density, could be inducing a reduction of the energetic cost for the oxygen jumping into an adjacent oxygen vacancy. Likewise, the oxygen pathway in an amorphous ‘pre’-perovskite structure is likely to be different and a lower activation energy for oxygen diffusion should then be expected. With this purpose a 10 × 10 mm2 ALSC8020 sample deposited at 400 °C by PLD was cut into four parts and isotopically exchanged at 325, 350, 375 and 400 °C respectively. The results are presented in Fig. 6(a). As discussed by De Souza et al. the activation energy can be described as ΔHD*, the sum of the vacancy migration enthalpy ΔHm and the oxygen vacancy generation enthalpy ΔHg; for an extended explanation refer to.31 Comparing the ΔHD* value calculated employing our amorphous films (black dots) with the one measured by De Souza on isotopically exchanged crystalline bulk samples (green dots), an enthalpy reduction of 73 kJ mol−1 for the amorphous LSC8020 is observed. As the ΔHm value reported in the literature for this class of perovskite is around 60–80 kJ mol−1 (ref. 31, 32 and 36) it is suggested that at least part of the reduction in ΔHD* is related to a reduction of the oxygen vacancy generation enthalpy.
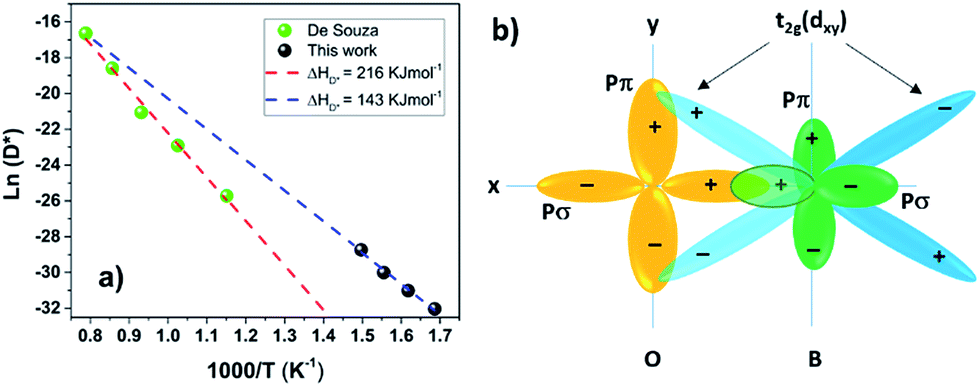 | ||
| Fig. 6 (a) Activation energy comparison between amorphous and crystalline LSC8020 (b) perovskite electron conducting orbitals configuration. (a) Amorphous samples show a decrease of the oxygen self-diffusion enthalpy compared with the crystalline LSC8020 (De Souza).31 This means a lower oxygen barrier for oxygen diffusion. (b) Covalent bonds between the oxygen ion pπ-orbitals and B-cation t2g (d) orbitals.39 | ||
Amorphous-LSC electronic conductivity
The properties that make a MIEC material an attractive cathode are good catalytic activity, high oxygen diffusivity and oxygen vacancy concentration at the cathode surface combined with high electronic conductivity. A reduction of the electronic conductivity of the metal-oxide cathode with the degree of amorphisation is expected. Before exploring the electronic conductivity difference between crystalline and amorphous LSC8020 films, let us discuss where the conducting electrons are localized/delocalized in a MIEC perovskite structure. Perovskite ABO3 cathode materials could be seen as three-dimensional networks of BO6 octahedra where the electronic conduction progresses via electrons or holes along the B–O–B bonds. The degree of covalence of the B–O–B bonds controls the electrical properties of the perovskite. Looking now in detail at the geometry of the B–O bond, Fig. 6(b), there is a directional sigma bond (σ) formed between the oxygen pσ strongly overlapped with the pσ of the B transition metal.38While this sigma collinear overlap is mainly responsible for the B–O bond, the electron/hole transport is related to the B–O π-orbital bonding. The t2g orbitals of the transition metal (dx–y, dx–z and dy–z) point in the direction of the pπ orbitals of the oxygen and form a π-orbital bond filled with the t2g transition metal valence electrons. Since oxygen is shared between neighbouring B cations (B–O–B), a π-orbital bonding network between B(t2g)–O(pπ)–B(t2g) has been proposed as the pathway for the itinerant electrons.39 This is the mechanism for the electron/hole transportation in metal oxide perovskites. In this scenario, where the conduction band electrons are moving through π-bonding between the metal t2g and the oxygen pπ, a difference in the electronic conductivity should be expected for the amorphous films in comparison with the crystalline ones.
The decrease of density of the amorphous LSC material implies an increase of the π-orbital bonding network distance between B(t2g)–O(pπ)–B(t2g) and consequently a decrease of the orbital overlapping area. The π orbital interaction should then be sensitive to changes in the cation-oxygen position on the contrary to what happens, for example, in amorphous semiconducting post-transition-metal oxides where the conduction is through neighbouring metal s orbital overlap, less sensitive to the distortion of metal–oxygen–metal (M–O–M) positions.9 As expected, the electronic conductivity of the amorphous LSC thick films measured by DC-resistivity is much lower compared with the crystalline material (Van Der Pauw Configuration; see ESI†). In the range of temperatures explored (250 °C max) the maximum difference in electronic conductivity was as high as two orders of magnitude. Despite the fact that there is a strong decrease in the macroscopic electronic conductivity, the overall catalytic properties of the amorphous LSC8020 improved. One can speculate that locally the electronic conductivity of the LSC material should not be dramatically affected. The increased oxygen mobility in the amorphous LSC is otherwise suggesting a mechanism where the oxygen adsorption, dissociation and diffusion from the surface into the bulk of the material is limiting the overall catalytic properties of the cathode despite the lower macroscopic electronic conductivity. The increase in k* can be explained by considering two different aspects. In the first place we observed, as in the amorphous film, the increasing distance between the cobalt and oxygen is responsible for the decrease of the macroscopic electronic conductivity. This is also causing the weakening of the Co–O bond responsible for the reduction of the formation enthalpy of oxygen vacancies at the film surface. A similar mechanism has been reported for epitaxial thin film growth of cathode materials. When the epitaxial film has an in plane tensile strain, the decrease in Co–O bond strength due to the bond enlargement distance produces an increase of the oxygen surface exchange coefficient of up to two orders of magnitude as published by Kushima et al.40 and Pavone et al.41 As reviewed by Yildiz,42 the higher the tensile strain, within the elastic limit, the lower the vacancy formation enthalpy and vacancy migration barrier. In addition, Cai et al. experimentally measured by XPS as the tensile strain induced a higher concentration of oxygen vacancies at the LSC8020 epitaxial film surface.43 In the case of the amorphous material the improvement of the catalytic activity of the cathode surface is not of the same magnitude but probably still driven by the weakening of the Co–O bond. Secondly, at such a low temperature the low oxygen diffusion is also limiting the surface oxygen exchange rate. Oxygen vacancies at the surface are occupied by adsorbed oxygen but do not regenerate quickly enough, limiting the surface exchange process. The decrease of the oxygen vacancy migration barrier could also play a role in the increase of the surface oxygen catalytic activity. In a self-diffusion experiment such as IEDP, the macroscopic low material electronic conductivity is not limiting the cathode surface oxygen reduction reaction (ORR) properties; locally the material maintains its catalytic characteristics. Conversely some effects will be observed when the amorphous cathode is under chemical and/or electrical potential.
IEDP experiments on different amorphous materials
To test the feasibility of the amorphous route as a general approach to low temperature SOFC devices, isotope exchange experiments on different amorphous cathode material thick films have been performed. A-La0.8Sr0.2CoO3, A-La0.6Sr0.4CoO3 (ALSC6040), A-(La0.5Sr0.5)2CoO4 (ALSC214) and A-La0.5Sr0.5Mn0.5Co0.5O3 (ALSMC) films have been prepared by PLD on silicon substrates at a nominal PLD deposition temperature of 200 °C. After the XRD analysis to check the level of crystallinity, the films were investigated by IEDP. The four different samples were exchanged at the same time in the same IEDP experiment in an attempt to minimize any experimental variables.The D* and k* values found by fitting the diffusion profile with the plane-sheet solution to Fick's law of diffusion are presented in Table 1. The difference in D* value between the ALSC8020, ALSC6040, the Ruddlesden–Popper ALSC214 and perovskite ALSMCO films are quite clear. Looking also at the k* surface exchange coefficient it is possible to conclude that overall the catalytic differences between different cathode materials have been maintained in the amorphous state. When the difference between the PLD deposition substrate temperature and the material melting temperature is very large (ratio value lower than 0.3), and the distance between adsorption sites on the substrate surface is higher than the material diffusion length, porosity can form. By TEM analysis no macroscopic porosity has been detected, but the presence of nano porosity cannot be excluded a priori. The different cathodes have been deposited in the same conditions, hence similar nano porosity should be expected. The distribution of D* and k* values in a range of different orders of magnitudes allows the exclusion of nano porosity as a cause of the observed oxygen diffusion. Amorphous cathode materials could have several advantages with respect to the crystalline ones that make this approach suitable for low temperature SOFC applications. First of all, in crystalline materials, grain boundaries (GB) due to their high-energy nature are generally detrimental for the long-term durability of SOFC devices.15–25 GB's can be fast diffusion pathways for the cation segregation to the material surface as demonstrated by Kubicek et al.25 in a Sr-isotope diffusion experiment on a LSC thick film. In the case of Sr-doped perovskites the Sr moves through the GB towards the surface where it accumulates reducing the catalytic activity of the cathode itself. To slow down the cathode degradation and reduce the detrimental metal cation migration through the grain boundaries, Liu et al. employed a new stabilization mechanism.44 They discovered that mixing nanoparticles with the Ba0.5Sr0.5Co0.8Fe0.2O3−δ phase before the material densification, the nanoparticles were naturally decorating the grain boundaries limiting the diffusion of metal ions and inhibiting the heterogeneous nucleation of a different phase in the GB which is detrimental for the cathode activity. Nevertheless this approach is only applicable in those cases where an undesired phase is forming at the GBs. At present we can speculate that since the amorphous cathode materials have no GBs a reduction of cation migration with a combined enhancement of the cathode lifetime could be expected. Secondly, the oxygen diffusion and surface activity of the amorphous Type 1 LSC8020 cathode materials are enhanced with respect to the crystalline ones. The work of Evans et al.12 on partially amorphous LSC6040 films used in a micro-SOFC device showed a notable power density of 200–250 mW cm−2 at 400–450 °C. Januschewsky et al. also studied by impedance analysis LSC6040 films prepared at low temperature by the PLD technique. They observed low polarization resistances of the amorphous LSC films and very little thermal deactivation within a few days of operation.11 Thermal deactivation in this perovskite is mainly related to Sr surface enrichment.23 The observation that the samples with no/lower crystallinity do not show the same polarization resistance increase with time compared to the crystalline ones is in good agreement with the hypothesis that the absence of grain boundaries is reducing the strontium segregation at the surface.
| Material cathode | Exchange conditions | D* [cm2 s−1] | k* [cm s−1] |
|---|---|---|---|
| La0.8Sr0.2CoO3−δ | T ex = 400 °C | 3.4 × 10−13 | 1.2 × 10−8 |
| La0.6Sr0.4CoO3−δ | pO2 = 200 mbar | 5.2 × 10−13 | 4.4 × 10−8 |
| (La0.5Sr0.5)2CoO4 | [18O2] ∼ 90% | 1.2 × 10−13 | 4.1 × 10−9 |
| La0.5Sr0.5Mn0.5Co0.5O3 | Timeex ∼ 15 min | 2.2 × 10−15 | 2.1 × 10−10 |
The fully amorphous route to cathode preparation could potentially be an alternative approach for developing low temperature SOFC applications. In a more broadened vision it can be expected that the electrolyte materials such as YSZ, CGO, LSGM etc. could also function in the amorphous state (Type-1) since oxygen diffusion is also driven through oxygen vacancies as in the LSC material.
Symmetric and fuel cell tests
As proof-of-concept, a LT-SOFC cell consisting of Ni-YSZ/YSZ/ALSC has been prepared as described in the experimental section. The IV curves of the cell have been collected in wet hydrogen at two different temperatures, 350 °C and 400 °C, as shown in Fig. 7(a).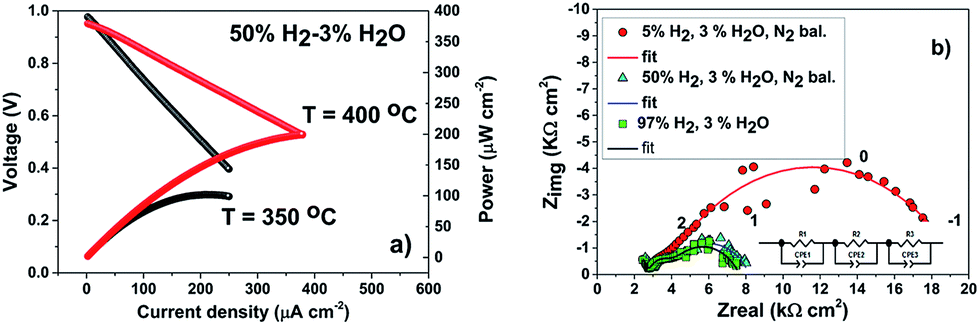 | ||
| Fig. 7 IV and EIS curves for the fuel cell Ni-YSZ/YSZ/ALSC. (a) I–V curves measured at 350 °C and 400 °C respectively. (b) Nyquist plot of the cell impedance at 350 °C for different H2 to N2 concentration. Nitrogen has been used as carrier gas and the water content maintained at 3%. The numbers close to the data points are the logarithm of the frequency. | ||
Even though the maximum power density is relatively low (200 μW cm−2 at 400 °C), mainly related to the large ohmic loss contribution corresponding to the thick YSZ electrolyte, it can be concluded that the deposited amorphous cathode can work in this range of temperatures. Power density has been calculated taking in account the free cathode surface area (75% of the area was occupied by the sputtered gold current collector, see ESI†). In Fig. 7(b) the impedance spectra for the fuel cell under OCV conditions at three different concentrations of hydrogen are presented. As can be seen, the impedance spectra can be described by three resistances in series with their coupled constant phase elements (3 semicircles). The high and intermediate frequency (>100 Hz) arcs correspond to the YSZ bulk and grain boundary contributions as the associated capacitances are ca. 197 pF (bulk) and 373 nF (grain boundaries). These values do not change dramatically with the hydrogen concentration. However, the low frequency resistance value that corresponds to the electrode contributions (<100 Hz), is particularly reduced when the hydrogen concentration increases from 3% to 50%. This decrease is mainly attributed to the anode. However, when the hydrogen concentration is further increased from 50% to 100% the resistance reduction is less pronounced. In these conditions on the contrary, the cathode polarization may dominate the low frequency electrode resistance. To have a better understanding of the influence of the amorphous cathode polarization, a symmetric cell of ALSC/YSZ/ALSC of the same commercial electrolyte ∼230 μm thick YSZ, the same ALSC film thickness (∼300 nm) deposited by PLD at room temperature on both sides, has also been prepared. From the symmetrical cell EIS results presented in Fig. 8(a) and (b), at least four different resistance contributions can be observed. For clarity, the lowest temperature measurement Fig. 8(a) (310 °C) and the highest temperature (415 °C) Fig. 8(b) have been presented. The first two arcs at higher frequencies correspond to the YSZ electrolyte bulk and grain boundaries respectively (see ESI†). One of the two main contributions to the total resistance of the symmetric cell are otherwise related to the middle-low frequency (Ri) arc associated with the YSZ–LSC interface. This resistance corresponds to the transfer of oxide ions O2− across the electrode-YSZ interface. Generally, the interface resistance between YSZ and LSC reported in the literature has been associated with the formation, due to the high sintering temperature, of an insulating resistive La2Zr2O7 (LZO) and/or SrZrO3 (SZO) layer at the electrolyte interface.45–49 This resistance can be greatly reduced simply by adding a different electrolyte thin film (CGO) in between the YSZ and the LSC layer. The introduction of a 1 μm CGO film for example reduced the interface resistance from 11.95 to 0.017 Ω cm2 at 800 °C as reported by Charojrochkul et al.50 In this study, the cause of this large interface resistance must be different since the LSC has been deposited at RT and the cell heated, during the EIS analysis to a maximum temperature of 415 °C with, theoretically, no LZO formation.
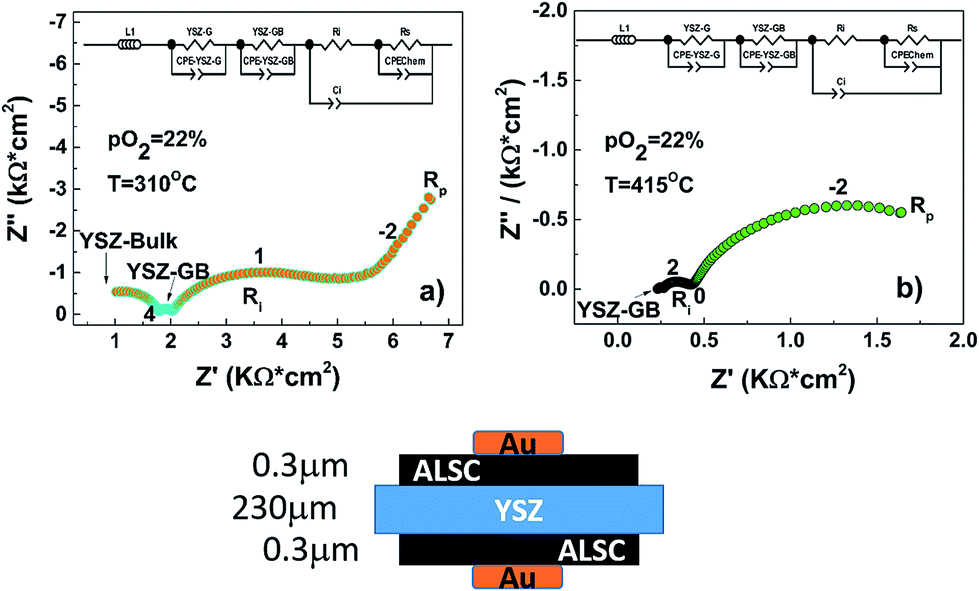 | ||
| Fig. 8 EIS curves at OCV for the symmetric cell ALSC/YSZ/ALSC. These particular spectra have been recorded at 310 °C (a) and 415 °C (b) respectively with a pO2 of 22% and an applied voltage of 10 mV. In total four different arcs are observable; from high to low frequencies they are associated respectively to: YSZ-grain, YSZ-grain boundaries, Ri-interfacial (YSZ–ALSC) resistance and Rp the cathode surface resistance. Numbers close to the data points represent the logarithm of the frequency. | ||
In addition the activation energy for the interfacial resistance when LZO and/or SZO form at the interface has been reported to be around 1.6 eV by Yang et al.51 In the EIS analysis presented in the ESI† an activation energy of 1.34 eV has been calculated. The measured interface capacitance value is generally associated with a Schottky barrier caused by the electronic carriers at the YSZ/LSC interface and in this case it is comparable with the one reported by Baumann et al.52 for a LSFC dense thick film. The experimental capacitance value, of the order of 40 μF cm−2, is too large to be purely related to an electrostatic double layer capacitance. A chemical change in the stoichiometry of the electrode/electrolyte interface must be at the origin of this capacitance, as argued by Baumann (ESI†). The oxygen dependence of the interfacial resistance with the oxygen pressure is close to zero as expected. To disclose the true nature of this interface amorphous-crystalline resistance further experiments replacing the electrolyte material for one with higher performance at such a low operational temperature (400 °C) such as CGO or LSGM should be implemented.
The second and main contribution to the total resistance is instead related to the dense thick film cathode surface. The dominant Rp resistance is associated with the oxygen exchange at the surface of the electrode. The critical thickness Lc calculated from the IEDP experiments and defined as D*/k* is of the order of 243 nm. Since the cathode thickness is not lower than the critical thickness any contribution from the chemical cathode bulk oxygen diffusion to the Rp cannot be excluded. The oxygen reduction reaction is also limited by surface diffusion of adsorbed oxygen and it can be further described by different sub-steps determining the overall rate-limiting step. The experimental oxygen partial pressure power dependence (m) of the cathode resistance Rp for the ALSC film decreases from m = 0.23 at 338 °C to m = 0.11 at 415 °C (see ESI†). At this very low temperature the rate determining step seems to be mostly controlled by atomic oxygen species charge transfer (pO2m with m ∼ 1/4 at 338 °C):
| Oad + e′ = O−ad |
On increasing the measurement temperature to 415 °C the electronic conductivity of the amorphous film increases and the dependence with pO2 decreases to a value close to 1/8 (0.11) probably related to the following charge-transfer process as limiting step:53
| O−ad + e− → O2−ad |
The sheet resistance defined as the electron migration resistance from the current collector to the active reaction site on the cathode surface is responsible for the large Rp observed. This is in agreement with both the loss of electronic conductivity of the amorphous ALSC8020 material and the usage as a current collector of a large gold mesh (∼1 mm). For an extended explanation on the EIS of the symmetric cell, refer to the ESI.† The limitation related to the lower electronic conductivity of the amorphous material should be considered in the design of the LT-SOFC current collectors. The area specific resistance (ASR) of the amorphous ALSC8020 cathode has been calculated to be 850 Ω cm2 at 415 °C in 22% of pO2. After the first set of EIS measurements at different temperatures and pO2 the ALSC symmetric sample has been analysed by XRD, Fig. 9(a). The only diffraction reflections observable correspond to the YSZ electrolyte and no sign of LSC crystallisation is visible (orange patterns). The same sample has been then remounted for EIS measurements and thermally treated at 600 °C for 3 h to induce the LSC material crystallisation. In Fig. 9(b) the impedance analysis measured at 364 °C and pO2 of 94% before (green) and after crystallisation (black) have been compared. At first sight the analyses seems to be inconsistent: the impedance values associated with the YSZ-bulk and the YSZ-GB are clearly lower after the crystallisation while they both should have remained unchanged. In these EIS symmetric cell measurements a round gold mesh current collector of 10 mm diameter has been employed while the deposited LSC cathode had instead a diameter of 18 mm. The area ratio between cathode and current collector was 3.24. A similar number can be obtained on calculating the ratio of the YSZ-bulk to the YSZ-GB impedance values before and after crystallisation, 3.5 and 3.3 respectively. It can be assumed that after crystallisation the LSC film electronic conductivity would have increased to its bulk value, allowing the current lines to be distributed through the full length of the LSC cathode as represented in the sketch in Fig. 9(c). The symmetric cell volume analysed before and after crystallization is effectively changing because the cathode, crystallizing, recovers its macroscopic electronic conductivity. In the Bode representation, Fig. 9(d), the characteristic frequencies associated with the Rp and Ri, change after crystallisation suggesting a change in the mechanism of oxygen diffusion and of the interface resistance as well. On the contrary the characteristic frequency associated with the YSZ-GB remains constant as expected. Further investigations will be necessary to obtain a better insight into the amorphous cathode oxygen reduction mechanism. After the crystallization and the EIS characterization, XRD analysis has revealed the appearance of the (020) diffraction peak of the LSC phase (black pattern) Fig. 9(a). It is worth emphasising that the lower electronic conductivity of the amorphous cathode has no detrimental effect on the IEDP experiments. The localised electronic conductivity is still able to promote the oxygen reduction reaction. Otherwise, considering an amorphous solid oxide fuel cell device, a limitation on the current collector maximum mesh dimension can be expected; the maximum mesh area will limit the cathode active surface available.
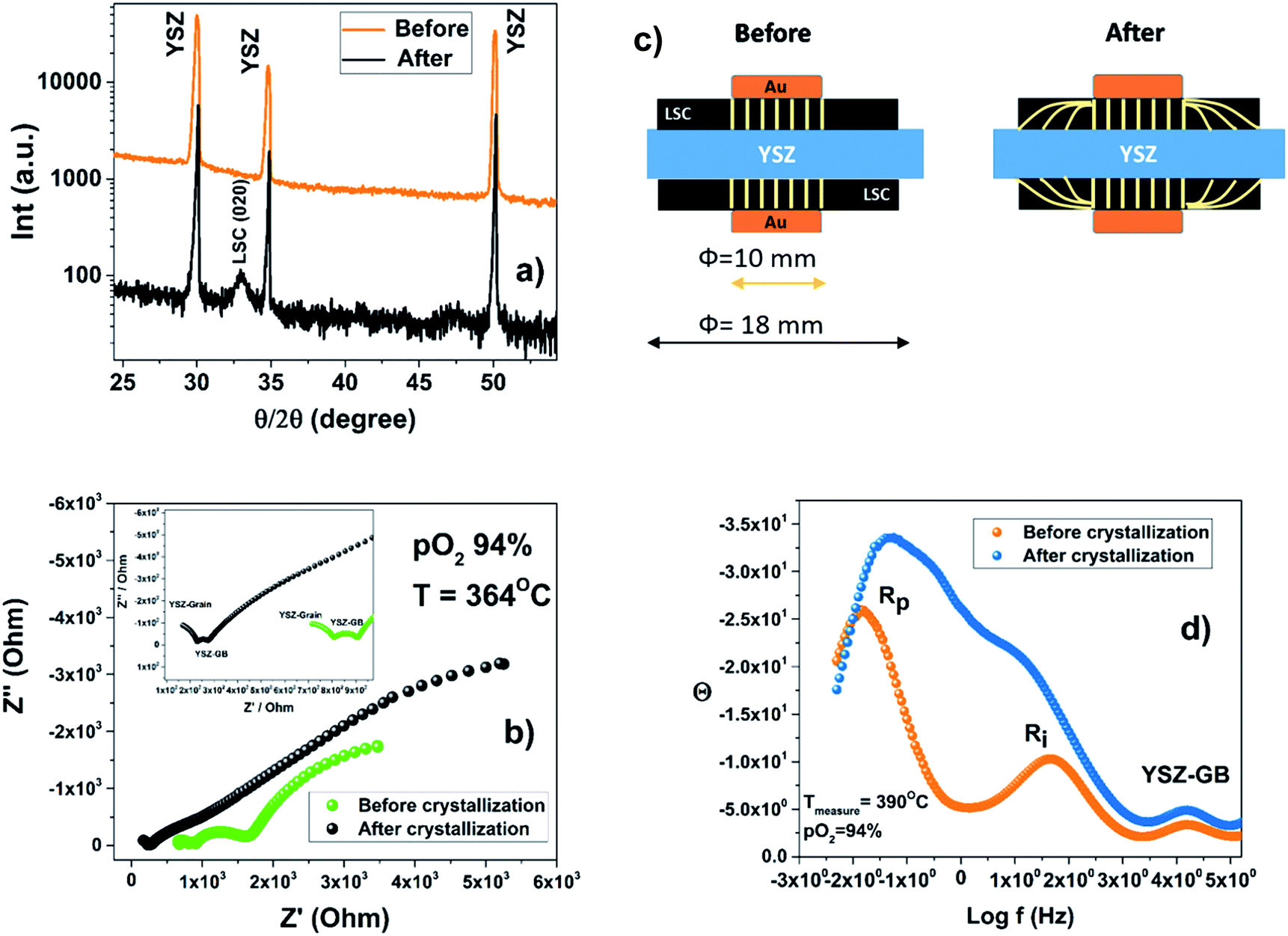 | ||
| Fig. 9 Comparison of the ALSC/YSZ/ALSC sample before and after crystallisation: (a) XRD theta two-theta scan: in the black pattern the diffraction peak of the LSC (020) reflection indicates the crystallization of the LSC film. (b) Impedance analysis at OCV at 364 °C and under a pO2 of 94%. (c) The sketch represents the current lines during the EIS analysis. After crystallization the current lines are not limited by the gold current collector area but go through the full LSC sample length. (d) Bode plot of the sample analysed at 390 °C at a pO2 of 94%: a change in the characteristic frequency related to Rp and Ri is observed when the LSC cathode crystallizes. | ||
Even though the ASR measured in the symmetric cell is high, at this stage of the work it was not a priority to pursue the best performance of the amorphous cathode in a fuel cell device but otherwise to prove its feasibility. To demonstrate that an amorphous cathode could be a solid technological alternative for LT-SOFC applications will require significant further development. A different electrolyte material, with higher performance at low temperature, a porous cathode with higher ionic and electronic conductivities and platinum anode would provide a better indication of the amorphous-cathode limitations. Tensile strained thin films have shown the best improvement in the cathode catalytic activity nevertheless due to the very strict growth conditions which require a single crystal, their implementation in devices is very unlikely. Amorphous route otherwise can be much more easily employed, not only in LT-SOFC but also in oxide based photocatalysts or redox-based memristive switches.
Conclusions
Low temperature PLD deposition has been demonstrated to be an effective method to produce amorphous cathode thick films (Type-1) to be employed in LT-SOFCs. Amorphous cathodes maintain their oxygen diffusion characteristics and oxygen surface activity that is important for an electrode to function in a SOFC device. In particular, the amorphous LSC8020 films have shown higher D* and k* values compared to the crystalline material. This performance could be related to the lower density of the amorphous film that might provoke a reduction of the thermodynamic energetic barrier for the oxygen diffusion and possibly even provide a different oxygen pathway from the ‘’saddle point’’ mechanism established for crystalline perovskite materials. This hypothesis is supported by a decrease, for the amorphous LSC8020 material, of the oxygen self-diffusion enthalpy compared to crystalline materials. Even though the amorphous ALSC8020 films have a much lower electronic conductivity compared to the crystalline ones, the increase in the oxygen mobility from one side and the increase in the surface oxygen vacancies related to the weakening of the Co–O bonds are the key factors in the enhancement of the cathode material catalytic properties. The general applicability of the amorphous route for low temperature SOFC applications has also been tested by preparing different amorphous cathode film materials: ALSC6040, ALSC214 and ALSMC. Each of these amorphous cathodes mainly maintain their characteristic surface activity and diffusivity crystalline properties. This research work has clearly highlighted a new route to cathode preparation for low temperature SOFC applications. Further tests will be needed to prove the real technological applicability of the amorphous route.Experimental
Film fabrication
The La0.8Sr0.2CoO3−δ, La0.6Sr0.4CoO3−δ, (La0.5Sr0.5)2CoO4 and La0.5Sr0.5Mn0.5Co0.5O3−δ PLD films were grown using a Complex Pro 201 KrF excimer laser (248 nm) at 10 Hz pulse repetition, at a laser fluence of around 1 J cm−2 (80 mJ in 8 mm2 spot size). During the film depositions a pO2 of 32 mTorr of pure oxygen was used. Substrates were commercial single crystals of Si (001) (Crystek, GmbH). Substrates temperature were set in the range between RT-750 °C. After the film deposition, an in situ film oxidation has been performed at a pO2 pressure of 600 Torr. The sample cooling rate was 10 °C min−1 after the PLD growth to reduce the surface defect formation as much as possible. The La0.8Sr0.2CoO3−δ target was prepared with a Praxair commercial powder, uniaxially and isostatically pressed in a 25 mm target before been densified at T = 1200 °C. The La0.6Sr0.4CoO3−δ and (La0.5Sr0.5)2CoO4 powders were instead prepared by citrate method and sintered in the same way as the LSC8020 target.General characterization
X-ray diffraction analyses, theta-two theta scan and reflectometry analysis, were performed in a four circle XRD PANAlytical Empyrean diffractometer with CuKα radiation (line-focus). The thick film surface characterization has been executed using a Veeco AFM instrument in tapping-mode.μ-Raman characterization
Raman spectroscopy was performed using a WiTEC 532 nm laser line, 2 mW power, and a 900 lines per mm grating coupled to a CCD detector. Mapping was performed using a 0.3 μm step size and a 0.5 s acquisition time per spot.Low energy ion scattering (LEIS): surface and sub-surface chemical composition
The Qtac100 LEIS instrument (ION-TOF GmbH) operated with a pure helium ion source at 3 keV was selected as the analysis primary beam. The primary beam scanning area was maintained at 750 μm2 in each analysis to ensure representative chemical information of the surface. This instrument is coupled with a secondary ion beam (Ar+) that allows the surface to be gently sputtered and provide an insight into the sub-surface composition as well (generally used to study the first 10 nm). The secondary argon beam has been employed with an energy of 0.5 keV and rastered on the sample surface over 1500 μm2. The LEIS depth profiling analysis has been run alternating the primary He+ beam with the Ar sputtering beam with a pause of one second in between. The He+ 3 keV ion current measured before and after the analysis by a Faraday cup was around 5000 pA, this is equivalent of an ion dose of around 2–3 × 1014 ions per cm2. In the case of the secondary argon beam the current was of the order of 100 nA corresponding to a total dose of around 4–5 × 1014 ions per cm2.Van Der Pauw electrical characterisation
To measure the thick film total conductivities a Linkam four probe chamber has been employed with the four points Van Der Pauw measurement configuration, more suitable for thin film measurements. Two of the tungsten probes, covered in gold, were connected to a DC current supply by a Keithley 220 current source. Voltage was measured with the other two probes by an Agilent 34401A multimeter. The applied current was adjusted to verify the linearity of the voltage measurement and to avoid overheating of the sample tip contacts.TEM specimen preparation
TEM cross-sections from the thick film samples were prepared using a focussed ion beam (FIB) system, FEI Helios nanolab 600, (2–30 keV Ga+ incident beam energy with currents of 16 pA to 21 nA). A 50 nm thick coating of Cr was evaporated onto the sample before FIB milling to protect the region of interest and prevent charging. Coarse milling was done at 30 kV and then samples were thinned to approximately 50 nm thickness. The specimens were FIB polished at the last stage with 5 keV and then 2 keV Ga+ to reduce the damage caused by the high energy Ga+ beam. Samples were examined in a JEOL JEM2100F operating at 200 kV. Field limiting aperture of 5 μm was used to obtain the selected area electron diffraction (SAED). The scales of SAED patterns and HRTEM images were calibrated against the external standards of gold particles on a carbon film. The energy dispersive X-ray (EDX) spectra (Oxford Instruments) were collected in the scanning transmission electron microscopy mode with a probe size of 1 nm.18O2 isotope exchange depth profile
The samples were inserted into a quartz tube connected to a high vacuum system. The system was first pumped down at around 10−7 mbar to reduce to the maximum the residual gas inside the system. Afterwards the sample was heated at Texchange in an ultra-pure 200 mbar of 16O2 atmosphere (99.9999%) with natural abundance of 18O2 (0.2%). This step lasts for ten times the isotope exchange time and is needed to neutralize any oxygen defect related to the PLD-deposition. The very reducing growth environment of the PLD chamber could have produced an oxygen understoichiometry of the films. Finally the sample was exchanged in 200 mbar of 18O2 enriched atmosphere (90%) for a fixed annealing time (ca. 15 min). The annealing time is measured and adjusted for the heating and cooling ramp using the Killoran method.54 Once the exchange was completed the sample was quenched to room temperature. The sample was finally analysed using a FEI-200-SIMS equipped with a Hidden mass spectrometer. The analysis area was first selected by SEM imaging. The FIB source was a 25 keV gallium gun and the current used in the analysis 300 pA. The analysis sputtered areas were maintained at 15 × 15 μm2 in all the analysis. Six analyses in different sample areas were collected to prove the homogeneity of the samples. The analyses were performed in SIMS-dynamic mode.Fuel cell test and EIS symmetric cell analysis
The sample electrolyte consisted of commercial 8YSZ (Fuel Cell materials, 230 μm thick, 20 mm diameter). In the case of the symmetric cell sample preparation, a tape with a circle of 18 mm diameter was applied to the YSZ electrolyte as a mask. The PLD cathode deposition was then carried out at room temperature on both sides. For the fuel cell test sample a tape cast anode (60 w% NiO–40 w% 8YSZ) was first deposited on one side of the electrolyte. The anode was then reduced for 2 hours at 700 °C, in 5% H2, 3% H2O with N2 as balance. The LSC amorphous cathode was finally deposited at room temperature on the other side. For the fuel cell measurements, silver wire and paint were used to make contact between the anode and the potentiostat. On the cathode side a gold grid was deposited on the surface and a silver wire contacted to it. Impedance spectroscopy was used to analyse the cell in open circuit conditions (OCV) under 3 different hydrogen concentrations, 5%, 50% and 100% with water humidifying the gas (3% H2O) and N2 as balance. For the impedance (10 mV signal) and the IV curves a potentiostat (Autolab PGSTAT 302) was used. Symmetric cell characterization has been performed in a Probostat station using a Solartron modulab impedance analyser. The impedance linear response with the voltage has been tested from 1 mV to 100 mV. A voltage of 10 mV was applied in all the EIS measurements. Frequency range was scanned between 1 MHz and 0.005 Hz. Gold circle grids have been used as electrodes to avoid any catalytic contribution. The oxygen partial pressure was controlled through two gas mass flow controllers connected to the Probostat station in the range of 0.5–94% of the total gas pressure and monitored with the programmable gas analyser (542 by Illinois Instruments, Inc (U.S)).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Authors acknowledge funding from the EPSRC grant (EP/M014142/1). AC and SP would like also to acknowledge the support of King Abdullah University of Science and Technology, who partially funded this work through the academic excellence alliance programme. We acknowledge Dr Antonio Bertei for the helpful discussion about the EIS analysis and Miss Celeste van den Bosch for the LSMCO target preparation.References
- J. H. Choi, Y. Mao and J. Chang, Development of hafnium based high-k materials-A review, Mater. Sci. Eng., R, 2011, 72, 97 CrossRef.
- M. Passlack, E. F. Schubert, W. S. Hobson, M. Hong, N. Moriya, S. N. G. Chu, K. Konstadinidis, J. P. Mannaerts, M. L. Schnoes and G. J. Zydzik, Ga2O3 films for electronic and optoelectronic applications, J. Appl. Phys., 1995, 77, 686 CrossRef CAS.
- H. Akinaga and H. Shima, Resistive Random Access Memory (ReRAM) Based on Metal Oxides, Proc. IEEE, 2010, 98, 2237 CrossRef CAS.
- H. Shima, F. Takano, H. Muramatsu, H. Akinaga, I. H. Inoue and H. Takagi, Control of resistance switching voltages in rectifying Pt/TiOx/PtPt/TiOx/Pt trilayer, Appl. Phys. Lett., 2008, 92, 043510 CrossRef.
- S. Clima, Y. Y. Chen, R. Degraeve, M. Mees, K. Sankaran, B. Govoreanu, M. Jurczak, S. De Gendt and G. Pourtois, First-principles simulation of oxygen diffusion in HfOx: Role in the resistive switching mechanism, Appl. Phys. Lett., 2012, 100, 133102 CrossRef.
- Y. Kim, J. Heo, T. Kim, S. Park, M. Yoon, J. Kim, M. S. Oh, G. Yi, Y. Noh and S. K. Park, Flexible metal-oxide devices made by room-temperature photochemical activation of sol–gel films, Nature, 2012, 489, 128 CrossRef CAS PubMed.
- F. Utsuno, H. Inoue, I. Yasui, Y. Shimane, S. Tomai, S. Matsuzaki, K. Inoue, I. Hirosawa, M. Sato and T. Honma, Structural study of amorphous In2O3 film by grazing incidence X-ray scattering (GIXS) with synchrotron radiation, Thin Solid Films, 2006, 496, 95 CrossRef CAS.
- M. A. Rafea and N. Roushdy, Determination of the optical band gap for amorphous and nanocrystalline copper oxide thin films prepared by SILAR technique, J. Phys. D: Appl. Phys., 2009, 42, 015413 CrossRef.
- K. Nomura, H. Ohta, A. Takagi, T. Kamiya, M. Hirano and H. Hosono, Room-temperature fabrication of transparent flexible thin-film transistors using amorphous oxide semiconductors, Nature, 2004, 432, 488 CrossRef CAS PubMed.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, A perspective on low-temperature solid oxide fuel cells, Energy Environ. Sci., 2016, 9, 1602–1644 CAS.
- J. Januschewsky, M. Ahrens, A. Opitz, F. Kubel and J. Fleig, Optimized La0.6Sr0.4CoO3−δ Thin-Film Electrodes with Extremely Fast Oxygen-Reduction Kinetics, Adv. Funct. Mater., 2009, 19, 3151–3156 CrossRef CAS.
- A. Evans, J. Martynczuk, D. Stender, C. W. Schneider, T. Lippert and M. Prestat, Low-Temperature Micro-Solid Oxide Fuel Cells with Partially Amorphous La0.6Sr0.4CoO3−δ Cathodes, Adv. Energy Mater., 2015, 5, 1400747 CrossRef.
- J. A. Kilner, B. C. H. Steele and L. Ilkov, Oxygen self-diffusion studies using negative-ion secondary ion mass spectrometry (SIMS), Solid State Ionics, 1984, 12, 89–97 CrossRef CAS.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Development of a novel SIMS technique for oxygen self-diffusion and surface exchange coefficient measurements in oxides of high diffusivity, Solid State Ionics, 1992, 53–56, 859–867 CrossRef CAS.
- J. Als-Nielsen and D. McMorrow, Elements of Modern X-Ray Physics, Wiley, New York, 2001 Search PubMed.
- http://www.panalytical.com/Xray-diffraction-software/Reflectivity.htm .
- J. F. Ziegler, The Stopping and Range of Ions in Matter, Pergamon Press, 1990, vol. 2–6, pp. 1977–1985 Search PubMed.
- H. Jalili, J. W. Han, Y. Kuru, Z. Cai and B. Yildiz, New Insights into the Strain Coupling to Surface Chemistry, Electronic Structure, and Reactivity of La0.7Sr0.3MnO3, J. Phys. Chem. Lett., 2011, 2, 801 CrossRef CAS.
- T. T. Fister, D. D. Fong, J. A. Eastman, P. M. Baldo, M. J. Highland, P. H. Fuoss, K. R. Balasubramaniam, J. C. Meador and P. A. Salvador, In situ characterization of strontium surface segregation in epitaxial La0.7Sr0.3MnO3 thin films as a function of oxygen partial pressure, Appl. Phys. Lett., 2008, 93, 151904 CrossRef.
- G. M. Rupp, H. Téllez, J. Druce, A. Limbeck, T. Ishihara, J. Kilner and J. Fleig, Surface chemistry of La0.6Sr0.4CoO3−δ thin films and its impact on the oxygen surface exchange resistance, J. Mater. Chem. A, 2015, 3, 22759–22769 CAS.
- E. Bucher, A. Egger, G. B. Caraman and W. Sitte, Stability of the SOFC Cathode Material (Ba, Sr) (Co, Fe)O3−δ in CO2-Containing Atmospheres, J. Electrochem. Soc., 2008, 155, B1218 CrossRef CAS.
- D. Oh, D. Gostovic and E. D. Wachsman, Mechanism of La0.6Sr0.4Co0.2Fe0.8O3 cathode degradation, J. Mater. Res., 2012, 27, 1992 CrossRef CAS.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chemical heterogeneities on La0.6Sr0.4CoO3−δ thin films correlations to cathode surface activity and stability, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- P. Hjalmarsson, M. Søgaard and M. Mogensen, Electrochemical performance and degradation of (La0.6Sr0.4)0.99CoO3−δ as porous SOFC-cathode, Solid State Ionics, 2008, 179, 1422–1426 CrossRef CAS.
- M. Kubicek, A. Limbeck, T. Frömling, H. Hutter and J. Fleig, Relationship between cation segregation and the electrochemical oxygen reduction kinetics of La0.6Sr0.4CoO3−δ thin film electrodes, J. Electrochem. Soc., 2011, 158, B727–B734 CrossRef CAS.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, Cation size mismatch and charge interactions drive dopant segregation at the surfaces of manganite perovskites, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- M. Kubicek, G. M. Rupp, S. Huber, A. Penn, A. K. Opitz, J. Bernardi, M. Stöger-Pollach, H. Hutter and J. Fleig, Cation diffusion in La0.6Sr0.4CoO3−δ below 800 °C and its relevance for Sr segregation, Phys. Chem. Chem. Phys., 2014, 16, 2715 RSC.
- M. Niania, R. Podor, B. Britton, S. Skinner and J. Kilner, High temperature and in situ study of SrO surface precipitation on perovskite ceramics, European Microscopy Congress 2016: Proceedings, 2016 Search PubMed.
- M. C. Verbraeken, E. Suard and J. T. S. Irvine, Structural and electrical properties of calcium and strontium hydrides, J. Mater. Chem., 2009, 19, 2766–2770 RSC.
- https://sourceforge.net/projects/trace-x/ .
- R. A. De Souza and J. A. Kilner, Oxygen transport in La1−xSrxMn1−yCoyO3±δ perovskites: Part I Oxygen tracer diffusion, Solid State Ionics, 1998, 106, 175–187 CrossRef CAS.
- T. Ishigaki, S. Yamauchi, K. Kishio, J. Mizusaki and K. Fueki, Diffusion of oxide ion vacancies in perovskite-type oxides, J. Solid State Chem., 1988, 73, 179–187 CrossRef CAS.
- W. Sitte, E. Bucher and W. Preis, Nonstoichiometry and transport properties of strontium-substituted lanthanum cobaltites, Solid State Ionics, 2002, 154–155, 517–522 CrossRef CAS.
- J. A. Duffy, Bonding, Energy Levels & Bands in Inorganic solids, Longman Scientific & Technical first printed in 1990 Search PubMed.
- J. A. Kilner and R. J. Brook, A Study of Oxygen Ion Conductivity in Doped Nonstoichiometric Oxides, Solid State Ionics, 1982, 6(3), 237–252 CrossRef CAS.
- M. Cherry, M. S. Islam and C. R. A. Catlow, Oxygen Ion Migration in Perovskite-Type Oxides, J. Solid State Chem., 1995, 118, 125–132 CrossRef CAS.
- M. S. Islam, M. Cherry and C. R. A. Catlow, Oxygen Diffusion in LaMnO3 and LaCoO3 Perovskite-Type Oxides: A Molecular Dynamics Study, J. Solid State Chem., 1996, 124, 230–237 CrossRef CAS.
- J. Richter, P. Holtappels, T. Graule, T. Nakamura and L. J. Gauckler, Materials design for perovskite SOFC cathodes, Monatsh. Chem., 2009, 140, 985–999 CrossRef CAS.
- K. Kamata, T. Nakamura and T. Sata, On the State of d-Electrons in Perovskite-Type Compounds ABO3, Bull. Tokyo Inst. Technol., 1974, 120, 73–79 CAS.
- A. Kushima, S. Yip and B. Yildiz, Competing strain effects in reactivity of LaCoO3 with oxygen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(11), 115435 CrossRef.
- M. Pavone, A. M. Ritzmann and E. A. Carter, Quantum-mechanics-based design principles for solid oxide fuel cell cathode materials, Energy Environ. Sci., 2011, 4(12), 4933 CAS.
- B. Yildiz, “Stretching” the energy landscape of oxides—Effects on electrocatalysis and diffusion, MRS Bull., 2014, 39, 147–156 CrossRef CAS.
- Z. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, Surface Electronic Structure Transitions at High Temperature on Perovskite Oxides: The Case of Strained La0.8Sr0.2CoO3 Thin Films, J. Am. Chem. Soc., 2011, 133(44), 17696 CrossRef CAS PubMed.
- Y. Liu, X. Zhu, M. Li, R. P. O'Hayreand and W. Yang, Nanoparticles at Grain Boundaries Inhibit the Phase Transformation of Perovskite Membrane, Nano Lett., 2015, 15, 7678–7683 CrossRef CAS PubMed.
- A. Mitterdorfer and L. J. Gauckler, La2Zr2O7 formation and oxygen reduction kinetics of the La0.85Sr0.15MnyO3, O2(g)|YSZ system, Solid State Ionics, 1998, 111, 185–218 CrossRef CAS.
- L. Dieterle, D. Bach, R. Schneider, H. Stormer, D. Gerthsen, U. Guntow, E. Ivers-Tiffee, A. Weber, C. Peters and H. Yokokawa, Structural and chemical properties of nanocrystalline La0.5Sr0.5CoO3−δ layers on yttria-stabilized zirconia analyzed by transmission electron microscopy, J. Mater. Sci., 2008, 43, 3135–3143 CrossRef CAS.
- A. Martínez-Amesti, A. Larrãnaga, L. M. Rodríguez-Martínez, A. T. Aguayo, J. L. Pizarro, M. L. Nó, A. Laresgoiti and M. I. Arriortua, Reactivity between La(Sr)FeO3 cathode, doped CeO2 interlayer and yttria-stabilized zirconia electrolyte for solid oxide fuel cell applications, J. Power Sources, 2008, 185, 401–410 CrossRef.
- H. Yokokawa, N. Sakai, T. Kawada and M. Dokiya, Thermodynamic Considerations on Reactions on Perovskite Oxide Electrode and Zirconia, Denki Kagaku, 1989, 57, 821–828 CAS.
- H. Kamata, A. Hosaka, J. Mizusaki and H. Tagawa, Chemical compatibility of perovskite-type oxide La0.7Ca0.3Cr1−yCoyO3 with Y2O3 stabilized ZrO2, Mater. Res. Bull., 1995, 30, 679–687 CrossRef CAS.
- S. Charojrochkul, K.-L. Choy and B. C. H. Steele, Cathode/electrolyte systems for solid oxide fuel cells fabricated using flame assisted vapour deposition technique, Solid State Ionics, 1999, 121, 107–113 CrossRef CAS.
- Y. L. Yang, C. L. Chen, S. Y. Chen, C. W. Chu and A. J. Jacobson, Impedance Studies of Oxygen Exchange on Dense Thin Film Electrodes of La0.5Sr0.5CoO3−δ, J. Electrochem. Soc., 2000, 147(11), 4001–4007 CrossRef CAS.
- F. S. Baumann, J. Fleig, H. U. Habermeier and J. Maier, Impedance spectroscopic study on well-defined (La,Sr)(Co,Fe)O3−δ model electrodes, Solid State Ionics, 2006, 177, 1071–1081 CrossRef CAS.
- Y. Gong, X. Li, L. Zhang, W. Tharp, C. Qin and K. Huanga, Promoting Electrocatalytic Activity of a Composite SOFC Cathode La0.8Sr0.2MnO3+δ/Ce0.8Gd0.2O2−δ with Molten Carbonates, J. Electrochem. Soc., 2014, 161(3), F226–F232 CrossRef CAS.
- D. R. Killoran, The Effective Duration of a Linear Slow-Cool, J. Electrochem. Soc., 1962, 190, 172 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00606c |
| ‡ Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2018 |