
Non-polymeric hybridization of a TEMPO derivative with activated carbon for high-energy-density aqueous electrochemical capacitor electrodes†
 *a
*a
Abstract
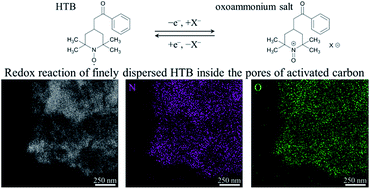
Organic compounds have great potential as electrode materials with high energy and power densities, together with long cycle lifetimes. These properties can be achieved by employing redox-active organic compounds and porous carbon substrates via well-suited hybridization methods. In this study, a 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) derivative, 4-hydroxy-TEMPO benzoate (HTB), was used as an electrode material for aqueous electrochemical capacitors due to its high redox potential characteristics to achieve high energy densities. The hybridization of HTB and activated carbon (AC) was accomplished simply by adsorbing HTB in AC via a solvent-free preparation. This procedure takes only one hour and enables achieving precise AC/HTB weight ratios, eliminating the excess use of HTB or the use of organic solvents. Of note in this method is that it is not necessary to introduce HTB molecules in polymer chains to prevent dissolution of HTB in an aqueous electrolyte with the aid of a hydrophobic group in HTB. HTB molecules are finely dispersed inside the AC pores and therefore present a huge contact area with a conductive carbon surface, allowing for fast redox reactions (i.e., high power densities) in aqueous 1 M H2SO4 despite HTB displaying poor electrical conductivity by itself. As a result, the obtained AC/HTB materials enable compatibility of high energy and power densities.
 Please wait while we load your content...
Please wait while we load your content...

