
Non-polymeric hybridization of a TEMPO derivative with activated carbon for high-energy-density aqueous electrochemical capacitor electrodes†
Hiroyuki
Itoi
 *a,
Hideyuki
Hasegawa
a,
Hiroyuki
Iwata
b and
Yoshimi
Ohzawa
a
*a,
Hideyuki
Hasegawa
a,
Hiroyuki
Iwata
b and
Yoshimi
Ohzawa
a
aDepartment of Applied Chemistry, Aichi Institute of Technology, Yachigusa 1247, Yakusa-cho, Toyota, 470-0392, Japan. E-mail: itoi-hiroyuki@aitech.ac.jp
bDepartment of Electrical and Electronics Engineering, Aichi Institute of Technology, Yachigusa 1247, Yakusa-cho, Toyota, 470-0392, Japan
First published on 8th January 2018
Abstract
Organic compounds have great potential as electrode materials with high energy and power densities, together with long cycle lifetimes. These properties can be achieved by employing redox-active organic compounds and porous carbon substrates via well-suited hybridization methods. In this study, a 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) derivative, 4-hydroxy-TEMPO benzoate (HTB), was used as an electrode material for aqueous electrochemical capacitors due to its high redox potential characteristics to achieve high energy densities. The hybridization of HTB and activated carbon (AC) was accomplished simply by adsorbing HTB in AC via a solvent-free preparation. This procedure takes only one hour and enables achieving precise AC/HTB weight ratios, eliminating the excess use of HTB or the use of organic solvents. Of note in this method is that it is not necessary to introduce HTB molecules in polymer chains to prevent dissolution of HTB in an aqueous electrolyte with the aid of a hydrophobic group in HTB. HTB molecules are finely dispersed inside the AC pores and therefore present a huge contact area with a conductive carbon surface, allowing for fast redox reactions (i.e., high power densities) in aqueous 1 M H2SO4 despite HTB displaying poor electrical conductivity by itself. As a result, the obtained AC/HTB materials enable compatibility of high energy and power densities.
Introduction
Organic compounds are widely employed as drugs, antibiotics, fine chemicals, agricultural chemicals, antiprotozoals, and in many other fields. Such chemicals are mainly composed of light elements and can thus be decomposed in a relatively facile manner even in Nature. Metals have also delivered significant benefits to our society due to their numerous characteristics, such as redox, conductive, semiconducting, radio-active, catalytic, and magnetic, together with their high melting point and rigid nature. The development of a variety of research areas has been drastically promoted by the diverse characteristics of metals. However, metallic elements are often toxic and thus an environmental burden. Moreover, many of them do not exist abundantly in Nature. Therefore, from environmental and cost perspectives, it is necessary to replace such precious metals with organic species. For this purpose, many organic compounds have been investigated as materials for semiconductors,1,2 batteries,3,4 catalysts,5,6 magnets,7 and many other applications. However, the high structural complexity of such organic compounds makes their use no longer practical in many cases.In contrast to the development of such complex organic compounds, the development of hybridization methods of simple organic compounds with suitable substrates may afford materials with properties superior to those of complex organic compounds.8 We have recently reported a hybridization method of redox-active organic compounds with porous carbon substrates for metal-free electrochemical capacitor electrodes through a solvent-free process.9 In this method, redox-active organic compounds are finely dispersed in the pores of porous carbon materials, and therefore, the contact area between the organic compounds and the conductive carbon surface is maximized. As a result, charge transfer between the carbon surface and the organic compounds, which typically exhibit poor electrical conductivity and cannot be used as electrodes by themselves, is significantly enhanced, thereby affording electrochemical capacitor electrodes with high power densities. Accordingly, the finely dispersed organic compounds exhibit improved properties compared to those in the bulk state.
Here, we demonstrate the hybridization of a 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) derivative, 4-hydroxy-TEMPO benzoate (HTB), and commercial activated carbon (AC) for aqueous electrochemical capacitor electrodes with high power and energy densities. TEMPO derivatives have indeed been previously studied as electrode materials due to their high redox potential characteristics10 and subsequent high energy densities, although they need to be introduced in polymer chains to prevent them from dissolution by the electrolyte.11,12 However, the resulting polymers display poor electrical conductivity3 and, therefore, achieving fast charging and discharging (i.e., high power densities) remains a challenge. In this study, HTB was finely dispersed inside the AC pores by simply adsorbing HTB in AC through a solvent-free preparation. Such hybridization was accomplished in a single step in 1 h with no organic solvents or purification of the electrode materials. The TEMPO derivative was completely adsorbed in AC at accurate weight ratios, eliminating any wastage of materials. As a result, the contact area between the HTB molecules and the conductive carbon surface was maximized, allowing for fast redox reactions at the interface despite HTB exhibiting poor electrical conductivity by itself. In addition, owing to the strong adsorption capability of AC and the hydrophobic group in the HTB structure, dissolution of HTB molecules in the aqueous electrolyte (1 M H2SO4) was prevented. Furthermore, HTB displays a relatively high redox potential (ca. 1.0 V vs. standard hydrogen electrode (SHE)) within the potential window of aqueous electrolytes, and inclusion of the high-energy-density material inside the AC pores was not accompanied by a volumetric expansion. Thus, the prepared composite materials afford electrochemical capacitor electrodes combining high volumetric energy density with high power density. The enhancement in the volumetric energy density is more advantageous for practical application in energy storage devices than improvements in the gravimetric energy density. This methodology paves the way for the application of versatile organic compounds in high-performance electrode materials with high volumetric energy and power density characteristics.
Experimental
Sample preparation
Commercial AC, MSC30 (Kansai Coke and Chemicals Co., Ltd.), and HTB were used as received. The adsorption of HTB in AC was performed in the gas phase. The preparation of the AC/HTB samples was conducted under N2 atmosphere. AC was dried at 150 °C for 6 h under vacuum to remove any adsorbed water and then weighed. The amount of HTB was accurately weighed using a microbalance (XPR2, METTLER TOLEDO) to achieve precise AC/HTB weight ratios of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, 70:30, 60:40, and 50:50. The dried AC and the TEMPO derivative were thoroughly mixed together and introduced in a glass ampule, which was then sealed under reduced pressure. The ampule was kept in an incubator at 130 °C for 1 h. The resulting samples were denoted as AC/HTB (X%), where X indicates the weight percent of HTB in the sample.
:20, 70:30, 60:40, and 50:50. The dried AC and the TEMPO derivative were thoroughly mixed together and introduced in a glass ampule, which was then sealed under reduced pressure. The ampule was kept in an incubator at 130 °C for 1 h. The resulting samples were denoted as AC/HTB (X%), where X indicates the weight percent of HTB in the sample.
Sample analysis
Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) analyses were performed on a JEM-2100Plus (JEOL) equipped with a Noran System 7 EDS system integrated with a scanning device comprising bright-field (BF) and annular dark-field (ADF) detectors. The accelerating voltage for TEM imaging was 200 kV. Nitrogen adsorption–desorption isotherms were obtained on a Micrometrics ASAP 2020 apparatus at −196 °C. For the removal of the adsorbed water, AC was dried at 150 °C for 6 h under vacuum. On the other hand, the AC/HTB samples could not be heated under vacuum because HTB would desorb under such conditions. Thus, the AC/HTB samples were treated under N2 atmosphere before measurement. The Brunauer–Emmett–Teller (BET) specific surface area was calculated from the adsorption isotherms in the P/P0 range of 0.05–0.20. The total pore volume (Vtotal) was determined from the amount of adsorbed N2 at P/P0 = 0.96. The micropore volume (Vmicro) was estimated by the Dubinin–Radushkevich (DR) method. The mesopore volume (Vmeso) was calculated by subtracting the micropore volume from the total pore volume (Vmeso = Vtotal − Vmicro). The pore size distribution was estimated by density functional theory (DFT) calculations. The cumulative DFT SSA was calculated on the basis of the slit model.Electrode preparation and electrode density measurement
The electrodes were prepared at a weight ratio of AC (i.e., excluding the weight of HTB), carbon black (Denka black, Denka Company Ltd.), and polytetrafluoroethylene (PTFE) of 18:1:1. For a fair comparison, the weight of AC in the working electrode (i.e., excluding the weight of HTB, carbon black, and PTFE) was fixed to 8.5 mg so as to make the electrode thickness constant in all the working electrodes, based on the fact that there is little HTB on the surface of the AC particles. The electrode was sandwiched with a stainless steel mesh and pressed at 30 MPa. A counter-electrode was prepared using 20 mg of the electrode prepared from AC. For the electrode density measurements, the electrode sheet was pressed at a precisely controlled pressure of 30 MPa for 5 min in a Compact Table-Top Universal/Tensile Tester (EZ-LX, Shimadzu) to form a pellet with a diameter of 13 mm. The thickness and weight of the pellet were accurately measured with an autocollimator (DIGIMICRO, MF-501, Nikon) and a microbalance, respectively, to determine the electrode density.
Electrochemical measurements
A Ag/AgCl (saturated KCl) electrode was used as the reference electrode in the three-electrode cell. Electrochemical analysis was performed with a potentiostat/galvanostat instrument (VMP3, Bio-logic) at 25 °C. Cyclic voltammetry measurements were carried out at a sweep rate of 1 mV s−1 in the potential range from −0.1 to 0.9 V (vs. sat. Ag/AgCl). Impedance analysis was conducted at the peak top potential of each sample to obtain Nyquist plots. The gravimetric capacity was measured by galvanostatic charge/discharge cycling (GC) in a potential range of −0.1 to 0.9 V. The capacity was calculated from the discharge curves from 0.9 to −0.1 V. The volumetric capacity was determined by multiplying the gravimetric capacity by the electrode density. Ragone plots were constructed from constant power discharge measurements at constant powers from 0.2 to 100 mW. The energy density was calculated from the constant power discharge curves from 0.9 to 0.4 V. The cycle lifetimes were determined using a two-electrode cell consisting of AC/HTB (30%) as the positive electrode and AC as the negative one with the same weight ratio at cell voltages from 0 to 1.0 V at 25 °C in 1 M H2SO4.Results and discussion
KOH-activated carbon (MSC30) was used as the porous carbon substrate. This AC has a high surface area of 3160 m2 g−1 derived from its large micropore (0.99 cm3 g−1) and mesopore (0.60 cm3 g−1) volumes. For the adsorption of redox-active organic compounds in porous carbon substrates, it is necessary to consider the sublimation temperature and molecular size of the organic compounds in addition to the pore size and pore volume of the substrates. The adsorption is performed at a little higher temperature than the sublimation temperature of the organic compound. The sublimation temperature of HTB was determined to be ca. 125 °C by thermogravimetric analysis (TGA) of HTB (Fig. S1, ESI†), and therefore, the adsorption of HTB was conducted at 130 °C. The weight ratio of HTB and AC was accurately adjusted by mixing previously dried and weighed AC with accurately weighed HTB. The adsorption procedure was accomplished in just 1 h in the gas phase without the need for any solvent and purification process. The possibility of precisely adjusting the weight ratio is advantageous to evaluate different loading amounts of HTB for the optimization of the electrochemical capacitor performance. This preparation method is indeed practical and free from any specialized technique or apparatus. We prepared a series of samples denoted as AC/HTB (X%), where X represents the weight percentage of HTB, ranging from 20% to 50%. As shown in Fig. 1, AC does not exhibit a carbon (002) diffraction peak due to its highly activated carbonaceous structure. In contrast, two broad peaks are observed at around 15° and 22° in the XRD patterns of the AC/HTB samples, and the intensity of each peak increases with the increasing amount of HTB in the sample (i.e., with X). This result indicates that HTB is finely dispersed inside the AC pores and that there are few HTB molecules on the surface of the AC particles since, if deposition of HTB existed on the particle surface of AC, distinct peaks derived from the crystalline structure of HTB would be detected in the XRD patterns.9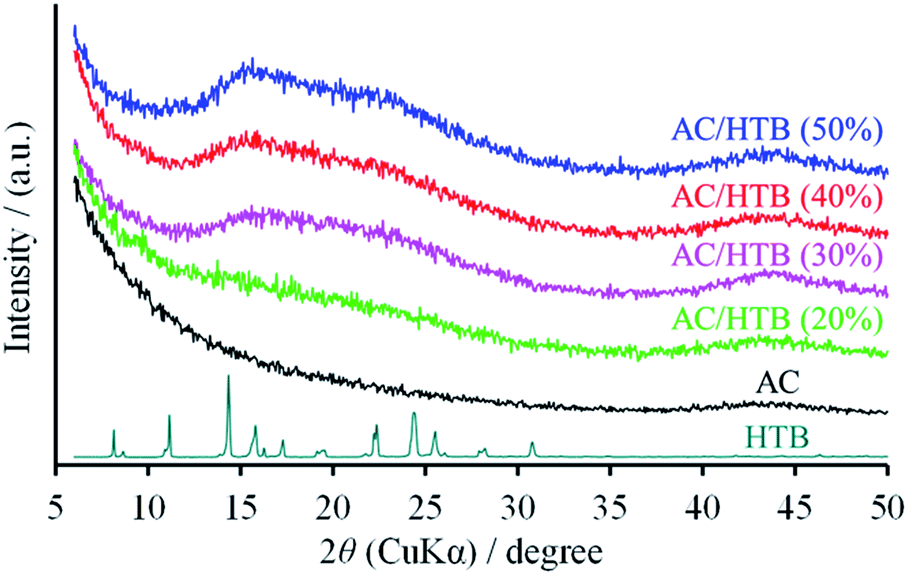 | ||
| Fig. 1 XRD patterns of the AC/HTB samples, AC, and HTB. | ||
The inclusion of HTB molecules inside the AC pores was also confirmed by the results obtained from the electrode density and N2 adsorption–desorption measurements of the samples. The experimental and theoretical electrode densities of the samples are summarized in Table 1. The theoretical electrode densities were calculated from the experimental electrode density of AC and the content of HTB (X), under the assumption that HTB is completely adsorbed inside the AC pores. As shown in the table, the experimental values are in good agreement with the theoretical ones. Such agreement also indicates that the AC particle volume does not expand upon adsorption of HTB. The pore filling by HTB was further evidenced in the N2 adsorption–desorption isotherms by the reduction of the adsorbed amount of N2 in a low relative pressure region (P/P0 < 0.25) (Fig. 2a). The BET specific surface areas (BET SSAs) and pore volumes are summarized in Table 2. In addition, the DFT SSAs were also calculated due to the reliability of DFT calculations for estimating the SSAs. Both the BET and DFT SSAs of the AC/HTB samples decrease with the increasing X, resulting from the HTB molecules filling the AC pores. The pore size distributions (PSDs) obtained from DFT calculations clearly indicate a reduction of the pore volume with the increasing X. As shown in Fig. 2b, AC possesses not only micropores but also small mesopores (<4 nm), which contribute to a large extent to the enhancement of the surface area. On the other hand, both micropore and mesopore volumes of the AC/HTB samples decrease with the increasing X, whereby a very small amount of pores remains in AC/HTB (50%). The remaining pores are necessary for electrolyte ion diffusion during charging and discharging (vide infra).
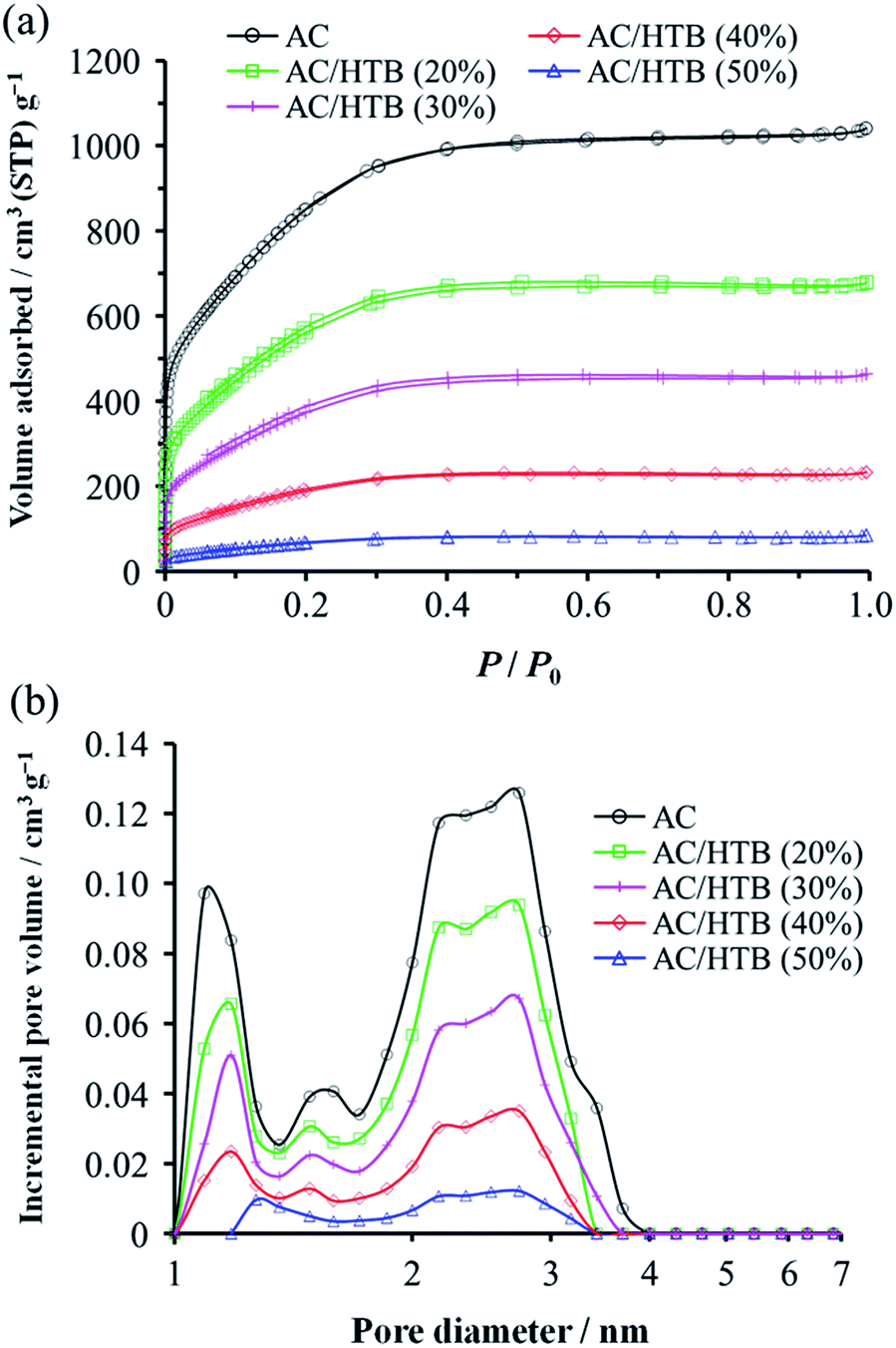 | ||
| Fig. 2 (a) Nitrogen adsorption–desorption isotherms and (b) PSDs for AC and the AC/HTB samples. | ||
| Sample | S BET (m2 g−1) | S DFT (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) |
|---|---|---|---|---|---|
| a BET SSAs. b DFT SSAs. c Total pore volumes estimated from the adsorbed nitrogen amount at P/P0 = 0.96. d Micropore volumes calculated using the DR method. e Mesopore volumes calculated from Vmeso = Vtotal − Vmicro. | |||||
| AC | 3160 | 1910 | 1.59 | 0.99 | 0.60 |
| AC/HTB (20%) | 2070 | 1050 | 1.04 | 0.64 | 0.40 |
| AC/HTB (30%) | 1410 | 640 | 0.70 | 0.42 | 0.28 |
| AC/HTB (40%) | 710 | 310 | 0.35 | 0.21 | 0.14 |
| AC/HTB (50%) | 250 | 100 | 0.12 | 0.07 | 0.05 |
From the TEM images, no differences are observed between AC and the AC/HTB (50%) sample (bearing the highest HTB loading), due in part to the absence of heavy elements (Fig. 3a and b). No deposition or agglomeration could be discerned after careful observation of AC/HTB (50%) (Fig. S2, ESI†). In contrast, the fine dispersion of HTB over the AC particles in AC/HTB (50%) was confirmed by scanning transmission electron microscopy (STEM) imaging and EDS analysis (Fig. 3c). From the electrode density and N2 adsorption–desorption results, in addition to TEM imaging, it is obvious that the HTB molecules are finely dispersed inside the AC pores and that inclusion of HTB in AC is not accompanied by a volumetric expansion of the AC particles. Even in the case of AC/HTB (50%), which contains the highest HTB loading of all the AC/HTB samples, no agglomeration or deposition exists on the surface of the AC particles (Fig. 1). This is one of the advantageous features of our solvent-free preparation. Conventional liquid phase adsorption methods involve the adsorption of solvent molecules inside the AC pores, and such liquid phase adsorption cannot achieve loadings as high as 50% without agglomeration or deposition of the adsorbate. From a practical perspective, the enhancement of the volumetric energy density is more important than that of the gravimetric energy density. Therefore, the adsorption of high-energy-density materials can be expected to enhance the volumetric energy density.
 | ||
| Fig. 3 Morphology of AC and AC/HTB (50%). TEM images of (a) AC and (b) AC/HTB (50%). (c) STEM image and EDS spectra of AC/HTB (50%). All scale bars in the images of panel c correspond to 250 nm. | ||
The electrochemical performance of the samples was examined in 1 M H2SO4 at 25 °C using a three-electrode cell. Fig. 4a shows the cyclic voltammograms of the AC/HTB samples collected at a sweep rate of 1 mV s−1 in the potential range from −0.1 to 0.9 V. For comparison, the voltammogram of AC is also shown. The voltammogram of AC is characterized by a rectangular shape; i.e., the typical electric double-layer behavior. In contrast, the voltammograms of the AC/HTB samples exhibit a distinct redox peak between 0.6 and 0.9 V (vs. Ag/AgCl). The anodic peak corresponds to the oxidation reaction of the neutral nitroxide radical to form an oxoammonium cation.12 The reversibility of this reaction suggests that the resulting positively charged oxoammonium cation forms an oxoammonium salt by incorporating a sulfate anion (Scheme 1).13,14 If the oxoammonium cation did not generate a salt, the positively charged cation would be attracted to the negatively charged counter-electrode during the oxidation process, resulting in almost complete desorption of HTB molecules from the AC pores after only several charge/discharge cycles. Therefore, it is inferred that counter-ion diffusion plays a crucial role during charge/discharge processes (vide infra). Counter-ion diffusion also plays an important role in conductive polymers such as polyaniline15 and polypyrrole,16 where counter-ions are inserted between the polymer chains during the redox reaction. Note that HTB itself does not show any apparent redox behavior in 1 M H2SO4 due to its insoluble nature in such an aqueous electrolyte, while a distinct peak was indeed observed in an organic electrolyte (for details, see the ESI†). TEMPO derivatives cannot be used as electrode materials themselves because they have poor electrical conductivity and most TEMPO derivatives are highly soluble in electrolyte solutions.3,11 However, in this study, the high dispersion of HTB molecules inside the AC pores enables the charge transfer between the HTB molecules and the conductive carbon surface. Furthermore, the hydrophobic benzoate group in the HTB molecule prevents HTB from dissolving in aqueous electrolytes.9 Accordingly, the resulting hybridized materials are able to function as electrodes in aqueous electrolytes. Thus, our method does not require the polymerization of the TEMPO derivative to anchor it to the conductive substrate. It must be noted in the voltammograms of the AC/HTB samples that they exhibit high redox potentials (ca. 1.0 V vs. SHE), higher than those of many other redox organic compounds17–19 and conductive polymers.20–23 This is advantageous for the desired energy density enhancements.
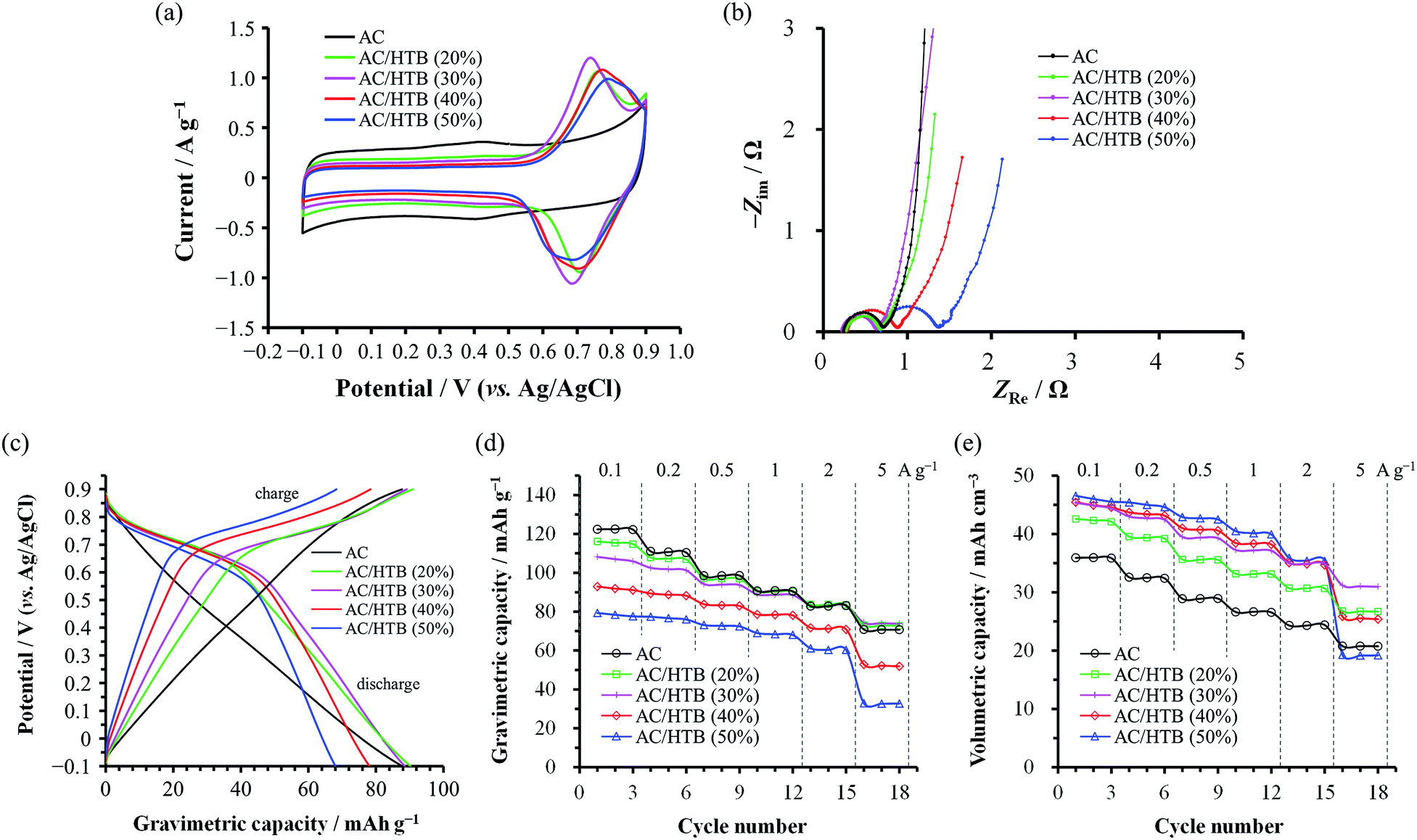 | ||
| Fig. 4 Electrochemical behavior of the AC/HTB and AC samples. All measurements were conducted using a three-electrode cell at 25 °C in 1 M H2SO4. (a) Cyclic voltammograms collected at a sweep rate of 1 mV s−1. (b) Nyquist plots collected at the potentials of the oxidation peak maxima for the AC/HTB samples and at 0.4 V for AC. (c) Discharge profiles of the AC/HTB samples and AC obtained from GC measurements at 1 A g−1. (d) Gravimetric and (e) volumetric capacities plotted at current densities from 0.1 to 5 A g−1. | ||
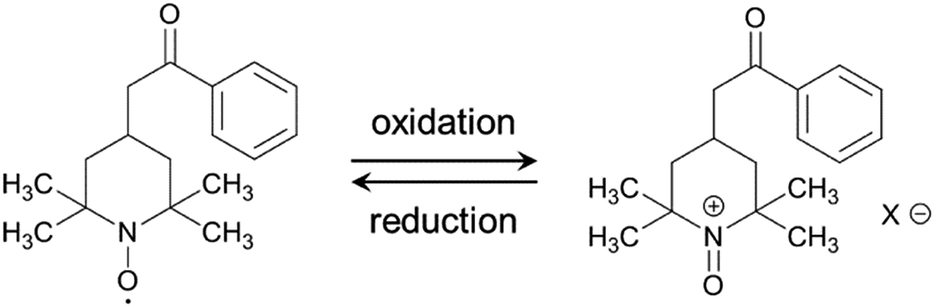 | ||
| Scheme 1 Possible redox reactions of HTB. | ||
The voltammograms of the AC/HTB samples suggest that there must be substantial charge transfer between the HTB molecules and the carbon surface. However, the Nyquist plots of the AC/HTB samples do not exhibit a significant enlargement of the semicircle in the high frequency region compared to that of AC (Fig. 4b). This has been ascribed to the reduced charge transfer distance between the highly dispersed HTB molecules and the conductive carbon surface. Similarly, in our previous study, polyaniline, which has been recognized as a low power density material,21,24 was synthesized inside the AC pores and the Nyquist plot of the resulting material exhibited a smaller semicircle than that of AC due to the shortened charge transfer distances and therefore reduced charge transfer resistance.20 However, regardless of the high dispersion of HTB molecules inside the AC pores, the semicircles of AC/HTB (40%) and AC/HTB (50%) are larger than that of AC. This is probably attributed to the HTB molecules inside the AC pores hampering the counter-ion diffusion, thus delaying the charge transfer and slowing down the charge transfer kinetics. It has been reported in the literature that the overall resistance is comprised of the bulk solution resistance (Rs), the interfacial resistance between AC particles and/or between AC particles and the current collector (Ri), and the charge transfer resistance (Rct).25–28 Since the particle size did not increase upon adsorption of HTB and all the electrodes and electrochemical cells were prepared in exactly the same manner,29Rs and Ri can be considered to be almost the same. Moreover, the semicircle in the Nyquist plot corresponds to Rct.30 Although some studies have associated Ri with this semicircle,25,31Ri must take the same value regardless of the X value due to the aforementioned reasons. Thus, differences in the size of the semicircle are ascribed to differences in the charge transfer resistance, which is influenced by the counter-ion diffusion in the case of the AC/HTB samples. This is in good agreement with the information obtained from the vertical line in the low frequency region, which indicates the existence of electric double-layer behavior.32 As is the case with AC, AC/HTB (20%) and AC/HTB (30%) show an almost vertical line in the low frequency region, corresponding to ideal capacitive behavior.33 In contrast, the deviation from the vertical line observed for AC/HTB (40%) and AC/HTB (50%) suggests an increase in the diffusion resistance attributed to difficulties in the counter-ion diffusion inside the AC pores due to the presence of adsorbed HTB molecules.
Fig. 4c shows the discharge profiles for AC and the AC/HTB samples, plotted in terms of the gravimetric (specific) capacity. The discharge profiles were obtained from the GC results collected at 1 A g−1. As shown in Fig. 4c, the potential of AC proportionally decreases with its gravimetric capacity (a linear change), typical of electric double-layer behavior.34 In contrast, the plots for the AC/HTB samples exhibit a plateau at ca. 0.6–0.8 V, characteristic of the redox behavior of HTB. Fig. 4d and e present the dependence of the gravimetric and volumetric capacities, respectively, on the current density. The volumetric capacity was calculated by multiplying the gravimetric capacity by the experimental electrode density (Table 1). Notably, the trends in the gravimetric and volumetric capacity enhancements are quite different. The gravimetric capacity of the AC/HTB samples does not exhibit any apparent enhancement in comparison with that of AC, but it rather decreases with the increasing X (Fig. 4d). However, the volumetric capacity of the AC/HTB samples clearly increases with the increasing X up to X = 30 (Fig. 4e). The HTB molecules inside the AC pores block the formation of an electric double-layer, thereby reducing the double-layer-derived (non-faradaic) capacity but inducing the faradaic one;35i.e., the inclusion of redox-active HTB molecules replaces the non-faradaic capacity with the faradaic one. Upon comparing the capacities per unit volume, the faradaic capacity induced by HTB can be concluded to be higher than the non-faradaic one. Moreover, the capacity of AC/HTB (20%) and AC/HTB (30%) was well retained up to a current density of 5 A g−1 despite the substantial contribution of the faradaic capacity. The commercial AC used in this study presents a high fraction of mesopores, which facilitate the ion diffusion inside the AC pores.36,37 Thus, the AC exhibits a high capacity retention of 58% at 5 A g−1 relative to that at 0.1 A g−1. However, based on the same comparison, AC/HTB (20%) and AC/HTB (30%) exhibit a capacity retention of 63–69%, higher than that of AC. These results are in good agreement with the Nyquist plots of the same samples and confirm that the huge contact area between the HTB molecules and the conductive carbon surface, together with the substantial retention of the number of pores, allows for very fast charge transfer affording power densities higher than that of AC; i.e., the faradaic capacity derived from HTB has a superior power density than the non-faradaic one, both induced inside the micro- and mesopores. Although the volumetric capacity of AC/HTB (40%) and AC/HTB (50%) increases with the increasing X up to a certain extent in the low current density region (<2 A g−1), they display inferior capacity retention at higher current densities (>2 A g−1) because, as expected from their Nyquist plots, the large amount of HTB molecules inside the AC pores disturbs not only the double-layer formation but the counter-ion diffusion inside the AC pores necessary for the redox reactions of HTB (Scheme 1). From a comprehensive perspective, AC/HTB (30%) provides excellent compatibility between high volumetric capacity and high power density at a relatively small HTB loading. Although the above discussion has focused on the capacity, the electrochemical behavior of the AC/HTB samples is characterized by their high redox potential and high volumetric capacity. Thus, high volumetric energy densities can be expected from the AC/HTB samples.
For the measurement of the energy density, a constant power discharge analysis was carried out to obtain Ragone plots for AC/HTB (30%) and AC. Note that the constant power discharge analysis was performed by using a three-electrode cell to measure the intrinsic energy densities of AC/HTB (30%) and AC. The AC/HTB samples are used as positive electrodes due to their high redox potential characteristics. Therefore, for a full two-electrode device using the AC/HTB samples, hybrid asymmetric devices must be constructed by using appropriate negative electrodes, where the resulting energy and power densities of the devices are strongly influenced by the types of negative electrodes (Fig. S5, ESI†).35Fig. 5a presents the Ragone plots based on the gravimetric energy and power densities. The AC used in this study shows a large gravimetric energy density of 18.0 W h kg−1 at 16.5 W kg−1 and also a high power density of 8.2 kW kg−1 at 1.1 W h kg−1. Since AC has a high surface area exceeding 3000 m2 g−1 and a large mesopore volume fraction, which reduces the diffusion resistance, AC exhibits superior energy and power densities than current conventional electrochemical capacitors.38 In contrast, AC/HTB (30%) exhibits higher gravimetric energy and power densities than AC in all the power density regions. The gravimetric energy density of AC/HTB (30%) reached 39.5 W h kg−1 at 16.5 W kg−1, which is 2.2 times higher than that of AC at the same power density. Moreover, the gravimetric energy density of AC/HTB (30%) remained at 14.1 W h kg−1 at 8.2 kW kg−1. The differences in the energy and power densities between AC/HTB (30%) and AC become remarkable after a volumetric comparison (Fig. 5b). AC/HTB (30%) exhibits a 3.1 time enhancement of the volumetric energy density (16.5 W h L−1) at 6.9 W L−1 compared to AC. At 3.5 kW L−1, AC/HTB (30%) retains a volumetric energy density of 5.9 W h L−1, surpassing by far that of AC (0.3 W h L−1). These results demonstrate that the inclusion of highly dispersed HTB inside the AC pores is advantageous for the enhancement of the volumetric energy and power densities of electrochemical capacitor electrodes. As shown in Fig. 5c, AC/HTB (30%) exhibits the capacity retention of 81 and 55% at the 2000th and 10000th cycle, respectively, which was determined by using a two-electrode cell consisting of AC/HTB (30%) as the positive electrode and AC as the negative one. Despite the non-polymeric hybridization of HTB, such a reversible redox characteristic is achieved by the insoluble nature of HTB (owing to its hydrophobic group) and the strong adsorption capability of AC. In addition, neutralization of the HTB molecules during the redox reaction also contributes to the redox reversibility. Redox-active materials are ionized and therefore charged during charge/discharge processes. If redox-active materials are not neutralized during these processes, the resulting species are attracted to the oppositely charged counter-electrode, resulting in dissolution in the electrolyte.20 However, the HTB molecules are maintained neutral by counter-anions, forming oxoammonium salts during the oxidation reaction (i.e., during charging) (Scheme 1),13,14 and such a neutralization impedes the undesired attraction by the electric field. The decrease of the capacity retention after the 10000th cycle is the result of desorption of non-neutralized HTB molecules due to the incomplete formation of oxoammonium salts at such a high current density measurement (1 A g−1), which was evidenced by the voltammogram of the negative electrode (AC) displaying a distinct redox peak after the cycle test (Fig. S6, ESI†). Only a small amount of HTB molecules in the electrode could not undergo the neutralization in each charge/discharge process due to the difficulty of counter ion diffusion at a high current density of 1 A g−1. The HTB molecules in the electrode were gradually desorbed and their continuous desorption resulted in the decrease of the capacity retention. However, it would be expected to enhance the capacity retention by the optimization of an electrolyte (e.g., the use of electrolyte ions with high ion mobility to reduce the diffusion resistance). Recently, we have reported the electrochemical capacitor performance of electrodes prepared from a porous carbon substrate and a hydrophobic quinone derivative in aqueous 1 M H2SO4 electrolyte.9 The quinone derivative underwent protonation in the proton acidic electrolyte, thus remaining neutral during the redox reactions.39 Moreover, its hydrophobic functional groups enabled the quinone derivative to remain inside the pores during the charge/discharge processes in an aqueous electrolyte.17 For the utilization of redox-active materials without the need for immobilization treatments (e.g., polymerization), their insolubility in the electrolyte and the neutralization of the redox-active compounds during the redox reactions are crucial aspects in order to achieve long cycle lifetimes. In addition, poorly conductive redox-active materials need to be in intimate contact with a conductive surface to enable successful charge transfer at their interface. A vast number of organic compounds have not been considered as potential electrode materials due to issues regarding their dissolution in the electrolyte or poorly conductive nature. However, upon suitable hybridization, they may become excellent electrode materials with high power and energy densities, together with long cycle lifetimes. Therefore, our methodology is expected to widen the potential of versatile organic compounds in the field of electrical energy storage.
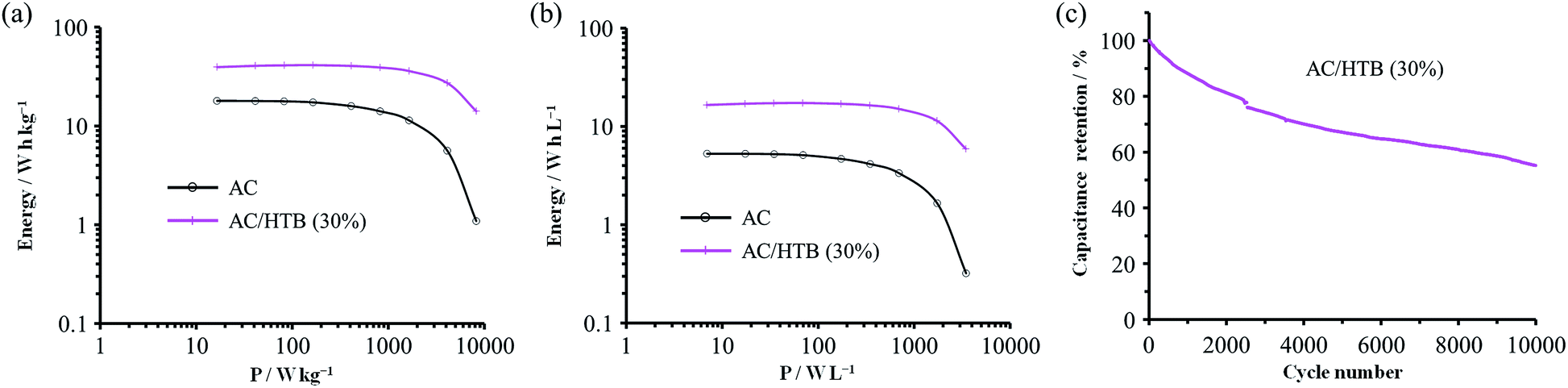 | ||
| Fig. 5 Ragone plots based on the (a) gravimetric and (b) volumetric comparison measured at constant power discharges from 0.9 to 0.4 V at constant powers from 0.2 to 100 mW. The measurements were conducted at 25 °C in 1 M H2SO4. (c) Dependence of the capacity retention on the cycle number measured using a two-electrode cell consisting of AC/HTB (30%) as the positive electrode and AC as the negative one at 25 °C in 1 M H2SO4. The measurements were performed by GC at 1 A g−1 and a cell voltage from 0 to 1.0 V. | ||
Conclusions
We have demonstrated a novel electrochemical capacitor electrode material employing a TEMPO derivative (HTB) and AC prepared by a solvent-free preparation. This method does not require any polymerization technique or organic solvent for the preparation of the electrode material. HTB was completely adsorbed in AC, eliminating the wastage of materials typical of conventional liquid phase adsorption methods. In addition, the preparation process consists of a single step, where cumbersome synthetic procedures or purification processes are not necessary. Although HTB displays poor electrical conductivity as most organic compounds, the huge contact area between the finely dispersed HTB molecules and the conductive AC surface enables fast redox reactions of HTB, resulting in high power densities. The fine dispersion of redox-active HTB inside the AC pores also effectively allows for a volumetric capacity enhancement. Moreover, the high redox potential of HTB provides both a high volumetric energy and power density enhancement. Furthermore, the redox reversibility was possible due to the neutralization of the active species via the formation of oxoammonium salts and the presence of a hydrophobic functional group in HTB. Although this study has focused on the preparation of high-performance electrochemical capacitor electrodes, we aim to widen our methodology beyond the preparation of electrode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant number 15K21478.References
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906–9907 CrossRef CAS PubMed.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS PubMed.
- L. M. Field and P. M. Lahti, Chem. Mater., 2003, 15, 2861–2863 CrossRef CAS.
- Y. Chen and J. Shi, Adv. Mater., 2016, 28, 3235–3272 CrossRef CAS PubMed.
- H. Itoi, Y. Yasue, K. Suda, S. Katoh, H. Hasegawa, S. Hayashi, M. Mitsuoka, H. Iwata and Y. Ohzawa, ACS Sustainable Chem. Eng., 2017, 5, 556–562 CrossRef CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- N. Kentaro, O. Kenichi and N. Hiroyuki, Chem. Lett., 2011, 40, 222–227 CrossRef.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- T. E. Herod and J. B. Schlenoff, Chem. Mater., 1993, 5, 951–955 CrossRef CAS.
- M. R. Warren and J. D. Madden, Synth. Met., 2006, 156, 724–730 CrossRef CAS.
- T. Tomai, S. Mitani, D. Komatsu, Y. Kawaguchi and I. Honma, Sci. Rep., 2014, 4, 3591 CrossRef PubMed.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC.
- S. Renault, J. Geng, F. Dolhem and P. Poizot, Chem. Commun., 2011, 47, 2414–2416 RSC.
- H. Itoi, S. Hayashi, H. Matsufusa and Y. Ohzawa, Chem. Commun., 2017, 53, 3201–3204 RSC.
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- X. Xia, D. Chao, X. Qi, Q. Xiong, Y. Zhang, J. Tu, H. Zhang and H. J. Fan, Nano Lett., 2013, 13, 4562–4568 CrossRef CAS PubMed.
- O. Mykhailiv, M. Imierska, M. Petelczyc, L. Echegoyen and M. E. Plonska-Brzezinska, Chem.–Eur. J., 2015, 21, 5783–5793 CrossRef CAS PubMed.
- Y.-k. Zhou, B.-l. He, W.-j. Zhou, J. Huang, X.-h. Li, B. Wu and H.-l. Li, Electrochim. Acta, 2004, 49, 257–262 CrossRef CAS.
- K.-P. Wang and H. Teng, J. Electrochem. Soc., 2007, 154, A993–A998 CrossRef CAS.
- C.-W. Huang and H. Teng, J. Electrochem. Soc., 2008, 155, A739–A744 CrossRef CAS.
- D. Qu, J. Power Sources, 2002, 109, 403–411 CrossRef CAS.
- Y. R. Nian and H. S. Teng, J. Electrochem. Soc., 2002, 149, A1008–A1014 CrossRef CAS.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165–1167 CrossRef CAS PubMed.
- J. Tong, H. Zhang, J. Gu, L. Li, C. Ma, J. Zhao and C. Wang, J. Mater. Sci., 2016, 51, 1966–1977 CrossRef CAS.
- C. Portet, P. L. Taberna, P. Simon and C. Laberty-Robert, Electrochim. Acta, 2004, 49, 905–912 CrossRef CAS.
- W.-C. Chen, T.-C. Wen and H. Teng, Electrochim. Acta, 2003, 48, 641–649 CrossRef CAS.
- M. D. Stoller, S. Park, Z. Yanwu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- K. Jurewicz, C. Vix-Guterl, E. Frackowiak, S. Saadallah, A. Reda, J. Parmentier, J. Patarin and F. Beguin, J. Phys. Chem. Solids, 2004, 65, 287–293 CrossRef CAS.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu, Z. F. Yan and H. M. Cheng, Carbon, 2006, 44, 216–224 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- K. Nueangnoraj, T. Tomai, H. Nishihara, T. Kyotani and I. Honma, Carbon, 2016, 107, 831–836 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00541e |
| This journal is © The Royal Society of Chemistry 2018 |