
Heteroatom-doped carbonaceous electrode materials for high performance energy storage devices
Afzal
Shah
 *ab,
Anum
Zahid
a,
Hanif
Subhan
a,
Azeema
Munir
a,
Faiza Jan
Iftikhar
a and
Muhammad
Akbar
a
*ab,
Anum
Zahid
a,
Hanif
Subhan
a,
Azeema
Munir
a,
Faiza Jan
Iftikhar
a and
Muhammad
Akbar
a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan
bDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, Toronto M1C 1A4, Canada. E-mail: afzal.shah@utoronto.ca; afzals_qau@yahoo.com
First published on 21st March 2018
Abstract
The development of cost affordable and efficient catalysts for speeding up electrode reactions is key for boosting up the commercial viability of fuel cells, metal air batteries and other energy production and storage devices. Although state-of-the-art platinum demonstrates excellent performance as an electrode material, its susceptibility to poisoning, high cost and limited availability have blocked the road for large scale commercialization of Pt based energy conversion/storage devices. In this connection carbon nanomaterials are attractive choices on account of their corrosion resistant properties, affordable cost, widespread availability, unique surface features, environmentally acceptable nature and practical capability for addressing the global energy challenge. Moreover, the catalytic role of carbon nanomaterials is expected to be further upgraded by the incorporation of dopants into their framework. Hence, the current review article is at the heart of underlining the recent progress achieved under the umbrella of heteroatom-doped carbons to accelerate reactions in advanced energy generation devices.
 Afzal Shah | Dr Afzal Shah is a tenured Associate Professor at Quaid-i-Azam University (QAU), Islamabad and a visiting professor at the University of Karachi, Pakistan. He earned his Ph.D. in 2010 under the supervision of Prof. Dr Rumana Qureshi. In 2014 he worked as a postdoctoral fellow in the research group of Prof. Kraatz at the University of Toronto. He is the recipient of several awards from QAU and Pakistan Council for Science and Technology including the best researcher award of 2013 as a gold medal. He has published 150 articles in peer reviewed journals. He has guided 5 Ph.D. and 30 M.Phil students. His research interests include designing of electrocatalysts for efficient water splitting, proton coupled electron transfer reactions of biologically important molecules, development of electrochemical sensors for the detection of cancer biomarkers, and designing of drug delivery systems. |
 Anum Zahid | Anum Zahid got her M.Phil degree (Physical Chemistry) from Quaid-i-Azam University, Islamabad in 2015. She is doing Ph.D. in the research group of Dr Afzal Shah. She has won a Higher Education Commission of Pakistan sponsored scholarship for doing a part of her Ph.D. research in Monash University Australia. Currently she is availing of a Tubitak fellowship at Ankara University, Turkey, under the supervision of Prof. Sibel Ozkan. Her research interests include surfactant chemistry, electrochemistry and nanomaterials. |
 Hanif Subhan | Hanif Subhan earned his M.Phil degree (Physical Chemistry) from Quaid-i-Azam University Islamabad, Pakistan, under the supervision of Dr Afzal Shah in 2014. He is currently doing his Ph.D. research at the University of Malakand, Pakistan. His research is focused on the development of heteroatom-doped carbonaceous materials and earth abundant non-noble metal based electrocatalysts for energy generation devices. |
 Azeema Munir | Azeema Munir received her M.Phil degree (Physical Chemistry) from Quaid-i-Azam University, Islamabad in 2013. She is doing Ph.D. in the research group of Dr Afzal Shah. Availing of a Higher Education Commission scholarship she completed a part of her Ph.D. research work in the group of Prof. Heinz-Bernhard Kraatz (University of Toronto) on a project related to the development of nanobiosensors for drug analysis and probing the mechanism of the electron transfer reaction in peptides. Currently she is availing of a Tubitak fellowship at Ankara University, Turkey, under the supervision of Prof. Sibel Ozkan. Her research interests include modification of electrode surfaces for the sensitive and selective detection of cancer biomarkers and wastewater contaminants. |
 Faiza Jan Iftikhar | Dr Faiza Jan Iftikhar is a Post Doc fellow in the Department of Chemistry, Quaid-i-Azam University (QAU) under the mentorship of Dr Afzal Shah, a tenured Associate Professor in QAU. She earned her Ph.D. in Dec., 2009 under the supervision of Prof. Guenter Grampp from the Institute of Physical and Theoretical Chemistry, Graz University of Technology, Austria with a foreign scholarship from the Higher Education Commission, Pakistan. Later she worked as a postdoctoral fellow in the research group of Prof. PGL Baker at the University of Western Cape, South Africa from 2010–2012 on electrochemical and biosensors harnessing sensor technology as a tool to employ conductive polymers immobilized with nano-sized particles for the sensing of environmental toxins. She has worked as an assistant professor from March 2013 to March 2016 at the International Islamic University Islamabad, Pakistan in the Environmental Sciences Department. Her research interests include biosensors, nanosensors, drug analysis and preparation of electrolysers. |
 Muhammad Akbar | Muhammad Akbar was born on 15th April 1990 in Multan (the oldest city of Pakistan). He did an M.Phil/MS in Physical Chemistry from Quaid-i-Azam University Islamabad under the supervision of Dr Afzal Shah. His research is focussed on the electrosynthesis of bimetallic and tri-metallic electrocatalysts for efficient water splitting with excellent results towards the oxygen evolution reaction. |
1. Introduction
The world is currently confronting a seemingly insurmountable bottleneck in terms of energy crisis due to rapid exhaustion of conventional energy reserves.1,2 Scientists are trying to achieve economical, sustainable, faster and safer production of energy via next-generation rechargeable and portable electronic devices.3,4 Their endeavours for rapid energy generation have led to the development of fuel cells which are viable electrochemical tools that hold great promise for substantially decreasing the global reliance upon fossil fuels.5,6 For a H2/O2 fuel cell in an acidic or a basic medium, oxidation of fuel in a membrane electrode assembly occurs at the anode from where electrons transfer via an electrical circuit to reach the cathode where reduction reactions take place.7,8 Researchers are trying to speed up the electrode reactions by using such electrocatalysts which could minimize the overpotential of the sluggish reactions in energy devices.9,10 Pt is an excellent material for catalysing reactions in fuel cells.11,12 However, the high cost, corrosion and reduction of the electrocatalytic activity of Pt by excess HO2− produced at its surface13,14 summons the search for other electrocatalysts. In this regard carbonaceous materials bestowed with good electrical conductivity and resistance to passivation hold great promise as electrode materials.Investigations have been steered in the direction of exploring cheaper, sustainable and durable catalysts for fuel cells. Hence, the pursuit of alternative catalysts that could outperform Pt in catalysing electrode reactions has accelerated. In this regard non-noble metal catalysts are promising options for satisfying the rigorous performance targets due to their higher activity, cost affordability and abundant availability. However, the development of improved low-cost non-Pt cathode catalysts as a sustainable solution for commercializing fuel cells and adoption of practical synthesis strategies to overcome the impediments to fabricating electrocatalytic materials by fine tuning their properties is a formidable task.15–17 The reason could be the high target of 300 A cm−3 volumetric current density at 0.8 V iR-free cell voltage set by the US Department of energy for the oxygen reduction reaction (ORR) catalysing activity of non-noble metal catalysts in fuel cells working below 80 °C.18 In this scenario non-metallic heteroatom-doped multifunctional carbon derived materials with tailored electronic and mechanical properties are promising options for use as electrode materials in futuristic high performance energy devices.19–21
No matter how useful conventional batteries and capacitors may have been in the past, these barely meet the challenging demands of today's market. Hence, one practically viable approach for enhancing the performance of batteries involves the development of new materials which could boost energy and power densities, cycle life and the charge/discharge rate along with overcoming the problem of polarization.22,23 The current energy density of lithium ion batteries (LIBs) must be increased by 2 to 5 times to meet the performance criteria of electric vehicles.24 Similar is the case of supercapacitors which have substantial power density but their energy density still lags behind as compared to present day batteries. Thus, in the search for tackling this issue, nanohybrids that hybridize current battery–capacitors have rekindled interest in the fabrication of new materials that could satisfy the requirements of high power and energy densities.3 Low cost fabrication strategies leading to a rational design and control in the structure are daunting technological issues of concern, which the current review undertakes by critically examining the modification of electrode materials for achieving excellent charge/discharge capacities and sustainable measures to meet the requirements for high-performance next generation LIBs and supercapacitors.
2. Fuel cells
The discovery of transition metal porphyrins and phthalocyanines as efficient cathode catalysts for fuel cell applications25 led to further exploration for the discovery of new ORR electrocatalysts.26–30 In this regard hetero-atom-doped carbons (X-doped carbons) bestowed with fascinating ORR-activity are the subject of extensive investigations owing to the ability of these electrode materials for possibly surpassing the performance of expensive platinum in energy storage devices. Elements such as N, P, B, S, Se etc., with electronegativity different from that of carbon are purposely incorporated into the framework of carbonaceous materials by various methods to speed up the inherent sluggish oxygen reduction reaction.31–34 Dual28,29 and triple doping in carbon materials30 have led to further improvement of their ORR activity. The subsequent section is therefore focused on the recent developments in the use of X-doped carbonaceous materials for improving the performance of fuel cells.2.1 Carbon doping with nitrogen
Nitrogen is an attractive choice for doping carbon due to its relatively high electronegativity, high electron affinity, and availability of five valence electrons for bonding with carbon. In all probability, tailoring carbonaceous materials with nitrogen guest atoms was the beginning of research in the realm of nanostructured carbons for catalyzing the ORR. The discovery by Jasinski et al., in 1964 (ref. 25) paved the way for the exploration of N-doped carbons as exciting electrocatalytic materials. | ||
| Fig. 1 (a) Scanning electron microscope photograph of the synthesized vertically aligned nitrogen containing carbon nanotubes (VA-NCNTs) over the surface of a quartz substrate; (b) RRDE voltammetric curves 1–3 showing the ORR in air saturated 0.1 M KOH for Pt–C/GC, VA-CCNT/GC, and VA-NCNT (curve 3) electrodes respectively. Reprinted with permission from ref. 26 copyright 2009 American Association for the Advancement of Science; (c) polarization curves obtained at 1600 rpm showing the superior ORR accelerating performance of EDA-NCNTs compared to Py-NCNTs and Pt/C. Reprinted with permission from ref. 43 copyright 2009 American Chemical Society. | ||
The CVD-method is an attractive route for high yield and relatively low-temperature synthesis of NCNTs, but the use of expensive and toxic precursors and metal catalysts (Fe, Ni, Co etc.) in this method45,46 demands further improvement regarding commercialization and environmental friendliness. Particularly, the catalyst residues in the product (NCNTs) of the CVD-method may be difficult to eliminate completely47–51 which ultimately influences the ORR electrocatalytic activity. For instance, the CVD-grown VA-CNTs,26 N-CNCs,42 and graphene nanoshells44 have been found to contain residual catalyst (Fe) particles even after screening through harsh purification steps. Therefore, further investigations were carried out to unfold the actual role of metal residues in the ORR catalytic activity of materials.52–57 Once the ORR suppresses the role of metal impurities, their non-compatibility in fuel cells or associated environmental snags are ensured, then the research endeavours must be directed towards adopting virtually metal-free routes for the preparation of N-doped carbon nanotubes to possibly replace Pt and traditional metal catalysts in high performance environmentally benign fuel cells.
The catalogue of the reported applied metal-free protocols for the synthesis of NCNTs includes an arc-discharge approach, thermal annealing methods, hydrothermal and solvothermal treatments.38,58–61 Rao et al., employed an alumina template technique for the synthesis of aligned NCNTs possessing ∼8.0% nitrogen content.59 Their ORR performance was found comparable to Pt/C in terms of onset potential (−0.05 V). An enhanced steady state diffusion current was observed for the NCNT catalyst which displayed 0.91 mA cm−2 current density compared to 0.95 mA cm−2 of Pt/C measured at 1600 rpm at 0.1 V. Likewise, Wang et al., adopted a metal-free route for the preparation of NCNTs possessing high N-content (20 atom%).38 The resultant electrode material (NCNT/GC electrode) showed slightly lower ORR-potential and comparable current density relative to the Pt/C-electrode. But regarding selectivity, stability and CO-tolerance, the electrode demonstrated a significant improvement. Similarly the research team of Dai adopted a metal-free route, plasma etched technology for the fabrication of NCNTs.60 Their electrocatalyst showed ORR-performance better than their undoped counterparts, but with far less catalytic activity than Pt. In contrast the NCNTs synthesised by Yang et al., via direct annealing of carbon nanotubes and tripyrrolyl[1,3,5]triazine under alkaline conditions62 registered ORR performance quite close to Pt/C as displayed in Fig. 2a. Overall, the catalytic potential of NCNTs annealed at 900 °C (NCNT-900) was found comparable to that of Pt/C. Moreover, NCNT-900 at high loading (280 mg cm−2) demonstrated a greater current density and less negative onset potential (Fig. 2b) indicating the superior performance of NCNT-900 compared to that of Pt/C. Besides these findings, Borghei et al., coated acid functionalised few walled carbon nanotubes (FWCNTs) with polyaniline followed by high temperature pyrolysis. The resultant N-doped electrocatalyst showed Pt-like performance when tested in an alkaline environment.61 However, in spite of adopting a metal-free route for NCNT synthesis using a CVD-grown FWCNT post-purification method, the material still possessed some metal residues. Therefore, it is important to know about the synthetic route adopted for precursor preparation, particularly CNTs, while acquiring it from a commercial trader. Only then the ORR-activity of NCNTs achieved via virtually metal-free protocols including precursor synthesis could exclusively be conceived to stem from N-incorporation into the framework of carbonaceous materials. Apart from metal residues, the influence of variation in morphology, N-content and the N-configuration of NCNTs synthesised via CVD and exclusive metal-free routes must also be considered while assessing their ORR catalytic activity.63,64
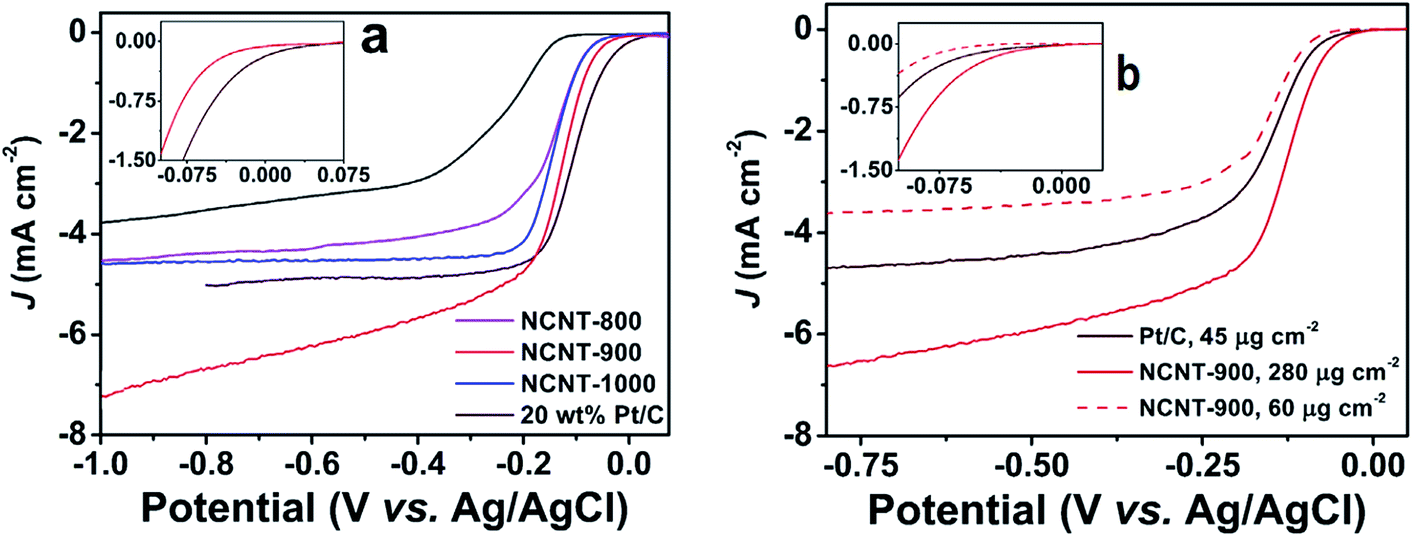 | ||
| Fig. 2 (a) Linear scan voltammograms recorded for NCNTs and Pt/C at 1600 rpm and a 10 mV s−1 scan rate in oxygen saturated 0.1 M KOH aq. solution; (b) LSVs of NCNT-900 obtained at 1600 rpm at a scan rate of 10 mV s−1 using different loadings in oxygen saturated 0.1 M KOH aq. solution. Reprinted with permission from ref. 62 copyright 2015 Elsevier. | ||
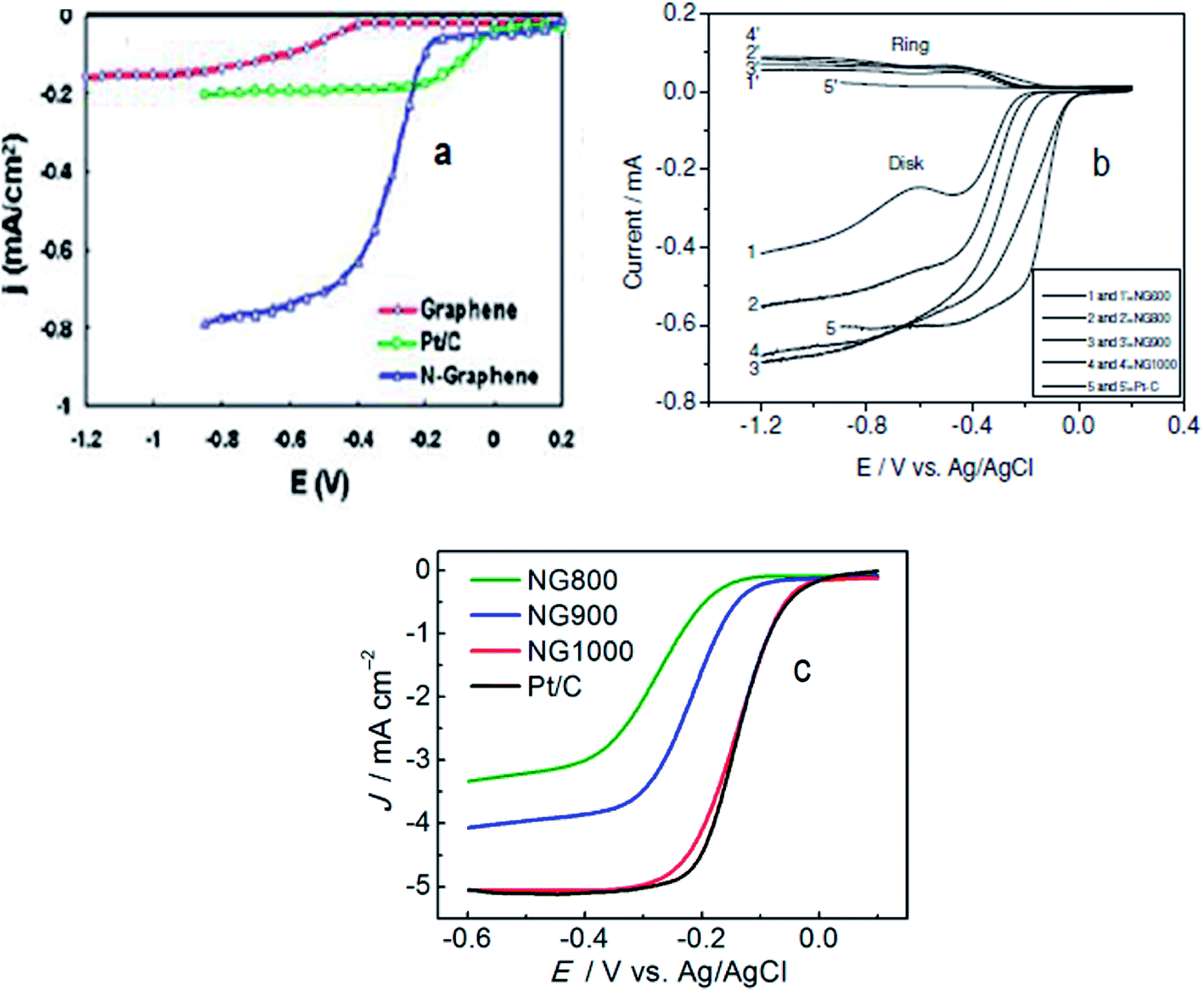 | ||
| Fig. 3 (a) Voltammetric curves obtained with a rotating ring-disk electrode (RRDE) maintaining the electrode rotation rate of 1000 rpm, sweep rate of 10 mV s−1 and 7.5 μg mass of graphene/N-graphene (blue curve) for the investigation of the oxygen reduction reaction in air-saturated 0.1 M KOH with the C-graphene electrode (red curve), Pt/C electrode (green curve), and N-graphene electrode (blue curve). Reprinted with permission from ref. 70 copyright 2010 American Chemical Society; (b) RRDE LSVs of NG600 (1 and 1), NG800 (2 and 2), NG 900 (3 and 3), NG1000 (4 and 4), and Pt–C (5 and 5) at a rotation rate of 1600 rpm. Reprinted with permission from ref. 77 copyright 2012 Wiley-VCH; (c) LSVs of different N-graphene samples and Pt/C at 1600 rpm. Reprinted with permission from ref. 27 copyright 2013 American Chemical Society. | ||
Wu et al., synthesized a metal-free electrocatalyst, pyridinic N-rich few-layer thick graphene, from methane and gaseous NH3 precursors by adopting a combined post-doped CVD method. Post doping was done with graphitic carbon nitride annealed at different temperatures (700–900 °C) resulting in varying concentration and configuration of N in the catalyst.72 The N-doped graphene based electrocatalyst annealed at 800 °C was found to have a Pt/C-like onset potential with the highest current density in comparison to other pyridinic-N configurations. Hence, N-graphenes prepared via the CVD approach are practically viable materials displaying ORR activities higher70,71 or comparable72 to Pt. The two inherent limitations of using costly and toxic precursors in the CVD-method compelled researchers to explore cheaper and environmentally friendly routes for the synthesis of heteroatom-doped graphene to catalyse the ORR.45,46 In this regard Yang et al., synthesized metal-free N-graphene by thermally treating graphene oxide and ammonia.77 Their electrocatalyst NG900 annealed at 900 °C displayed onset potentials comparable to Pt/C and current density (shown in Fig. 3b) higher than Pt/C suggesting its suitability for enhanced ORR-activity. Similarly, Zhang et al.,27 documented the preparation of an N-doped graphene based electrocatalyst (NG1000) from edible sugar and urea which demonstrated Pt/C-like electrochemical characteristics as shown in Fig. 3c. Moreover, the N-graphene reported by Feng et al.,78 prepared by a detonation technique using cheaper and readily available reactants such as cyanuric chloride and trinitrophenol, showed a markedly better performance (tolerance, crossover effect and long term stability) than Pt/C. The superior ORR-performance of N-graphenes achieved via metal-free routes can be ascribed to –C–N type catalytic sites originating from the insertion of N in the graphene skeleton. The reported virtually metal-free fabricated N-graphenes have shown comparable or even better fuel cell cathode performance in terms of stability and anti-crossover effect with respect to the commercial Pt/C catalyst.27,77,78 However, improvement in their catalytic activity towards the ORR in comparison to the gold-standard Pt catalyst is still under extensive investigation.79–88 The catalyst at an elevated annealing temperature originating from an appropriate level of N-doping and high degree of graphitization along with increased conductivity shows ORR-activity equal to or higher than platinum in comparison to the same catalyst at lower temperatures. For instance, the electrocatalyst NG900 annealed at 900 °C (ref. 77) and NG1000 (ref. 27) have ORR-activity comparable to Pt but NG600 and NG800 have lower activity than platinum. A thorough comparison between catalysts of high and low ORR activity is expected to underline the underlying factors responsible for influencing their activity. These results suggest that materials of higher activity are to be selected for further investigations as effective catalysts. Moreover, examining the effect of various parameters (T, pH, concentration, etc.) on the catalytic performance (particularly ORR-activity) of materials is expected to provide further insights into their selection and development for use in high performance energy generation and storage devices.
The N-doped composite form of CNTs and graphene has been practically achieved by several research groups. For instance, Ma et al., synthesised 3D-N-doped CNTs/graphene composite by pyrolysing pyridine in the presence of a Ni catalyst supported on graphene sheets.93 The resultant tangled NCNTs of quasi-aligned arrays were found to be sparsely distributed yet strongly linked to graphene sheets as depicted in Fig. 4a. The composite markedly enhanced activity and selectivity towards the ORR as depicted by the anodic shift in the onset and peak potentials coupled with high peak current density compared to its undoped counterpart (CNTs/G). However, the performance of the doped composite is still not on a par with the commercial Pt/C material. Moreover, an observation of the TEM image of the material (Fig. 4b) reveals the presence of Ni-residues encapsulated in carbon shells which do not contribute towards ORR activity. The work of Li et al., demonstrates that few walled CNTs (FW-CNTs) after oxidation and annealing with ammonia at high temperatures exhibit enhanced ORR catalytic activity under alkaline as well as acidic conditions.94 The oxidation occurs under drastic conditions which partially unzip and exfoliate the outer walls of FW-CNTs, resulting in a unique composite of graphene nano-sheets that adhere to the inner tubes. Their estimated catalytic potential was found to be very close to Pt/C, most probably due to many defect sites, edges and nitrogen incorporation and residual-Fe as a result of metal seeds trapped in the matrix.95 However, as mentioned earlier, complete removal of Fe residues from CVD-grown NCNTs/graphene composites or adopting essentially metal-free protocols for the synthesis of the catalyst is undeniably important in order to estimate the exact ORR catalytic potential of the composite materials.
 | ||
| Fig. 4 (a) Scanning electron microscope photograph of the NCNTs/G composite; (b) arrows point to the catalyst nanoparticles coated with carbon shells. Reprinted with permission from ref. 93 copyright 2011 American Chemical Society. | ||
To date, some fascinating metal-free routes aimed at overcoming the limitations of the CVD method have been developed. One particular example is the sequential doping method (Scheme 1) adopted for the preparation of a nitrogen and sulphur-doped graphene/CNT composite in alkaline medium.96 The application of this composite shows a prominent increase in the onset potential by 0.05 and 0.07 V associated with N- and S-doped graphene/CNT ORR catalysts as compared to individual graphene and CNTs co-doped separately with nitrogen and sulphur respectively where co-doping can significantly improve catalytic activity and stability. This finding highlights the importance of the synergistic contribution of the unique complimentary arrangement of graphitized CNTs over graphene sheets. Similarly Ma et al., documented the preparation of carbon–nitrogen (CNx)/graphene composites via a carbonization process of polypyrrole (PPy) and reduced graphene oxide (rGO) sheets which showed excellent metal-free electrocatalytic activity towards the ORR.97 In the same streak, Ratso et al., fabricated N-doped graphene/CNTs by subjecting multiwalled CNTs and graphene oxide to pyrolysis in the presence of N precursors (urea and dicyandiamide) under alkaline conditions.98 The ORR-activity of this material was found to match with Pt/C mainly due to pyridinic and quaternary-N species. Hence, the observed performance of the composites may exclusively stem from N-incorporation and synergistic enhancement induced by the composite arrangement.
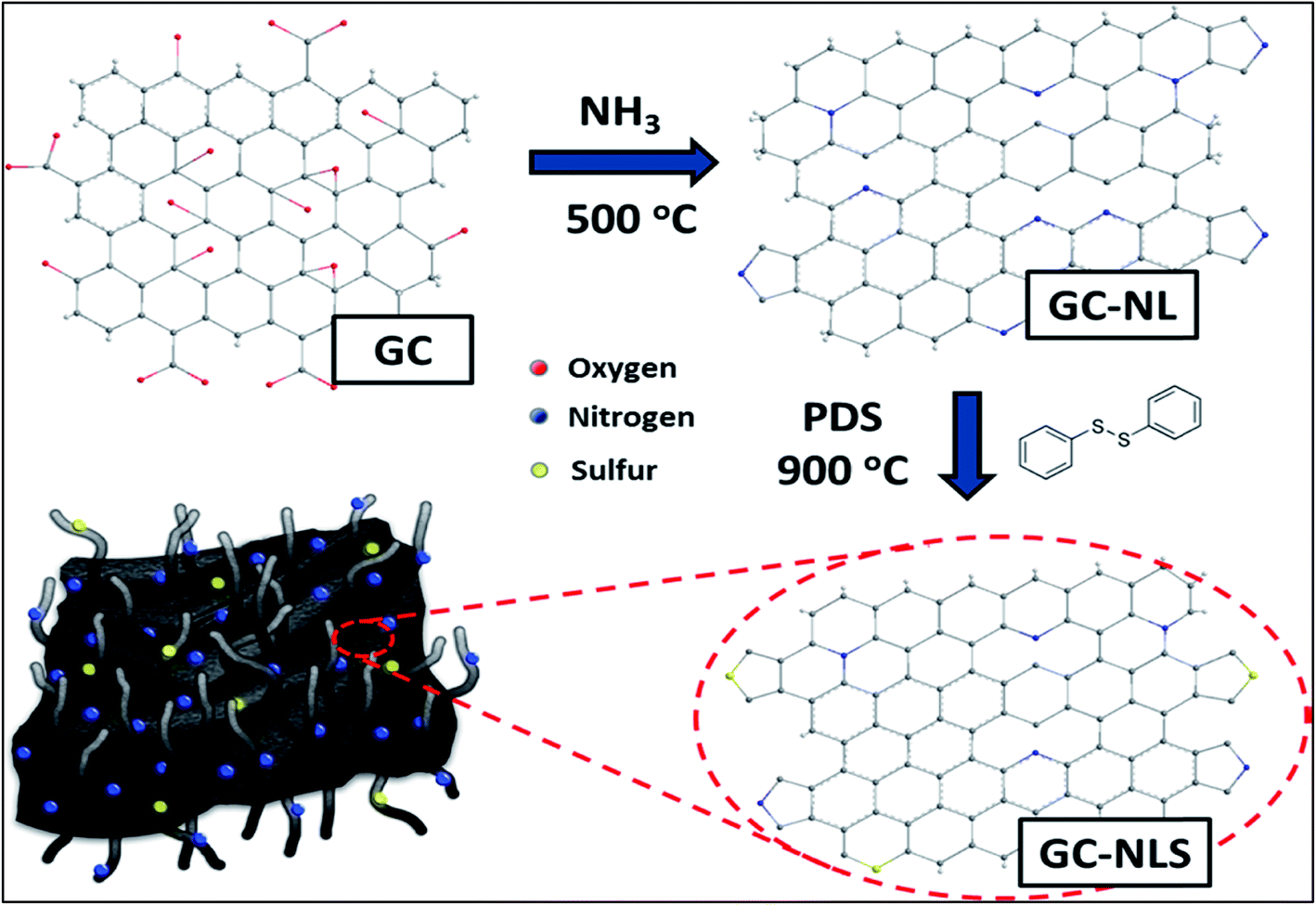 | ||
| Scheme 1 Schematic illustration of the sequential doping process for synthesis of GC-NLS nanocomposites. Reprinted with permission from ref. 96 copyright 2014 American Chemical Society. | ||
2.2 Carbon doping beyond nitrogen
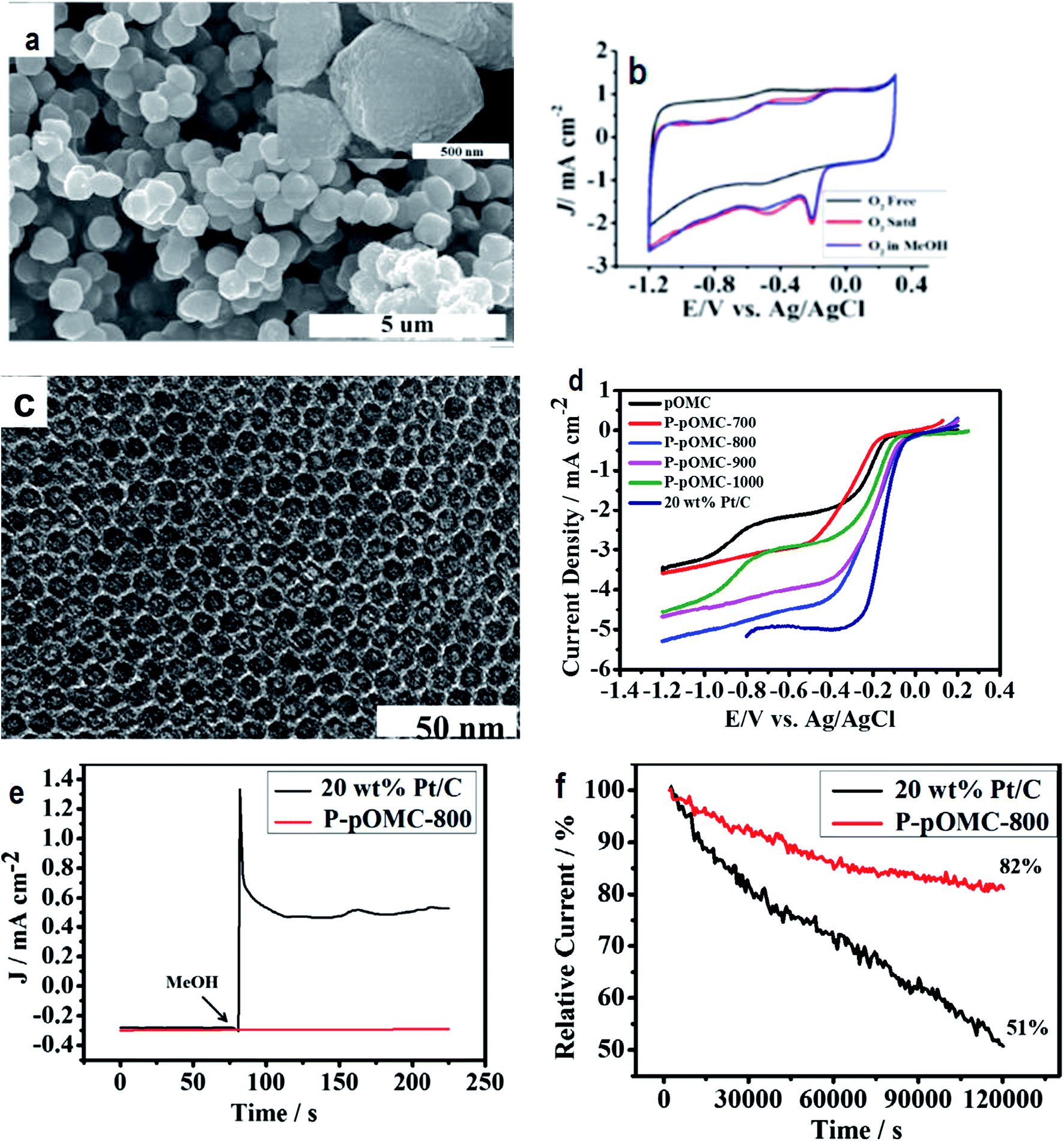 | ||
| Fig. 5 (a) Field emission scanning electron microscope photo of POMC-3 demonstrates the uniform distribution of spherical shaped carbon microstructures with an average thickness and length of about 0.6 μm and 0.7 μm, respectively; (b) cyclic voltammograms showing the ORR performance of POMC-3 at 100 mV s−1; (c) TEM image of P-POMC-800 showing hexagonally arranged carbon fibers. Reprinted with permission from ref. 100 copyright 2012 American Chemical Society; (d) steady-state RRDE experiments carried out using P-POMC, POMC and 20 wt% Pt/C (E-TEK) catalysts for the investigation of the oxygen reduction reaction at an electrode rotation rate of 1600 rpm and a 10 mV s−1 sweep rate; (e) chronoamperometry (CA) curves recorded under conditions of −0.3 V in O2-saturated 0.1 M KOH solution before and after the addition of 3.0 M methanol; (f) relative J–t response as a function of time. Reprinted with permission from ref. 110 copyright 2014 Elsevier. | ||
Charge transfer or polarization must not occur in C–S bonding as both sulphur (2.58) and carbon (2.55) have almost the same electronegativity. Even then, S substitution in the carbon framework in the S–C–S-like configuration introduces edge defects for side-on oxygen adsorption by enhancing the charge and spin density which facilitate the ORR performance of S-doped carbonaceous materials. In one particular study, sulphur-doped ordered mesoporous carbon (OMC–S-X) was synthesized using SBA-15 as a template, benzyl di-sulphide and sucrose as precursors of different sulphur contents (X =1, 2, 3) and carbon, respectively. The as-synthesized OMC–S-3 demonstrated stable and remarkable electrocatalytic performance in comparison to Pt–C/GCE, suggesting the suitability of this material as a metal-free electrocatalyst.111 OMC–S-3/GCE was found to have a more negative onset potential than Pt–C/GCE and comparable current density at lower potentials. Among different ratios of sulphur-doped mesoporous carbon (OMC–S-1, OMC–S-2, OMC–S-3), the sulphur in a thiophenic configuration in OMC–S-3 exhibited the most remarkable ORR performance and resistance to the alcohol crossover effect. Working in this area, another research group proposed sulphur-doped carbon spheres of high surface areas and pore channels synthesized via an in situ doping approach to facilitate unhindered mass transport of O2 during the ORR.112 The higher ORR performance was related to the induction of active sites in the carbon spheres by the incorporation of S doping that causes distortion in the hexagonal carbon network. Inamdar et al.,113 proposed sulphur-containing carbon soot synthesized by a flame pyrolysis method as a promising ORR catalyst. The synthesized material exhibited better properties such as electrocatalytic activity, resistance to methanol oxidation and higher stability than Pt/C with a preference for a 4e reduction route. The ORR electrocatalytic performance registered improvement with the increase in sulphur content, thus, highlighting the importance of S as a promoter of active sites. Clues for the role of S in improving the ORR efficiency were obtained from two pieces of evidence: (i) lone pairs present on the S atom for interaction with the surrounding oxygen molecules and (ii) more prominent defects and strains in the carbon framework by the doped S, augment charge localization for chemisorption of oxygen due to the larger nuclear radius of S (102 pm) in contrast to nitrogen and carbon having radii of 65 pm and 77 pm.
The origin of the enhanced ORR performance of N, B or P-doped carbonaceous materials can be related to the disruption of electroneutrality due to the electronegativity difference between dopants and carbon that induces positive sites for oxygen adsorption and facilitates its reduction to OH−via a 4e− process.114,115 Theoretical calculations of Zhang et al.,116 suggest that alteration in spin density and charge distribution can determine the catalytic sites for accelerating ORR activity. S doping induces changes in spin density and the distribution of charge density on C atoms in the neighbourhood of dopants which is the main reason for ORR activity enhancement. Boron doping in graphenes improves their ORR performance due to high spin density achieved through injection of a positive hole into the valence band of the sp2-hexagonal graphene network inducing p-type conductivity which can favour oxygen adsorption.117–120 Similarly Jo et al., suggested an economical and single step green approach for the synthesis of boron-doped carbon (BDC) electrocatalysts as a promising alternative to precious-metal catalysts for accelerating oxygen reduction reaction under alkaline conditions.121 The ORR performance of BDC was found to be further enhanced upon increasing the pyrolysis temperature from 700 °C to 900 °C. The authors also observed superior durability and stability of BDC compared to commercial Pt/C with complete insusceptibility to methanol oxidation and CO poisoning. In another study boron-doped mesoporous carbons (BOMCs)122 synthesized by co-impregnation and carbonization of 4-hydroxyphenylboronic acid and sucrose into a silica template (SBA-15) were tested for catalysing the ORR. The results revealed the excellent ORR activity of BOMCs via a 4 electron pathway and improved selectivity and stability compared to commercial Pt/C. The boron content and surface area were identified as essential features for promoting ORR activity. Another research group suggested a high temperature diffusion method for fabricating onion-like carbon (OLC) with a high content of boron.123 The boron content ratio in OLC substituted for carbon approached 29% which is the highest amongst the previously reported boron-doped carbon materials.124,125 It is worth mentioning that like commercial Pt/C, boron-doped OLC followed a perfect 4 electron pathway for the ORR and showed improved stability in terms of resistance to CH3OH and CO poisoning. On the basis of experimental findings it was suggested that the higher charge density around Ef, lower work function and higher electronic density of states of boron-doped OLC are indispensable features for improving the ORR catalytic behaviour. Carbon based materials doped with halogens have also been reported.126–128 Owing to the higher electronegativity and larger atomic size of halogens (Cl and Br) than C, they polarize the neighbouring carbon that further augments the ORR performance of these materials.126 Jeon et al., proposed iodination of activated charcoal by a mechanochemical method.127 The proposed material exhibited electrocatalytic performance on a par with the commercial Pt/C. Working in this area of research, Sun et al., proposed fluorine-doped carbon black (CB–F) as an inexpensive and sustainable catalyst for excellent ORR performance in basic medium.128 The doped F atoms improved the electronic properties of CB with concomitant advancement in its ORR activity. The results of electrochemical studies revealed that the ideal BP-18F with optimum amounts of NH4F and BP (Black Pearls 2000) as precursors exhibits higher ORR activity and longer operational stability with improved resistivity than commercial Pt/C catalysts and exceeds in performance the best non-Pt based cathode catalysts. These metal-free electrocatalysts are useful alternatives for catalysing ORR in power devices as a consequence of their low cost and high performance.
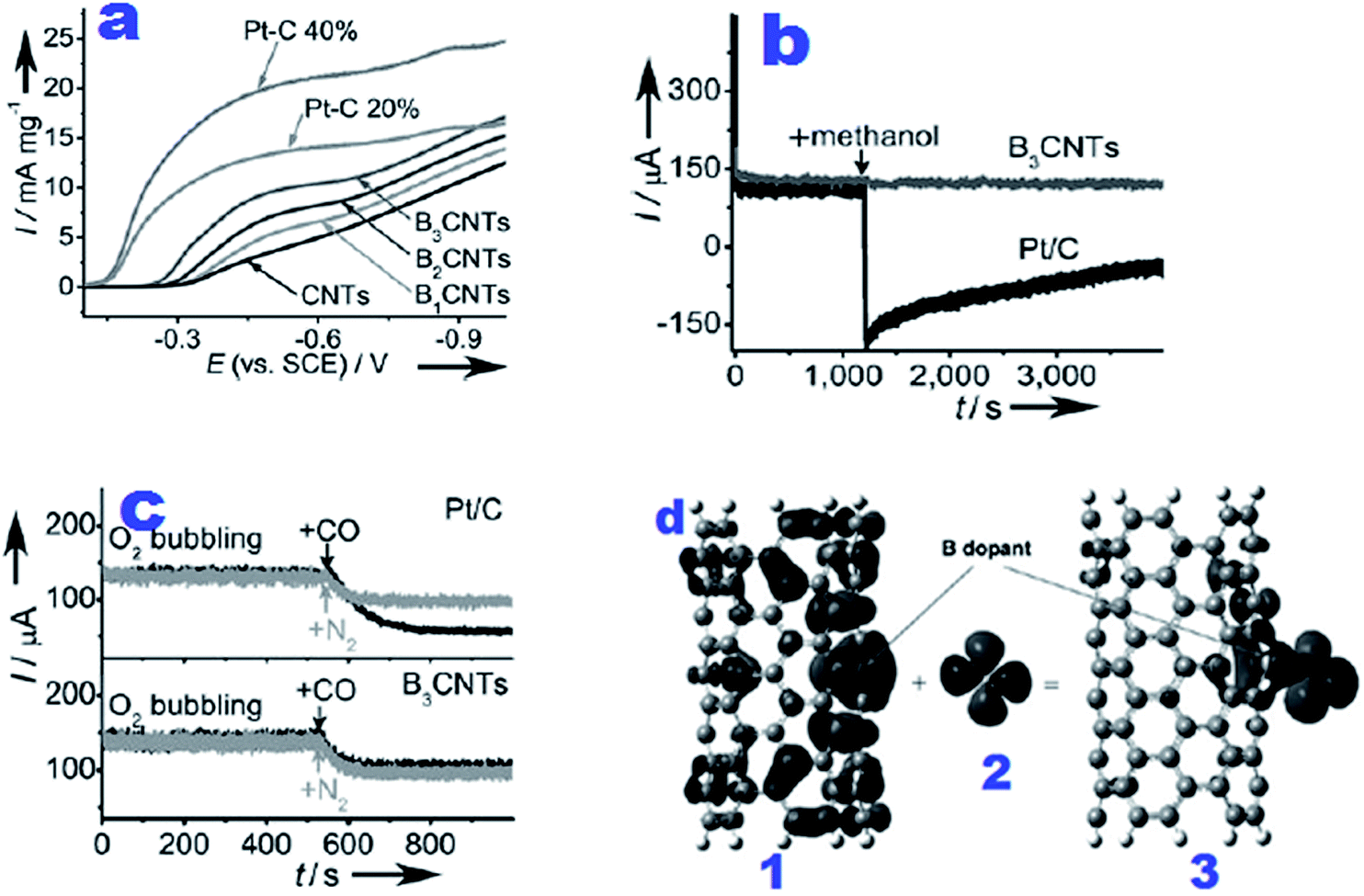 | ||
| Fig. 6 (a) RDE voltammograms obtained at a rotation speed of 2500 rpm and 10 mV s−1 using BCNT electrocatalysts for accelerating the ORR in O2-saturated 1 M NaOH electrolyte; (b) chronoamperometry curves of Pt/C (black) and B3CNT (dark gray) catalysts in the O2-saturated electrolyte for the investigation of methanol crossover after introducing 1.5 mL methanol into the electrolyte at 1200 s; (c) CA curves for testing CO poisoning after injection of additional CO with the same flow of O2 into the electrolyte at 520 s for the same electrode as in b; (d) involvement of important molecular orbitals in the adsorption of O2 on BCNT(5,5). (1) spin-down HOMO-1 of BCNT(5,5). (b) LUMO of triplet O2. (c) Spin-down HOMO-2 of O2-BCNT(5,5).33 Reprinted with permission from ref. 33 copyright 2011 Wiley-VCH. | ||
 | ||
| Scheme 2 Schematic illustration of BCNP synthesis. Reprinted with permission from ref. 133 copyright 2015 Elsevier. | ||
| Electrode material | Synthetic method | ORR performance vs. Pt/C | electrons | Ref. |
|---|---|---|---|---|
| VA-NCNTs | Pyrolysis of iron phthalocyanine in the presence or absence of NH3 | Slightly more positive onset potential and significantly enhanced (∼0.8 mA) steady state diffusion current | 4 | 26 |
| N-graphene | Pyrolysis of sugar in the presence of urea | NG1000 has comparable overpotential and slightly higher kinetic current density than Pt/C | 3.8 | 27 |
| B-CNTs | Chemical vapor deposition with benzene and triphenylborane (TPB) used as precursor | Good electrocatalytic activity with immunity to CO poisoning and methanol crossover | 2.5 | 33 |
| Metal-free NCNTs | DACVD process using melamine as a C/N precursor | Lower ORR-potential and comparable current density relative to Pt/C | 2.6 | 38 |
| Stacked NCNCs | CVD-method using MeCN, EtOH, and ferrocene as liquid precursors | Similar catalytic activity as Pt–CNTs | 2.6 | 42 |
| Essentially metal-free NCTs | Alumina template technique using poly(2-methyl-1-vinylimidazole) | Close onset potential and enhanced steady state diffusion current compared to Pt/C | ∼3.8 | 59 |
| Metal-free NCNTs | Plasma etching technology | Improved ORR-activity compared to pristine CNT | 3.52–3.96 | 60 |
| N-graphene | CVD method using methane and NH3 | 3 times higher catalytic current density and onset potential more negative than Pt | 3.6–4 | 70 |
| N-graphene | Hard templating approach | The catalyst named PNGF has a comparable onset potential and high current density to Pt/C | 3.9 | 71 |
| N-graphene | Denotation technique using cyanuric chloride and trinitrophenol | Comparable ORR-activity to Pt | 2–4 | 78 |
| NGCA | Heat treatment of an N-doped graphene–CNT self-assembly (NGCA) with diphenyldisulfide | High electrocatalytic activity with more stability and durability than Pt/C | — | 92 |
| NCNTs/graphene | Pyrolysis of pyridine over graphene supported Ni-catalysts | Lower onset potential and low current density than Pt/C | 3.47 | 93 |
| Few walled CNTs–graphene complex | Exfoliation of few walled CNTs via oxidation followed by reaction with NH3 at high T | Comparable onset potential and slightly less current density than Pt/C | — | 94 |
| FLG-CNT composite catalyst | Hummers' method using urea and dicyanamide, graphene oxide and MWCNTs | Slightly negative and high catalytic current density | — | 98 |
| POMC | Casting method | Higher electrocatalytic activity; more stability and tolerance than Pt/C | 4 | 100 |
| P-POMC | Pyrolysis method using platelet SBA-15 furfuryl alcohol and triphenyl phosphine as a precursor | Comparable onset potential and higher current density than Pt/C | 3.8 | 110 |
| OMC–S-3 | SBA-15, benzyl disulphide and sucrose as a precursor | More negative potential and higher current density than Pt/C. High resistance to the cross over effect, and C–S–C played crucial role in ORR activity | 3.8 | 111 |
| S–CS | Sulphur-doped carbon spheres synthesized via an in situ doping approach | Improved ORR activity with more stability | 3.8 | 112 |
| S–C | Sulphur-doped carbon synthesized via a flame pyrolysis method | Better tolerance and more stability to methanol oxidation as compared to Pt/C | — | 113 |
| S-OGnPs | S-OGnPs in the presence of H2O2 | SOGnPs showed more effective electrocatalytic activity than S-GnPs following a 4 e transfer mechanism | 3.6 | 116 |
| PN-MWCNTs | Synthesized by a pyrolysis method | Durability and stability better than Pt/C | 3.73 | 121 |
| BOMCs | Co-impregnation and carbonization of 4-hydroxy phenyl boronic acid and sucrose into a silica template | Better stability and selectivity | 4 | 122 |
| BOLC | Synthesized by a high temperature diffusion method | High tolerance to methanol oxidation and efficient electrocatalysts with more stability than Pt/C | 3.96 | 123 |
| IAcC | Mechanochemical ball-milling activated charcoal in the presence of iodine | Excellent electrocatalytic activity and higher stability against CO | 4 | 127 |
| CB-F | Pyrolysis of CB and NH4F | Higher current density than Pt/C | 3.96 | 128 |
| P-MCNTs | Thermolysis method using toluene solution containing ferrocene and TPP as a precursor | Higher electrocatalytic activity with better tolerance and durability as compared to Pt/C | 3 | 131 |
| B-MWCNTs | Thermal annealing method using MWCNTs in the presence of boric acid | Efficient and active electrocatalysts and activity depends on the boron content. 20@B-MWCNTs possessed high electrocatalytic activity and more efficient than all | 2 | 132 |
| B-CNP | Synthesized via a solution plasma process using triphenyl borate and benzene as a precursor | Long term durability and stability toward methanol oxidation | 2.47–3.15 | 133 |
| P-TRG | Thermal annealing method using graphene oxide and BmimPF6 as a precursor | Electrocatalytic performance is comparable to that of Pt/C | 3.6 | 138 |
| S-GN | Annealing GO and benzyl disulfide in argon | Comparable onset potential and higher current density at −0.8 V | 3.82 | 139 |
| I-GN | Thermal annealing of GO and iodine | Comparable onset potential and higher current density | 3.86 | 141 |
| X-GnPs | Ball milling graphite in the presence of Cl2, Br2 and I2 | Efficient electrocatalytic activity, good tolerance toward methanol crossover and excellent cycling stability | 3.5–3.9 | 142 |
| X-RGO | GO is synthesized via an electrochemical method followed by the halogenation of RGO in H2SO4 medium | Excellent electrocatalytic activity and higher current density than Pt/C | 4 | 143 |
| B-G | Thermal annealing in the presence of GO and B2O3 | Excellent electrocatalytic activity similar to Pt/C with more stability and tolerance toward CO | 3.5 | 144 |
| (3D S GNs) | Ion exchange method using 732-type sulphonic acid ion exchange resin as a precursor | 4e pathway with more tolerance and stability than Pt/C with 12.8 wt% of sulphur | 3.45–3.80 | 146 |
where * represents free sites on the surface. This conversion is a manifestation of the associative 4 electron pathway where OOH(ads) can be desorbed by the reaction; OOH(ads) + e → OOH− leading to an associative 2 electron pathway. In one particular study, it has been reported that desorption of the adsorbed species OOH* and OH* leads to selection of either an associative 2 electron or 4 electron mechanism.160 The predominant overall 4 electron pathway of an electrocatalytic ORR under basic conditions is given by: O2 + 2H2O + 4e → 4OH−. However, the dissociative 4 electron route is initiated by
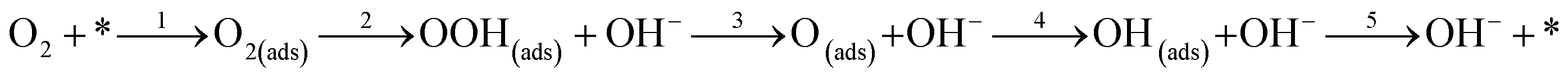
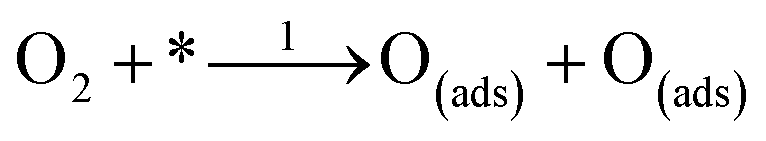 followed by steps 4 and 5.
followed by steps 4 and 5.
Generally, a strong interaction between the catalyst and reactant is crucial for excellent catalytic activity. Pristine CNTs and graphene exhibit inert behaviour toward O2-adsorption due to their high adsorption barrier compared to pure platinum.161,162 In contrast, NCNTs161 and X (N, B, and P)-doped graphene163 have better ORR catalytic activity due to their high affinity for adsorption of O2 and OOH, respectively. This is highlighted by a continuous increase in O–O distance with a concomitant decrease in C–O distance pointing towards C–O bond formation at the doped carbon surfaces. Okamoto et al., simulated the ORR process on N-graphene in acidic medium using first-principles molecular dynamics calculations164 where they argued that by analogy with Pt, the associative mechanism is favourable for reduction of O2 to H2O. Moreover, the results revealed that for alkaline fuel cells at lower working temperatures, the energetically favoured associative reduction step forming OOH(ads) has an effective energy barrier of 0.51 eV which is remarkably lower than the dissociation barrier (1.56 eV) of O2(ads) in the presence of water, thus, prohibiting the dissociation of O2(ads) from occuring with a practical rate on N-graphene surfaces.158 These research findings are helpful for the basic understanding of the ORR mechanism on N-graphene surfaces following a “pseudo” 4-electron process during electrochemical generation of a HO2− intermediate via a catalytic regenerative route (step 1) followed by a chemical disproportionation reaction to form OH− and O (step 3) known as an electrochemical-chemical (EC) reduction mechanism catalyzed by N.165,166
An experimental and theoretical approach was adopted for the investigation of the origin of the electrocatalytic action of doped graphene-based materials for accelerating reduction of oxygen.159 The electrocatalytic ORR performance of a perfect metal-free graphene-based catalyst (X-graphene) showed a current density of 2.1 × 10−6 A cm−2, an on-set potential of 0.33 V (versus the standard hydrogen electrode, SHE) and a 4e mechanistic route. These parameters are practically identical to or even better than those of a Pt catalyst. These outcomes of corroborated computational and experimental studies are useful for uncovering an efficient route for ORR electrocatalysis using graphene-based materials. DFT calculations indicated that for all heteroatom-doped graphene models, the first step of electron transfer where OOH* is physically adsorbed at an active site reflects the highest resistance to the ORR while the second and third steps leading to chemisorbed O* and physisorbed OH* are exothermic steps in the overall reaction O2 → OOH* → O* → OH* → OH−. However, the last electron transfer step has an easily surmountable energy barrier that leads to desorption of OH*. The results suggest that for a 100% 4e− selective ORR mechanism, desorption of OOH* from the electrode surface must be endothermic while OH* desorption should be exothermic. Fan et al., also employed a theoretical model (DFT B3LYP) for exploring the active site induction role of dopants in promoting the ORR activity of heteroatom-doped carbon nanomaterials.155 The calculations pointed to the occurrence of the ORR by an efficient four-electron pathway on the selected material leading to the production of OH−(aq.) under basic conditions. While in acidic electrolyte either O2 is directly reduced to water by a 4e pathway or to an undesirable metastable state hydrogen peroxide153via a 2e process; O2 + 2H+ + 2e → H2O2
The electrocatalysts following a 4e mechanism are considered to be better than those involving a two-electron mechanism. It is observed that the site near the dopant (B or N) offers strong interaction and binding energy for the ORR by adsorption of OOH. There might be another possibility for protonation at the active site which results in lowering the efficiency of ORR by passivation of the active centre. However, the formation energy calculated for OOH* over the surface of graphene was found larger than the formation energy of HC, HN and HB with graphene, therefore OOH* adsorption seems more favourable for playing a crucial role in catalysing the ORR.155,159 Upon physical adsorption of OOH to the active site, either the bond with the active site C–O or the O–O bond in OOH is broken leading to metastable H2O2 or stable H2O, respectively, by the formation of energetically favourable epoxide on graphene by addition of H+. From these results it can be inferred that strong adsorption between the active sites of doped graphene and OOH* may result in a charge transfer of electrons from graphene to H+via radical *OOH according to the following mechanism.
| *OOH + e− + H+(aq.) → H2O + *O |
| H+(aq.) + e− → *H(aq.) |
| *OOH + *H(aq.) → H2O + *O |
On a little more delving exclusively into the details of the active sites for the ORR, it is noted that the substitutional incorporation of N into the carbon framework can result in different nitrogen configurations including graphitic, pyridinic and pyrrolic nitrogen.78,99 No doubt, N-incorporation into carbon materials leads to improved ORR-activity but specifically assigning catalytic activity to graphitic, pyridinic or pyrrolic-N is still debatable. In this regard theoretical investigations have revealed that graphitic-N drastically lowers the energy barrier for O2 dissociation as compared to other N-configurations.83 The experimental results of Liu et al. have also validated that the enhanced ORR-potential of the N-doped ordered mesoporous graphitic arrays is related to the presence of a high graphitic proportion of nitrogen.167 Therefore, these findings coupled with the results of other research groups85,86 suggest the key role of graphitic-N in boosting the ORR performance of N-doped carbon materials. In contrast, conflicting results about the role of pyridinic-N in improving the ORR-potential of the N-doped material have also been reported. For instance Park et al., linked the higher activity of X-doped graphene nano-sheets to the presence of pyridinic-N.84 In another study the substantially larger limiting current density of N-graphene compared to Pt/C (20 wt%) was ascribed to the abundance of pyridinic as well as pyrrolic N for accelerating ORR activity.70 Surprisingly, Luo et al., reported that N-doped graphene having pyridinic nitrogen as the principal dopant could not exhibit promising results to catalyze the ORR.73 Thus, different configurations of bonded N in the carbon framework result in different ORR activity enhancement mechanisms following either a 2 or 4 electron mechanistic route. Moreover, the presence of pyridinic- and pyrrolic-N at the edge planes confers uncertainty to these functionalities as to whether they act as the proper active sites or highly probable active edges and/or defect sites for favouring ORR activity.32 Hence, the challenges in determining the exact location of N-atoms in the carbon skeleton, chemical nature of catalytic centers and the electrochemical kinetics of N-doped carbon electrodes for their role in enhancing ORR activity are yet to be overcome.168 Similarly graphitic-N dominant materials display higher ORR-potentials than pyridinic- and pyrrolic-N containing catalysts. This demands in-depth analysis of the subject for establishing an accurate nexus between different N-configurations and catalytic activity. The disagreement in the results of different research groups can be resolved if similar experimental conditions are maintained during the synthesis and ORR-performance evaluation of the catalysts. Apart from these, N-doped carbon materials bearing diverse C–N bonding configurations may further be thoroughly investigated for establishing a convincing relationship between N-configurations and catalytic activity. A combined theoretical and experimental work in this domain is highly indispensable for probing the underlying factors, responsible for controlling the ORR performance of the catalysts.
Also nitrogen incorporation into the CNTs skeleton induces a substantial positive charge at the adjacent carbon atoms as displayed in Fig. 7a. This facilitates O2-adsorption at the active sites. Simulation of several positions where adsorption of oxygen takes place in NCNTs, indicates a carbon atom (C1) with positive charge density located adjacent to electronegative nitrogen as the preferred O2-adsorption site.169 Conversely, the theoretical study of the ORR-mechanism at the surface of N-graphene nanoribbons does not support the presumption of polarization of C–N bonds at NCNTs.170 Instead, the barrier for O2-adsorption decreases as the nitrogen dopant moves toward inner sites from O2-adsorption locations in the direction from N0 to N4 as shown in Fig. 7b. Furthermore, it was proposed that the mode of O2-adsorption to alter from end-on at CNTs to side-on attachment at NCNTs causes considerable weakening of the O–O bond, thus, maintaining the enhanced activity of the N-doped counterpart.171 In contrast, the theoretical work of Kim et al., does not substantiate alteration in the mode of O2-adsorption at N-graphene nanoribbons or simply at the graphene edges.170 The tubular symmetry of NCNTs has been reported to be responsible for alteration in the mode of O2-adsorption while the planar symmetry of N-graphene may not have the same effect.59 The role of nitrogen doping in fine-tuning the properties of NCNTs and N-graphene in different manners might be another reason. Additionally, the variation in morphology, nitrogen content and nitrogen configuration of the catalyst material cannot be excluded from causing inconsistency in the results obtained. Therefore, optimization of properties is crucial for a clear understanding of the ORR mechanism at the surface of X-doped carbon materials.
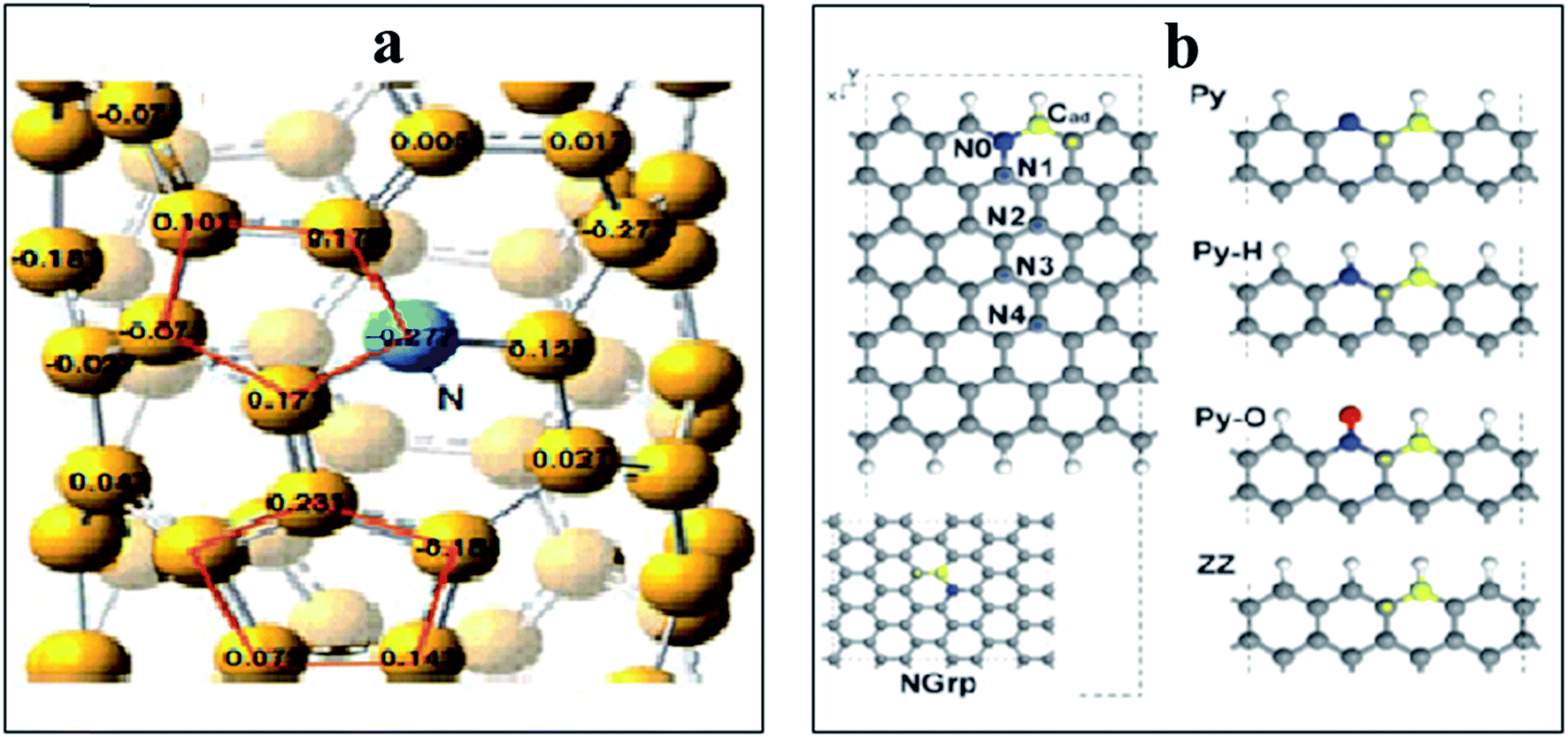 | ||
| Fig. 7 (a) Distribution of charge density on the atoms of NCNTs, showing the higher positive charge on C atoms in the vicinity of pyrrole-like nitrogen compared to C atoms close to pyridine-like N atoms. Reprinted with permission from ref. 26 copyright 2009 American Association for the Advancement of Science. (b) Representation of model structures along with N-doping sites, and edge states. The oxygen molecule is adsorbed at Cad (yellow atom). The N, O and H atoms are shown in blue, red, and white colours with dotted lines denoting a periodic boundary. Reprinted with permission from ref. 170 copyright 2011 the Royal Society of Chemistry. | ||
N-doped VA-CNTs,26 N-doped nanocages,63 N-graphene,70,79,172 X(P, N, and B)-doped graphene163 and three dimensional nitrogen-doped graphene foams71 have been found to promote the ORR through a 4e reduction pathway. Nevertheless some investigators have proposed an intermixed two- and four-electron mechanism. In fact Y. Tang and coworkers42 documented a combined two- and four-electron route for the ORR at the surface of N-doped carbon nanotube cups as opposed to Pt–CNTs that follow a 4 electron pathway. Similarly, Wang et al. proposed a two- and four-electron combined route for the ORR at NCNTs, suggesting the transfer of 2.6 electrons in the process38 similar to the work reported by Tang et al.42 Similarly Dai et al., proposed a 4 electron pathway for oxygen reduction on VA-NCNTs26 and thus no sound consensus seems to be available on the mechanism of the ORR. Additionally, the research team of Stevenson documented a dual site mechanism with regards to carbon and nitrogen active sites for the reduction process to take place at a trace metal Fe site, bound to the surface of NCNTs.166 The initial two-electron O2-reduction occurred at an N–C type active site which led to the formation of HO2− following either a two-electron electrochemical reduction of the intermediate to OH− or chemical disproportionation at FexOy/Fe type sites to give O2 and OH1− species (Scheme 3). Although, the four-electron route for N-doped carbon is extensively reported,26,27,43,59 still some researchers favour the successive two step 2 and 4 electron combined route.38,42,78,166 Therefore, the research to date on unfolding the exact number of electrons involved in the ORR has not yet come to a conclusive end and further investigations are required to resolve the controversy of the involvement of either 2e or 4e or their combination.
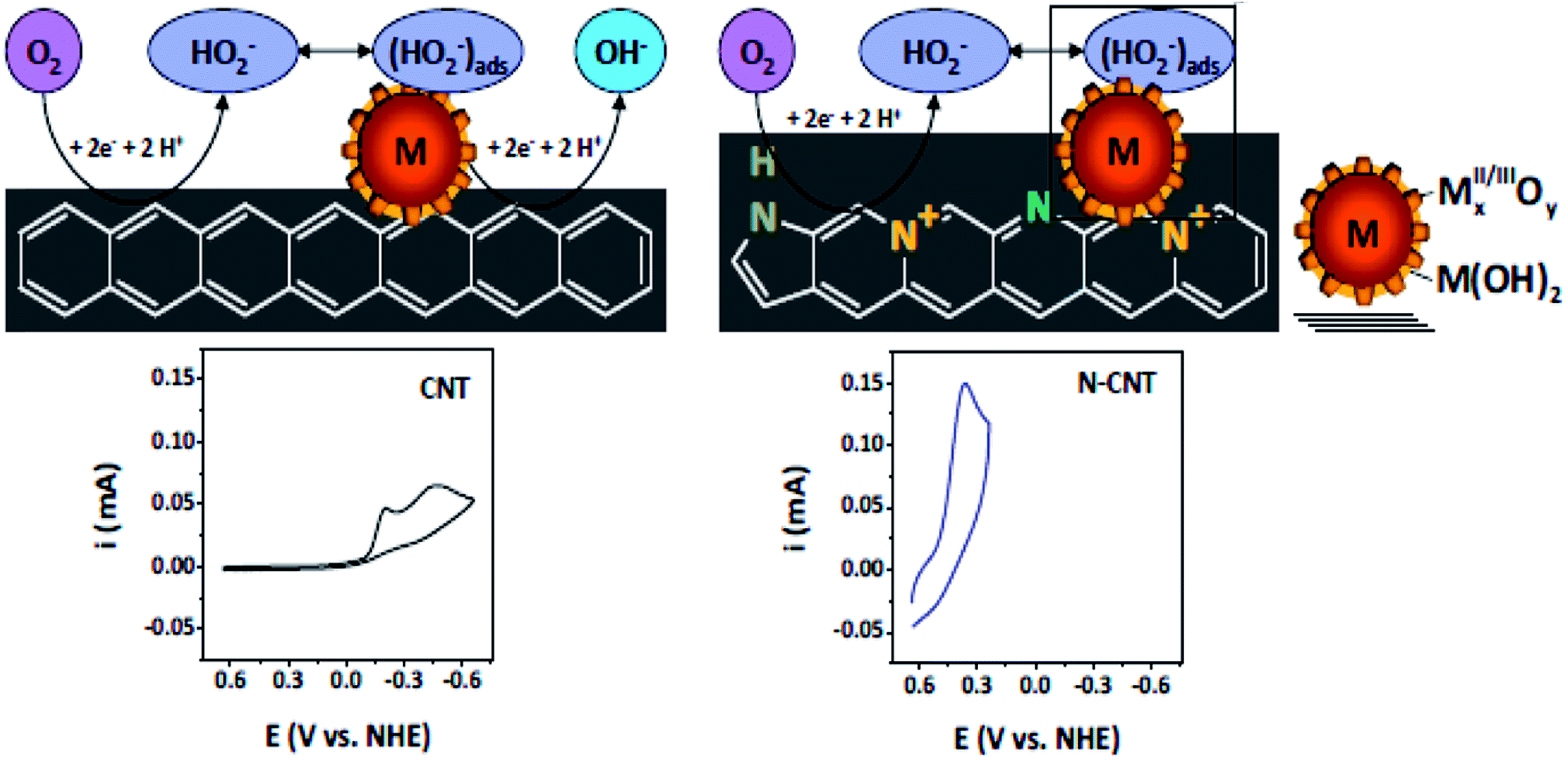 | ||
| Scheme 3 Schematics of the proposed ORR mechanistic pathways in CNT and N-CNT electrodes along with representative CVs. Reprinted with permission from ref. 166 copyright 2011 American Chemical Society. | ||
3. Lithium ion batteries (LIBs)
Lithium ion batteries find extensive use in personal digital assistants (PDAs), laptops, iPods and cell phones due to their unique features such as rechargeability, lightweight, enhanced energy density and increased cycle life.173,174 Nevertheless, their power density is reduced due to large polarization originating at a high charge/discharge rate from slow diffusion rates of Li ions and lower thermal/electrical conduction across the interface of the electrolyte and electrode. For overcoming this issue, it is imperative to either modify the existing electrode materials or design new electrode materials specifically with small diffusion paths for efficient transport of ions to meet the requirements for commercializing high-performance LIBs.175,1763.1 Graphitic materials
Among the graphitic materials (graphene, graphite, carbon nanotubes, and activated carbon), graphite is commonly used in LIBs. However, its theoretical specific capacity with a magnitude of 372 mA h g−1 is critically lower than that desired for efficient LIB technology.177 Hence in this scenario, for the last two decades researchers have been working on substituting graphite with other nonconventional anode materials such as SnO2, Sn, and Si with respective specific capacity values of 782 mA h g−1,178 994 mA h g−1 (ref. 179) and 4200 mA h g−1 (ref. 180) which are much higher as compared to graphite. However, on the downside, exorbitant volume changes as a result of electrochemical processes of insertion and desertion of Li ions cause poor cycle life and hence their application in LIBs is circumscribed.181 It is argued that if the carbonaceous material is changed from graphite to graphene, the capacity will be significantly increased up to 500–1100 mA h g−1. Graphene has a calculated capacity of 740 mA h g−1 (ref. 182) which is much higher than the theoretical value of graphite making it a viable anode substitute in electrochemical energy storage devices. Owing to the non-layered structure facilitating Li storage at both the surface and edge planes,183 high electron mobility66 and high surface to mass ratio favouring enhanced Li storage,184–186 graphene is a proven useful anode material for LIBs.187–189 However, to surpass the existing features of LIBs, the amount of Li adsorbed onto graphene and its adsorbing energy demand further exploration for the realization of high energy density and reduction of charging time.190–192 Working in this domain Yoo et al., reported a specific capacity of 540 mA h g−1 for graphene nanosheets (GNS) by exploring tuning of their layered structures for lithium accumulation.184 For increasing the Li ion storage capacity, carbon nanotubes and C60 were incorporated in GNS for enhanced specific capacities approaching 730 mA h g−1 and 784 mA h g−1, respectively. Dahn et al., suggested that insertion of Li ions on both sides of disorganized carbons in graphene can increase the material's reversible capacity and hence in the same vein, GNS composed of disordered graphene would find potential applications as anode materials in LIBs.182 In fact the graphene nanosheets prepared by Guo et al., possessing a disordered structure of graphene carbons exhibited good cycle life and a 672 mA h g−1 reversible capacity.193In order to increase the rate capability of Li ions, restacking of the graphene layer is suggested to be controlled by creating a 3D structure that opposes restacking.194–196 On the other hand the performance of LIBs in terms of power capability can be improved when an in-plane defect by a carbon vacancy in the form of a hole/pore is introduced into the graphene sheets. This holey graphene gives access to more active sites for Li accumulation and a shorter path for diffusion of Li through the layers during the lithiation/delithiation process, thus, overcoming the drawback of the huge aspect ratio for graphene.197 It displays a 2.3 times higher reversible capacity of 742 mA h g−1 after 80 cycles at a current density of 0.1 Ag−1 than pristine graphene and retains higher capacities at 10 mA g−1. Thus, holey graphene due to its improved rate capability finds applications in high power EES devices. However, care must be taken during graphene preparation as its desired anode properties can be affected by modification in its structure, functionalities and surface chemistry. In fact the reversible capacity of graphene has been reported to vary from 672 to 1264 mA h g−1 depending on the method chosen e.g., chemical oxidation, thermal exfoliation, chemical reduction methods, and chemical vapour deposition methods.189,193,198,199
For the graphite model it was found that the Li trapped in a disordered carbon on a covalent site inside the benzene ring prefers the stable edge site over the bulk.200,201 Extending this finding, Kheirabadi et al., mapped out interactions between Li and circular graphene by DFT calculations to elucidate the storage sites of lithium ions at different locations in the graphene clusters and unravel the mechanism with special emphasis on the edge effects of large and small clusters. A number of parameters such as the dipole moment, spin polarization, electronic gap, etc. of the graphene model were calculated for each cluster. The results202 related the relative stability of a cluster to the increase in the number of intercalated Li ions and an increase in absorption energy where Li preferred adsorption around the edge. The results also suggested that a smaller cluster between two larger clusters can lead to improvement in graphene based Li-ion batteries. In one particular study graphene nanoflakes with a controlled morphology were prepared from the liquid phase exfoliation of undoped graphite as an ink203 which was deposited by conventional printing technology. This class of battery has the potential of offering a solution to current technological electrical energy storage issues.186 Indeed the graphene nanoflakes were found to possess high crystalline order for fast electron transfer188 and a small lateral size for offering storage of Li ions in the active sites of their edges. These edges are crucial since they provide 50% of the binding energies for Li+ in comparison to the graphene basal plane. Furthermore, theoretical calculations have also predicted that edges on graphene nanoribbons (GNRs) provide smaller energy barriers for diffusion of Li, with regard to the graphene basal plane.204 Such a graphene nanoflake ink supported on Cu has shown a specific capacity of ∼1500 mA h g−1 at 100 mA g−1 for about 150 cycles well over the theoretical specific capacity of freestanding graphene which suffers from restacking during its preparation and capacity reduction as anode materials due to cluster formation of Li ions on its surface and their dendrite nucleation.205–209
Endeavours in the direction of exploiting chemically modified graphenes (CMGs) that favour mass production include graphene oxide (GO) and reduced graphene oxide (RGO), for use as additives of both the anode117,184,188,198 and the cathode.210,211 However, limited conductivity and slow Li ion diffusion restrict their utility.212 To date, a specific capacity of ∼1200 mA h g−1 at 100 mA g−1 has been achieved for the best anodes with CMGs.198 RGO with its few-layered graphene characteristics and a high specific surface area is useful for employing directly as an anode material and can deliver improved performance in LIBs compared to graphite.213 The results of Pan et al.,191 reveal that graphene nanosheets with a disordered structure can act as more suitable candidates for applications in LIBs contributing towards good cyclability and excellent reversible capacity due to active defects and edge planes providing additional sites for enhanced capacitance. The presence of defects in multilayered and monolayered graphene sheets prepared by the CVD method on Ni and Cu as the substrate surface were also found to facilitate the diffusion of Li ions.214 This finding was validated by DFT calculations exhibiting a moderate barrier height for various defects especially highly ordered defects for the diffusion of Li ions. Some researchers have reported that defects in graphenes lead to the formation of a solid electrolyte interphase film which lowers the electrochemical performance of graphene based electrodes.191,215 Thus, it is important to consider the role of defects as these influence the electrochemical performance of graphene based LIBs.216–218
Hard carbon based composites can act as potential electrode materials for use in future high performance LIBs.219 In one particular study nanograined Sn particles were evenly distributed in the carbon framework220 and the use of the as-synthesized nano-Sn/C composite as an anode in LIBs showed superior discharge capacity whilst maintaining an initial capacity of 710 mA h g−1 at 0.25C (where C = rate performance) even after 130 cycles. Even at a higher rate of 20C, the discharge capacity of the anode remained considerable (∼600 mA h g−1). This excellent role of the nano-Sn/C composite anode can be accredited to the exceptional mechanical support of the carbon matrix that buffers the stress due to the significant volume change of nano-Sn, prevention from agglomeration of nano-Sn by the carbon matrix and facilitation of a continuous path for electrons and Li ions in the spheres of the nano-Sn/C composite.220 In the same vein, Zhang and co-workers prepared a porous Si/C electrode material composed of conductive carbon-coated sheets and interlinked porous silicon via the Rochow reaction under mild reaction conditions i.e. 1 atm pressure, 325–450 °C. This demonstrated excellent reversible capacity and long cycle life which are essential for emerging next generation LIBs. These characteristics also enable porous nanostructured Si materials to be used in nanoelectronics, sensors, photonics and solar cells, under specific conditions.221 Furthermore, lithium titanate nanofibers (LTO NFs) were synthesized by coating these uniformly with a carbon layer via hydrothermal synthesis while wrapping LTO NF in a graphene oxide (GO) layer by electrostatic interaction with positively charged LTO NFs. This unique material led to an improved rate capability of 110 mA h g−1 at 20C and an over two times greater diffusion coefficient i.e. ∼1.04 × 10−11 cm2 s−1 as compared to virgin LTO NFs. These results reveal that the LTO NFs coated with carbon and wrapped with GO facilitate electron and lithium ion transportation, thus, showing remarkable electrochemical performance.222,223
Carbon nanotubes (CNTs) can work as useful electrode materials in LIBs owing to their exceptional 1D tubular structure, greater thermal and electrical conductivities and high surface area. These nanostructured electrode materials present many benefits over other carbon based materials in the context of reversible intercalation/extraction of Li without damage in their structure, offering a shorter path for efficient electron transfer and exhibiting reduced volume changes as a result of greater contact with the electrolyte. Thus, most of the advanced electrode materials for LIBs are mainly driven by hybrid nanostructures comprised of CNTs and Li storage materials.224 In order to compare the performance of acetylene carbon black (CB), carbon fibres (CF) and multiwall carbon nanotubes, Guoping et al., investigated the behaviour of these materials as conductive additives in LIBs. Fig. 8 shows that the incorporation of MWCNTs into the LiCoO2/MWCNT composite electrode material leads to uniformity of the capacity retention ratio as compared to 10% and 30% reduction in the ratio of composites possessing CF and CB, respectively, after 20 cycles.225 The importance of MWCNTs can also be judged by the fact that despite the capability of various electrode materials for Li storage, cracks are developed in these materials during the charge/discharge process which is the major reason for fast degradation of their rate capacities resulting from drastic volume changes in the lithiation/de-lithiation processes. But combining MWCNTs as cathodic fillers endows the electrode with the ability to obstruct pulverization as well as to increase their electronic conductivity. In this regard, Liu et al., studied the electrochemical parameters of various anode materials with and without additives of CNTs.224 The results of their experiments revealed the superior cycle performance of the MWCNT based nanocomposite electrode materials in comparison to a neat anode.
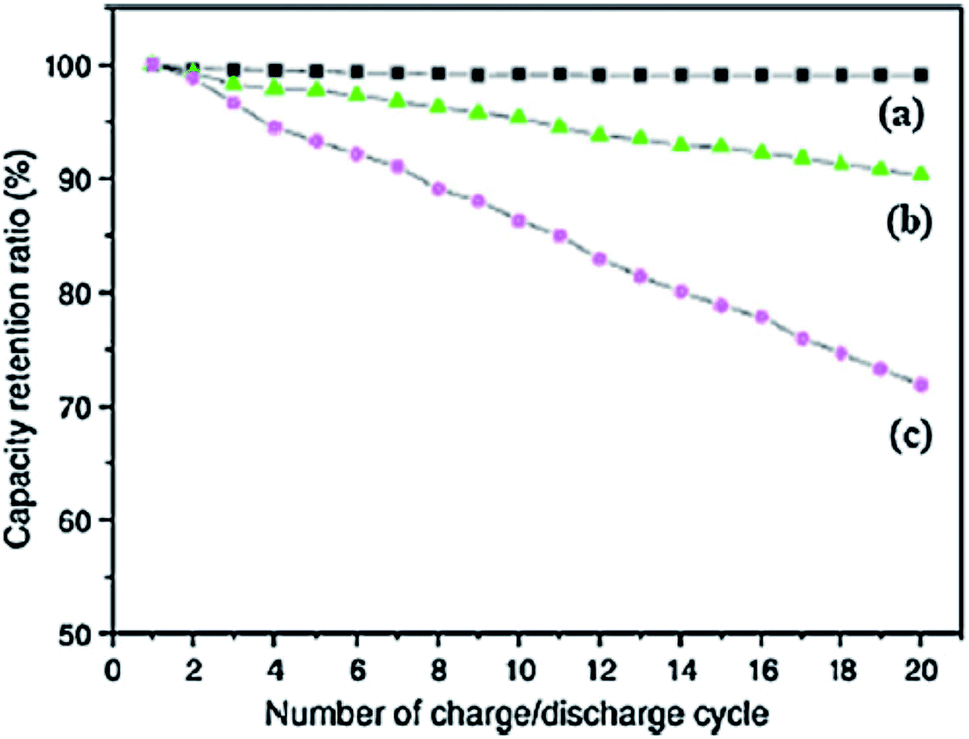 | ||
| Fig. 8 Influence of conductive additives on the capacity retention ratio of (a) MWCNTs, (b) carbon fibers, and (c) carbon black at a rate of 2C. Reprinted with permission from ref. 225 copyright 2008 Elsevier. | ||
3.2 Heteroatom-doped graphene materials
Because of the shortcomings of virgin graphene and chemically derived graphenes, several research groups have explored effective ways to enhance the storage capacity of Li by doping graphene with heteroatoms.125,224,226 The vital role of N-doping in catalysing the ORR can be extended to several other carbon materials for the expansion of their characteristics in LIBs. In this context, Qie et al., reported the doping of nitrogen in carbon nanofiber webs (CNFWs) as electrode materials for LIBs after carbonization-activation of polypyrrole with nitrogen and KOH. The unique porous CNFWs showed high rate capability even after 600 cycles and a reversible capacity of 943 mA h g−1 at a current density of 2 A g−1.227 N-graphene was also interlinked with SnO2 to form N-graphene–SnO2 sandwich paper for use as an electrode material. The improved rate capacity and cyclic performance of N-doped G–SnO2 sandwich papers compared to pure SnO2 nanoparticles can be seen in Fig. 9. The modified SnO2 papers demonstrated well maintained reversible capacity with an initial capacity retention of about 63% (910 mA h g−1) after 50 cycles, followed by only a 0.02% capacity loss per cycle in contrast to a mere 7% retention (117 mA h g−1) after 30 cycles, leading to a 2.9% capacity loss per cycle for unmodified SnO2 nanoparticles.228 For comparison, Zhang et al., documented the synthesis of graphene/SnO2 composites with 32 wt% of SnO2 content and assessed their electrochemical performance. The results of their experiments showed a reversible capacity of 786 mA h g−1 in the first cycle, with a 45% capacity retention (588 mA h g−1) after 50 cycles, resulting in a greater per cycle capacity loss (2.7%) than G–SnO2 sandwich papers.229 Thus, in the context of superior cycling performance and rate capacity, N-doped G–SnO2 sandwich papers are considered much better electrode materials than graphene/SnO2 composites.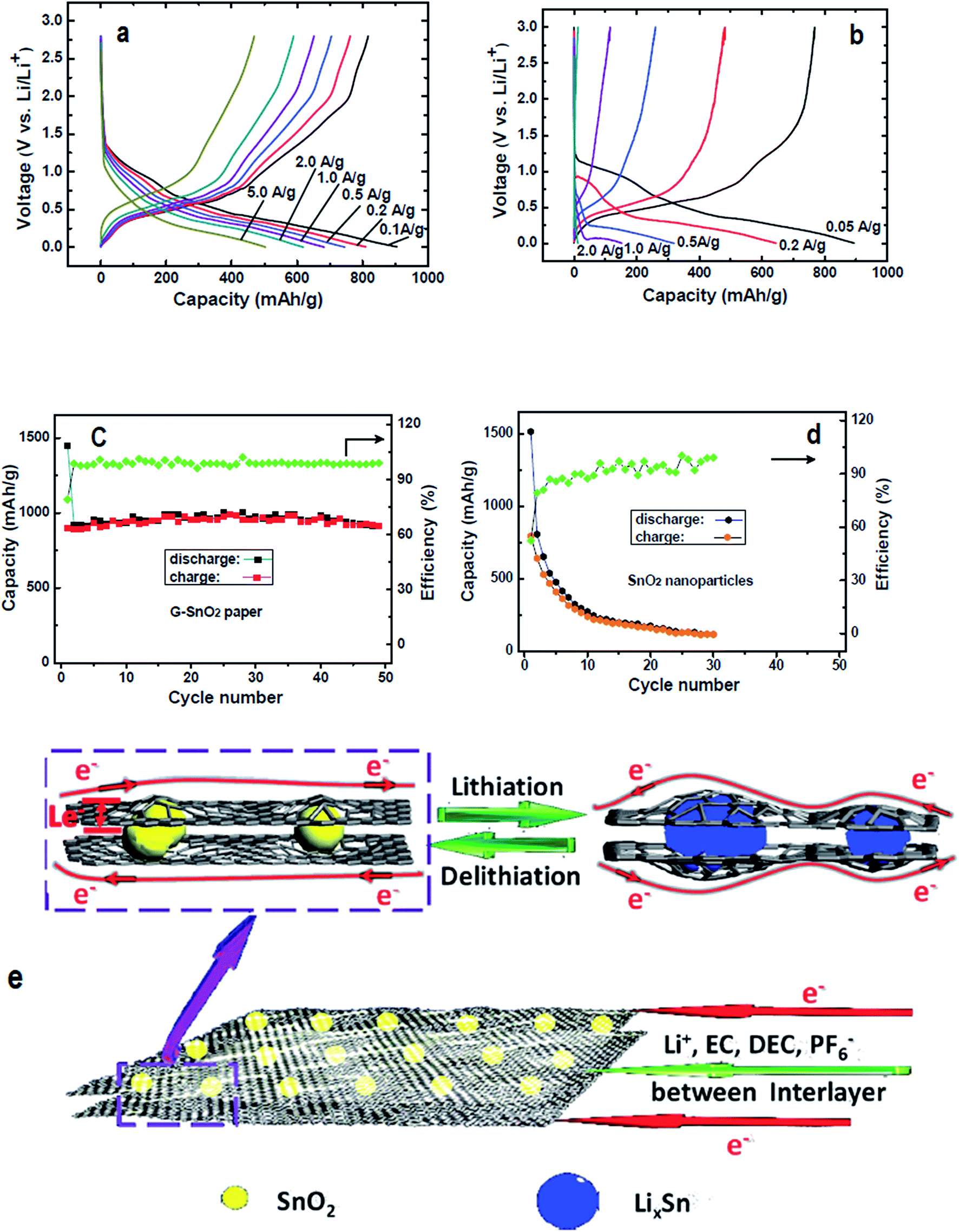 | ||
| Fig. 9 Charge/discharge curves of (a) N-doped G–SnO2 papers and (b) commercial SnO2 nanoparticles (20–50 nm), along with their cycling performance (c) and (d) at a 50 mA g−1 current density; (e) schematic representation of the transport of electrons and lithium-ions in N-doped G–SnO2. Reprinted with permission from ref. 228 copyright 2012 Wiley-VCH. | ||
3.3 Coupled inorganic/graphene hybrid materials
Apart from heteroatom-doped graphene based electrode materials, the use of compatible inorganic/graphene hybrids as advanced energy-storage materials has always posed an age old debate. A number of inorganic/graphene hybrid materials have been reported so far with the objective of enhancing the electrochemical performance of LIBs. For example, SnO2-based nanomaterials are considered suitable candidates for use as electrode materials in LIBs owing to their environmental friendliness and multifold increase in theoretical capacity (1494 mA h g−1) in contrast to traditional graphite.230 Despite these intriguing properties, SnO2-based anodes suffer from capacity loss due to agglomeration of tin particles during insertion of lithium along with the development of a thick solid electrolyte interphase (SEI). Electrode pulverization during the Li insertion/extraction cycle phase, depletion of the electrolyte and formation of an electrochemically inactive Li2O layer are their other snags.228 To tackle these issues, Zhou and co-workers introduced an in situ N2H4·H2O vapour reduction method to produce a SnO2 nanocrystal hybrid material with nitrogen-doped reduced graphene oxide (SnO2 NC@N–RGO). In this material SnO2 nanocrystals are confined uniformly between the graphene sheets to facilitate electronic transport, to impede the growth of the SEI layer, to offset the large volume change and to limit the aggregation of tin nanoparticles generated during lithiation. The application of SnO2 NC@N–RGO as anode material in LIBs showed high rate capability and a capacity of 1352 mA h g−1 (approximately 1021 mA h g−1 in terms of the total mass of the hybrid) and extended cycle life.231 Similarly another inorganic/graphene hybrid was prepared by employing a chemical deposition method for uniformly depositing Co3O4 nanoparticles (NPs) of 10–30 nm sizes on graphene sheets to form Co3O4@graphene composites (CGCs) as promising anode materials in LIBs by invoking the synergistic effects of graphene and NPs. The graphene sheets enhanced the stability of the CGC framework and electrical conductivity leading to a reversible capacity of 941 mA h g−1 in the first cycle along with increased current density and a pronounced cyclic performance of 740 mA h g−1 after 60 cycles as compared to the theoretical value of CGCs (which is 88.3% of it).232 Similarly, in another study a hybrid material fabricated from MnO nanocrystals deposited on conductive nanosheets of graphene was explored to tackle the issue of low cycling stability and inferior rate capability of the MnO based anode material in LIBs. The as-prepared MnO/graphene hybrid electrode displayed a reversible capacity of 2014.1 mA h g−1 even after 150 cycles of charging/discharging at a current density of 200 mA g−1, a high cyclability of 843.3 mA h g−1 after 400 cycles of charging/discharging at 2000 mA g−1 with a loss of only 0.01% capacity per cycle and a superior rate capability of 625.8 mA h g−1 at 3000 mA g−1 due to ultrafine MnO NPs supporting the longevity of the anode material and co-existence of conductive graphene reducing the transportation path for electrons and Li ions.233 Li et al., prepared sandwiched hierarchical graphene–MnO2–GNRs (GMG) using wrapped porous MnO2 in graphene coated over graphene nanoribbons (GNRs). The good contact of MnO2 between graphene and GNRs increased the electrical conductivity of GMG, buffered the changes in volume and avoided the loss of MnO2 with Li. Its specific discharge capacity maintained a magnitude of 890 mA h g−1 after 180 cycles at a current density of 0.1 A g−1 which increased by almost about 24% relative to the initial capacity at 0.4 A g−1 after 245 cycles. Thus, the GMG composite demonstrated its suitability for application as an electrode material of LIBs.234The unique properties of hybrid materials have spurred researchers on for devising novel candidates of this class as electrode materials of LIBs. In one particular study, a sol–gel method was employed for the deposition of ultra-dispersed TiO2 nanoparticles on the surface of reduced graphene oxide to form TiO2/RGO sheets. The resultant hybrid material possessing ∼5 nm sized ultra-dispersed anatase nanoparticles, ≤3 layered ultrathin thickness, and a surface area of 229 m2 g−1 displayed around 94 mA h g−1 specific capacity at ∼59C.235 The ability of ultralight 3D-graphene aerogels (GAs) to facilitate multidimensional electron transport suggests their potential candidature for application in LIBs. In another study a novel hydrothermal method was used to grow mesoporous ultra-dispersed TiO2 nanocrystals on GAs. The resultant TiO2/GAs were found to show large specific capacity, enhanced rate capability, and stable cycling due to interaction between GAs and TiO2, good electrical conductivity, and the porous structure of these hybrids.236
In the search for high performance electrode materials for LIBs, Zhou and coworkers fabricated nanocomposites of silicon NPs dispersed in graphene by developing electrostatic attraction between Si NPs adsorbed onto polyelectrolyte poly(diallylmethylammonium chloride) and GO by a self-assembly approach.237 The as-synthesized Si-NP@G nanocomposites displayed an ∼1205 mA h g−1 reversible capacity after 150 cycles. Zhou and coworkers also prepared micrometre-sized aggregates of silicon nanoparticles embedded in graphene sheets by combining thermal reduction and freeze-drying methods and found the as-synthesized Si NPs@thermally reduced graphene composites (Si/TRG)238 to have a cycling performance of 1153 mA h g−1 after 100 cycles. The research of these nanocomposites was further extended by introducing graphene coated silicon nanoparticles into the 3D ultrathin-graphite foam (UGF) via a drop casting method to prepare a Si/graphene/UGF 3D electrode design. The gravimetric capacity in terms of the whole mass of the electrode was determined to be 983 mA h g−1 in the first cycle up to 370 mA h g−1 was retained after 100 cycles, showing improvement in the performance compared to traditional graphite and Si deposited on metal foil.239 Taking the simplicity of the synthesis and the improved performance of Si/graphene composites into account, this class of materials can be selected for further tuning their properties (lower volume change and higher electronic conductivity) as electrode materials.
3.4 Future advances in LIB technology
Currently available LIBs are suitable for small-scale energy devices but not yet immaculate enough in terms of energy or lifetime that could meet the requirements to replace internal combustion engines for use in vehicles. They must achieve at least 2–5 times higher energy density to fulfil the goal of ideal performance in futuristic vehicles i.e. plug-in hybrid-electric vehicles of all-electric range 40–80 miles and all-electric vehicles (EVs) in the range of 300–400 miles. Over the past few decades, lithium-battery technology has made breakthroughs in the discovery of efficient electrode materials.24 The most recent high energy density Li–O2 cell introduced by Bruce et al., can achieve a specific gravimetric energy of magnitude 1752 W h kg−1. However, challenges regarding the safety limits, slow charging, limited cyclability, polarization issues, and discharge rates in the advancement of lithium battery technology are yet to be overcome.240 Regardless of the indisputable importance of LIBs in recent rechargeable worldwide battery market and their role in swapping oil driven vehicles with electric vehicles for limiting the indiscriminate use of conventional fuels and emission of greenhouse gases, efforts are being made to replace lithium in LIBs with the earth's most abundant element sodium to commercialize Na ion batteries (NIBs). In the past few years, simultaneous investigations of both Li- and Na-ion electrodes have revealed that Li-ion cells are better suited for use in pocket-sized portable energy storage devices. However, interest in Na-ion cells has recently revived in spite of the quite low voltage and larger change in volume on Na+ removal/insertion compared to Li+ because its power density based on different crystal structures can be manipulated for application in large-scale storage devices.241 Investigating Na as an alternative for Li, the research group of Y. Xu systematically studied the electrochemical performance of mesoporous tin/carbon anodes in Li-ion and Na-ion batteries. The Na-ion battery with mesoporous Sn/C anodes displayed similar stability in cycling but decreased capacity and lower rate capability compared to the Li-ion battery mainly due to the comparatively larger size of the Na ion, leading to its slow diffusion and a larger change in volume of the porous Sn/C composite anode during alloying/dealloying processes. Thus, insights into the reaction mechanism between Sn and Na+ ions are expected to pave the way for exploring and devising new alloy based anode materials for Na-ion batteries.242 Moreover, a biodegradable and naturally available compound i.e. polydopamine (PDA) has been used as an electrode material for LIBs and NIBs. Surprisingly, these optimized PDA derived LIBs and NIBs displayed excellent electrochemical performance with higher rate capacity (LIBs display 1818 mA h g−1 and NIBs show 500 mA h g−1) and very stable cyclability (LIBs have 93% retention in capacity after 580 cycles and NIBs have 100% retention in capacity after 1024 cycles) as compared to their equivalents with conventional binders.243 From these investigations, it is inferred that low cost NIBs can partially be used as an alternative for LIBs; however, further explorations are required to endorse this outcome.4. Supercapacitors
Supercapacitors (SCs) are promising substitutes for the currently available energy storage systems due to their ability of rapid charging and delivering high power densities within a few seconds.244 Presently the most common and commercially accessible electrochemical capacitors (ECs) are electrochemical double-layer capacitors (EDLCs) which store electrical energy by accretion of electrostatic charges in the region of the electric double-layer at the interface of the electrode/electrolyte. The comparison between the energy and power densities of various energy storage devices given in Fig. 10 indicates that SCs exceed the level of efficiency of their conventional predecessors i.e., capacitors and batteries.245,246 Electrical double layer capacitors based on emergent carbon nanomaterials have attracted the attention of many researchers owing to their high power density, stability and cost viability features.247–250 Porous activated carbons (ACs)251 and heteroatom-doped carbonaceous materials252 are desirable electrode materials for EDLCs. Therefore, many researchers have used these materials in advanced EDLCs.253–259 By employing AC as an electrocatalyst Wei, et al., achieved specific capacitance with a value almost twice that of the commercially used carbons.247 Carbon based materials derived from TiC exhibit a broad distribution of pore size and thus showcase themselves as a promising electrode for electrochemical double layer supercapacitors with a specific capacity of 125 F g−1 in acetonitrile and tetramethylethylammonium tetrafluoroborate AN/[TEMA][TFB4] solution.260 Computational simulations of carbon based EDLCs are performed for offering interpretation of the experimental results. In this regard Yang et al., carried out molecular dynamics calculations for modelling the specific capacitance of CNT-forest as an electrode material for EDLCs.259 The computed capacitance was found to be in agreement with the experimental capacitance of EDLCs utilizing negatively (71 F g−1) and positively (82 F g−1) charged SWCNT electrodes in the same solvent and same electrolyte. These computed capacitance values also matched well with the experimental values obtained for carbide-derived and CNF electrodes.261,262 For a good agreement between theoretical and experimental findings of carbon based EDLCs, solvent properties, ion type, its hydration sphere, orientation and distribution on the electrode are important factors to be considered.263–267 | ||
| Fig. 10 Ragone plot of several energy storage devices showing specific energy density as a function of specific power density. Reprinted with permission from ref. 245 copyright 2008 Nature publishing group. | ||
Quantum capacitance (QC) of the nanoscale materials acts as a limiting factor of the total capacitance.268,269 QC originates due to the shortage of states near Fermi levels. To elucidate the origin of suppressed capacitance, Ji et al., measured the EDL capacitance in an ultrathin carbonaceous architecture with one to five layers at different potentials by using a frequency response analysis method.270 Several other researchers have also attempted to investigate the basis of suppressed capacitance.271–273 Dröscher et al., suggested that a low density of states (DOS) at the point of zero charge (PZC) results in lowering of capacitance.274 N-doped carbons with improved DOS and increased charge carrier density can act as promising materials in EDLCs.275 This is validated by simulations through molecular dynamics that consider interaction of the mobile carriers with the locations of ions.270 EDL capacitance studies by MD simulations suggest that it is valuable to consider the introduction of defects into the framework of carbonaceous materials upon doping with N or B or even transition metals that can significantly reduce the limitations of quantum capacitance.276–278 Plane wave DFT calculations also show that the quantum capacitance of active carbons can be enhanced to an impressive value of 486.32 μF cm−2 when doped with 6.38% N.279 The DFT calculations of Khoshdel et al., offer another piece of evidence about the increase in the QC of active carbons due to codoping of P and N, S and N and Si and N.273 These theoretical results also corroborate the experimental findings.280,281
Chen et al., documented a new state near the Fermi level of N/S codoped graphene by DFT calculations and found an enhanced QC of the codoped material to be about 50%.282 As a representative class, negative curvature carbon (NCC) with a 3D saddle-like network of one sp2 atom thick walled carbons has great potential as electrode materials and these are expected to rival batteries due to their high specific surface area and energy density.283–285 The theoretical and the experimental work relates quantum capacitance with doping and the density of states at the Fermi level.286,287 The research team of Targholi also found quantum capacitance enhancement upon doping CNTs with N, P or B. N-doping was found to shift the Fermi levels to positive energy while boron incorporation caused electron deficiency and shifted the Fermi levels towards the valence band, consequently producing new states below the Fermi levels. However, phosphorus incorporation into CNTs as a dopant induced new states around the Fermi levels with no observable shift.288 Despite the promising results of the theoretical and computational investigations summarized in Table 2, the energy density of commercial EDLCs (<12 W h kg−1)245 still lags behind that of Li-ion batteries and requires enhancement by optimization of electrode materials without sacrificing their in-hand desirable features such as good stability and high power density.270 Therefore, new strategies and theoretical models may be developed in light of the above mentioned critical parameters for a deeper understanding of capacitance in carbon based electrodes in order to come up with a rational design of EDLCs.
| Electrode material | Synthetic method | Surface area (m2 g−1) | Specific capacitance (F g−1) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| N-doped activated carbons | Synthesis of polypyrrole followed by activation | 2095–3432 | 300 | Ionic liquid (EMImBF4) | 247 |
| N-doped cheese-like carbon | Carbonization of pyrrole modified lignin followed by KOH-activation | 2661 | 248 | KOH | 251 |
| N-doped carbon nanofibers | Carbonization of polypyrrole coated nanofibers | — | 202 | KOH | 252 |
| N-doped activated carbons | Activated carbons from Australian brown coal followed by K2CO3 activation | 2000 | 36 | K2CO3 | 253 |
| N-doped activated carbons | Carbonization of waste medium fibreboard followed by K2CO3 activation | 569 to 1027 | 147 to 223 | KOH | 254 |
| C(TiC) | — | 1505 | 99.1 | (C2H5)4NBF4 in acetonitrile | 256 |
| C(Al4C3) | 1470 | 82.3 | (C2H5)3CH3NBF4 in acetonitrile | ||
| C(Mo2C) | 1490 | 120 | (C2H5)3CH3NBF4 in acetonitrile | ||
| C(B4C) | 1525 | 70.9 | (C2H5)3CH3NBF4 in acetonitrile | ||
| Three dimensional CNTs/graphene sandwich | Chemical vapour deposition method | 612 | 385 | KOH | 258 |
| Graphene electrode | Molecular dynamics simulations | — | 100–280 (theoretical) | Aqueous monovalent electrolytes | 265 |
| 130–300 (experimental) | |||||
| Zigzag graphene nanoribbons | Molecular dynamics simulation | — | C q C d C t | [BMIM][PF6] | 277 |
| 378.1 101.8 80.2 | |||||
| 300.6 103 76.8 |
Graphene, as mentioned vide supra, is an affordable cost and readily available electrode material for accelerating reactions in energy storage devices. This material is used in supercapacitors owing to its high electrical conductivity and improved mechanical properties. Moreover, the uniformity of its surface area does not undergo alteration with pore size variation and its easy accessibility to electrolyte from both sides makes this material perfectly ideal for supercapacitor applications. Graphene–polymer composites, graphene–nanoporous carbon, and graphene–metal oxides find applications in symmetric and asymmetric supercapacitors.68 Moreover, these are used as electrodes in electrochemical double-layer capacitors. Current research in this domain chronicles critical factors such as the pore size distribution, surface area, interlayer distance, surface functionality, heteroatom doping and conductivity of these materials as well as their impact on the rate performance and capacitance of graphene-based EDLCs in order to resolve the issues regarding the establishment of standard measurement of capacitors, price reduction, missing steps in the ion sorption mechanism, the self-discharge mechanism on graphene and storage procedures.289
A number of research teams have explored the effect of graphene modification on its properties. For instance Lai and co-workers investigated the synergetic interaction of polyaniline (PANI) and the porous carbonaceous matrix in functionalized graphene–PANI composites for use in EDLCs. This surface modification of graphene led to the induction of features such as an increase in capacitance and stability in cyclability which are required for the development of next generation capacitors. This study suggests that incorporation of an amine group into graphene not only increases the capacitance of graphene–PANI composites but also sustains this increase (119%) even up to 680 cycles. Fig. 11 shows the improvement in specific capacitance before and after 10 wt% loading of PANI onto various surface-functionalized graphenes in 1 M H2SO4 electrolyte.290 Zhang and co-workers prepared porous 3D graphene-based materials in the bulk state by combining the two important properties of crystallinity and the amorphous character of carbon using an environmentally friendly approach. Their synthesized 3D material displayed excellent bulk conductivity, a specific surface area (SSA) of 3523 m2 g−1, an energy density of 98 W h kg−1 and a specific capacitance of 231 F g−1 as required for various supercapacitor applications.291 The research group of Zhang also devised an experimentally supported theoretical method for systematically studying the influence of capacitance on the 3D graphene-based materials in two types of ionic liquids by controlling the specific surface area, pore size distribution (PSD) and activation parameters. The shift in PSD of the as-synthesized 3D graphene-based materials (cork-HA, GO-HA, and PF24GO-HA) in the micropore range indicated improved accessibility and a higher effective-SSA (E-SSA) as shown in Fig. 12a and b. Furthermore, the E-SSA value of carbon material PF24G-HA was found to be much larger (1657 m2 g−1) while using 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIMBF4) as the electrolyte which is 93.1% of the net DFT-SSA i.e. 1779 m2 g−1 compared to RP20 (740 m2 g−1) in EMIMBF4 electrolyte (Fig. 12c). Thus, among all the 3D porous graphene-based materials, PF24G-HA demonstrated excellent supercapacitor performance showing a 220 F g−1 specific capacitance in EMIMBF4.292 Besides graphene based materials, Zhang and co-workers introduced a unique material via a method in which vertically adjusted multiwalled carbon nanotube (VA-CNT) carpets were longitudinally split by potassium vapours resulting in the production of a carpet of partially split graphene nanoribbons (GNRs) and MWCNTs. Upon splitting, the surface area of VA-CNTs increased the ion accessibility. Vertical conductive paths related to preserved GNR alignment led to long lasting conductivity. Moreover, SEM, TEM, AFM, X-ray and Raman spectroscopy results revealed that the split VA-CNT carpet has excellent cyclability, improved specific capacitance with a 103 kW kg−1 maximum power density (at 5.2 W h kg−1 energy density) and a 9.4 W h kg−1 energy density, thus, offering options for its use as a promising supercapacitor electrode material.293 A 3D sandwich of CNT/graphene has been successfully fabricated achieving a specific capacitance of 385 F g−1 in a KOH solution with a SSA of 612 m2 g−1.258 Efforts to stitch CNTs with graphene for enhancing its properties have been very challenging.
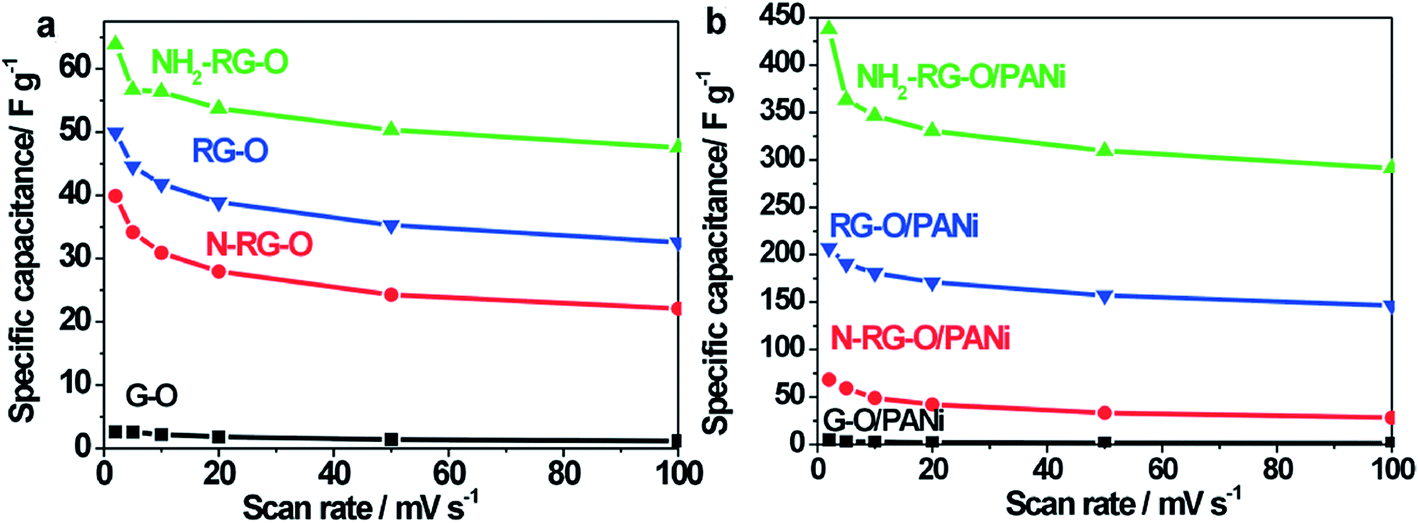 | ||
| Fig. 11 Plots of specific capacitance as a function of scan rate before (a) and after (b) 10 wt% loading of various surface-functionalized graphene materials. Reprinted with permission from ref. 290 copyright 2012 American Chemical Society. | ||
 | ||
| Fig. 12 (a) Pore size distribution of the optimized materials; (b) effective specific surface area of the synthesized materials using 1-ethyl-3-methylimidazolium tetrafluoroborate as the electrolyte; (c) bars showing specific capacitance as a function of effective specific surface area. Reprinted with permission from ref. 292 copyright 2013 American Chemical Society. | ||
A number of carbon derived materials have been synthesized for testing their electrode performance in supercapacitors. In this regard, Wang et al., documented cheese-like N-doped mesoporous carbon as an EDLC-electrode of specific capacitance equal to 248 F g−1 and 211 F g−1 in 6 M KOH solution at current densities of 0.1 A g−1 and 10 A g−1, respectively.251 Chemical activation of carbons for porosity via KOH is well known; however, due to the formation of hydrogen gas and metallic K it poses difficulties. A specific capacitance of 36 F g−1 with a specific surface area (SSA) of 2000 m2 g−1 has been reported by Yoshikawa et al., who have developed a new method of chemical activation through K2CO3 by adding together melamine or urea and Brown Coal as raw materials for N and C.253 Likewise, waste from fibreboard was used to prepare N-doped AC by K2CO3 activation for improved performance of EDLC electrodes. Depending on the conditions of activation, the ratio of K2CO3 to coke and % N doping, the specific capacitance varied from 147–233 F g−1 with a SSA value ranging from 569–1027 m2 g−1.254 Kumar et al., sonochemically modified an activated carbon (AC) matrix with carbon nanodots (C-dots) and investigated the effect of porosity of AC/C-dots on their performance as electrode materials.294 The results revealed their three-fold greater specific capacitance (0.185 F cm−2 per specific area of electrode) than an untreated AC electrode. The stunning results of using AC/C-dot electrodes were their 100% coulombic efficiency and stability in the electrochemical behaviour up to several thousand cycles. Taking advantage of the role of porosity, another porous carbon material denoted as PCPAC-2 (1D nanorods) was synthesized with a specific BET surface of 679 m2 g1 and a net pore volume of magnitude 1.63 cm3 g1. As an active material for supercapacitors, it displayed a 306 F g−1 maximum specific capacitance at 1 A g−1 and 96% capacitance retention even after 5000 cycles at 1 A g−1.295 Thus, carbon based electrode materials perform well in supercapacitors and for further improvement in their electrochemical performance, research on the modification of the structure of these materials is in progress. For achieving high specific capacitance, methods for alternative materials are also developed. In this connection Lu et al., offered a simple, low cost and efficient electrodeposition method for the fabrication of 3D α-Fe2O3@PANI core–shell nanowire arrays as shown in Fig. 13. The material prepared by this method displayed an increase in specific capacitance from 33.93 mF cm−2 to 103 mF cm−2.296,297 Similarly, a facile solution method followed by post annealing was developed for the synthesis of single-crystalline NiCo2O4 nanoneedle arrays fabricated onto conductive substrates such as Ti foil and Ni foam as binder-free electrodes. The Ni foam embedded NiCo2O4 nanowire arrays showed longer cycling stability and much better capacitance standardized with area/mass in contrast to porous free-standing NiCo2O4 nanowires and other arrays.298 Working in this research area, Niu and coworkers documented the synthesis and performance of a supercapacitor cathodic electrode material i.e. amorphous nickel tungstate nanostructure (NiWO4).299 Its specific capacitance of 586.2 F g−1 as a cathode and activated carbon as the anode led to a maximum energy density of 25.3 W h kg−1 at a 200 W kg−1 power density. Moreover, NiWO4 demonstrated long term stability as evident from 91.4% specific capacitance retention after 5000 successive scans.299 Hence, some non-carbon electrode materials also possess the ability of replacing expensive Pt in supercapacitors.
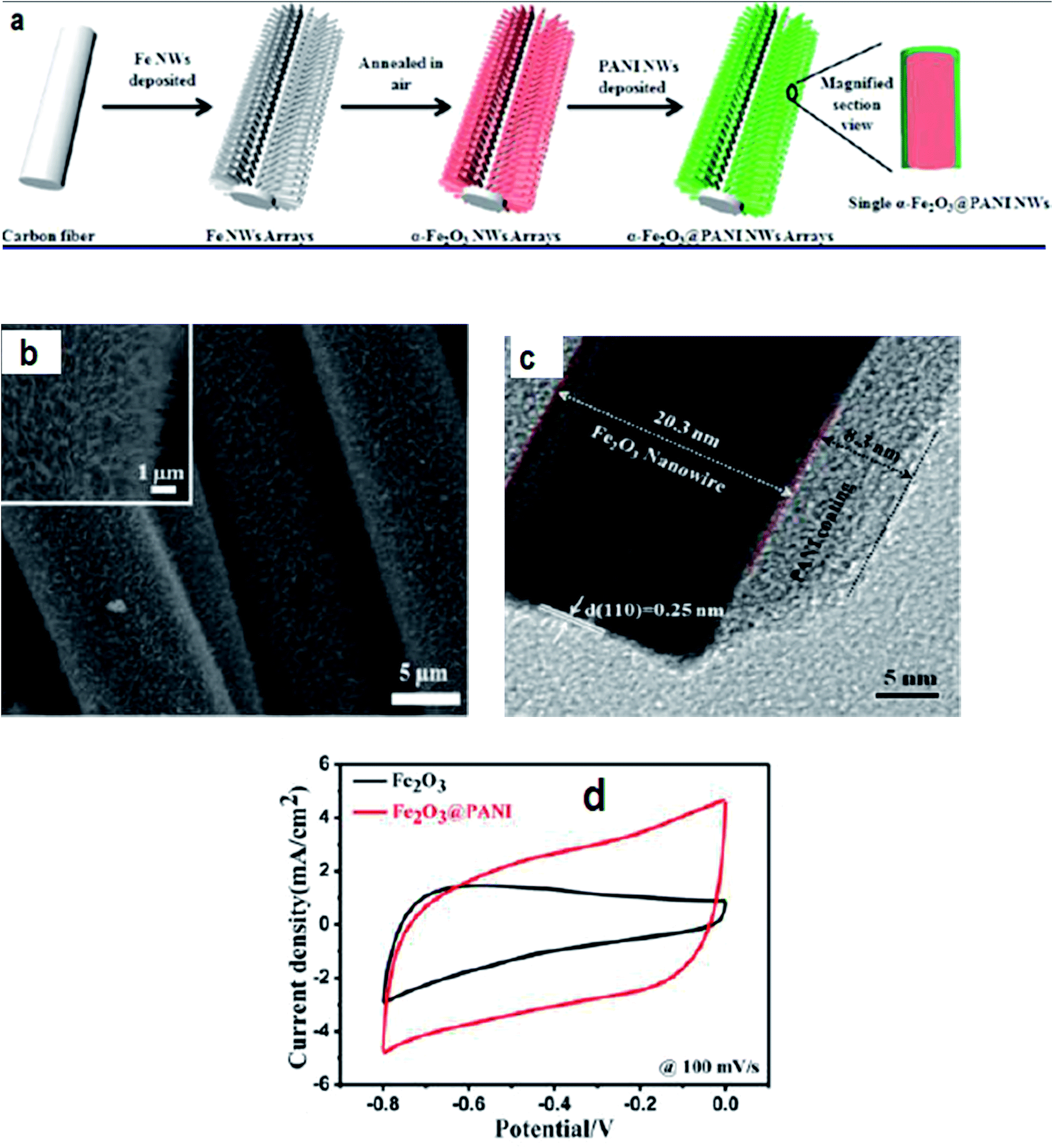 | ||
| Fig. 13 (a) Schematic illustration of electrode fabrication and a magnified view of the unit structure of α-Fe2O3@PANI core–shell nanowires; (b) SEM and (c) TEM images showing the structure of α-Fe2O3@PANI core–shell nanowires; (d) cyclic voltammograms obtained at a 100 mV s−1 scan rate using α-Fe2O3@PANI and α-Fe2O3 electrodes. Reprinted with permission from ref. 296 copyright 2015 American Chemical Society. | ||
Dual heteroatom doping boosts the unique features of nanostructured carbon materials for their advanced applications.300 Recent research involves doping of carbon structures with nitrogen ad-atoms along with other dopants such as boron, fluorine, iodine, phosphorus, etc. For dual doping, hierarchical porous nitrogen-doped carbon nanosheets (HPNC-NS) are suitable supercapacitor electrode materials due to their excellent electrocatalytic performance quantified as a 242 F g−1 capacitance and 102 W h kg−1 energy density accompanied with a loss of only 9% in cycling stability after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The distinctive characteristic of the HPNC-NS electrode is its use as the same material in both supercapacitors and high-performance LIBs showing the highest lithium storage capacity of 1865 mA h g−1.301 Phosphorus nitrogen-doped carbon (PNDC) is a catalytically active dual doped material for use in supercapacitors. Similarly, Bairia and co-workers also reported a phosphorus and nitrogen co-doped porous carbon material for the storage of charge in supercapacitors. The surface area of porous PNDC was calculated to be 433 m2 g−1 with a specific capacitance of 161 F g−1.302 Moreover, an N-doped carbon fibre aerogel (N-CFA) modulated with NiS nanoparticles was synthesized through a combination of carbonization, polymerization and solvothermal processes to prevent aggregation and enhance the conductivity of NiS NPs by the presence of nanofibers on the N-CFA material to ensure excellent electrochemical properties of the nanocomposite. The N-CFA in the synthesized nanostructured material was found to provide sufficient active sites for NiS NP growth to avoid their aggregation and to offer a shorter route for the efficient transport of ions and electrons. The NiS/N-CFA nanocomposite was found to exhibit a 1612.5 F g−1 specific capacitance at a current density of 1 A g−1 and 66.7% rate capacitance retention at 20 A g−1.303 Thus, dual dopants in carbon materials have resulted in a multifold increase of their catalytic performance. This rapid advancement in the designing of high performance electrode materials has led to a rise in the demand for microscale supercapacitors.304 Hence, future endeavours towards improvement in power and energy density by the synergistic coupling of electroactive pseudo-capacitive and electrical double layer capacitive materials are expected to lead to promising results for advanced applications of SCs.
000 cycles. The distinctive characteristic of the HPNC-NS electrode is its use as the same material in both supercapacitors and high-performance LIBs showing the highest lithium storage capacity of 1865 mA h g−1.301 Phosphorus nitrogen-doped carbon (PNDC) is a catalytically active dual doped material for use in supercapacitors. Similarly, Bairia and co-workers also reported a phosphorus and nitrogen co-doped porous carbon material for the storage of charge in supercapacitors. The surface area of porous PNDC was calculated to be 433 m2 g−1 with a specific capacitance of 161 F g−1.302 Moreover, an N-doped carbon fibre aerogel (N-CFA) modulated with NiS nanoparticles was synthesized through a combination of carbonization, polymerization and solvothermal processes to prevent aggregation and enhance the conductivity of NiS NPs by the presence of nanofibers on the N-CFA material to ensure excellent electrochemical properties of the nanocomposite. The N-CFA in the synthesized nanostructured material was found to provide sufficient active sites for NiS NP growth to avoid their aggregation and to offer a shorter route for the efficient transport of ions and electrons. The NiS/N-CFA nanocomposite was found to exhibit a 1612.5 F g−1 specific capacitance at a current density of 1 A g−1 and 66.7% rate capacitance retention at 20 A g−1.303 Thus, dual dopants in carbon materials have resulted in a multifold increase of their catalytic performance. This rapid advancement in the designing of high performance electrode materials has led to a rise in the demand for microscale supercapacitors.304 Hence, future endeavours towards improvement in power and energy density by the synergistic coupling of electroactive pseudo-capacitive and electrical double layer capacitive materials are expected to lead to promising results for advanced applications of SCs.
5. Conclusion
The replacement of exorbitant platinum by alternative catalysts possessing improved features of cost affordability and sustainability bedecked with dopants for achieving exceptional electrocatalytic performance is a hotspot in the realm of energy production and application. In this regard heteroatom-doped nanocarbon based catalysts have shown great promise for replacing costly platinum in the context of stability, selectivity and immunity towards CO-poisoning and crossover effects. In the same vein, some heteroatom-doped carbon frameworks have shown ORR activities comparable to or even higher than those of platinum but most of the studies conducted so far reveal that in terms of onset potential and current density they are still far from outperforming the conventional material, platinum at a commercial level. Some studies have shown the higher ORR activity of CVD-grown heteroatom-doped carbons than nanocarbons which may stem from the residual catalyst particles in the carbon matrix; however, unravelling all the underlying factors influencing the ORR performance demands a thorough comparison between high and low activity catalysts and their testing under the same experimental conditions. It is presumed that charged sites are induced in carbon based materials upon doping with electron rich or electron deficient species which favour oxygen adsorption over undoped carbon materials. The results of experimental and theoretical studies reveal that the electron transfer number is governed by changes in the charge densities of carbon atoms upon doping. However, the exact nature of active sites which control ORR-activity in nanostructured carbons is still a debatable topic probably due to the unavailability of direct techniques for active site determination and problems confronted while locating dopants in the carbon framework. Thus, before capitalizing on the large scale development of energy storage devices using doped carbon frameworks, there is a dire need for further studies focused on the determination of active sites and the underlying mechanism via which electrode reactions are catalyzed. In this regard, combined theoretical and experimental work is expected to prove pivotal in the development of efficient and affordable heteroatom-doped carbonaceous materials for next generation commercial energy storage devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support of the Higher Education Commission of Pakistan (6815/NRPU/R&D/HEC) and Quaid-i-Azam University, Islamabad (URF 2017).References
- B. C. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- X. Yao and Y. Zhao, Chem, 2017, 2, 171–200 CAS.
- N. G. Sahoo, Y. Pan, L. Li and S. H. Chan, Adv. Mater., 2012, 24, 4203–4210 CrossRef CAS PubMed.
- N. Popovich, 2015 Annual Progress Report: DOE Hydrogen and Fuel Cells Program, National Renewable Energy Laboratory (NREL), Golden, CO, United States, 2015 Search PubMed.
- H. Lv, D. Li, D. Strmcnik, A. P. Paulikas, N. M. Markovic and V. R. Stamenkovic, Nano Energy, 2016, 29, 149–165 CrossRef CAS.
- M. E. Scofield, H. Liu and S. S. Wong, Chem. Soc. Rev., 2015, 44, 5836–5860 RSC.
- F. Si, Y. Zhang, L. Yan, J. Zhu, M. Xiao, C. Liu, W. Xing and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, Amsterdam, 2014, pp. 133–170 Search PubMed.
- L. W. Liao, Y. L. Zheng, J. Wei and Y. X. Chen, Electrochem. Commun., 2015, 58, 73–75 CrossRef CAS.
- M. Markiewicz, C. Zalitis and A. Kucernak, Electrochim. Acta, 2015, 179, 126–136 CrossRef CAS.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- L. Genies, R. Faure and R. Durand, Electrochim. Acta, 1998, 44, 1317–1327 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. T. Goh, T. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- L. Feng, X. Sun, S. Yao, C. Liu, W. Xing and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, Amsterdam, 2014, pp. 67–132 Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- C. Guo, Y. Wu, Z. Li, W. Liao, L. Sun, C. Wang, B. Wen, Y. Li and C. Chen, Nanoscale Res. Lett., 2017, 12, 144 CrossRef PubMed.
- O. T. Holton and J. W. Stevenson, Platinum Met. Rev., 2013, 57, 259–271 CrossRef.
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CAS.
- M. Kaukonen, R. Kujala and E. Kauppinen, J. Phys. Chem. C, 2011, 116, 632–636 Search PubMed.
- T. Ikeda, M. Boero, S.-F. Huang, K. Terakura, M. Oshima and J.-i. Ozaki, J. Phys. Chem. C, 2008, 112, 14706–14709 CAS.
- J. Zheng, J. Lu, K. Amine and F. Pan, Nano Energy, 2017, 33, 497–507 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- F. Pan, J. Jin, X. Fu, Q. Liu and J. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 11108–11114 CAS.
- J. Xu, G. Dong, C. Jin, M. Huang and L. Guan, ChemSusChem, 2013, 6, 493–499 CrossRef CAS PubMed.
- Y. Xue, D. Yu, L. Dai, R. Wang, D. Li, A. Roy, F. Lu, H. Chen, Y. Liu and J. Qu, Phys. Chem. Chem. Phys., 2013, 15, 12220–12226 RSC.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787–1793 CrossRef CAS PubMed.
- Z. Yang, H. Nie, X. a. Chen, X. Chen and S. Huang, J. Power Sources, 2013, 236, 238–249 CrossRef CAS.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., 2011, 123, 7270–7273 CrossRef.
- Z. Jin, H. Nie, Z. Yang, J. Zhang, Z. Liu, X. Xu and S. Huang, Nanoscale, 2012, 4, 6455–6460 RSC.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS PubMed.
- H. Dai, in Carbon nanotubes: Synthesis, structure, properties, and applications, ed. M. S. Dresselhaus, G. Dresselhaus and P. Avouris, Springer Berlin Heidelberg, Berlin, Heidelberg, 2001, pp. 29–53 Search PubMed.
- P. Ajayan, Chem. Rev., 1999, 99, 1787–1800 CrossRef CAS PubMed.
- Z. Wang, R. Jia, J. Zheng, J. Zhao, L. Li, J. Song and Z. Zhu, ACS Nano, 2011, 5, 1677–1684 CrossRef CAS PubMed.
- M. Terrones, A. G. Souza Filho and A. M. Rao, in Carbon nanotubes, Springer, 2007, pp. 531–566 Search PubMed.
- S. N. Goubert-Renaudin and A. Wieckowski, J. Electroanal. Chem., 2011, 652, 44–51 CrossRef CAS.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- Y. Tang, B. L. Allen, D. R. Kauffman and A. Star, J. Am. Chem. Soc., 2009, 131, 13200–13201 CrossRef CAS PubMed.
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2009, 113, 21008–21013 CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- Z. Yang, Y. Xia and R. Mokaya, Chem. Mater., 2005, 17, 4502–4508 CrossRef CAS.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C. P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS.
- M. Lefèvre, J. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef.
- J. Maruyama, J. Okamura, K. Miyazaki, Y. Uchimoto and I. Abe, J. Phys. Chem. C, 2008, 112, 2784–2790 CAS.
- J. Maruyama and I. Abe, Chem. Mater., 2006, 18, 1303–1311 CrossRef CAS.
- F. Jaouen, S. Marcotte, J. P. Dodelet and G. Lindbergh, J. Phys. Chem. B, 2003, 107, 1376–1386 CrossRef CAS.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- D. Geng, H. Liu, Y. Chen, R. Li, X. Sun, S. Ye and S. Knights, J. Power Sources, 2011, 196, 1795–1801 CrossRef CAS.
- A. Bouwkamp-Wijnoltz, W. Visscher, J. Van Veen, E. Boellaard, A. Van der Kraan and S. Tang, J. Phys. Chem. B, 2002, 106, 12993–13001 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CAS.
- P. H. Matter, E. Wang, J.-M. M. Millet and U. S. Ozkan, J. Phys. Chem. C, 2007, 111, 1444–1450 CAS.
- J. L. Lyon and K. J. Stevenson, Langmuir, 2007, 23, 11311–11318 CrossRef CAS PubMed.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- C. Venkateswara Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340–4346 CAS.
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127–15129 CrossRef CAS PubMed.
- M. Borghei, P. Kanninen, M. Lundahl, T. Susi, J. Sainio, I. Anoshkin, A. Nasibulin, T. Kallio, K. Tammeveski and E. Kauppinen, Appl. Catal., B, 2014, 158, 233–241 CrossRef.
- M. Yang, D. Yang, H. Chen, Y. Gao and H. Li, J. Power Sources, 2015, 279, 28–35 CrossRef CAS.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed.
- G. Tuci, C. Zafferoni, A. Rossin, A. Milella, L. Luconi, M. Innocenti, L. Truong Phuoc, C. Duong-Viet, C. Pham-Huu and G. Giambastiani, Chem. Mater., 2014, 26, 3460–3470 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed.
- Y. B. Tan and J. M. Lee, J. Mater. Chem. A, 2013, 1, 14814–14843 CAS.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- X. Zhou, Z. Bai, M. Wu, J. Qiao and Z. Chen, J. Mater. Chem. A, 2015, 3, 3343–3350 CAS.
- J. Wu, L. Ma, R. M. Yadav, Y. Yang, X. Zhang, R. Vajtai, J. Lou and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 14763–14769 CAS.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- S. Yasuda, L. Yu, J. Kim and K. Murakoshi, Chem. Commun., 2013, 49, 9627–9629 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- L. Feng, Y. Chen and L. Chen, ACS Nano, 2011, 5, 9611–9618 CrossRef CAS PubMed.
- Y. Zhang, J. Ge, L. Wang, D. Wang, F. Ding, X. Tao and W. Chen, Sci. Rep., 2013, 3, 2771 CrossRef PubMed.
- D. Qazzazie, M. Beckert, R. Mülhaupt, O. Yurchenko and G. Urban, J. Energy Power Eng., 2015, 9, 886–895 CrossRef PubMed.
- M. Liu, Y. Song, S. He, W. W. Tjiu, J. Pan, Y.-Y. Xia and T. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 4214–4222 CAS.
- L. Chen, R. Du, J. Zhu, Y. Mao, C. Xue, N. Zhang, Y. Hou, J. Zhang and T. Yi, Small, 2015, 11, 1423–1429 CrossRef CAS PubMed.
- B. Zheng, J. Wang, F. B. Wang and X. H. Xia, Electrochem. Commun., 2013, 28, 24–26 CrossRef CAS.
- M. Park, T. Lee and B. S. Kim, Nanoscale, 2013, 5, 12255–12260 RSC.
- H. P. Cong, P. Wang, M. Gong and S. H. Yu, Nano Energy, 2014, 3, 55–63 CrossRef CAS.
- Z. Lin, M.-k. Song, Y. Ding, Y. Liu, M. Liu and C. P. Wong, Phys. Chem. Chem. Phys., 2012, 14, 3381–3387 RSC.
- J. Masa, A. Zhao, W. Xia, Z. Sun, B. Mei, M. Muhler and W. Schuhmann, Electrochem. Commun., 2013, 34, 113–116 CrossRef CAS.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 CAS.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- C. H. Choi, M. W. Chung, H. C. Kwon, J. H. Chung and S. I. Woo, Appl. Catal., B, 2014, 144, 760–766 CrossRef CAS.
- Y. Ma, L. Sun, W. Huang, L. Zhang, J. Zhao, Q. Fan and W. Huang, J. Phys. Chem. C, 2011, 115, 24592–24597 CAS.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- Y. Zhang, J. S. Hu, W. J. Jiang, L. Guo, Z. D. Wei and L. J. Wan, J. Electrochem., 2014, 20, 401–409 CAS.
- D. C. Higgins, M. A. Hoque, F. Hassan, J. Y. Choi, B. Kim and Z. Chen, ACS Catal., 2014, 4, 2734–2740 CrossRef CAS.
- Y. W. Ma, L. R. Zhang, J. J. Li, H. T. Ni, M. Li, J. L. Zhang, X. M. Feng, Q. L. Fan, Z. Hu and W. Huang, Chin. Sci. Bull., 2011, 56, 3583–3589 CrossRef CAS.
- S. Ratso, I. Kruusenberg, M. Vikkisk, U. Joost, E. Shulga, I. Kink, T. Kallio and K. Tammeveski, Carbon, 2014, 73, 361–370 CrossRef CAS.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- H. L. Poh, P. Simek, Z. Sofer and M. Pumera, Chem.–Eur. J., 2013, 19, 2655–2662 CrossRef CAS PubMed.
- T. Wu, H. Shen, L. Sun, B. Cheng, B. Liu and J. Shen, New J. Chem., 2012, 36, 1385–1391 RSC.
- L. Panchakarla, K. Subrahmanyam, S. Saha, A. Govindaraj, H. Krishnamurthy, U. Waghmare and C. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- P. A. Denis, Chem. Phys. Lett., 2011, 508, 95–101 CrossRef CAS.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., 2011, 123, 3315–3319 CrossRef.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- M. Zhang and L. Dai, Nano Energy, 2012, 1, 514–517 CrossRef CAS.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- D. S. Yang, D. Bhattacharjya, M. Y. Song and J. S. Yu, Carbon, 2014, 67, 736–743 CrossRef CAS.
- H. Wang, X. Bo, Y. Zhang and L. Guo, Electrochim. Acta, 2013, 108, 404–411 CrossRef CAS.
- Y. Sun, J. Wu, J. Tian, C. Jin and R. Yang, Electrochim. Acta, 2015, 178, 806–812 CrossRef CAS.
- S. Inamdar, H.-S. Choi, P. Wang, M. Y. Song and J. S. Yu, Electrochem. Commun., 2013, 30, 9–12 CrossRef CAS.
- J. Dai and J. Yuan, J. Phys.: Condens. Matter, 2010, 22, 225501 CrossRef PubMed.
- E. Cruz-Silva, D. A. Cullen, L. Gu, J. M. Romo-Herrera, E. Muñoz-Sandoval, F. López-Urías, B. G. Sumpter, V. Meunier, J.-C. Charlier and D. J. Smith, ACS Nano, 2008, 2, 441–448 CrossRef CAS PubMed.
- I. Y. Jeon, S. Zhang, L. Zhang, H. J. Choi, J. M. Seo, Z. Xia, L. Dai and J. B. Baek, Adv. Mater., 2013, 25, 6138–6145 CrossRef CAS PubMed.
- S. Yu, W. Zheng, C. Wang and Q. Jiang, ACS Nano, 2010, 4, 7619–7629 CrossRef CAS PubMed.
- X. Li, L. Fan, Z. Li, K. Wang, M. Zhong, J. Wei, D. Wu and H. Zhu, Adv. Energy Mater., 2012, 2, 425–429 CrossRef CAS.
- Y. B. Tang, L. C. Yin, Y. Yang, X. H. Bo, Y. L. Cao, H. E. Wang, W. J. Zhang, I. Bello, S. T. Lee and H.-M. Cheng, ACS Nano, 2012, 6, 1970–1978 CrossRef CAS PubMed.
- X. Lü, J. Wu, T. Lin, D. Wan, F. Huang, X. Xie and M. Jiang, J. Mater. Chem., 2011, 21, 10685–10689 RSC.
- G. Jo and S. Shanmugam, Electrochem. Commun., 2012, 25, 101–104 CrossRef CAS.
- X. Bo and L. Guo, Phys. Chem. Chem. Phys., 2013, 15, 2459–2465 RSC.
- Y. Lin, Y. Zhu, B. Zhang, Y. A. Kim, M. Endo and D. S. Su, J. Mater. Chem. A, 2015, 3, 21805–21814 CAS.
- J. Han, L. L. Zhang, S. Lee, J. Oh, K. S. Lee, J. R. Potts, J. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2012, 7, 19–26 CrossRef PubMed.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- R. Ma, Y. Ma, Y. Donga and J.-M. Lee, Nano Adv., 2016, 1, 50–61 CrossRef.
- I.-Y. Jeon, C. Kim, G. Kim and J. B. Baek, Carbon, 2015, 93, 465–472 CrossRef CAS.
- X. Sun, Y. Zhang, P. Song, J. Pan, L. Zhuang, W. Xu and W. Xing, ACS Catal., 2013, 3, 1726–1729 CrossRef CAS.
- F. Navarro-Pardo, A. L. Martinez-Hernandez and C. Velasco-Santos, J. Nanomater., 2016, 2016, 1–16 CrossRef.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J.-G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1, 195–220 CrossRef CAS.
- Z. Liu, F. Peng, H. Wang, H. Yu, J. Tan and L. Zhu, Catal. Commun., 2011, 16, 35–38 CrossRef CAS.
- Y. Cheng, Y. Tian, X. Fan, J. Liu and C. Yan, Electrochim. Acta, 2014, 143, 291–296 CrossRef CAS.
- G. Panomsuwan, N. Saito and T. Ishizaki, Electrochem. Commun., 2015, 59, 81–85 CrossRef CAS.
- Y. J. Sa, C. Park, H. Y. Jeong, S. H. Park, Z. Lee, K. T. Kim, G. G. Park and S. H. Joo, Angew. Chem., 2014, 126, 4186–4190 CrossRef.
- A. G. Güell, N. Ebejer, M. E. Snowden, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 7258–7261 CrossRef PubMed.
- D. Boukhvalov and M. Katsnelson, Nano Lett., 2008, 8, 4373–4379 CrossRef CAS PubMed.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- R. Li, Z. Wei, X. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 RSC.
- J.-e. Park, Y. J. Jang, Y. J. Kim, M.-s. Song, S. Yoon, D. H. Kim and S.-J. Kim, Phys. Chem. Chem. Phys., 2014, 16, 103–109 RSC.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a. Chen and S. Huang, ACS Nano, 2011, 6, 205–211 CrossRef CAS PubMed.
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027–1029 RSC.
- J. Xu, I. Y. Jeon, J. M. Seo, S. Dou, L. Dai and J. B. Baek, Adv. Mater., 2014, 26, 7317–7323 CrossRef CAS PubMed.
- K. Kakaei and A. Balavandi, J. Colloid Interface Sci., 2016, 463, 46–54 CrossRef CAS PubMed.
- Z. H. Sheng, H. L. Gao, W.-J. Bao, F. B. Wang and X. H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC.
- Z. Ma, S. Dou, A. Shen, L. Tao, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2015, 54, 1888–1892 CrossRef CAS PubMed.
- Y. Zhang, M. Chu, L. Yang, W. Deng, Y. Tan, M. Ma and Q. Xie, Chem. Commun., 2014, 50, 6382–6385 RSC.
- X. a. Chen, X. Chen, X. Xu, Z. Yang, Z. Liu, L. Zhang, X. Xu, Y. Chen and S. Huang, Nanoscale, 2014, 6, 13740–13747 RSC.
- M. Seredych, K. László, E. Rodríguez-Castellón and T. J. Bandosz, J. Energy Chem., 2016, 25, 236–245 CrossRef.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- B. Blizanac, P. N. Ross and N. Marković, J. Phys. Chem. B, 2006, 110, 4735–4741 CrossRef CAS PubMed.
- T. Schmidt, V. Stamenkovic, M. Arenz, N. Markovic and P. Ross, Electrochim. Acta, 2002, 47, 3765–3776 CrossRef CAS.
- N. Markovic and P. N. Ross, in Interfacial electrochemistrys theory, experiments and applications, ed. A. Wieckowski, Marcel Dekker, Inc., New York, 1999, pp. 821–841 Search PubMed.
- T. Zhang and A. B. Anderson, Electrochim. Acta, 2007, 53, 982–989 CrossRef CAS.
- B. Grgur, N. Marković and P. Ross, Can. J. Chem., 1997, 75, 1465–1471 CrossRef CAS.
- X. Fan, W. Zheng and J.-L. Kuo, RSC Adv., 2013, 3, 5498–5505 RSC.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Norskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 2948–2951 CrossRef CAS PubMed.
- X. Hu, Y. Wu, H. Li and Z. Zhang, J. Phys. Chem. C, 2010, 114, 9603–9607 CAS.
- G. L. Chai, Z. Hou, D. J. Shu, T. Ikeda and K. Terakura, J. Am. Chem. Soc., 2014, 136, 13629–13640 CrossRef CAS PubMed.
- M. Del Cueto, P. Ocón and J. Poyato, J. Phys. Chem. C, 2015, 119, 2004–2009 CAS.
- Y. Okamoto, Appl. Surf. Sci., 2009, 256, 335–341 CrossRef CAS.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707–4716 CrossRef CAS PubMed.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2011, 115, 20002–20010 CAS.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., 2010, 122, 2619–2623 CrossRef.
- A. Serov and C. Kwak, Appl. Catal., B, 2009, 90, 313–320 CrossRef CAS.
- W. Wong, W. R. W. Daud, A. Mohamad, A. Kadhum, K. Loh and E. Majlan, J. Eng. Sci. Technol., 2015, 10, 68–77 Search PubMed.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- Z. Shi, J. Zhang, Z.-S. Liu, H. Wang and D. P. Wilkinson, Electrochim. Acta, 2006, 51, 1905–1916 CrossRef CAS.
- Z.-H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- G. G. Wallace, J. Chen, A. J. Mozer, M. Forsyth, D. R. MacFarlane and C. Wang, Mater. Today, 2009, 12, 20–27 CrossRef CAS.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. D. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef PubMed.
- Y. Yu, L. Gu, A. Dhanabalan, C.-H. Chen and C. Wang, Electrochim. Acta, 2009, 54, 7227–7230 CrossRef CAS.
- A. Wilson, B. Way, J. Dahn and T. Van Buuren, J. Appl. Phys., 1995, 77, 2363–2369 CrossRef CAS.
- N. Dimov, S. Kugino and M. Yoshio, Electrochim. Acta, 2003, 48, 1579–1587 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CAS.
- N. Kurita and M. Endo, Carbon, 2002, 40, 253–260 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 CAS.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- M. Kim, H. M. Hwang, G. H. Park and H. Lee, FlatChem, 2017, 6, 48–76 CrossRef CAS.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS.
- P. Guo, H. Song and X. Chen, Electrochem. Commun., 2009, 11, 1320–1324 CrossRef CAS.
- J. Luo, X. Zhao, J. Wu, H. D. Jang, H. H. Kung and J. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS PubMed.
- L. Fan, X. Li, B. Yan, X. Li, D. Xiong, D. Li, H. Xu, X. Zhang and X. Sun, Appl. Energy, 2016, 175, 529–535 CrossRef CAS.
- C. H. Wu, N. W. Pu, P. J. Wu, Y. Y. Peng, C. N. Shih, C. Y. Chen, Y. M. Liu and M. D. Ger, Microelectron. Eng., 2015, 138, 47–51 CrossRef CAS.
- C. H. Wu, N. W. Pu, Y. M. Liu, C. Y. Chen, Y. Y. Peng, T. Y. Cheng, M. H. Lin and M. D. Ger, J. Taiwan Inst. Chem. Eng., 2017, 80, 511–517 CrossRef CAS.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 55, 3909–3914 CrossRef CAS.
- F. Xiang, R. Mukherjee, J. Zhong, Y. Xia, N. Gu, Z. Yang and N. Koratkar, Energy Storage Materials, 2015, 1, 9–16 CrossRef.
- A. Márquez, A. Vargas and P. B. Balbuena, J. Electrochem. Soc., 1998, 145, 3328–3334 CrossRef.
- M. Nakadaira, R. Saito, T. Kimura, G. Dresselhaus and M. Dresselhaus, J. Mater. Res., 1997, 12, 1367–1375 CrossRef CAS.
- N. Kheirabadi and A. Shafiekhani, J. Appl. Phys., 2012, 112, 124323 CrossRef.
- F. Torrisi, T. Hasan, W. Wu, Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. Hsieh, S. Jung, F. Bonaccorso and P. J. Paul, ACS Nano, 2012, 6, 2992–3006 CrossRef CAS PubMed.
- C. Uthaisar and V. Barone, Nano Lett., 2010, 10, 2838–2842 CrossRef CAS PubMed.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero and V. Pellegrini, Nano Lett., 2014, 14, 4901–4906 CrossRef CAS PubMed.
- K. Chen, S. Song, F. Liu and D. Xue, Chem. Soc. Rev., 2015, 44, 6230–6257 RSC.
- A. Abouimrane, O. C. Compton, K. Amine and S. T. Nguyen, J. Phys. Chem. C, 2010, 114, 12800–12804 CAS.
- E. Pollak, B. Geng, K. J. Jeon, I. T. Lucas, T. J. Richardson, F. Wang and R. Kostecki, Nano Lett., 2010, 10, 3386–3388 CrossRef CAS PubMed.
- X. Fan, W. Zheng, J.-L. Kuo and D. J. Singh, ACS Appl. Mater. Interfaces, 2013, 5, 7793–7797 CAS.
- H. Kim, S. W. Kim, J. Hong, H.-D. Lim, H. S. Kim, J.-K. Yoo and K. Kang, J. Electrochem. Soc., 2011, 158, A930–A935 CrossRef CAS.
- H. Wang, Y. Yang, Y. Liang, L. F. Cui, H. Sanchez Casalongue, Y. Li, G. Hong, Y. Cui and H. Dai, Angew. Chem., Int. Ed., 2011, 50, 7364–7368 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- X. Lu, X. Jin and J. Sun, Sci. China: Technol. Sci., 2015, 58, 1829–1840 CrossRef CAS.
- F. Yao, F. Güneş, H. Q. Ta, S. M. Lee, S. J. Chae, K. Y. Sheem, C. S. Cojocaru, S. S. Xie and Y. H. Lee, J. Am. Chem. Soc., 2012, 134, 8646–8654 CrossRef CAS PubMed.
- K. H. Park, D. Lee, J. Kim, J. Song, Y. M. Lee, H.-T. Kim and J.-K. Park, Nano Lett., 2014, 14, 4306–4313 CrossRef CAS PubMed.
- H. S. Song, M. G. Park, S. J. Kwon, K. B. Yi, E. Croiset, Z. Chen and S. C. Nam, Appl. Surf. Sci., 2013, 276, 646–652 CrossRef CAS.
- H. S. Song, C. H. Ko, W. Ahn, B. J. Kim, E. Croiset, Z. Chen and S. C. Nam, Ind. Eng. Chem. Res., 2012, 51, 10259–10264 CrossRef CAS.
- W. Ahn, H. S. Song, S. H. Park, K.-B. Kim, K. H. Shin, S. N. Lim and S. H. Yeon, Electrochim. Acta, 2014, 132, 172–179 CrossRef CAS.
- E. Irisarri, A. Ponrouch and M. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, W. Ren, Q. Tan, Y. Chen, H. Li, Z. Zhong and F. Su, Angew. Chem., 2014, 126, 5265–5269 CrossRef.
- J. Kim, J. Y. Kim, D. Pham-Cong, S. Y. Jeong, J. Chang, J. H. Choi, P. V. Braun and C. R. Cho, Electrochim. Acta, 2016, 199, 35–44 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- X.-M. Liu, Z. Dong Huang, S. Woon Oh, B. Zhang, P.-C. Ma, M. M. Yuen and J.-K. Kim, Compos. Sci. Technol., 2012, 72, 121–144 CrossRef CAS.
- W. Guoping, Z. Qingtang, Y. Zuolong and Q. MeiZheng, Solid State Ionics, 2008, 179, 263–268 CrossRef.
- H. Chakraborti and S. K. Pal, Chem. Phys. Lett., 2014, 600, 118–122 CrossRef CAS.
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D. M. Tang, H. Li, T. Zhai and L. Li, Adv. Funct. Mater., 2012, 22, 2682–2690 CrossRef CAS.
- L. S. Zhang, L. Y. Jiang, H. J. Yan, W. D. Wang, W. Wang, W. G. Song, Y. G. Guo and L. J. Wan, J. Mater. Chem., 2010, 20, 5462–5467 RSC.
- Z. Chen, M. Zhou, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2012, 2, 95–102 CrossRef CAS.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- B. Li, H. Cao, J. Shao, G. Li, M. Qu and G. Yin, Inorg. Chem., 2011, 50, 1628–1632 CrossRef CAS PubMed.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Adv. Funct. Mater., 2013, 23, 2436–2444 CrossRef CAS.
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298–6302 CrossRef CAS PubMed.
- W. Li, F. Wang, S. Feng, J. Wang, Z. Sun, B. Li, Y. Li, J. Yang, A. A. Elzatahry and Y. Xia, J. Am. Chem. Soc., 2013, 135, 18300–18303 CrossRef CAS PubMed.
- B. Qiu, M. Xing and J. Zhang, J. Am. Chem. Soc., 2014, 136, 5852–5855 CrossRef CAS PubMed.
- X. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS.
- X. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC.
- J. Ji, H. Ji, L. L. Zhang, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, Adv. Mater., 2013, 25, 4673–4677 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- S. W. Kim, D. H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- Y. Xu, Y. Zhu, Y. Liu and C. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
- T. Sun, Z. j. Li, H. g. Wang, D. Bao, F. l. Meng and X. b. Zhang, Angew. Chem., 2016, 128, 10820–10824 CrossRef.
- K. Naoi, W. Naoi, S. Aoyagi, J.-i. Miyamoto and T. Kamino, Acc. Chem. Res., 2012, 46, 1075–1083 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Funct. Mater., 2012, 22, 827–834 CrossRef CAS.
- S. Gao, Y. Chen, H. Fan, X. Wei, C. Hu, H. Luo and L. Qu, J. Mater. Chem. A, 2014, 2, 3317–3324 CAS.
- H. Zhu, X. Wang, X. Liu and X. Yang, Adv. Mater., 2012, 24, 6524–6529 CrossRef CAS PubMed.
- Y. Lu, F. Zhang, T. Zhang, K. Leng, L. Zhang, X. Yang, Y. Ma, Y. Huang, M. Zhang and Y. Chen, Carbon, 2013, 63, 508–516 CrossRef CAS.
- K. Wang, Y. Cao, Z. Gu, P. Ahrenkiel, J. Lee and Q. H. Fan, RSC Adv., 2016, 6, 26738–26744 RSC.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- M. Yoshikawa and H. Fujimoto, Meeting Abstracts, The Electrochemical Society, 2016, p. 977 Search PubMed.
- M. Y. Zhang, X. J. Jin and Q. Zhao, N. Carbon Mater., 2014, 29, 89–95 CrossRef CAS.
- D. A. Iozzo, M. Tong, G. Wu and E. P. Furlani, J. Phys. Chem. C, 2015, 119, 25235–25242 CAS.
- A. Janes, H. Kurig and E. Lust, Carbon, 2007, 45, 1226–1233 CrossRef.
- N. Fattah, H. Ng, Y. Mahipal, A. Numan, S. Ramesh and K. Ramesh, Materials, 2016, 9, 450 CrossRef PubMed.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- L. Yang, B. H. Fishbine, A. Migliori and L. R. Pratt, J. Am. Chem. Soc., 2009, 131, 12373–12376 CrossRef CAS PubMed.
- J. Leis, M. Arulepp, A. Kuura, M. Lätt and E. Lust, Carbon, 2006, 44, 2122–2129 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P.-L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- G. Jiang, C. Cheng, D. Li and J. Z. Liu, Nano Res., 2016, 9, 174–186 CrossRef CAS.
- C. Cheng, J. Uhe, X. Yang, Y. Wu and D. Li, Prog. Nat. Sci.: Mater. Int., 2012, 22, 668–672 CrossRef.
- P. Lu, Q. Dai, L. Wu and X. Liu, Appl. Sci., 2017, 7, 939 CrossRef.
- Y.-W. Tan, Y. Zhang, K. Bolotin, Y. Zhao, S. Adam, E. Hwang, S. D. Sarma, H. Stormer and P. Kim, Phys. Rev. Lett., 2007, 99, 246803 CrossRef PubMed.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- H. Ji, X. Zhao, Z. Qiao, J. Jung, Y. Zhu, Y. Lu, L. L. Zhang, A. H. MacDonald and R. S. Ruoff, Nat. Commun., 2014, 5, 3317 Search PubMed.
- M. Hahn, M. Baertschi, O. Barbieri, J.-C. Sauter, R. Kötz and R. Gallay, Electrochem. Solid-State Lett., 2004, 7, A33–A36 CrossRef CAS.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303–1310 CrossRef CAS.
- M. Mousavi-Khoshdel, E. Targholi and M. J. Momeni, J. Phys. Chem. C, 2015, 119, 26290–26295 CAS.
- S. Dröscher, P. Roulleau, F. Molitor, P. Studerus, C. Stampfer, K. Ensslin and T. Ihn, Phys. Scr., 2012, 2012, 014009 CrossRef.
- Z. Jin, J. Yao, C. Kittrell and J. M. Tour, ACS Nano, 2011, 5, 4112–4117 CrossRef CAS PubMed.
- E. Paek, A. J. Pak and G. S. Hwang, J. Electrochem. Soc., 2013, 160, A1–A10 CrossRef CAS.
- A. J. Pak, E. Paek and G. S. Hwang, J. Phys. Chem. C, 2014, 118, 21770–21777 CAS.
- E. Paek, A. J. Pak and G. S. Hwang, ACS Appl. Mater. Interfaces, 2014, 6, 12168–12176 CAS.
- B. SanthiBhushan, M. S. Khan, V. K. Bohat and A. Srivastava, IEEE Trans. Nanotechnol., 2018, 17(2), 205–211 CrossRef.
- C. Wang, Y. Zhou, L. Sun, Q. Zhao, X. Zhang, P. Wan and J. Qiu, J. Phys. Chem. C, 2013, 117, 14912–14919 CAS.
- T. Wang, L. X. Wang, D. L. Wu, W. Xia and D. Z. Jia, Sci. Rep., 2015, 5, 9591 CrossRef CAS PubMed.
- L. Chen, X. Li, C. Ma, M. Wang and J. Zhou, J. Phys. Chem. C, 2017, 121, 18344–18350 CAS.
- R. S. Ruoff, MRS Bull., 2012, 37, 1314–1318 CrossRef CAS.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- Z. Zhang, J. Chen and B. Li, Nanoscale, 2017, 9, 14208–14214 RSC.
- J. Dai, J. Li, H. Zeng and X. Cui, Appl. Phys. Lett., 2009, 94, 093114 CrossRef.
- V. Parkash and A. K. Goel, Nanoscale Res. Lett., 2010, 5, 1424 CrossRef CAS PubMed.
- M. Mousavi-Khoshdel, P. Jahanbakhsh-bonab and E. Targholi, Phys. Lett. A, 2016, 380, 3378–3383 CrossRef.
- W. Yang, M. Ni, X. Ren, Y. Tian, N. Li, Y. Su and X. Zhang, Curr. Opin. Colloid Interface Sci., 2015, 20, 416–428 CrossRef CAS.
- L. Lai, H. Yang, L. Wang, B. K. Teh, J. Zhong, H. Chou, L. Chen, W. Chen, Z. Shen and R. S. Ruoff, ACS Nano, 2012, 6, 5941–5951 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma and A. Yu, Sci. Rep., 2013, 3, 1408 CrossRef PubMed.
- L. Zhang, X. Yang, F. Zhang, G. Long, T. Zhang, K. Leng, Y. Zhang, Y. Huang, Y. Ma and M. Zhang, J. Am. Chem. Soc., 2013, 135, 5921–5929 CrossRef CAS PubMed.
- C. Zhang, Z. Peng, J. Lin, Y. Zhu, G. Ruan, C.-C. Hwang, W. Lu, R. H. Hauge and J. M. Tour, ACS Nano, 2013, 7, 5151–5159 CrossRef CAS PubMed.
- V. B. Kumar, A. Borenstein, B. Markovsky, D. Aurbach, A. Gedanken, M. Talianker and Z. Porat, J. Phys. Chem. C, 2016, 120, 13406–13413 CAS.
- Y. Tan, Q. Gao, J. Xu and Z. Li, RSC Adv., 2016, 6, 51332–51336 RSC.
- X. Lu, X. Chen, W. Zhou, Y. Tong and G. Li, ACS Appl. Mater. Interfaces, 2015, 7, 14843–14850 CAS.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M. S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 CrossRef CAS PubMed.
- G. Q. Zhang, H. B. Wu, H. E. Hoster, M. B. Chan-Park and X. W. D. Lou, Energy Environ. Sci., 2012, 5, 9453–9456 CAS.
- L. Niu, Z. Li, Y. Xu, J. Sun, W. Hong, X. Liu, J. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 8044–8052 CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef CAS PubMed.
- V. G. Bairi, U. B. Nasini, S. K. Ramasahayam, S. E. Bourdo and T. Viswanathan, Electrochim. Acta, 2015, 182, 987–994 CrossRef CAS.
- Y. Zhang, L. Zuo, L. Zhang, J. Yan, H. Lu, W. Fan and T. Liu, Nano Res., 2016, 9, 2747–2759 CrossRef CAS.
- M. F. El-Kady and R. B. Kaner, Nat. Commun., 2013, 4, 1475 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |